

Abstract
Herpesviruses are DNA viruses harboring the capacity to establish lifelong latent-recurrent infections. There is currently limited knowledge on viruses targeting the innate DNA sensing pathway and also on how the innate system impacts on the latent reservoir of herpesvirus infections. Here we report that murine gammaherpesvirus MHV68, in contrast to alpha- and beta-herpesviruses, induce very limited innate immune responses through DNA-stimulated pathways, which correspondingly played only a minor role in control of MHV68 infections in vivo. Similarly, Kaposi's sarcoma-associated herpesvirus also did not stimulate immune signaling through the DNA sensing pathways. Interestingly, a MHV68 mutant lacking the deubiquitinase (DUB) activity, embedded within the large tegument protein ORF64, gained the capacity to stimulate the DNA-activated STING pathway. We found that ORF64 targeted a step in the DNA-activated pathways upstream of the bifurcation into the STING and AIM2 pathways, and lack of the ORF64 DUB was associated with impaired delivery of viral DNA to the nucleus, which instead localized to the cytoplasm. Correspondingly, the ORF64 DUB active site mutant virus exhibited impaired ability to establish latent infection in wild type but not STING-deficient mice. Thus, gammaherpesviruses evade immune activation by the cytosolic DNA sensing pathway, which in the MHV68 model facilitates establishment of infections.
Introduction
Herpesviruses are capable of establishing latent-recurrent infections, and can give rise to disease in both the acute and recurrent phase, mainly in immuno-suppressed individuals. Herpesviruses are DNA viruses that enter cells through the plasma membrane or an endosomal route and shuttle through the cytoplasm for delivery of DNA to the nucleus where productive replication takes place (1). Three sub-classes of herpesviruses exist, termed alpha-, beta-, and gamma-herpesviruses, which have a common set of properties as well as functions specific for the different subfamilies. Advances in the field of innate immunology have revealed important roles for the innate immune system in host control of herpesvirus infections (2-4). In corroboration with this notion, knowledge on herpesviruses evading innate immune responses are now starting to emerge (5), but information on how this impacts on establishment of acute and latent infections is still lacking. Therefore, in this study we hypothesized that herpesviruses target key elements of the innate immune system with the purpose of facilitating establishment of latent infection.
Nucleic acids are believed to be the main inducers of innate immunity during viral infection (6), with type I IFN production and inflammasome-dependent IL-1β maturation being two important biological responses evoked by these viral molecules. For herpesviruses it is known that viral RNA and DNA are detected by endosomal TLRs as well as cytoplasmic RNA sensors (3,7-9). Recently, much focus has been given towards intracellular DNA, and it is now emerging that viral DNA in the cytoplasm and the nucleus can also trigger the innate immune system (4,10-15). In vivo studies have revealed important roles for DNA sensing pathways in host protection against herpesvirus infections (2,4). Common to the IFN-stimulating pathways triggered by intracellular DNA is the requirement for Stimulator of IFN genes (STING) (2). STING is a tetra-membrane-spanning endoplasmic reticulum protein, which translocates to specific vesicular structures after DNA recognition, where signaling is believed to take place (2). Cytosolic DNA also stimulates inflammasome activation through a pathway dependent on absent in melanoma 2 (AIM2), and in this manner leads to assembly of a caspase 1-containing complex capable of cleaving pro-IL-1β and pro-IL-18 into the biologically active forms (16).
We recently demonstrated that genomic DNA of alpha-herpesviruses (HSV) and beta-herpesvirues (cytomegalovirus (CMV)) is detected by DNA sensors in the cytosol after degradation of the capsid (17). Detection of HSV and CMV DNA has been reported to involve the DNA sensors IFN inducible protein (IFI) 16 and cyclic-GMP-AMP (cGAMP) synthetase (cGAS), the latter stimulating cGAMP-mediated STING-dependent signaling and induction of type I IFN expression (15,17). It is also documented that CMV stimulates inflammasome activation in a manner dependent on AIM2 (4). The subfamily of gammaherpesviruses includes the human pathogenic viruses Epstein-Barr virus (EBV) and Kaposi's sarcoma-associated herpesvirus (KSHV), and the murine gammaherpesvirus 68 (MHV68). All these viruses are detected by TLR9 in plasmacytoid dendritic cells (9,18,19), and it has been reported that IFI16 detects KSHV DNA in the nucleus in endothelial cells to induce caspase 1 activation and pro-IL-1β maturation (14). However, despite the importance of STING and AIM2 in the response to alpha- and beta-herpesvirus infections, there is limited information on the role of the DNA-activated pathways in innate responses to gammaherpesvirus infections. Moreover, information on the impact of innate immune activation on the establishment of the latent reservoir of herpesvirus infections is still sparse.
In this work we report that the gammaherpesviruses MHV68 and KSHV, in contrast to alpha- and beta-herpesviruses, only weakly induce innate immune responses through DNA-stimulated pathways. We identified that an ORF64 deubiquitinase (DUB)-deficient MHV68 mutant gained the capacity to induce type I interferon through the DNA-activated STING pathway, hence suggesting an essential role for the DUB in evading host immune activation. Importantly, the MHV68 ORF64 mutant virus exhibited impaired ability to establish latent infection in wild type but not in STING-deficient mice. Collectively, these data suggest that gammaherpesviruses evade immune activation by the cytosolic DNA sensing pathway via the DUB activity embedded in the viral tegument and that this contributes to the ability of these viruses to establish latent infections.
Materials and Methods
Mice
For this study we used C57BL/6, TLR3-/-, TLR2/9-/-, STINGgt/gt, ASC-/-, NLRP3-/-, and AIM2-/- mice. The C57BL/6, TLR3-/-, TLR2/9-/-, and STINGgt/gt mice were kept in the animal facility of The Faculty of Health, Aarhus University, while the ASC-/-, NLRP3-/-, and AIM2-/- mice were kept in the animal facility of UMASS Medical School, MA, USA. All mice were housed in a specific pathogen-free environment.
Viruses
MHV68 used in this study was the WUMS strain. MHV68 was propagated and titrated in BHK-21 hamster kidney cells in Glasgow modified essential medium (G-MEM). For select experiments we used BAC-derived MHV68-GFP (20). The ORF64 C33A mutant and the ORF36 N36S mutant, we well as the corresponding revertant viruses were produced as previously described (21,22). KSHV was produced as described previously (9). Briefly, baculovirus ORF50 was amplified in Sf9 cells. Vero cells stably expressing KSHV-GFP were infected with baculovirus ORF50 and treated with 2 mM sodium butyrate to allow reactivation of the latent KSHV. At 72 h supernatant was harvested and filtered through a sterile 0.45μM filter. Virus concentration was performed by layering virus supernatant over a cushion of 20% sucrose. The virus was pelleted by ultracentrifugation for 2 h at 4°C at 24,000 rpm. Virus pellets were resuspended in endotoxin-free sterile DPBS.
Herpes simplex virus 1 (HSV-1, KOS strain) and the VP1-2 C65A mutant (23) was amplified in Vero-cells. MCMV mutagenesis, diagnosis, and propagation were performed essentially as previously described (24). MCMV-K181 bacterial artificial chromosome (accession AM886412.1; (25)) was used to generate MCMV-M48(C23S) by recombineering using a two-step allelic exchange. Briefly, a selection/counter-selection cassette was used to replace nucleotides 67098-67111 of the WT MCMV BAC, which was in turn replaced with a PCR amplicon containing synonymous substitutions to introduce a unique diagnostic restriction site and a T to A substitution mutation at genomic position 67109, changing codon 23 of M48 from cysteine (TGC) to serine (AGC). Recombinant BACs were selected and confirmed by PCR and RFLP analysis. Infectious virus was reconstituted as previously described (24), amplified by growth in STO cells (CRL 1503; ATCC, USA) in the presence of 25 μg/ml 6-thioguanine (Sigma, USA), and plaque purified by limiting dilution. Parental and mutant viral stocks were then generated as previously described (24) in NIH3T3 fibroblasts or IFNAR1-/- MEFs. Virus preparations were tested for endotoxin content, and found all to contain similar low levels (∼ 0.75 endotoxin units/ml).
Reagents
For in vitro stimulation experiments we used dsDNA (HSV60mer, 2 μg/ml), cGAMP (2 μM) (InvivoGen), Poly (I:C) (25 μg/ml) (InvivoGen), ODN1826 (5 μM) (InvivoGen), Pam3CSK4 (200 ng/ml)(InvivoGen), Lipopolysaccharide (200 ng/ml) Poly (dA:dT) (1 μg/ml) (Sigma-Aldrich), Nigericin (10 μM) (Sigma-Aldrich), recombinant murine IFN-β and TNF-α (both R&D Systems). The HSV-1 60mer is derived from the HSV-1 genome nt. 144107-144166 and was obtained from DNA Technology (26). For transfection, the DNA was complexes with lipofectamin2000 (Invitrogen) and transfected into the cells according to manufacturer's instructions.
