

Abstract
The neurotransmitter acetylcholine (ACh) can regulate neuronal excitability by acting on the cys-loop cation-conducting ligand-gated nicotinic ACh receptor channels (nAChRs). These receptors are widely distributed throughout the central nervous system, being expressed on neurons and non-neuronal cells, where they participate in a variety of physiological responses such as anxiety, the central processing of pain, food intake, nicotine seeking behavior, and cognitive functions. In the mammalian brain, nine different subunits have been found thus far, which assemble into pentameric complexes with much subunit diversity; however the α7 and α4β2 subtypes predominate in the CNS. Neuronal nAChR dysfunction is involved in the pathophysiology of many neurological disorders. Here we will briefly discuss the functional makeup and expression of the nAChRs in the mammalian brain, and their role as targets in neurodegenerative diseases (in particular Alzheimer’s disease), neurodevelopmental disorders (in particular autism and schizophrenia), and neuropathic pain.
Keywords: α7 receptor, α4β2 receptor, nAChR, cys-loop receptor
FUNCTIONAL ROLE of NICOTINIC ACh RECEPTORS IN THE BRAIN
The nicotinic acetylcholine receptors (nAChRs) belong to the superfamily of cys-loop receptors (Fig. 1), which also includes the serotonin 5-HT3, GABAA and GABAC, and glycine receptors, and participate in a variety of physiological functions, including the regulation of neuronal excitability and neurotransmitter release [1–4]. The nAChRs are widely distributed throughout the peripheral and central nervous systems, as well as the immune system and various peripheral tissues [5–9]. In the mammalian brain, nine different nAChR subunits are known to exist (α2–7 and β2–4), which combine as either homo- or heteromeric complexes into multiple functionally diverse pentameric receptors [3, 10, 11]. The predominant subtypes functionally expressed in the brain are categorized as α7* subunit-containing receptors (either homo- or heteromeric), or those composed of both α and β subunits, including the α4β2* and α3β4* subtypes [12–17]; the * denotes that these nAChRs can contain other α and β subunits as well.
Figure 1. Nicotinic ACh receptor channel; basic structure and functional properties.
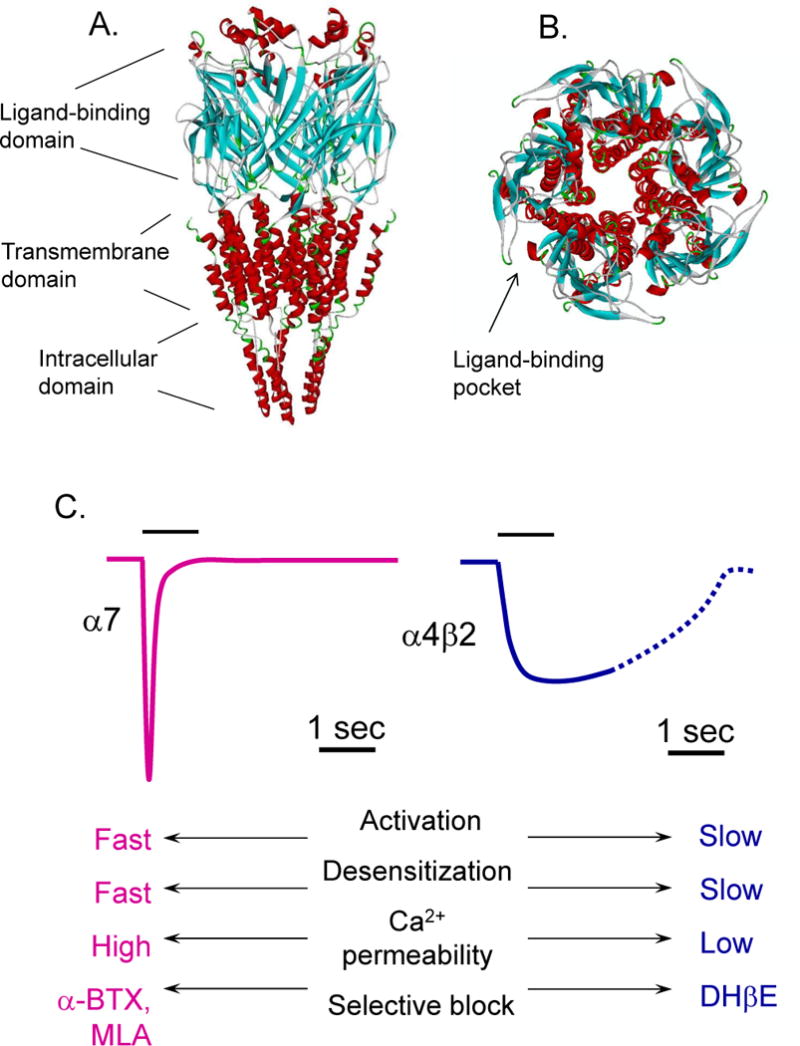
Molecular model of the rat α7 nAChR with ligand-binding from a side view showing the ligand-binding, transmembrane, and intracellular domain (A), and a top down view (B) showing the pentameric nature of the receptor with the ligand-binding pocket at the interface between two subunits. C, The basic functional and pharmacological properties of the α7 and α4β2 nAChR subtypes.
The α4β2* receptor subtype was initially found to be the major nAChR subtype in the brain (where it comprises 90% of the high affinity nicotine binding sites [16, 18]), while the α3β4* nAChR is known primarily as a ganglionic receptor in the peripheral nervous system. The α3β4* nAChR is also expressed in a variety of brain areas, including (but not limited to) the interpeduncular nucleus and medial habenula [16, 17]. In addition, the α2, α5, α6 and β3 subunits participate in nAChRs expressed in various brain regions, although they represent a minority population of the total.
The α7* nAChR subunit is a particularly intriguing subunit as these receptors are expressed on a variety of cell types in the periphery, including immune cells [19–23] and neurons [24], as well as in brain regions that underlie learning and memory [25, 26]. Furthermore, these receptors are highly permeable to calcium, implicating them as significant modulators of intracellular signaling and neurotransmitter release from neurons. In the brain, the α7* receptors are expressed on both neurons and non-neuronal cells [20], including (but not limited to) astrocytes, microglia, oligodendrocyte precursor cells, endothelial cells, and chondroitin sulfate proteoglycan NG2-expressing (NG2) cells [21–23, 27–30]. Expression of α7* receptors in these non-neuronal cells suggests a possible role in brain innate immunity, inflammation, and neuroprotection [31, 32]. Immune cell expression of α7* receptors has been shown to modulate inflammatory responses by regulating the production of inflammatory cytokines and chemokines [33, 34].
While the α7* nAChR was initially thought to be functionally expressed as homomeric receptors, it has recently been shown to be capable of co-assembling with other subunits, which provides an explanation for the incongruent properties of in situ α7-containing receptors and in vitro expressed homomeric α7 receptors [35–39]. Initially, we found that α7 and β2 subunits co-assembled in vitro [14, 15, 40]; subsequently it was found that basal forebrain cholinergic neurons express functional α7β2 receptors with an enhanced sensitivity to the amyloid-β (Aβ) peptide associated with Alzheimer’s disease [41] (see below).
In the brain, the nAChRs are expressed and function at the synapse (both pre- and postsynaptically) as well as extrasynaptically [13, 16, 42–44], and participate in nAChR-mediated postsynaptic responses [45]. While α7* nAChR-mediated synaptic responses have previously been observed in hippocampal interneurons utilizing electrophysiological techniques [46, 47], optogenetic stimulation of direct cholinergic inputs to the hippocampus revealed new evidence for α4β2* nAChR-mediated postsynaptic responses from interneurons [48] and pyramidal cells [45]. Various subtypes of nAChRs have been shown to modulate synaptic transmission in other areas of the brain, including (but not limited to) the visual cortex, cortical interneurons, supraoptic nuclei, and thalamic nuclei [49–52]. In addition to presynaptic and postsynaptic locations in which they function to promote neurotransmitter release and excitability, nAChRs are also functionally expressed extrasynaptically where they participate in non-synaptic communication [4, 53]. Although most cholinergic presynaptic neurotransmitter terminals do not make direct postsynaptic contacts, they are able to release ACh. However the precise role that extrasynaptic nAChRs play in brain circuit excitability and plasticity remains to be determined.