Primary murine bone-marrow-derived cells
Bone marrow cells were harvested from the hind legs of mice. Cells were cultured in growth media (RPMI-1640 medium with 10 % heat-inactivated fetal calf serum (FCS), 600 μg/ml Glutamine, 200 IU/ml of penicillin and 100 μg/ml streptomycin (Gibco)) and stimulated with GM-CSF (R&D Systems) in the concentration of 40 ng/ml. On day 3 and day 5 fresh GM-CSF was added. On day 7 the non-adherent bone-marrow derived dendritic cells (BMDC), and the adherent cells bone marrow derived macrophages (BMMs) were harvested. For experiments, cells were seeded out in RPMI-1640 medium with 10 % FCS at a density of 200.000 cells per cm2.
Human monocyte/macrophage cell line
THP1 cells (ATCC) were cultured as non-adherent monocyte like cells in growth media (RPMI containing 10% FCS, 600 μg/ml Glutamine, 200 IU/ml of penicillin and 100 μg/ml streptomycin (Gibco)). The cells were differentiated into macrophage-like cells by addition of 150 nM Phorbol 12-myristate 13-acetate (PMA) (Sigma-Aldrich) 16-20 h prior to use in experiments.
shRNA mediated silencing
The lentiviral shRNA expression plasmid pLKO.1 was utilised for generating stable gene expression knockdown in THP1 cells (OpenBiosystems, Huntsville, AL). The targeting shRNA sequence was: IFI16 clone ID # TCRN0000019079; Sting clone ID # TCRN000163296 and AIM2 clone ID # TRCN0000107503. The control shRNA vector was an empty vector pLKO.1 with an 18nt shuttle sequence instead of the hairpin sequences. Virus-like particles were produced using Fugene 6 (Roche) transfection of HEK293T cells with the packaging system pMDIg/p-RRE, pRSV-rev, pMG.2 and shRNA vector plasmid. Virus supernatants were harvested after 48 hours and filtrated through a 0.45um membrane. Undifferentiated THP1 cells were infected with increasing amounts of virus supernatant and after two days post infection placed under selection with puromycin at 1ug/ml. Levels of knockdown on IFI16 and STING were determined by Western blotting on extracts from differentiated THP1 as described (27). For detection of AIM2 we used anti-human AIM2 (Adipogen, clone 3B10) and anti-β-Actin.
Murine intranasal MHV68 infection model
Mice were infected intranasally with 5×104 pfu of MHV68 in 25 μl while under anesthesia. On day 3, 6, 16 or 90 post infection, the mice were euthanized by cervical dislocation, and use for further analysis.
Isolation of single cells from the lung
The lung was removed and followed by an injection of 1 ml digestion media (Collagenase/Dispase 1 mg/ml DNAse 0.5 mg/ml in PBS) into the tissue. The lung was subsequently digested in a digestion media in 10% FCS in IMEM at the concentration mentioned above for 30 minutes in 37°C incubator. Single cells of the lungs were obtained by grinding the tissues passing through the strainer 70 μm. The cell suspension was incubated with ACK lysis buffer for 3 minutes to eliminate the red blood cells, washed and supernatant was discarded. Single cells were resuspended in 0.5% BSA/PBS for subsequent analysis.
Flow cytometry analysis
Single cells were blocked with Fc blocking antibody (2.4G2), and subsequently stained with the following antibodies: CD11b (M1/70), Ly6g (1A8), CD3 (17A2), CD19 (1D3), I-Ab (AF6-120.1) and CD11c (HL3) from BD: CD4(GK1.5) and CD8a (53-6.7) from eBioscience: F4/80 (Cl:A3-1) from Serotec. Dead cells were excluded by staining with Aqua (Invitrogen). Data were collected with a FC500 instrument (Beckman Coulter) and were analyzed using FlowJo software (Tree Star).
Gene expression analysis by nCounter (Nanostring)
RNA isolated from cells that were mock infected or infected with MCMV and MHV68 as indicated. Cells were harvested into RLT buffer containing 2-βME for subsequent processing with the RNeasy Mini kit (Qiagen). Each RNA sample was adjusted to contain the same quantity by using the Nanodrop ND-1000 spectrophotometer (Thermo Scientific). RNA was then hybridized and quantified with the NanoString nCounter analysis system (NanoString Technologies, Seattle, WA) using a customized probeset containing probes for innate immune genes per the manufacturer's protocol. The gene expression data were first normalized to an internal positive control, then to an internal negative control and finally to seven housekeeping genes, (GAPDH, β-glucuronidase, β-actin, hypoxanthine phosphoribosyltransferase 1, tubulin β, phosphoglycerate kinase 1, and clathrin H chain 1). All values were log10-transformed and a heat map was generated using the open source R-based software at UMMS.
ELISA
Supernatants or spleen homogenates from cells cultures and mice, respectively, were analyzed for cytokine levels by ELISAs for human IL-1β and the murine cytokines IL-1β, CXCL10, TNF-α, IFN-γ, or IL-6 using matched antibody pairs obtained R&D Systems, as described previously (17,28).
Type I IFN Bioassay
IFN-α/β levels were measured by a cell based assay. In brief supernatant harvested from BMDCs or BMMs was UV-treated for 6 minutes to inactivate residual virus in the samples. Afterwards, samples were plated in successive 2-fold dilutions on a 96-well plate, with 20.000 L929-cells per well in 100 μl MEM with 5 % FCS. After overnight incubation, the L929 cells were infected with vesicular stomatitis virus (VSV/V10). After 2-3 days of infection, the results were evaluated by microscopic examination of each well. A 50 % protection of cells in a well from virus-induced cell death was used defined as 1 U/ml of IFN- α/β.
Quantitative real-time polymerase chain reaction (PCR) analysis
RNA was purified from homogenates of lung tissues using RNeasy Plus Mini Kit (Qiagen) following the manufacturer protocol. The RNA was treated with DNase I to eliminate contaminating genomic DNA. RNA was isolated from cells in culture using High Pure RNA Isolation Kit (Roche). Expression or mRNAs for human and murine cytokines was determined by real time PCR, using TaqMan detection systems (Applied Biosystems). Expression levels were normalized to β-Actin expression and data presented as the fold induction over un-treated controls for each phenotype. The TaqMan assays used were: hIFN-β, Hs01077958_s1; hTNF-α, Hs01113624_g1; hβ-Actin Hs99999903_m1; mIFN-β, Mm00439546_s1; mCXCL10, Mm00445235_m1; mβ-actin, Mm01205647_g1. For detection of viral genomic material, DNA was isolated from lungs and spleens using ISOLATE II Genomic DNA Kit (Bioline). For amplification of MHV68 glycoprotein B DNA we used the primers: Forward: 5′-CCGCTCATTACGGCCCAAATTCAA-3′; 5′-Reverse: GGCAGCGACAGGCTTTCCATAAAT-3′. SYBR green was used a fluorescent dye to quantitate DNA products in real time.
Confocal microscopy
For visualization of IFI16 and STING, following infection, THP1-cells were fixed and permeabilized with methanol at -20°C and labeled with antibodies against IFI16 (N-terminal Santa Cruz sc-8023) or STING (Imgenex IMG-6485A). Images were acquired on Zeiss LSM 710 confocal microscope, using 63× 1.4 oil lense. Image processing was performed using Zen 2010 (Zeiss) and ImageJ.
Propagation of deoxy-5-ethynylcytidine (EdC)-labeled MHV68 and visualization of capsid-free DNA
EdC (deoxy-5-ethynylcytidine) labeled MHV68 was propagated in BHK-21 cells in presence of 3 uM EdC (Sigma, St. Louis), which was added at 6 hours post infection. The supernatant was collected 4 days later, and debris was removed by centrifugation at 4500× g for 1 hour. Virus in the supernatant was pelleted at 26700× g for 5 hours and resuspended in PBS, followed by determination of the viral titers on BHK-1 cells. To visualize capsid-free viral DNA in the infected cells the Click-iT Imaging Kit (Invitrogen, Carlsbad) was used. Briefly, the infected cells were fixed with methanol for 5 min at -20 °C, and stained with click staining mix, freshly prepared according to the manufacture, at room temperature for 30 min in the dark. Nucleoli of the cells were stained with DAPI. Images were acquired on a Zeiss LSM 710 confocal microscope, using a 63× 1.4 oil-immersion objective; Image processing was performed using Zen 2010 (Zeiss).
Statistics
The data are shown as mean ± st.dev. Statistically significant difference between observations was determined by 2-tailed Student's t test or Wilcoxon rank sum test. For p values of less than 0.05, data were considered to be significantly different.
Ethics
Protocols for the animal experiments were approved by the Local Ethical committees and conducted in accordance with institutional guidelines for animal care and use.
Results
Induction of innate immune responses by alpha-, beta-, and gamma-herpesviruses in leukocytes
In a first series of experiments we wanted to characterize the ability of MHV68 to induce cytokines associated with innate immune responses and compare with MCMV and HSV-1. BMDC were seeded and infected with increasing doses of MHV68, HSV-1 and MCMV. The doses of MCMV and HSV-1 were chosen based on experiments where CXCL10 induction by different doses of these viruses were examined (Supplemental Fig. 1A). Despite clear induction of type I IFN expression by MCMV and HSV-1 infections, we were not able to detect IFN bioactivity in the supernatants from cells infected with MHV68 (Fig. 1A). This was not due to lack of infection of the cells as measured by expression of GFP in cells infected with an MHV68-GFP virus (Supplemental Fig. 1B). By contrast to the lack of type I IFN induction, MHV68 infection in DCs did stimulate expression of TNF-α, IL-6 and IL1β, although high doses of virus were required to evoke these responses (Fig. 1B-1D). A similar picture was observed in BMMs with no detectable IFN bioactivity in the supernatants from MHV68-infected cells but clear induction of IL-6 and TNF-α and modest induction of IL-1β (Supplemental Fig. 1C-1F).
Figure 1.
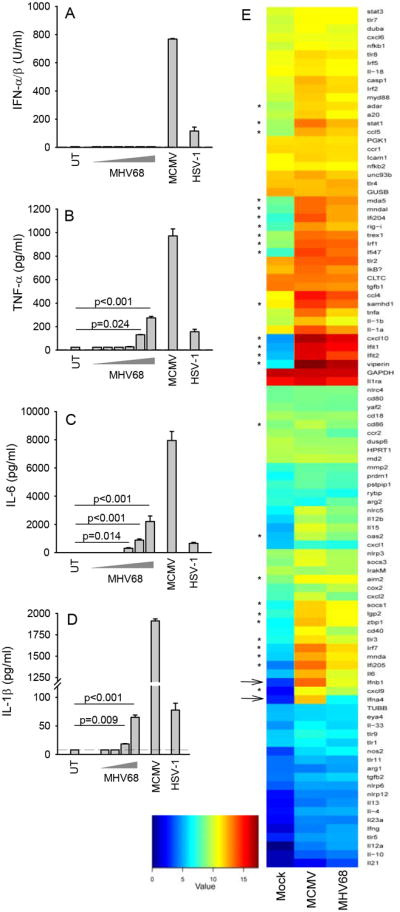
MHV68 stimulates a blunted innate immune response in dendritic cells. (A-D) Mouse bone marrow-derived dendritic cells (BMDCs) from C57BL/6 mice were seeded and infected with MHV68 at increasing doses between MOI 1 and 1000 (in panel D between 10 and 100), MCMV (MOI 1), and HSV-1 (MOI 3). Supernatants were harvested 24 hr post infection and used for measurement of cytokines by type I IFN bioassay (A) or ELISA (B-D). Data are presented as means of measurements from triplicate cultures +/- st.dev. (E) BMDCs were treated with MCMV (MOI 10) and MHV68 (MOI 10) for 6 h, prior to harvest of total RNA, which was subjected to analysis for specific transcripts using NanoString Technology. Data are presented as heat maps. Arrows indicate type I IFN genes, while stars indicate IFN-stimulated genes. Data are representative of three independent experiments.
To get a more complete picture of the innate response evoked by MHV68 infection in DCs, we subjected RNA isolated from mock or virus infected cells to analysis using nCounter (Nanostring), a fluorescent barcoded technique that allows multiplex analysis of mRNA's important in innate immunity. Infection with MHV68 did stimulate expression of most mRNAs transcripts examined for (Fig. 1E). However, the response to MHV68 infection was generally much lower than what was induced by MCMV infection. In particular, transcripts for type I IFNs (IFN-β and IFN-α4) and IFN-stimulated genes (ISGs) accumulated at much lower levels in cells infected with MHV68 as compared to infection with MCMV. Consistent with this, we found induction of the ISG CXCL10 at the protein level only after MHV68 infection at high MOI (Supplemental Fig. 1G).
Altogether, infection of macrophages and DCs with the gammaherpesvirus MHV68 induced expression of genes associated with innate immunity to a limited extent compared to what was observed after infection with other classes of herpesviruses. In particular, the IFN response to MHV68 infection was weak indicating this virus possesses immune evasion strategies targeting the IFN pathway.
Role for TLRs and DNA sensing pathways in the innate immune response to herpesviruses in dendritic cells
In order to characterize the pathways through which herpesviruses induced innate immune responses, we first examined whether lack of STING expression affected activation of innate immune responses by MHV68. For comparison, we included alpha- and beta-herpesviruses in the analysis. As expected, induction of type I IFN bioactivity by HSV-1 and MCMV was strongly reduced in cells from STINGgt/gt mice (Fig. 2Ai-Bi), whereas the IL-1β response was unaffected (Fig. 2Aii-Bii). In addition, the induction of TNF-α and IL-6 by the alpha- and beta-herpesviruses was significantly impaired in BMDCs lacking STING (Fig. 2Aiii-2Aiv, 2Biii-2Biv), clearly demonstrating an essential role for STING in immune activation beyond the IFN pathway. In sharp contrast to the strong dependence on STING for induction of innate immune responses by HSV-1 and MCMV, we found that the ability of MHV68 to evoke expression of TNF-α and IL-6 was not affected by the absence of STING (Fig. 2C), but was significantly compromised in cells from TLR2/9-/- mice (Supplemental Fig. 2A-2D). As controls we measured cytokine levels in supernatants from BMDCs treated with synthetic dsDNA or poly I:C. These data confirmed an essential role for STING in DNA-induced expression of type I IFNs, IL-6 and TNF-α, but not IL-1β (Fig. 2D), and that RNA-induced innate immune responses occur independent of STING (Fig. 2E). Collectively, these data show that MHV68 stimulates expression of inflammatory cytokines in myeloid cells in a manner independent of STING but dependent on TLRs.
Figure 2.
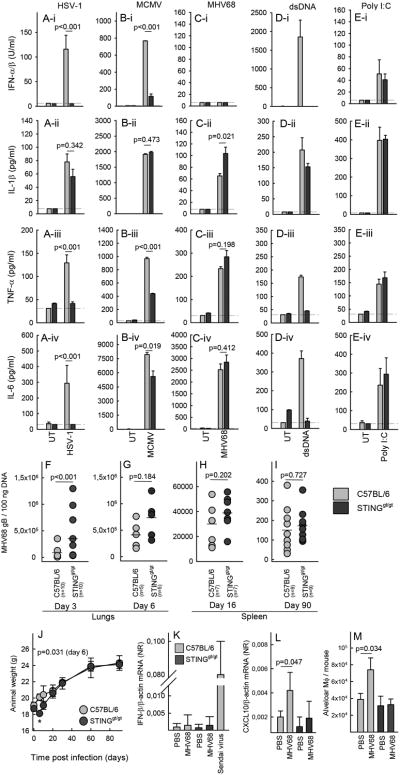
A limited role for STING in host control of MHV68 infection. (A-E) BMDCs from C57BL/6 and STINGgt/gt mice were treated with (A) HSV-1, MOI 3, (B) MCMV, MOI 3, (C) MHV68, MOI 100, (D) dsDNA, 2 μg/ml, (E) Poly I:C. Supertantants were harvested 18 h post treatment for measurement of levels of (row i) type I IFN bioactivity, (row ii) IL-1β, (row iii) TNF-α, (row iv) IL-6. Data are presented as means of measurements from triplicate cultures +/- st.dev. Data presented in panel A-E are representative of three independent experiments. (F-I) C57BL/6 and STINGgt/gt mice were infected intranasally with 5×104 pfu of MHV68. Lungswere isolated from mice infected for (F) 3 or (G) 6 days and spleens were isolated from mice infected for (H) 16 or (I) 90 days. Organs were homogenized and analyzed for the presence of MHV68 DNA by PCR. Each data point presented represents one individual mouse. (J) Weights of C57BL/6 and STINGgt/gt mice infected with MHV68. Data are presented as the mean weight of each group of animals +/- st.dev. Statistically significant difference between C57BL/6 and STINGgt/gt mice on day 6 is indicated with a star and the specific p value is given. (K-L) RNA was isolated from lung homogenates from C57BL/6 and STINGgt/gt mice (5 mice per group) 3 days after intranasal treatment with PBS or MHV68 (5×104 pfu). The RNA was analyzed for levels of IFN-β and CXCL10 mRNA by RT-qPCR. Data are presented as means +/- st.dev. (M) Single cell suspensions from lungs of C57BL/6 and STINGgt/gt mice (5 mice per group) treated for 3 days as in panel J and K, and analyzed by flow cytometry for levels of macrophages using CD45+ autofluorescence+ CD11blow CD11c+ as markers. The data are presented as average number of alveolar macrophages per mouse +/- st.dev.