Several layers of complexity contribute to the challenge of deciphering the role of nAChRs in brain circuit excitability and plasticity, as well as in disorders and diseases of the brain. Subunit composition directly contributes to nAChR channel permeability and kinetics, whereas subcellular localization within the neural network determines the precise contribution of a given nAChR population in a spatial- and time-dependent manner. The unique anatomical distribution of each nAChR subtype within the brain implicates particular ones in brain disease (Table 1). As such, the α7* and non-α7 nAChR subtypes are active targets for therapeutic development in neurodegenerative disease, neurodevelopmental disorders, and chronic pain. Below we will discuss these receptors as they are understood in the etiology and treatment of Alzheimer’s disease, autism, schizophrenia, and neuropathic pain.
Table 1. Summary table for nAChRs as targets in CNS diseases.
Categorized by the CNS diseases discussed in the present review, brain regions implicated in each disease, and the nAChR subunits implicated in each disease as well as potential pharmacological interventions currently identified using preclinical and clinical trial design.
| CNS Disease | Brain Regions Implicated | nAChR Subunits Implicated | Potential Pharmacological Strategies |
|---|---|---|---|
| Alzheimer’s disease | Basal forebrain cholinergic nuclei, hippocampus, cerebral cortex | α7, α4, β2 | Agonists, partial agonists and PAMs for α7* nAChRs, α4β2* agonists |
| Autism, Autism Spectrum Disorders | Cerebral cortex, hippocampus, amygdala, basal ganglia, cerebellum | α3, α4, β2 | agonists or PAMs for α4β2* nAChRs |
| Schizophrenia | Hippocampus, cortex | α7, α4β2 | agonists, partial agonists, or PAMs for α7* nAChRs or partial agonists for α4β2* nAChRs |
| Pain | Dorsal root ganglia, Brainstem rostral ventromedial medulla, midbrain periaqueductal gray, spinal cord dorsal horn | α3, α4, α5, α7, α9, α10, β2 | agonists or PAMs for α4β2* nAChRs or type II or PAMs for α7* nAChRs |
ALZHEIMER’S DISEASE
Neuronal nAChR dysfunction is involved in the pathophysiology of various neurodegenerative diseases, including Alzheimer’s (AD) and Parkinson’s (PD) diseases. Here we will focus solely on AD since there are excellent recent reviews discussing the complexity between nAChR distribution within the nigro-striatal pathway, how nAChR functional contributions to this network, and PD pathophysiology [54].
Prevalence and causes
Alzheimer’s disease (AD) is a neurodegenerative disease characterized by memory and cognitive loss, and represents the leading cause of dementia in people aged >60 years [55]. Sporadic, or non-inherited, forms of AD comprise the majority of diagnosed cases of the disease. However, identification of the genetic mutations that underlie the familial, or inherited, forms of AD have provided enormous insight into fundamental mechanisms, namely that the trigger for synaptic and neural network dysfunction is the aberrant accumulation of misfolded amyloid-β (Aβ).
Aβ is formed through proteolytic cleavage of its precursor protein, a type 1 membrane protein called amyloid precursor protein (APP). Aβ peptide length can vary from 38–43 amino acids in length depending on the γ-secretase C-terminal cleavage site and N-terminal posttranslational truncation events (Fig. 2A).
Figure 2.
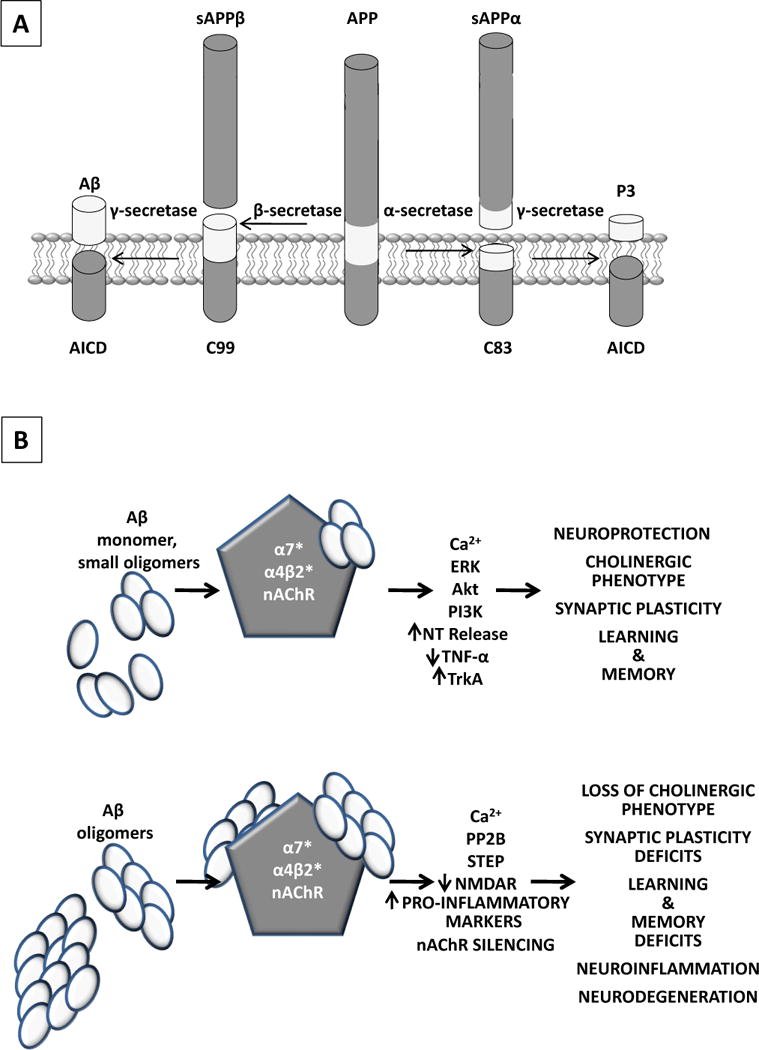
A, Aβ is formed through proteolytic cleavage of its precursor protein, a type 1 membrane protein called amyloid precursor protein (APP). APP cleavage by β- and γ-secretases lead to the formation of the N- and C-terminal residues of Aβ, respectively, in addition to soluble APP (sAPPβ) and a C-terminal fragment (CTF) of 99 amino acids (C99). If APP is cleaved by the α-secretase, then Aβ formation is precluded as this enzyme cleaves APP within the Aβ sequence to generate sAPPα and CTF83. When cleaved by γ-secretase, CTFs generate the AICD (APP intracellular domain) and either P3 or Aβ resulting from α- and β-secretase cleavage, respectively. B, As Aβ accumulates one can postulate that at lower concentration and smaller oligomer aggregates (upper panel), transient nAChR activation may occur depending on the nAChR subunit composition (Table 2). If activation ensues, it can lead to membrane depolarization, increased intracellular/presynaptic calcium and synaptic transmission, activation of kinases important for synaptic plasticity, learning and memory as well as the induction of genes and proteins necessary for the cholinergic phenotype and neuroprotection. As disease progresses and Aβ concentration and oligomer status increase (lower panel), nAChR function is lost either through loss of receptor protein (α4β2*) or function (α7*) leading to synaptic dysfunction and loss of the cholinergic phenotype, learning and memory deficits, increased inflammatory status and progressive neurodegeneration.
The nAChRs are integral to early AD cholinergic hypofunction. In addition to the complex biochemical processes involved in neuronal degeneration due to misfolded Aβ accumulation and development of neurofibrillary tangles comprised of misfolded tau, the cholinergic deficit due to the loss of basal forebrain cholinergic neurons and production of ACh significantly contributes to early AD dementia [56]. The cholinergic deficit is evident by reduced choline acetyltransferase (ChAT) protein and activity, and vesicular ACh transporter (vAChT) protein. In presynaptic nerve terminals, ChAT synthesizes ACh, and the vAChT is responsible for the transport of ACh into synaptic vesicles for storage until exocytotic release into the synapse [57]. Acetylcholinesterases (AChE), resident within the synaptic cleft, hydrolyse ACh to rapidly terminate the availability of neurotransmitter and nAChR activation. Acetylcholinesterase inhibitors (Aricept, Exelon, Razadyne, Cognex) were the first FDA-approved therapies to treat the cognitive symptoms (memory loss, confusion, and problems with thinking and reasoning) of early AD that are thought to be due in part to the cholinergic deficit and are a first line of therapy today.
Brain regions associated with attention, spatial, and episodic memory lose cholinergic innervation in early AD; thus, cholinergic hypofunction is most evident in the neocortex and temporal lobes inclusive of the hippocampus [58]. The current model for the cholinergic deficit in AD posits that the septo-hippocampal pathway preferentially accumulates misfolded Aβ and tau, which leads to loss of the cholinergic phenotype, e.g., loss of cholinergic markers (ChAT, VAChT) and eventually cholinergic neurons from the basal forebrain nuclei through altered nAChR function and disruption of the nerve growth factor (NGF) trophic support system [59]. A compelling overlap exists between NGF-mediated signaling and the α7* nAChR subtype as α7 nAChR activation also promotes the cholinergic phenotype [31]; thus, loss of α7* nAChR function in particular likely contributes to early AD cholinergic hypofunction and cognitive deficits.
As briefly discussed above, nAChRs (primarily α4β2* and α7* subtypes) are expressed within the cholinergic forebrain nuclei, as well as at pre- and postsynaptic locations within their projection areas. For example, in the hippocampus, α4β2- and α7-containing nAChRs have been localized presynaptically and somato-dendritically on glutamatergic principal cells that drive synaptic plasticity; however, these nAChRs are also enriched on GABAergic interneurons that play a significant role in modulating principal cell activity [60]. Furthermore, nAChR subtypes within the basal forebrain are important players in modulating synaptic transmission and plasticity, learning and memory [45, 61].
Distinct nAChR subtypes are differentially affected in AD. The observation that Aβ preferentially accumulates in brain regions that are also enriched for α4β2* and α7* nAChRs may provide an important clue for the selective vulnerability of the hippocampus and neocortex to Aβ toxicity given the high affinity interaction between Aβ and these nAChRs [62, 63]. Importantly, α7* nAChRs exhibit an exceptionally high Aβ affinity (picomolar range) [64], suggesting a physiological interaction that may influence synaptic transmission and plasticity [65–67], as well as contribute to Aβ-mediated synaptic neural network dysfunction (Fig. 2B) [66, 68]. While the extant literature appears contradictory, if one considers the stunning complexity of nAChR subunit stoichiometry, brain region, and subcellular localization, one must acknowledge that the most parsimonious interpretation of nAChR-Aβ experimental results is that Aβ can either activate or antagonize α4β2* and α7* nAChRs, depending on the anatomical microenvironment as well as the concentration, length, and conformation of Aβ peptides[41, 69–76]. Given that in vivo Aβ constituents and conformations will be highly heterogeneous, while concentration will increase with disease state, it is likely that nAChR responses to the Aβ microenvironment will vary and be in constant flux. The next challenge is to decipher these dynamics to refine therapeutic strategies.
In advanced AD, it is unequivocal that muscarinic AChR levels remain relatively intact [77–79], and upwards of 50% of α4β2* nAChR binding sites are lost from neocortex and hippocampus compared to unaffected humans at similar age, and α7* nAChRs remain relatively stable in neocortex, although some reports vary [80, 81]. The status of nAChRs in mild cognitive impairment (MCI) due to probable AD [82] reflects a different dynamic in that ChAT activity and α7* nAChR expression levels have been reported to be elevated in the hippocampus and frontal neocortex [83–85]. In fact, individuals’ levels inversely correlated with neuropsychological tests that are used to help diagnose AD (e.g., Global Cognitive Score and Mini-Mental State Examination) [85], suggesting that α7* nAChR expression in early AD is a reflection of cognitive reserve; (a term that refers to superior memory function for the stage of AD pathology seen with currently accepted biomarkers for AD staging [86, 87]).
With few exceptions [76], α4β2* nAChRs are antagonized by Aβ [41] and lost in early AD, whereas α7* nAChR-Aβ interaction may result in transient receptor activation [88, 89] and downstream signal transduction cascades that promote neuronal survival and function [67, 90] (however see [91]); a potential explanation for recent studies reporting that α7* nAChRs are preserved in early AD. However as Aβ concentration increases, receptor desensitization and functional down-regulation likely ensues with concomitant receptor upregulation to transiently repopulate the functional receptor pool [31, 62, 92]. This model is supported by reports that α7* nAChRs are upregulated in several AD patient cell types, including astrocytes and neurons [93–96], as well as in AD animal models [97], and neuronal cell cultures chronically exposed to Aβ [98]. Furthermore, the majority of α7* nAChR protein in AD brain is associated with Aβ [99, 100], which upon dissociation, can resurrect α7* nAChR function. While this suggests a potential therapeutic intervention, a recent in vitro study indicates that neuronal hyperexcitability may be an unintended consequence of therapeutic interventions that reverse neuronal Aβ-α7 nAChR interaction [98].
An additional level of complexity for the role of α7* nAChRs in the pathophysiology of AD stems from work focused on these receptors expressed by astrocytes. It is now recognized that astrocytes express α7* nAChRs [21, 22, 27] and participate in synaptic communication through intimate interactions with neurons [101–103]. A principal mechanism is through the release of gliotransmitters, such as glutamate, in response to astrocytic calcium elevations that can activate neuronal glutamate receptors. Recently, two independent studies [66, 68] demonstrated that biologically-relevant concentrations of Aβ elicited α7* nAChR-dependent calcium elevations in astrocytes and induced glutamate gliotransmission to activate neuronal glutamate receptors. Furthermore in AD mouse models for Aβ over-production and accumulation, tonic glutamate receptor [68] and spontaneous astrocytic calcium elevations [66] were of higher frequency compared to controls; observations that laid the foundation for the newly explored hypothesis that a significant aspect of AD pathophysiology is epileptiform dysfunction [104, 105].
The cholinergic deficit in early AD has led to the development of drugs able to prevent ACh hydrolysis (e.g. AChE inhibitors). While these drugs do not change the course of the disease, AChE inhibitors improve memory and other cognitive functions throughout most of the duration of the disease. The pharmacological activity of these drugs suggests an effect beyond the mere increase of ACh levels; e.g. galantamine (Razadyne) has been shown to act also as a positive allosteric modulator (PAM) on α7* and α4β2* nAChRs [106, 107], and both donepezil (Aricept) and galantamine increase nAChR density [108]. Nonetheless, long-term clinical assessments indicate that the main effect of AChE drugs is symptomatic treatment with limited disease modifying actions [109].