In a next series of experiments we challenged mice intranasally with MHV68 and examined viral load in the lungs during the acute phase of the infection (day 3 and 6) and in the spleen during latency (day 16 and 90). Interestingly, we observed elevated levels of viral DNA in the lungs of STINGgt/gt mice on day 3 post infection (Fig. 2F). However, this difference did not reach statistical significance on day 6 post infection (Fig. 2G), and on day 16 and 90, no significant difference in splenic latent viral load was observed between C57BL/6 and STINGgt/gt mice (Fig. 2H, 2I). In corroboration with this, the STING-deficient mice exhibited an early weight loss after MHV68 infection, which was not seen in wild type mice, but at later time points the two mice strains followed indistinguishable weight curves (Fig. 2J). A flow cytometric characterization of spleen cells from C57BL/6 and STINGgt/gt mice revealed no significant difference in the number of B cells between the two strains (Table 1). B cells are required for establishment of latency after intranasal MHV68 infection (29). However, we did observe that several leukocytes cell types were suppressed in STINGgt/gt but not C56BL/6 mice infected with MHV68 for 90 days (Table 1). For TLR2/9-/- mice, we also observed elevated viral load in the lungs on day 3, but not on day 90 in the spleen (Supplemental Fig. 2E-2F). Despite the early STING-dependent control of MHV68 in the lungs, we did not find elevated expression of IFN-β mRNA in the lungs of infected mice (Fig. 2K). By performing a wider screen for MHV68-induced gene expression in the lungs, CXCL10 was the only mRNA found to be elevated in the lungs after infection, and this was dependent on expression of STING (Fig. 2L). This correlated with early recruitment of macrophages to the lungs through a STING-dependent process (Fig. 2M). Thus, STING plays a limited role in activation of the pathways mediating control of MHV68, which failed to induce type I IFN responses in vitro and in vivo. However, we did observe a very early role for STING in vivo correlating with low induction of CXCL10 and recruitment of macrophages.
Table 1. Flow cytometric characterization of spleens from C57BL/6 and STINGgt/gt mice after MHV68 infection.
| WT | STINGgt/gt | ||||
|---|---|---|---|---|---|
| PBS (n=5) | MHV68 (n=9) | PBS (n=6) | MHV68 (n=7) | ||
| Cell number (×106) | Total splenocytes | 156.2 ± 12.5 | 189.5 ± 11.1* | 126.8 ± 22.4 | 98.8 ± 5.7 |
| CD11c+ | 5.8 ± 0.6 | 5.6 ± 0.5 | 4.2 ± 0.5 | 2.8 ± 0.2* | |
| CD11b+CD11c+ | 3.5 ± 0.5 | 3.15 ± 0.4 | 2.4 ± 0.3 | 1.5 ± 0.1* | |
| I-Ab+ in CD11c+ | 3.1 ± 5.9 | 2.6 ± 4.2 | 2.1 ± 0.3 | 1.3 ± 0.2* | |
| CD11b+CD11c- | 14.1 ± 1.1 | 13.5 ± 1.8 | 10.2 ± 2.7 | 5.1 + 0.7 | |
| CD19+ | 72.1 ± 10.7 | 82.9 ± 6.5 | 61.6 ± 7.5 | 60.7 ± 2.9 | |
| CD3+ | 46.0 ± 3.0 | 56.1 ± 5.7 | 43.8 ± 4.1 | 33.6 ± 3.2* | |
| F4/80+ | 1.6 ± 0.5 | 14.9 ± 6.6* | 1.5 ± 0.2 | 1.6 ± 7.2 | |
p value < 0.05. Compared between PBS and MHV68 infection within the same genotype
The finding that MHV68-induced cytokine expression occurred independent of STING indicated that this virus, in contrast to alpha- and beta-herpesviruses, triggers innate immune responses independent of intracellular DNA sensing. Another major pathway of DNA-driven innate immune activation is the AIM2 inflammasome pathway. Therefore, we examined the role for AIM2 in production of IL-1β in lipopolysaccharide -primed BMMs. We first showed that these cells did produce high amounts of IL-1β in response to the potent inflammasome activator poly(dAdT) and this was dependent on AIM2 and the inflammasome adaptor protein ASC, but not NLRP3 (Fig. 3A). For MCMV, production of IL-1β was strongly reduced in AIM2-/- and ASC-/- cells as previously reported (4) (Fig. 3A). By contrast, MHV68 induced IL-1β secretion in a manner independent of AIM2 but dependent on NLRP3 and ASC (Fig. 3B). The AIM2 independent nature of the MHV68-driven response was also observed in vivo, since AIM2 deficiency did not reduce the levels of IL-1β or IFN-γ, the latter produced through an IL-18 dependent pathway (4), in mice infected with MHV68 (Fig. 3C, 3D). In further support of the lack of a role for AIM2 in immune sensing of MHV68, AIM2-deficiency did not affect viral load in mice latently infected with MHV68 (Fig. 3E). By contrast, ASC-/- and NLRP3-/- mice exhibited significantly elevated viral load, demonstrating that the inflammasome was involved in control of MHV68 infection (Fig. 3E). Collectively, these data demonstrate that MHV68 activates the inflammasome pathway via the NLRP3-ASC pathway and independent of the DNA-sensing AIM2 inflammasome.
Figure 3.

The AIM2 inflammasome is not involved in immune activation during MHV68 infection. (A-B) BMMs from C57BL/6, ASC-/-, NLRP3-/-, and AIM2-/- mice were stimulated with lipopolysaccharide (200 ng/ml) for 3 h prior to treatment with MCMV (MOI 10), Poly dA:dT (1 μg/ml), nigericin (10 μM) or MHV68 (MOI 10). Supertantants were harvested 24 h post treatment for measurement of IL-1β. Data are presented as means of measurements from triplicate cultures +/- st.dev. (C-E) C57BL/6 and AIM2-/- mice were infected with 5×105 pfu of MHV68. Spleens were isolated from mice infected for 6 and 60 days. The organs were homogenized and analyzed for levels of IL-1β and IFN-γ by ELISA (day 6), and MHV68 DNA by PCR (day 60). (C, D) The ELISA data are presented as means of measurements from 6-8 mice per group +/- st.dev. (E) Each data point presented represents measurement of MHV68 DNA from one individual mouse. Data are representative of three independent experiments.
KSHV induces innate immune responses independent of known DNA-stimulated pathways
KSHV is a human gammaherpesvirus closely related to MHV68 and the cause of Kaposi's Sarcoma (30). In order to determine whether KSHV also stimulates innate immune responses independent of known intracellular sensing systems, we first examined the effect of STING knock-down on virus-induced cytokine expression. The STING shRNA knock-down cell line has been reported previously (27), and was tested to have more than 80 % reduced expression of STING as compared to the empty vector control cell line. As shown in Fig. 4A and 4B, knock-down of STING in THP1 cells reduced expression of IFN-β and TNF-α induced by HSV-1 or dsDNA but did not affect the cytokine response to KSHV. This was not due to inability of KSHV to infect the cells as evidenced by green fluorescence in cells infected with a GFP-encoding KSHV (Supplemental Fig. 3). Consistent with a limited role for STING in the response to KSHV, we found that in contrast to what was observed after HCMV infection, neither STING nor IFI16, were mobilized after infection with this virus (Fig. 4C). To test the role for AIM2 in KSHV-induced IL-1β expression, we generated a stable THP1 cell with shRNA-mediated knock-down of AIM2 (Fig. 4D). However, despite efficient knock-down of AIM2, the ability of KSHV to induce IL-1β production was not affected. It has been reported that KSHV activates an IFI16-dependent inflammasome after nuclear sensing of the virus in endothelial cells(14). To test the role for IFI16 in IL-1β production during KSHV infection, we measured IL-1β levels in supernatants from THP1 cells expressing control shRNA or IFI16 shRNA. The IFI16 shRNA knock-down cell line cell line has been reported previously (27), and was tested to have more than 80 % reduced expression of IFI16 as compared to the empty vector control cell line. In contrast to the previous data from endothelial cells, we did not observe reduced KSHV-induced IL-1β expression in monocytic cells with IFI16 knock-down (Fig. 4E). We also found that the induction of IFN-β by KSHV was independent of IFI16, in contrast to HSV-1, which relied on this DNA sensor for stimulation of IFN-β expression as previously reported (17). All together, these data show that KSHV, like MHV68, stimulates innate immune responses independent of the DNA-driven pathways via STING and AIM2.