The α4β2* and α7* nAChRs within the basal forebrain cholinergic system are important for the types of cognitive performance that are impaired in early AD. Thus, several subtype-selective agonists and partial agonists that target α4β2* as well as α7* nAChRs have been tested [110]. While study design and placebo effects may account for some of the variability, it is now evident that many AD clinical trials likely ‘fail’ due to enrollment of patients at advanced disease stages. There is an emerging consensus that the best strategy for AD clinical efficacy is to treat people during the earliest stages of disease [111]; e.g., prodromal Alzheimer’s or MCI due to AD [112]. The_current challenge is to reliably diagnose at such early stages using a combination of biomarkers, brain imaging, and cognitive testing [86, 113]. Currently in trial development (clinicaltrials.gov) targeting nAChRs for mild to moderate AD is EVP-6124 (FORUM Pharmaceuticals), also known as MT-4666 (Mitsubishi Tanabe Pharma Corporation), an α7 nAChR partial agonist, varenicline (Pfizer), an α4β2* nAChR partial agonist, and AZD-3480 (AstraZeneca), a partial agonist for α4β2 and α2β2 nAChRs [114]. Nicotine and acetylcholinesterase inhibitors (e.g. galantamine and donepezil) continue to be evaluated for efficacy in AD as well. Furthermore, since α4β2* nAChRs are lost early in AD, α4β2* nAChR single photon emission computed tomography (SPECT) and positron emission tomography (PET) radioligands are being developed and tested in clinical trials as potential diagnostic tools and biomarkers for AD staging. For example [123I]5-I-A-85380 (sponsored by the National Institutes of Health) is being tested for use in SPECT and [18F]XTRA (Johns Hopkins University) is currently being tested for use in PET.
The continued design and synthesis of nAChR ligands with the desired pharmacokinetics and pharmacodynamics to target the desired receptor population, in combination with continued elucidation of the disease-stage properties of nAChRs, may ultimately achieve the goal of developing an interventional tool to combat the loss of cholinergic basal forebrain connectivity in this most prevalent of the devastating neurodegenerative disorders.
NEURODEVELOPMENTAL DISORDERS
The nAChRs are widely expressed on both differentiated and undifferentiated CNS cells, and are known to influence development and modulate multiple pathways of neurogenesis during brain development [115, 116]. Here we discuss the role of nAChRs in two of the most common neurodevelopmental disorders, autism and schizophrenia.
Autism
Autism is a neurodevelopmental disorder, also known as autism spectrum disorders (ASD), which is characterized by deficits in socialization and a lack of verbal as well as non-verbal communication with pronounced cognitive impairment [117, 118]. Neuropathological data suggests that abnormalities in the cholinergic system may be related to autism [119]. In a study carried out on adult post-mortem autistic brains using quantitative RT-PCR for measuring mRNA expression together with protein level expression and specific radioligand receptor binding, it was found that there was a decrease of 40–50% in the expression of the α3, α4, and β2 nAChR subunits in the cerebral cortex, while there was no change in the expression of the α7 subunit [120]. The reduced gene expression of the α4 and β2 receptor subunits in the cerebral cortex is a major feature of the neurochemical pathology of ASD [120]. If there is a loss of cholinergic tone in the autistic brain, then nAChR ligands that restore or improve cholinergic transmission may be used to compensate for such loss [121, 122]. Therefore agonists and PAMs for α4β2* nAChRs may be employed to compensate for the loss of nAChR function in the autistic brain.
In contrast to this, it has also been suggested that nicotinic cholinergic antagonism may provide symptomatic relief in ASD [123]. This was based primarily on the low prevalence of smoking in the ASD population [124, 125], although this may due to the loss of expression of the α4β2* nAChRs early in development and presumably loss of sensitivity to the rewarding properties of nicotine since the α4β2* receptors are primarily responsible for the addictive properties of nicotine [126, 127]. In support of this notion, in a placebo-controlled pilot study with a nonselective and noncompetitive antagonist of the nAChRs, mecamylamine, it was found that while this drug was well tolerated, it lacked efficacy in treating autistic symptoms [128].
Recently it has been reported that AChE inhibitors (e.g. donepezil and galantamine), which, in general, increase cholinergic tone at the synapse [129], cause a pronounced improvement in typical symptoms associated with autism (e.g. verbal communication, eye to eye contact, and emotional responsiveness) in a single case report [130]. Similarly in a randomized, double-blind, placebo-controlled clinical trial, galantamine was shown to be safe and relatively effective in alleviating some autism-related symptoms (e.g. irritability and lethargy/social withdrawal) [131]. The lack of efficacy of the nAChR antagonist mecamylamine, combined with the effectiveness of galantamine and donepezil, suggest that nAChR agonists and PAMs (rather than antagonists) are more effective in providing symptomatic relief in autism and ASD. While further studies are needed to investigate and develop nAChR drugs as a treatment option for symptoms related to autism and ASD, the FDA-approved AChE inhibitors may provide symptomatic relief in the interim.
Schizophrenia
Schizophrenia is a neurodevelopmental disorder that is characterized by cognitive deficits and disordered thoughts that manifest as hallucinations, delusions, and paranoia. Schizophrenia has both genetic and environmental etiologies, and has been previously genetically linked to dysfunction in the hippocampal nAChR system [132]. In contrast to ASD, the schizophrenia patient population exhibits a much higher prevalence for smoking than the general population which some consider to be a form of self-medication as it may normalize some of the cognitive and sensory deficits [133, 134]. Nicotine exposure (either through smoking, nicotine gum, nasal spray, or the patch) appears to improve or normalize sensory deficits in schizophrenia [134]; however due to the adverse health effects and toxicity of nicotine, other ligands acting on nAChRs may prove useful as a treatment option for symptoms related to schizophrenia.
Indeed the nAChR partial agonist, varenicline, improves cognition in schizophrenic patients [135, 136]. For example in a randomized, double-blind, placebo-controlled clinical trial for patients diagnosed with schizophrenia, varenicline improved some aspects of cognitive function (e.g. Digital Symbol Substitution and the Wisconsin Card Sorting tests) compared to controls [137]. However it is unclear which nAChR subtypes may be involved since it is a partial agonist for the a4β2 nAChRs and a full agonist for the α7 nAChRs [138].
A robust feature of the schizophrenic brain is a decrease in the number of α7* nAChRs (particularly in the hippocampus) [132]. In fact, a direct genetic linkage to the α7 subunit was first reported in 1997 where it was found that a polymorphism existed at chromosome site 15q13–14, the site of the α7 nAChR subunit gene [139, 140]. In support of a mechanistic role for α7* nAChRs in schizophrenia, studies on auditory gating (investigated using the P50 auditory-evoked response), in which deficits are known to be associated with schizophrenia, have been done. The systemic administration of the type II α7 nAChR PAMs (PNU-120596 or JNJ-1930942) improved auditory gating deficits caused by amphetamine or MK-801 in rodent models that reflect circuit level disturbances associated with schizophrenia [141–143]. Furthermore several clinical trials support the use of α7 nAChR agonists to treat cognitive deficits in schizophrenia. For example, cognitive and/or sensory deficits were improved with the α7 partial agonists GTS-21[144], DMXB-A [145], or EVP-6124 [146] in schizophrenia cohorts. Additionally in a randomized, double-blind, placebo-controlled study of schizophrenic patients, the novel and selective α7 nAChR agonist, EVP-6124, provided significant improvement in a number of areas of cognitive function that are compromised in schizophrenia [146].
At the moment, α7* nAChR agonists and PAMs appear to be the most effective hope for the future in the treatment of schizophrenia. What is needed next is a determination of the mechanism of synaptic failure in schizophrenia and the potential cellular mechanisms of the positive cognitive actions of these selective α7* nAChR ligands in these patients. This information will provide further insight into the development of therapeutic treatments for the cognitive impairments associated with schizophrenia. Along these lines, we recently found that presynaptic α7* nAChRs, via a mechanism involving PKA, enhanced hippocampal CA3 mossy fiber glutamatergic transmission [147]. These types of studies may help to provide a mechanistic explanation for the hypothesis that nicotine enhances the mossy fiber glutamatergic transmission in the hippocampal CA3 region to compensate for a loss of synaptic connection in schizophrenic patients [148].