Figure 4.

KSHV induces expression of IFN-β and production of IL-1β independent of the STING and AIM2 pathways. (A-B) PMA-differentiated THP1-derived cells stably transduced with lentiviral shRNAs (control and STING) were infected with HSV-1 (MOI 3) or KSHV (30 genomes/cell) or transfected with dsDNA (2 μg/ml). Total RNA was harvested 6 h post treatment and analyzed for levels of IFN-β and TNF-α. Data are presented as means of normalized ratios relative to untreated using β-actin as internal reference +/- st.dev. (C) MDMs were infected with CMV (MOI 3) or KSHV (30 genomes/cell) for 4 h. Subcellular distribution of IFI16 and STING was determined by confocal microscopy. White box indicates area displayed in zoom column (zoom is magnified 5× from original image). Scale bar, 10 μm. (D) Western blot of AIM2 and β-actin on extracts from PMA-differentiated THP1-derived cells stably transduced with lentiviral shRNAs (control and AIM2). The shRNA THP1 cells were left untreated or infected with KSHV. Supernatants were harvested 18 h post and levels of IL-1β were measured by ELISA. Data are presented as means of normalized ratios relative to untreated +/- st.dev. (E-F) PMA-differentiated THP1-derived cells stably transduced with lentiviral shRNAs (control and IFI16) were infected with HSV-1 (MOI 3) or KSHV (30 genomes/cell). Total RNA and culture supernatants were harvested 6 h and 18 h post treatment, respectively and analyzed for levels of IFN-β mRNA and IL-1β protein. Data are presented as means of normalized ratios relative to untreated +/- st.dev. All data presented in this figure are representative of four or more independent experiments. NR, normalized ratio.
The MHV68 ORF64 DUB antagonizes innate immune responses activated by viral DNA
Given the finding that MHV68 infection in myeloid cells did not lead to detectable innate immune activation by the DNA-sensing pathway, we were interested in identifying the step in the pathway targeted by the virus. In a first series of experiments, we pretreated murine DCs with MHV68 followed by infections and stimulations as indicated in Fig. 5A. Supernatants were collected for measurement of type I IFN activity. Interestingly, we observed that HSV-1-induced expression of type I IFN, CXCL10 and IL-6 was abrogated in cells pretreated with MHV68 (Fig. 5A, Supplemental Fig. 4A-4B). By contrast, MHV68 infection did not affect the ability to activate the pathway via dsDNA or cGAMP, nor was the virus able to modulate the intracellular RNA sensing pathway. The inhibition of HSV-induced cytokine expression by MHV68 was not due to a blockage of HSV-1 entry, since accumulation of the HSV major capsid protein VP5 in the cytoplasm of infected cells was not affected by pretreatment with MHV68 (Supplemental Fig. 4C). Similar to the inhibition of HSV-1-induced IFN expression, MHV68 also inhibited IL-1β expression induced by MCMV but not synthetic DNA (Fig. 5B). Upon examination of the assembly of STING signalsomes, as measured by STING foci formation and recruitment of TBK1, we found that although HSV-1 infection did stimulate this activity, MHV68 infection did not (Fig. 5C). Despite the lack of response to MHV68 infection, purified MHV68 DNA did induce IFN-β expression if transfected into cells (Fig. 5D). These findings suggest that MHV68 infection inhibits herpesvirus-driven DNA-mediated immune activation and suggests that this occurs upstream of the bifurcation into the STING-dependent IFN pathway and the AIM2-dependent IL-1β pathway.
Figure 5.

The DUB activity of MHV68 mediates evasion of herpesvirus-induced production of type I IFN and IL-1β. (A-B) BMDCs from C57BL/6 mice were infectedwith MHV68 (MOI 10) 1 hour prior to treatment with HSV-1 (MOI 3), MCMV (MOI 1), ODN1826 (1 μM), poly I:C (25 μg/ml), dsDNA (2 μg/ml), cGAMP (2 μM), and lipofectamine. Supernatants were harvested 16 h later for measurement of type I IFN bioactivity and IL-1β. (C) BMDCs from C57BL/6 mice were infected with HSV-1 (MOI 3) and MHV68 (MOI 20) for 5 h and stained with DAPI and anti-STING and anti-TBK1 antibodies. (D) BMDCs were stimulated with genomic DNA from HSV-1 and MHV68 (0.25 μg/ml). Supernatants were isolated 18 h post stimulation for measurement of IFN bioactivity. (E-F) BMDCs from C57BL/6 mice were treated with MHV68 ORF64 C33A or the revertant virus at MOIs of 1, 10 and 100. Supernatants were harvested 16 h later for measurement of type I IFN bioactivity and IL-1β (only MOI 10). (G) BMDCs from C57BL/6 and STINGgt/gt mice were infected with MHV68 ORF64 C33A or the revertant virus (MOI 50). Supernatants were harvested 18 h post infection and IFN bioactivity was measured. (H) BMMs were infected with EdC-labeled MHV68 ORF64 C33A or the revertant virus (MOI 20) for 3 h and fixed. Accessible DNA was visualized using copper(I)-catalyzed Azide Alkyne Cyccoaddiotion staining. Nucleic acids were visualized using DAPI. Scale bar, 10 μm. (I) Quantification of cytosolic MHV68 DNA versus total MHV68 genomes in BMMs infected for 3 h with EdC-labeled MHV68 ORF64 C33A or the revertant virus (MOI 10 and 20). (J-K) BMDCs from C57BL/6 and STINGgt/gt mice were infected with DUB active site mutants of (J) MCMV (MOI 1) and (K) HSV-1 (MOI 3). Supernatants were harvested 18 h post infection and IFN bioactivity was measured. All cytokine data are presented as means of measurements from triplicate cultures +/- st.dev. All data presented in this figure are representative of three or more independent experiments.
The findings above indicate that gammaherpesviruses target a step general for all virus-activated DNA-dependent innate pathways tested. We recently reported that alpha- and beta-herpesvirus capsids become ubiquitinated and degraded in the cytoplasm in macrophages, hence exposing the viral DNA for cytosolic DNA sensors (17). Since herpesviruses encode a conserved DUB activity embedded in the large tegument protein (21,31,32), we hypothesized that herpesviruses utilize this activity to counteract DNA-driven innate immune activation. To address this question, DCs were stimulated with a MHV68 mutant with a point mutation in the active site of the DUB as well as with a revertant virus (21). Interestingly, in contrast to the revertant virus, infection with the DUB active site mutant virus did stimulate type IFN activity and led to elevated IL-1β production (Fig. 5E and 5F). The IFN response was dependent on STING, thus demonstrating that deletion of the DUB activity of ORF64 exposes an intrinsic capacity of gammaherpesviruses to stimulate DNA-driven innate immune responses (Fig. 5G). The MHV68 kinase ORF36, previously reported to inhibit IFN regulatory factor 3 activation in fibroblasts (22), did not seem to be essential for evasion of DNA-driven IFN expression in myeloid cells (Supplemental Fig. 4D). In order to evaluate whether infection with the ORF64 C33A mutant differed from WT virus with respect to subcellular localization of viral genomes, we generated MHV68 with EdC incorporated into the viral genomes. In infected cells the viral genomes were visualized using click-chemistry (33). Interestingly, while the viral genome was found mainly in the nucleus of cells infected with the revertant virus, the viral DNA was localized preferentially to the cytoplasm in cells infected with the ORF64 C33A virus (Fig. 5H, 5I).
In order to evaluate whether the DUB activity of alpha- and beta-herpesviruses also plays a role in immune evasion, DCs were infected with WT or DUB mutant HSV-1 and MCMV followed by measurement of IFN bioactivity in the culture supernatants. For both HSV-1 and MCMV we found that although the WT viruses did induce IFN expression, this was further elevated in the absence of the tegument DUB activity (Fig. 5J and 5K). Together, these data demonstrate that deletion of the DUB activity of the large MHV68 tegument protein ORF64 leads to augmented cytosolic localization of viral DNA, and renders MHV68 capable of stimulating DNA-driven innate immune responses. Moreover, the data also reveal the herpesvirus tegument DUB activity to be a conserved mechanism to counteract immune activation through the DNA-driven STING pathway.