PAIN
Pain is an unpleasant sensory feeling that includes neuropathic, nociceptive and psychogenic pain [149]. If the pain persists for longer than a few months, the condition is said to be chronic (as opposed to acute) pain [150]. The most common reason for neuropathic pain is a disruption in the normal functioning of the somatosensory system [151]. The nAChRs have been known for some time to be involved in the process of mediating pain, and as such the analgesic effects of nAChR agonists as well as positive allosteric modulators (PAMs) have long been investigated [152, 153]. The microinjection of nicotine into different regions of the brainstem produced an antinociceptive effect [154]. In rats, the antinociception induced by nicotine was further shown to be inhibited by the administration of the general nAChR antagonist mecamylamine [155]. Apart from nicotine, the frog skin-derived alkaloid epibatidine, which acts as a non-selective agonist for nAChRs, was shown to have analgesic properties [156, 157]. When epibatidine and morphine were tested in the mouse straub tail response, both compounds were able to reduce pain [158, 159]. As expected, the analgesic effect of morphine was blocked by naloxone, but not that of epibatidine [158, 160]. Upon further investigation it was found that while epibatidine caused anti-nociception in animal models of pain, it also produced several major adverse effects like dose-dependent decreases in locomotor activity, and a similar decrease in body temperature [156]. Epibatidine is a non-selective agonist since it activates multiple subtypes of the neuronal nAChRs, as well as the neuromuscular receptor [161], which accounts for its various adverse effects [162], limiting its use as a therapeutic.
Based upon these observations, the research focus has since shifted to develop compounds that would selectively target specific neuronal nAChR subtypes with the purpose of maintaining the analgesic effect while reducing or eliminating the adverse effects that are seen with non-selective agonists such as epibatidine. The α4β2 subtype-selective ligands were the first to be developed. ABT-594, which is a structural analogue of epibatidine, was shown to be a potent full agonist for the α4β2 nAChRs with a strong analgesic effect [158, 163, 164]. In various animal models of pain, the analgesic activity of ABT-594 was inhibited by the administration of the general antagonist mecamylamine [162, 165]. At lower doses, ABT-594 does not induce any significant analgesic effects, while at higher doses (150 to 300 mg), the analgesic effects of ABT-594 were accompanied by significant adverse effects like nausea, dizziness, and vomiting [166], indicating that ABT-594 has a narrow therapeutic window and may act upon peripheral nAChRs at higher doses. Subsequently other selective agonists for the α4β2 nAChRs (e.g. TC-2696 and TC-6499) were developed [167]. While both TC-2696 and TC-6499 showed significant analgesic effects in animal models for neuropathic pain, they were not developed further because both had a narrow therapeutic window (i.e. the difference in doses at which they produce analgesic effects and adverse effects was small) [167, 168], similar to that seen with ABT-594. Recently, another drug that targets the α4β2 nAChRs, A-366833, was been reported to cause significant analgesia in a variety of models for neuropathic pain [169, 170]. In this scenario, it is thought that the activation of presynaptic α4β2* receptors within the spinal cord that modulate GABA and glycinergic neurons may lead to an inhibitory effect on nociception [153, 168, 171–175]. However, while the activation of α4β2* receptors may be required, it may not be sufficient to induce an analgesic effect; the activation of other subtypes of nAChRs, specifically the α3-containing nAChRs, may also be required [168, 176].
In addition to α4β2* and α3-containing nAChRs, compounds that act on the α7 nAChRs have also been studied for their effects on pain. Choline, a by-product of ACh enzymatic degradation by AChE and a selective agonist for the α7 receptors, has analgesic effects in rodent models of pain [177, 178]. However other agonists that target α7 nAChRs, such as SSR-180711 and tropisetron, fail to induce analgesia when tested in the formalin model for pain [168], suggesting that targeting α7* nAChRs for pain relief is modality selective. Based on these contradictory results, further work needs to be done to clearly define the role of α7 nAChR agonists in analgesia.
A variety of α4β2 and α7 nAChR PAMs have been evaluated in preclinical models for pain [122, 179–181]. For instance the α7 nAChR PAM PNU-120596 caused a significant reduction in mechanical hyperalgesia in rats [182]. Additionally, PNU-120596 is able to decrease formalin-induced pain by itself, and in combination with the α7 nAChR agonists choline, nicotine and PHA-543613 [183]. PNU-120596 is also able to reduce edema induced by a hind paw infusion of carrageenan in mice [184]. Another α7 nAChR PAM, NS-1738, has also been tested for its effects on pain. Both NS-1738 and PNU-120596 were able to reduce heat-induced hyperalgesia [184]. In a chronic constriction injury model for neuropathic pain, an anti-hyperalgesic and anti-allodynic effect of PNU-120596 was observed, however NS-1738 did not have any effect in the same study [184]. One possible mechanism for this may be the ability of PNU-120596 to enhance α7 nAChR activation over a certain threshold, which would indirectly stimulate inhibitory neurons located in the neural circuits for neuropathic pain.
The α4β2 nAChR PAM, NS-9283, administered alone did not affect mechanical allodynia in the spinal nerve ligation test (a model of neuropathic pain), nor on nociception and inflammation in various rodent models of pain [185, 186]. However, when NS-9283 was coadministered with the α4β2 receptor agonist ABT-594, the presence of the α4β2 nAChR PAM increased the anti-allodynic effects of the agonist alone [185]. In addition NS-9283 was able to strengthen the antinociceptive actions of the α4β2 nAChR partial agonist NS-3965 [187]. Therefore combining an α4β2 receptor PAM with a nAChR full agonist may be a viable strategy in therapeutic drug development for treatment of acute as well as chronic pain to avoid the major adverse effects commonly seen with a nAChR agonist alone.
Besides the α4β2* and α7* receptors, other nAChRs are also known to be involved in the processing of pain [153]. For example, it is emerging that the α5 nAChR subunit plays a significant role in nociception. The α5 subunit can participate in α4β2*, α3β2* or α3β4* nAChRs [188]. The anti-nociception effect of nicotine in α5 subunit knockout mice was reduced compared to wildtype mice, indicating that the α5 subunit may play an important role in the processing of pain [188]. In addition there may be a role for the α9/α10 nAChRs in pain. Dorsal root ganglion neurons express the α9/α10 nAChR subtypes [189].
There is accumulating evidence, based upon pharmacological testing and behavioral studies, which strongly implicates nAChRs in the process of nociception and the treatment of pain. However given the complexity of the nAChR subtypes expressed within the CNS and PNS, as well as the complexity and plasticity of the neuronal circuits involved in the processing of pain, developing therapeutic strategies that target nAChRs to treat acute and chronic pain poses significant challenges.
CONCLUDING REMARKS
The distribution of nAChRs in the nervous system is extensive, yet discrete, and this has made them an active target of drug development; historically for treating neurodegenerative diseases such as AD. However, the nAChRs are increasingly appreciated for their roles in neurodevelopment and sensory processing, and thus have been identified as therapeutic targets in autism, schizophrenia, and neuropathic pain. Unfortunately, no nAChR compounds have demonstrated disease-modifying properties for any of the disorders in which these receptors are implicated to date. Thus we contend that a new generation of nAChR ligands is needed for studies on their ability to treat neurodegenerative or neurodevelopmental disorders, as well as neuropathic pain.
What are the prospects for the future? Currently there are at least two main obstacles. First, more mechanistic details about the stoichiometry, expression, and developmental regulation of the various subtypes of nAChRs in the brain and nervous system are needed, both in neurons and non-neuronal cells, and how these receptors are regulating brain circuit function, excitability, plasticity, and development. It is only through this knowledge that we can know which nAChRs we need to target for the design of therapeutics in order to treat disorders and diseases linked with nAChRs. This will also help us to understand how we want to alter receptor function; e.g. in some instances we may need to enhance function (either competitively or via allosteric modulation), while in other instances we may need to block or reduce function (either short- or long-term). Great strides are now being made and will continue, in part through the use of optogenetics, in vivo physiological studies in live animals, and genetic methods to remove or modify specific nAChR subtypes from specific brain regions, cell types, and at specific developmental stages.