ORF64 DUB deficiency enables a STING-dependent antiviral pathway controlling establishment of a latent MHV68 reservoir
Given the finding that the ORF64 DUB active site mutant gained the capacity to induce IFN expression in a STING-dependent manner, we were interested in examining whether this MHV68 mutant had an altered course of infection in wild type and STINGgt/gt mice. A previous report has shown that the MHV68 ORF64 C33A mutant was not attenuated with respect to the ability to establish latent infection after intraperitoneal infection (21). In this study we used the intranasal infection model, and it has previously been reported that the route of infection affects the role of innate immune pathways in control of MHV68 infection (19). We first examined whether the MHV68 ORF64 DUB mutant was capable of inducing IFN-β expression in vivo and also whether treatment with recombinant IFN-β affected the acute viral load in the lungs and the latent viral reservoir in the spleen. As shown in Fig. 6A, RNA in lung homogenates from mice infected for 3 days with the MHV68 ORF64 C33A mutant contained elevated levels of IFN-β mRNA, whereas the IFN-β mRNA levels in homogenates from mice infected with the revertant virus were indistinguishable from the mock treated mice. This finding prompted us to evaluate whether an elevated innate immune response could affect viral load in infected organs at different time points during the infection. Therefore, mice were treated with recombinant IFN-β and TNF-α before the infection and subsequently once every week during the course of the infection. When evaluating the viral load in the lungs, we found that IFN-β treatment led to reduced accumulation of viral DNA on day 6 post infection (Fig. 6B). Interestingly, in the spleens harvested 90 day after infection, treatments with single cytokines had not affected the latent viral load, but this was indeed the case in spleens from mice treated with both IFN-β and TNF-α (Fig. 6C). Thus, the elevated expression of proteins associated with innate immune responses during infection with the MHV68 ORF64 DUB mutant could potentially affect viral load in the lung and the spleen.
Figure 6.

The ORF64 DUB activity facilitates establishment of latency by targeting a STING-dependent pathway. (A) RNA was isolated from lung homogenates from C57BL/6 mice (5 mice per group) 3 days after intranasal treatment with PBS, MHV68 ORF64 C33A, or the revertant virus (5×104 pfu). The RNA was analyzed for levels of IFN-β mRNA by RT-qPCR and presented as a normalized ratio using β-actin as internal reference. Data are presented as means +/- st.dev. (B, C) C57BL/6 mice were treated intraperitoneally with recombinant IFN-β and TNF-α (2 μg of each cytokine), at the time of intranasal MHV68 infection (5×104 pfu) and every 7 days during infection. (B) Lungs were isolated from mice infected for 6 days and (C) spleens were isolated from mice infected for 90 days. Organs were homogenized and analyzed for the presence of MHV68 DNA by PCR. Each data point presented represents one individual mouse. (D-F) C57BL/6 and STINGgt/gt mice were infected intranasally with 5×104 pfu of MHV68 ORF64 C33A or the revertant virus. (D) Lungs were isolated from mice infected for 3 days and (E, F) spleens were isolated from mice infected for 16 and 90 days. Organs were homogenized and analyzed for the presence of MHV68 DNA by PCR. Each data point presented represents one individual mouse.
In a final series of experiments, we challenged C57BL/6 and STINGgt/gt mice with the MHV68 ORF64 C33A mutant or the revertant virus. When measuring viral load in the lung on day 3 post infection, significantly elevated levels of virus were found in STINGgt/gt mice as compared to wild type mice after infection with either virus (Fig. 6D). However, no significant difference between the viral loads in the lungs of mice infected with the DUB mutant or revertant viruses were observed, regardless of the mouse strain (Fig. 6D). Thus, lack of ORF64 DUB activity does not affect the early stages of MHV68 infection. By contrast, when we looked for viral DNA in the spleen from mice infected for 16 or 90 days, we observed significantly lower levels of viral DNA in C57BL/6 mice infected with the MHV68 ORF64 DUB mutant as compared to the revertant virus (Fig. 6E and 6F). Interestingly, the impaired ability of the MHV68 ORF64 DUB mutant to establish latent infection was rescued when the infection occurred in STINGgt/gt mice. Similar experiments in TLR2/9-/- mice revealed that the ORF64 DUB mutation did not confer mouse-genetics-dependent differential latent viral load in this model (Supplemental Fig. 2G, 2H). Altogether, MHV68 antagonizes the STING-dependent pathway stimulated by viral genomic DNA through a mechanism dependent on the DUB embedded in ORF64, and this facilitates establishment of a latent reservoir of MHV68.
Discussion
KSHV and EBV are important human pathogens, and are among the small group of viruses capable of causing tumors (30). These viruses belong to the family of gammaherpesviruses, and have the capacity to establish lifelong latent-recurrent infections. The ability of the host to control infections with herpesviruses is important for prevention of disease development, as exemplified by the frequent development of Kaposi's sarcoma in individuals with acquired immunodeficiency syndrome (30). It is now known that herpesviruses are controlled by both the innate and adaptive immune system, but despite this there is limited knowledge on how the innate immune system detects infections with gammaherpesviruses. Recently much attention has been given to DNA as a major stimulator of innate immune responses (34), and it has been reported that alpha-herpesviruses (HSV), and beta-herpesviruses (CMV) are detected by the DNA sensing machinery (4,10,17). At this stage, two major pathways of immune activation by DNA are known: DNA-induced IFN responses relying on the adaptor protein STING (2), and DNA-stimulated inflammasome activation occurring via AIM2 (16). Here we show that wild type MHV68 is a poor inducer of innate immune responses and the responses evoked by MHV68 and also by KSHV are independent of described intracellular DNA sensing systems known to be essential for host detection of alpha- and beta-herpesviruses. In line with this, STINGgt/gt and AIM2-/- mice did not exhibit major defects in control of MHV68 infection, and in particular the establishment of latent viral reservoirs was unaffected by defective DNA sensing pathways. An MHV68 mutant with a point mutation in the tegument DUB ORF64, which is conserved among herpesviruses, induced significant type I IFN expression through a STING-dependent mechanism. Moreover, wild type mice infected with the DUB mutant harbored a lower latent viral load than mice infected with the revertant, but this impaired activity of the DUB mutant was rescued in STINGgt/gt mice. Altogether, we propose that gammaherpesviruses target the DNA sensing system and this contributes to the ability of this class of viruses to establish latent infections.
In myeloid cells MHV68 did not stimulate detectable IFN bioactivity but did stimulate expression of IL-1β, IL-6 and TNF-α to significant levels. However, the IL-1β response was independent of AIM2, but dependent on NLRP3 and ASC, and the induction of TNF-α and IL-6 was dependent on TLR2 and TLR9. This was in contrast to the responses evoked by HSV-1 and MCMV where important roles were ascribed to STING and AIM2 for induction of type I IFN expression and secretion of IL-1β, respectively. Similar to what we found with MHV68 in murine myeloid cells, the induction of IFN-β and IL-1β evoked by KSHV infection in a human monocytic cell line, was independent of STING and AIM2. Thus, unlike alpha- and beta-herpesvirues, gammaherpesviruses are detected independent of the intracellular DNA sensing system, at least in the cell types tested, where particularly the TLR pathway is important for induction of inflammatory genes and also for early control of viral load in vivo. The role for TLRs in sensing of gammaherpesviruses is in line with previous work demonstrating that TLR2 and 9 can recognize gammaherpesviruses, with particularly the recognition by TLR9 in plasmacytoid DCs being well documented (9,18,19,35). For TLR2, a role has been reported in fibroblast and in early control in the lung after intranasal MHV68 infection (35). It should be noted that we did find elevated viral load in the lungs of STINGgt/gt mice at early time points after MHV68 infection correlating with weight loss and impaired recruitment of macrophages. At this stage, we cannot explain this early phenotype but possible explanations include (i) activation of DNA sensing in other cell types than the ones studied here, (ii) low grade sensing of MHV68 DNA and activation of signaling in the myeloid cells at early time points, and (iii) activation of STING-dependent signaling independent of DNA sensing, such as virus-cell fusion (28). A recent study demonstrated modest elevation of MHV68 load in lungs and spleen of cGAS-deficient mice on day 7 after intraperitoneal MHV68 infection (36). These results are consistent with our data, and suggest a minor role for the cGAS-STING pathway early during MHV68 infection despite the presence of ORF64.
It has been reported that KSHV and EBV can be sensed by IFI16 in the nucleus to stimulate an IFI16-ASC-caspase 1 inflammasome in permissive and latently infected cells (14,37). In our system using a human monocytic cell line, we found that IFI16 was not essential for production of IL-1β during KSHV infection, indicating that this DNA sensor is not involved in KSHV-driven inflammasome activation in these cells. However, the data presented in this work do not address the question of where in the cell the DNA sensing takes place. Moreover, it is also important to note that the proposed nuclear IFI16-ASC-caspase 1 inflammasome pathway is not abrogated in STINGgt/gt or AIM2-/- mice. Therefore, a potential role for nuclear DNA sensing during herpesvirus infections is not excluded by the data presented in this work.