Second, a more targeted approach to develop therapeutics acting selectively and effectively on the various nAChR subtypes is needed. For example, more structural and biophysical information on the properties of the nAChRs and their binding site will help in the targeting of potential therapeutic drugs to various regions of the extracellular domain of the receptor (e.g. binding site, pore, or putative allosteric site). The various ACh-binding proteins (AChBPs), which are a family of pentameric and soluble protein analogous to the extracellular ligand-binding domain of nAChRs and have been crystalized with several nAChR ligands including varenicline [190], will certainly open new perspectives for the design of new drugs targeting nAChRs. Furthermore the recent determination of the crystal structures of the neurotransmitter-gated members of the cys-loop receptor family (e.g. the 5-HT3 and GABAA receptors; [191, 192]), and the ligand-binding domain of the α7 nAChR [193]), will not only enhance our understanding of the structure and possible function of mammalian cys-loop receptors, but will also further aid in the targeted development of new compounds that one day may be used to treat various disorders and diseases linked with nAChRs.
Table 2. In situ and heterologous expression studies have revealed varied nAChR responses to Aβ.
nAChR responses to Aβ appear to depend upon the species of receptor, subunit composition, have revealed receptor activation and receptor inhibition for the α7 nAChR subtype
| nAChR Subtype | Experimental Preparation | Type of Interaction | Downstream Consequences | Reference |
|---|---|---|---|---|
| human α7* | human brain: control & AD | co-localization & co-IP | n/a | Wang et al., 2000a |
| human α7* | human & rat cell lines | high-affinity binding | competitive binding: BTX vs. Aβ | Wang et al., 2000a, 2000b |
| rat α7 | Xenopus oocytes | receptor activation | Ca2+ influx, ERK activation | Dineley et al., 2001, 2002 |
| rat α7*, non-α7* | isolated presynaptic terminals: hippocampus, neocortex | receptor activation | increased presynaptic Ca2+ | Dougherty et al., 2003 |
| rat α4* | diagonal band nucleus | receptor activation | membrane depolarization, increased mEPSC frequency | Fu & Jhamandas, 2003; Chin et al., 2007 |
| mouse α7*, β2* | NG108-15 cells, isolated presynaptic terminals: hippocampus, neocortex | receptor activation | increased presynaptic Ca2+ | Khan & Nichols, 2007 |
| rat α7*, non-α7* | acute hippocampal slice, GABAergic interneurons | functional antagonism, reversible | decreased open po | Pettit et al., 2001 |
| rat α7β2* | medial septum/diagonal band cholinergic neurons | functional antagonism, reversible | noncompetitive (choline) | Liu et al., 2009 |
| rat α7* | cultured hippocampal neurons | functional antagonism, reversible | noncompetitive (ACh) | Liu et al., 2001 |
| human α7 | Xenopus oocytes | functional antagonism, reversible | noncompetitive (ACh) | Grassi et al., 2003; Pym et al., 2005 |
| rat α7 | Xenopus oocytes | no effect | n/a | Lamb et al., 2005 |
| human α4β2 | SH-EP1 cells | functional antagonism, reversible | noncompetitive (nicotine) | Wu et al., 2004 |
| human α4β2 | Xenopus oocytes | agonist potentiation (ACh) | membrane depolarization | Pym et al., 2005 |
| rat α4β2, α2β2, α4α5β2 | Xenopus oocytes | functional antagonism, reversible | n/a | Lamb et al., 2005 |
| human α3β4 | Xenopus oocytes | no effect | n/a | Pym et al., 2005 |
| mouse muscle γ- or ɛ-nAChR | BOSC 23 cells | functional antagonism, reversible | noncompetitive | Grassi et al., 2003 |
Neuronal nicotinic ACh receptor (nAChR) dysfunction is involved in the pathophysiology of many neurological disorders
nAChRs are increasingly appreciated for their roles in neurodevelopment and sensory processing
α4β2 and α7 nAChRs in the basal forebrain cholinergic system are important for cognitive performance
Acknowledgments
FUNDING
This work was supported by the Intramural Research Program, NIEHS/NIH to JLY and AAP, a kind gift to KTD from J&W Mohn, and an Independent Investigator Research Grant from the Alzheimer’s Association to KTD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yakel JL. Gating of nicotinic ACh receptors: latest insights into ligand binding and function. J Physiol. 2010;588:597–602. doi: 10.1113/jphysiol.2009.182691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gay EA, Yakel JL. Gating of nicotinic ACh receptors; new insights into structural transitions triggered by agonist binding that induce channel opening. J Physiol. 2007;584:727–733. doi: 10.1113/jphysiol.2007.142554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dani JA, Bertrand D. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu Rev Pharmacol Toxicol. 2007;47:699–729. doi: 10.1146/annurev.pharmtox.47.120505.105214. [DOI] [PubMed] [Google Scholar]
- 4.Albuquerque EX, et al. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiological reviews. 2009;89:73–120. doi: 10.1152/physrev.00015.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seguela P, et al. Molecular cloning, functional properties, and distribution of rat brain alpha 7: a nicotinic cation channel highly permeable to calcium. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rubboli F, et al. Distribution of nicotinic receptors in the human hippocampus and thalamus. The European journal of neuroscience. 1994;6:1596–1604. doi: 10.1111/j.1460-9568.1994.tb00550.x. [DOI] [PubMed] [Google Scholar]
- 7.Rubboli F, et al. Distribution of neuronal nicotinic receptor subunits in human brain. Neurochemistry international. 1994;25:69–71. doi: 10.1016/0197-0186(94)90055-8. [DOI] [PubMed] [Google Scholar]
- 8.Drago J, et al. Neuronal nicotinic receptors: insights gained from gene knockout and knockin mutant mice. Cellular and molecular life sciences : CMLS. 2003;60:1267–1280. doi: 10.1007/s00018-003-2259-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dominguez del Toro E, et al. Immunocytochemical localization of the alpha 7 subunit of the nicotinic acetylcholine receptor in the rat central nervous system. The Journal of comparative neurology. 1994;349:325–342. doi: 10.1002/cne.903490302. [DOI] [PubMed] [Google Scholar]
- 10.Yakel JL. Cholinergic receptors: functional role of nicotinic ACh receptors in brain circuits and disease. Pflugers Archiv : European journal of physiology. 2013;465:441–450. doi: 10.1007/s00424-012-1200-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hurst R, et al. Nicotinic acetylcholine receptors: From basic science to therapeutics. Pharmacol Ther. 2012 doi: 10.1016/j.pharmthera.2012.08.012. [DOI] [PubMed] [Google Scholar]
- 12.Jones S, et al. Nicotinic receptors in the brain: correlating physiology with function. Trends Neurosci. 1999;22:555–561. doi: 10.1016/s0166-2236(99)01471-x. [DOI] [PubMed] [Google Scholar]
- 13.Jones S, Yakel JL. Functional nicotinic ACh receptors on interneurones in the rat hippocampus. J Physiol. 1997;504(Pt 3):603–610. doi: 10.1111/j.1469-7793.1997.603bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khiroug SS, et al. Rat nicotinic ACh receptor alpha7 and beta2 subunits co-assemble to form functional heteromeric nicotinic receptor channels. J Physiol. 2002;540:425–434. doi: 10.1113/jphysiol.2001.013847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sudweeks SN, Yakel JL. Functional and molecular characterization of neuronal nicotinic ACh receptors in rat CA1 hippocampal neurons. J Physiol. 2000;527(Pt 3):515–528. doi: 10.1111/j.1469-7793.2000.00515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Colombo SF, et al. Biogenesis, trafficking and up-regulation of nicotinic ACh receptors. Biochem Pharmacol. 2013 doi: 10.1016/j.bcp.2013.06.023. [DOI] [PubMed] [Google Scholar]
- 17.Quick MW, et al. Alpha3beta4 subunit-containing nicotinic receptors dominate function in rat medial habenula neurons. Neuropharmacology. 1999;38:769–783. doi: 10.1016/s0028-3908(99)00024-6. [DOI] [PubMed] [Google Scholar]
- 18.Millar NS, Harkness PC. Assembly and trafficking of nicotinic acetylcholine receptors (Review) Molecular membrane biology. 2008;25:279–292. doi: 10.1080/09687680802035675. [DOI] [PubMed] [Google Scholar]
- 19.Wang H, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–388. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- 20.Wessler I, Kirkpatrick CJ. Acetylcholine beyond neurons: the non-neuronal cholinergic system in humans. Br J Pharmacol. 2008;154:1558–1571. doi: 10.1038/bjp.2008.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen JX, Yakel JL. Functional alpha7 nicotinic ACh receptors on astrocytes in rat hippocampal CA1 slices. J Mol Neurosci. 2012;48:14–21. doi: 10.1007/s12031-012-9719-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharma G, Vijayaraghavan S. Nicotinic cholinergic signaling in hippocampal astrocytes involves calcium-induced calcium release from intracellular stores. Proc Natl Acad Sci U S A. 2001;98:4148–4153. doi: 10.1073/pnas.071540198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shytle RD, et al. Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J Neurochem. 2004;89:337–343. doi: 10.1046/j.1471-4159.2004.02347.x. [DOI] [PubMed] [Google Scholar]
- 24.Si ML, Lee TJ. Alpha7-nicotinic acetylcholine receptors on cerebral perivascular sympathetic nerves mediate choline-induced nitrergic neurogenic vasodilation. Circulation research. 2002;91:62–69. doi: 10.1161/01.res.0000024417.79275.23. [DOI] [PubMed] [Google Scholar]
- 25.Levin ED. alpha7-Nicotinic receptors and cognition. Current drug targets. 2012;13:602–606. doi: 10.2174/138945012800398937. [DOI] [PubMed] [Google Scholar]
- 26.Leiser SC, et al. A cog in cognition: how the alpha 7 nicotinic acetylcholine receptor is geared towards improving cognitive deficits. Pharmacology & therapeutics. 2009;122:302–311. doi: 10.1016/j.pharmthera.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 27.Velez-Fort M, et al. Functional alpha 7-containing nicotinic receptors of NG2-expressing cells in the hippocampus. Glia. 2009;57:1104–1114. doi: 10.1002/glia.20834. [DOI] [PubMed] [Google Scholar]
- 28.De Simone R, et al. Activation of alpha7 nicotinic acetylcholine receptor by nicotine selectively up-regulates cyclooxygenase-2 and prostaglandin E2 in rat microglial cultures. Journal of neuroinflammation. 2005;2:4. doi: 10.1186/1742-2094-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suzuki T, et al. Microglial alpha7 nicotinic acetylcholine receptors drive a phospholipase C/IP3 pathway and modulate the cell activation toward a neuroprotective role. J Neurosci Res. 2006;83:1461–1470. doi: 10.1002/jnr.20850. [DOI] [PubMed] [Google Scholar]
- 30.Hawkins BT, et al. Modulation of cerebral microvascular permeability by endothelial nicotinic acetylcholine receptors. American journal of physiology. Heart and circulatory physiology. 2005;289:H212–219. doi: 10.1152/ajpheart.01210.2004. [DOI] [PubMed] [Google Scholar]
- 31.Hernandez CM, Dineley KT. alpha7 nicotinic acetylcholine receptors in Alzheimer’s disease: neuroprotective, neurotrophic or both? Curr Drug Targets. 2012;13:613–622. doi: 10.2174/138945012800398973. [DOI] [PubMed] [Google Scholar]
- 32.Shen JX, Yakel JL. Nicotinic acetylcholine receptor-mediated calcium signaling in the nervous system. Acta Pharmacol Sin. 2009;30:673–680. doi: 10.1038/aps.2009.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gahring LC, et al. Nicotinic receptor alpha7 expression identifies a novel hematopoietic progenitor lineage. PLoS One. 2013;8:e57481. doi: 10.1371/journal.pone.0057481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olofsson PS, et al. Rethinking inflammation: neural circuits in the regulation of immunity. Immunol Rev. 2012;248:188–204. doi: 10.1111/j.1600-065X.2012.01138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anand R, et al. Homomeric and native alpha 7 acetylcholine receptors exhibit remarkably similar but non-identical pharmacological properties, suggesting that the native receptor is a heteromeric protein complex. FEBS letters. 1993;327:241–246. doi: 10.1016/0014-5793(93)80177-v. [DOI] [PubMed] [Google Scholar]
- 36.Yu CR, Role LW. Functional contribution of the alpha7 subunit to multiple subtypes of nicotinic receptors in embryonic chick sympathetic neurones. J Physiol. 1998;509(Pt 3):651–665. doi: 10.1111/j.1469-7793.1998.651bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Girod R, et al. Heteromeric complexes of alpha 5 and/or alpha 7 subunits. Effects of calcium and potential role in nicotine-induced presynaptic facilitation. Ann N Y Acad Sci. 1999;868:578–590. doi: 10.1111/j.1749-6632.1999.tb11331.x. [DOI] [PubMed] [Google Scholar]
- 38.Shao Z, Yakel JL. Single channel properties of neuronal nicotinic ACh receptors in stratum radiatum interneurons of rat hippocampal slices. J Physiol. 2000;527(Pt 3):507–513. doi: 10.1111/j.1469-7793.2000.00507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palma E, et al. Nicotinic acetylcholine receptors assembled from the alpha7 and beta3 subunits. J Biol Chem. 1999;274:18335–18340. doi: 10.1074/jbc.274.26.18335. [DOI] [PubMed] [Google Scholar]
- 40.Murray TA, et al. alpha7beta2 nicotinic acetylcholine receptors assemble, function, and are activated primarily via their alpha7-alpha7 interfaces. Mol Pharmacol. 2012;81:175–188. doi: 10.1124/mol.111.074088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu Q, et al. A novel nicotinic acetylcholine receptor subtype in basal forebrain cholinergic neurons with high sensitivity to amyloid peptides. J Neurosci. 2009;29:918–929. doi: 10.1523/JNEUROSCI.3952-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wonnacott S. Presynaptic nicotinic ACh receptors. Trends Neurosci. 1997;20:92–98. doi: 10.1016/s0166-2236(96)10073-4. [DOI] [PubMed] [Google Scholar]
- 43.Gotti C, et al. Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol Sci. 2006;27:482–491. doi: 10.1016/j.tips.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 44.Jones S, et al. Nicotinic receptors in the brain: correlating physiology with function. Trends Neurosci. 1999;22:555–561. doi: 10.1016/s0166-2236(99)01471-x. [DOI] [PubMed] [Google Scholar]
- 45.Gu Z, Yakel JL. Timing-dependent septal cholinergic induction of dynamic hippocampal synaptic plasticity. Neuron. 2011;71:155–165. doi: 10.1016/j.neuron.2011.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alkondon M, et al. alpha-bungarotoxin- and methyllycaconitine-sensitive nicotinic receptors mediate fast synaptic transmission in interneurons of rat hippocampal slices. Brain Res. 1998;810:257–263. doi: 10.1016/s0006-8993(98)00880-4. [DOI] [PubMed] [Google Scholar]