The phenomenon of viral evasion of the innate immune response is well-described and known to impact the pathogenicity of viruses (38). Herpesviruses have been reported to target all steps in the pathway, including modulation of PAMPs (39), avoidance of sensing(40), inhibition of signaling (41), and inhibition of transcription of key genes, including IFN-β (5,42). Here we found that wild type MHV68 largely prevented expression of type I IFN expression in macrophages and myeloid DCs, and induced only modest levels of IL-1β. By contrast, in plasmacytoid dendritic cells and MEFs it has previously been shown that MHV68 induces type IFN expression and this is stimulated through TLR-dependent pathways (19,35). Altogether, such data suggest that MHV68 utilizes cell-type specific means to evade innate immune responses and that ORF64 is of importance in cell types sensing this virus through the DNA sensing pathway. Other reported innate immune evasion mechanisms of MHV68 include targeting of TBK1 and IFN regulatory factor 3, by ORF11 and ORF36, respectively (22,43). As to the known roles of herpesvirus tegument DUBs in innate immune evasion, most information has been gathered through work in HEK293 cell-based systems. It has been reported that KSHV ORF64 can target RIG-I through deubiquitination of RIG-I (44), and that the EBV DUB BPLF1 inhibits the TLR2 pathway by deubiquitination of TRAF6, NEMO, and IκBα(45). Using a similar system it has also been demonstrated that HSV-1 UL36 can target TRAF3 to prevent type I IFN expression through the RIG-I pathway (46). However, in contrast to the studies mentioned above, which were mainly performed in overexpression systems (44-46), we found no inhibitory effects of MHV68 infection on induction of type I IFN and IL-6 by Sendai virus and TLR ligands. Moreover, the MHV68 DUB mutant induced elevated expression of both type I IFN and IL-1β as compared to the wild type virus, and the IFN response was evoked through the DNA-stimulated STING pathway. Our data therefore do not support a model where the MHV68 DUB targets a specific TRAF protein during viral infection in myeloid cells, but rather that the virus targets a step at the level of DNA sensing upstream of bifurcation between the STING and AIM2 pathways. The herpesvirus DUB activity embedded in the N terminal part of the large tegument protein is highly conserved (31). Since this activity can cleave either K48- or K63-linked polyubiquitin chains it therefore seems likely that herpesvirus DUBs can serve a broad role in targeting of intracellular innate sensing and signaling pathways as already reported (44-46), and that this occurs in a manner dependent on a range of factors including cell types and the stage of the viral replication cycle.
The establishment of latent gammaherpesvirus infections involves a complex series of events governed by viral proteins with a particularly important role for the latency-associated nuclear antigen (47). In addition, several host systems are positively and negatively involved in establishment of latency, including IFNs, apoptosis, autophagy, and TLRs (47). We found that the DUB mutant virus, which activated STING-dependent innate immune responses, exhibited impaired ability to establish latent infection in the spleen. The mechanism mediating this STING-dependent restriction of latency was not identified in this study, and further work is needed to describe the pathway capable of controlling DUB-deficient MHV68. Previous work has revealed that the route of viral infection impacts on the apparent role for specific proteins in the host response to MHV68 infection (19). The same may be the case for the ability of MHV68 to facilitate establishment of latency through targeting of the DNA sensing machinery. Published data on latent viral load in the spleen of C57BL/6 mice after intraperitoneal infection with the MHV68 ΔDUB mutants did not reveal an attenuated phenotype of the virus (21).
Collectively, in this work we report that gammaherpesviruses evade sensing of viral genomic DNA, which facilitates establishment of a latent viral reservoir in the host. These data are consistent with a role for the gammaherpesvirus DUB in prevention of innate immune activation by the viral DNA, and suggests the molecular target to be upstream of bifurcation into the AIM2 and STING pathways. The STING-dependent antiviral program impaired by ORF64 works together with TLR-mediated activities to control acute and latent MHV68 infection. We propose that the ability of gammaherpesviruses to deubiquitinate viral and cellular substrates plays an important role in viral immune evasion and hence for establishment of infection.
Supplementary Material
Acknowledgments
We thank Kirsten Stadel Petersen for technical assistance.
Footnotes
This work was funded by The Danish Medical Research Council (grant no 09-072636, 12-124330), The Novo Nordisk Foundation, The Velux Foundation, The Lundbeck Foundation (grant no R34-3855), Kathrine og Vigo Skovgaardes Fond, Elvira og Rasmus Riisforts almenvelgørende Fond, and Fonden til Lægevidenskabens Fremme (all awarded to S.R.P.). C.S. was funded by a China Scholarship Council Scholarship (201206170055). K.A.H. was funded by a Marie Curie Incoming International Fellowship. This work is supported by grants from the NIH (AI083713 and AI093752 awarded to K.A.F) and NIH grants AI109965, AI107810, CA019014 and DE018281 awarded to BD. Cancer Prevention & Research Institute of Texas (CPRIT) grant R1202 (J.W.U.)
Abbreviations: AIM, absent in melanoma; ASC, apoptosis-associated speck-like protein containing a carboxy-terminal CARD; cGAMP, cyclic-GMP-AMP; cGAS cyclic-GMP-AMP synthetase; CMV, cytomegalovirus; DUB, deubiquitinase; EdC, deoxy-5-ethynylcytidine; IFI, IFN inducible protein; KSHV, Kaposi's sarcoma-associated herpesvirus; MHV 68, murine gammaherpesvirus 68; ORF, pen reading frame; STING, Stimulator of IFN genes, TBK Tank-binding kinase.
References
- 1.Pellet PE, Roizman B. The Family Herpesviridae: A brief introduction. In: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE, editors. Fiel's Virology. 5th. Lippincott-Williams and Wilkins; New York: 2007. pp. 2479–2499. [Google Scholar]
- 2.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reinert LS, Harder L, Holm CK, Iversen MB, Horan KA, Dagnaes-Hansen F, ULhoj B, Holm T, Mogensen TH, Owens T, Nyengaard JR, Thomsen AR, Paludan SR. TLR3-deficiency renders astrocytes permissive to HSV infection and facilitates establishment of CNS infection in mice. J Clin Invest. 2012;122:1368–1376. doi: 10.1172/JCI60893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, Hornung V, Vogel SN, Szomolanyi-Tsuda E, Fitzgerald KA. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. 2010;11:395–402. doi: 10.1038/ni.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paludan SR, Bowie AG, Horan KA, Fitzgerald KA. Recognition of herpesviruses by the innate immune system. Nat Rev Immunol. 2011;11:143–154. doi: 10.1038/nri2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kawai T, Akira S. Toll-like Receptors and Their Crosstalk with Other Innate Receptors in Infection and Immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 7.Tabeta K, Georgel P, Janssen E, Du X, Hoebe K, Crozat K, Mudd S, Shamel L, Sovath S, Goode J, Alexopoulou L, Flavell RA, Beutler B. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Natl Acad Sci U S A. 2004;101:3516–3521. doi: 10.1073/pnas.0400525101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lund J, Sato A, Akira S, Medzhitov R, Iwasaki A. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J Exp Med. 2003;198:513–520. doi: 10.1084/jem.20030162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.West JA, Gregory SM, Sivaraman V, Su LS, Damania B. Activation of Plasmacytoid Dendritic Cells by Kaposi's Sarcoma-Associated Herpesvirus. Journal of Virology. 2011;85:895–904. doi: 10.1128/JVI.01007-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois C, Jin T, Xiao T, Fitzgerald P, Paludan SR, Bowie AG. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol. 2010;11:997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang ZQ, Yuan B, Bao MS, Lu N, Kim T, Liu YJ. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nature Immunology. 2011;12:959–965. doi: 10.1038/ni.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, Ohba Y, Taniguchi T. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- 13.Li T, Diner BA, Chen J, Cristea IM. Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. PNAS. 2012;109:10558–10563. doi: 10.1073/pnas.1203447109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P, Chandran B. IFI16 Acts as a Nuclear Pathogen Sensor to Induce the Inflammasome in Response to Kaposi Sarcoma-Associated Herpesvirus Infection. Cell Host Microbe. 2011;9:363–375. doi: 10.1016/j.chom.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horan KA, Hansen K, Jakobsen MR, Holm CK, Waggoner L, West JA, Unterholzner L, Iversen MB, Soby S, Thomson M, Jensen SB, Rasmussen SB, Ellermann-Eriksen S, Kurt-Jones EA, Landolfo S, Melchjorsen J, Bowie AG, Damania B, Fitzgerald KA, Paludan SR. Proteasomal degradation of herpes simplex virus capsids in macrophage releases DNA to the cytosol for recognition by DNA sensors. J Immunol. 2013;190:2311–2319. doi: 10.4049/jimmunol.1202749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fiola S, Gosselin D, Takada K, Gosselin J. TLR9 Contributes to the Recognition of EBV by Primary Monocytes and Plasmacytoid Dendritic Cells. J Immunol. 2010;185:3620–3631. doi: 10.4049/jimmunol.0903736. [DOI] [PubMed] [Google Scholar]
- 19.Guggemoos S, Hangel D, Hamm S, Heit A, Bauer S, Adler H. TLR9 contributes to antiviral immunity during gammaherpesvirus infection. J Immunol. 2008;180:438–443. doi: 10.4049/jimmunol.180.1.438. [DOI] [PubMed] [Google Scholar]
- 20.Adler H, Messerle M, Wagner M, Koszinowski UH. Cloning and mutagenesis of the murine gammaherpesvirus 68 genome as an infectious bacterial artificial chromosome. J Virol. 2000;74:6964–6974. doi: 10.1128/jvi.74.15.6964-6974.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gredmark-Russ S, Isaacson MK, Kattenhorn L, Cheung EJ, Watson N, Ploegh HL. A gammaherpesvirus ubiquitin-specific protease is involved in the establishment of murine gammaherpesvirus 68 infection. J Virol. 2009;83:10644–10652. doi: 10.1128/JVI.01017-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hwang S, Kim KS, Flano E, Wu TT, Tong LM, Park AN, Song MJ, Sanchez DJ, O'Connell RM, Cheng G, Sun R. Conserved herpesviral kinase promotes viral persistence by inhibiting the IRF-3-mediated type I interferon response. Cell Host Microbe. 2009;5:166–178. doi: 10.1016/j.chom.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bolstad M, Abaitua F, Crump CM, O'Hare P. Autocatalytic activity of the ubiquitin-specific protease domain of herpes simplex virus 1 VP1-2. J Virol. 2011;85:8738–8751. doi: 10.1128/JVI.00798-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Upton JW, Kaiser WJ, Mocarski ES. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe. 2010;7:302–313. doi: 10.1016/j.chom.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Redwood AJ, Messerle M, Harvey NL, Hardy CM, Koszinowski UH, Lawson MA, Shellam GR. Use of a murine cytomegalovirus K181-derived bacterial artificial chromosome as a vaccine vector for immunocontraception. J Virol. 2005;79:2998–3008. doi: 10.1128/JVI.79.5.2998-3008.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi MK, Wang Z, Ban T, Yanai H, Lu Y, Koshiba R, Nakaima Y, Hangai S, Savitsky D, Nakasato M, Negishi H, Takeuchi O, Honda K, Akira S, Tamura T, Taniguchi T. A selective contribution of the RIG-I-like receptor pathway to type I interferon responses activated by cytosolic DNA. Proc Natl Acad Sci U S A. 2009;106:17870–17875. doi: 10.1073/pnas.0909545106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jakobsen MR, Bak RO, Andersen A, Berg RK, Jensen SB, Jin T, Laustsen A, Hansen K, Ostergaard LJ, Fitzgerald KA, Xiao T, Mikkelsen JG, Mogensen TH, Paludan SR. IFI16 senses DNA forms of the retroviral replication cycle and controls HIV-1 replication. PNAS. 2013;110:E4571–E4580. doi: 10.1073/pnas.1311669110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holm CK, Jensen SB, Jakobsen MR, Cheshenko N, Horan KA, Moller HB, Gonzalez-Dosal R, Rasmussen SB, Christensen MH, Yarovinsky TO, Rixon FJ, Herold BC, Fitzgerald KA, Paludan SR. Virus-cell fusion as a trigger of innate immunity dependent on the adaptor STING. Nat Immunol. 2012;13:737–743. doi: 10.1038/ni.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Usherwood EJ, Stewart JP, Robertson K, Allen DJ, Nash AA. Absence of splenic latency in murine gammaherpesvirus 68-infected B cell-deficient mice. J Gen Virol. 1996;77(Pt 11):2819–2825. doi: 10.1099/0022-1317-77-11-2819. [DOI] [PubMed] [Google Scholar]
- 30.Wen KW, Damania B. Kaposi sarcoma-associated herpesvirus (KSHV): molecular biology and oncogenesis. Cancer Lett. 2010;289:140–150. doi: 10.1016/j.canlet.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kattenhorn LM, Korbel GA, Kessler BM, Spooner E, Ploegh HL. A deubiquitinating enzyme encoded by HSV-1 belongs to a family of cysteine proteases that is conserved across the family Herpesviridae. Mol Cell. 2005;19:547–557. doi: 10.1016/j.molcel.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 32.Gonzalez CM, Wang L, Damania B. Kaposi's Sarcoma-Associated Herpesvirus Encodes a Viral Deubiquitinase. Journal of Virology. 2009;83:10224–10233. doi: 10.1128/JVI.00589-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang IH, Suomalainen M, Andriasyan V, Kilcher S, Mercer J, Neef A, Luedtke NW, Greber UF. Tracking viral genomes in host cells at single-molecule resolution. Cell Host Microbe. 2013;14:468–480. doi: 10.1016/j.chom.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 34.Paludan SR, Bowie AG. Immune sensing of DNA. Immunity. 2013;38:870–880. doi: 10.1016/j.immuni.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Michaud F, Coulombe F, Gaudreault E, Kriz J, Gosselin J. Involvement of TLR2 in recognition of acute gammaherpesvirus-68 infection. PLoS One. 2010;5:e13742. doi: 10.1371/journal.pone.0013742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schoggins JW, Macduff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, Mar KB, Richardson RB, Ratushny AV, Litvak V, Dabelic R, Manicassamy B, Aitchison JD, Aderem A, Elliott RM, Garcia-Sastre A, Racaniello V, Snijder EJ, Yokoyama WM, Diamond MS, Virgin HW, Rice CM. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature. 2013 doi: 10.1038/nature12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ansari MA, Singh VV, Dutta S, Veettil MV, Dutta D, Chikoti L, Lu J, Everly D, Chandran B. Constitutive interferon-inducible protein 16-inflammasome activation during Epstein-Barr virus latency I, II, and III in B and epithelial cells. J Virol. 2013;87:8606–8623. doi: 10.1128/JVI.00805-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bowie AG, Unterholzner L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat Rev Immunol. 2008;8:911–922. doi: 10.1038/nri2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pezda AC, Penn A, Barton GM, Coscoy L. Suppression of TLR9 Immunostimulatory Motifs in the Genome of a Gammaherpesvirus. Journal of Immunology. 2011;187:887–896. doi: 10.4049/jimmunol.1003737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Orzalli MH, DeLuca NA, Knipe DM. HSV-1 ICP0 redistributes the nuclear IFI16 pathogen sensor and promotes its degradation. PNAS. 2012;109:E3008–E3017. doi: 10.1073/pnas.1211302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Verpooten D, Ma Y, Hou S, Yan Z, He B. Control of TANK-binding kinase 1-mediated signaling by the gamma(1)34.5 protein of herpes simplex virus 1. J Biol Chem. 2009;284:1097–1105. doi: 10.1074/jbc.M805905200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu FX, King SM, Smith EJ, Levy DE, Yuan Y. A Kaposi's sarcoma-associated herpesviral protein inhibits virus-mediated induction of type I interferon by blocking IRF-7 phosphorylation and nuclear accumulation. Proc Natl Acad Sci U S A. 2002;99:5573–5578. doi: 10.1073/pnas.082420599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kang HR, Cheong WC, Park JE, Ryu S, Cho HJ, Youn H, Ahn JH, Song MJ. Murine Gammaherpesvirus 68 Encoding Open Reading Frame 11 Targets TANK Binding Kinase 1 To Negatively Regulate the Host Type I Interferon Response. J Virol. 2014;88:6832–6846. doi: 10.1128/JVI.03460-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Inn KS, Lee SH, Rathbun JY, Wong LY, Toth Z, Machida K, Ou JH, Jung JU. Inhibition of RIG-I-mediated signaling by Kaposi's sarcoma-associated herpesvirus-encoded deubiquitinase ORF64. J Virol. 2011;85:10899–10904. doi: 10.1128/JVI.00690-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gent MV, Braem SGE, de Jong A, Delagic N, Peeters JGC, Boer IGJ, Moynagh PN, Kremmer E, Wiertz EJ, Ovaa H, Griffin BD, Ressing ME. Epstein-Barr virus large tegument protein BPLF1 contributes to innate immune evasion through interference with Toll-like receptor signaling. PLoS Pathog. 2014;10:e1003960. doi: 10.1371/journal.ppat.1003960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang S, Wang K, Li J, Zheng C. Herpes simplex virus 1 ubiquitin-specific protease UL36 inhibits beta interferon production by deubiquitinating TRAF3. J Virol. 2013;87:11851–11860. doi: 10.1128/JVI.01211-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barton E, Mandal P, Speck SH. Pathogenesis and host control of gammaherpesviruses: lessons from the mouse. Annu Rev Immunol. 2011;29:351–397. doi: 10.1146/annurev-immunol-072710-081639. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.