- 47.Frazier CJ, et al. Synaptic potentials mediated via alpha-bungarotoxin-sensitive nicotinic acetylcholine receptors in rat hippocampal interneurons. J Neurosci. 1998;18:8228–8235. doi: 10.1523/JNEUROSCI.18-20-08228.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bell KA, et al. Nicotinic excitatory postsynaptic potentials in hippocampal CA1 interneurons are predominantly mediated by nicotinic receptors that contain alpha4 and beta2 subunits. Neuropharmacology. 2011;61:1379–1388. doi: 10.1016/j.neuropharm.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roerig B, et al. Fast synaptic signaling by nicotinic acetylcholine and serotonin 5-HT3 receptors in developing visual cortex. J Neurosci. 1997;17:8353–8362. doi: 10.1523/JNEUROSCI.17-21-08353.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun YG, et al. Biphasic cholinergic synaptic transmission controls action potential activity in thalamic reticular nucleus neurons. J Neurosci. 2013;33:2048–2059. doi: 10.1523/JNEUROSCI.3177-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hatton GI, Yang QZ. Synaptic potentials mediated by alpha 7 nicotinic acetylcholine receptors in supraoptic nucleus. J Neurosci. 2002;22:29–37. doi: 10.1523/JNEUROSCI.22-01-00029.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bennett C, et al. Mechanisms generating dual-component nicotinic EPSCs in cortical interneurons. J Neurosci. 2012;32:17287–17296. doi: 10.1523/JNEUROSCI.3565-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lendvai B, Vizi ES. Nonsynaptic chemical transmission through nicotinic acetylcholine receptors. Physiological reviews. 2008;88:333–349. doi: 10.1152/physrev.00040.2006. [DOI] [PubMed] [Google Scholar]
- 54.Quik M, Wonnacott S. alpha6beta2* and alpha4beta2* nicotinic acetylcholine receptors as drug targets for Parkinson’s disease. Pharmacological reviews. 2011;63:938–966. doi: 10.1124/pr.110.003269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thies W, et al. 2013 Alzheimer’s disease facts and figures. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2013;9:208–245. doi: 10.1016/j.jalz.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 56.Auld DS, et al. Alzheimer’s disease and the basal forebrain cholinergic system: relations to beta-amyloid peptides, cognition, and treatment strategies. Progress in neurobiology. 2002;68:209–245. doi: 10.1016/s0301-0082(02)00079-5. [DOI] [PubMed] [Google Scholar]
- 57.Prado VF, et al. Regulation of cholinergic activity by the vesicular acetylcholine transporter. The Biochemical journal. 2013;450:265–274. doi: 10.1042/BJ20121662. [DOI] [PubMed] [Google Scholar]
- 58.Mesulam MM. Cholinergic circuitry of the human nucleus basalis and its fate in alzheimer’s disease. The Journal of comparative neurology. 2013 doi: 10.1002/cne.23415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mufson EJ, et al. Cholinergic system during the progression of Alzheimer’s disease: therapeutic implications. Expert Rev Neurother. 2008;8:1703–1718. doi: 10.1586/14737175.8.11.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yakel JL. Nicotinic ACh receptors in the hippocampus: role in excitability and plasticity. Nicotine & tobacco research : official journal of the Society for Research on Nicotine and Tobacco. 2012;14:1249–1257. doi: 10.1093/ntr/nts091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Levin ED. Complex relationships of nicotinic receptor actions and cognitive functions. Biochemical pharmacology. 2013;86:1145–1152. doi: 10.1016/j.bcp.2013.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Parri HR, et al. Research update: Alpha7 nicotinic acetylcholine receptor mechanisms in Alzheimer’s disease. Biochemical pharmacology. 2011;82:931–942. doi: 10.1016/j.bcp.2011.06.039. [DOI] [PubMed] [Google Scholar]
- 63.Dineley KT. Beta-amyloid peptide–nicotinic acetylcholine receptor interaction: the two faces of health and disease. Front Biosci. 2007;12:5030–5038. doi: 10.2741/2445. [DOI] [PubMed] [Google Scholar]
- 64.Wang HY, et al. Amyloid peptide Abeta(1–42) binds selectively and with picomolar affinity to alpha7 nicotinic acetylcholine receptors. J Neurochem. 2000;75:1155–1161. doi: 10.1046/j.1471-4159.2000.0751155.x. [DOI] [PubMed] [Google Scholar]
- 65.Puzzo D, et al. Endogenous amyloid-beta is necessary for hippocampal synaptic plasticity and memory. Ann Neurol. 2011;69:819–830. doi: 10.1002/ana.22313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pirttimaki TM, et al. alpha7 Nicotinic receptor-mediated astrocytic gliotransmitter release: Abeta effects in a preclinical Alzheimer’s mouse model. PloS one. 2013;8:e81828. doi: 10.1371/journal.pone.0081828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dineley KT, et al. Beta-amyloid activates the mitogen-activated protein kinase cascade via hippocampal alpha7 nicotinic acetylcholine receptors: In vitro and in vivo mechanisms related to Alzheimer’s disease. J Neurosci. 2001;21:4125–4133. doi: 10.1523/JNEUROSCI.21-12-04125.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Talantova M, et al. Abeta induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci U S A. 2013;110:E2518–2527. doi: 10.1073/pnas.1306832110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Moretti M, et al. The Novel alpha7beta2-nicotinic Acetylcholine Receptor Subtype is Expressed in Mouse and Human Basal Forebrain: Biochemical and Pharmacological Characterisation. Molecular pharmacology. 2014 doi: 10.1124/mol.114.093377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu Q, et al. beta-Amyloid peptide blocks the response of alpha 7-containing nicotinic receptors on hippocampal neurons. Proc Natl Acad Sci U S A. 2001;98:4734–4739. doi: 10.1073/pnas.081553598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pettit DL, et al. beta-Amyloid(1–42) peptide directly modulates nicotinic receptors in the rat hippocampal slice. J Neurosci. 2001;21:RC120. doi: 10.1523/JNEUROSCI.21-01-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Colon-Saez JO, Yakel JL. The alpha7 nicotinic acetylcholine receptor function in hippocampal neurons is regulated by the lipid composition of the plasma membrane. J Physiol. 2011;589:3163–3174. doi: 10.1113/jphysiol.2011.209494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lamb PW, et al. Inhibition of neuronal nicotinic acetylcholine receptor channels expressed in Xenopus oocytes by beta-amyloid1–42 peptide. J Mol Neurosci. 2005;27:13–21. doi: 10.1385/JMN:27:1:013. [DOI] [PubMed] [Google Scholar]
- 74.Chin JH, et al. Amyloid beta protein modulates glutamate-mediated neurotransmission in the rat basal forebrain: involvement of presynaptic neuronal nicotinic acetylcholine and metabotropic glutamate receptors. J Neurosci. 2007;27:9262–9269. doi: 10.1523/JNEUROSCI.1843-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dougherty JJ, et al. Beta-amyloid regulation of presynaptic nicotinic receptors in rat hippocampus and neocortex. J Neurosci. 2003;23:6740–6747. doi: 10.1523/JNEUROSCI.23-17-06740.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fu W, Jhamandas JH. Beta-amyloid peptide activates non-alpha7 nicotinic acetylcholine receptors in rat basal forebrain neurons. J Neurophysiol. 2003;90:3130–3136. doi: 10.1152/jn.00616.2003. [DOI] [PubMed] [Google Scholar]
- 77.Court J, et al. Nicotinic receptor abnormalities in Alzheimer’s disease. Biol Psychiatry. 2001;49:175–184. doi: 10.1016/s0006-3223(00)01116-1. [DOI] [PubMed] [Google Scholar]
- 78.Aubert I, et al. Comparative alterations of nicotinic and muscarinic binding sites in Alzheimer’s and Parkinson’s diseases. J Neurochem. 1992;58:529–541. doi: 10.1111/j.1471-4159.1992.tb09752.x. [DOI] [PubMed] [Google Scholar]
- 79.Martin-Ruiz CM, et al. Alpha4 but not alpha3 and alpha7 nicotinic acetylcholine receptor subunits are lost from the temporal cortex in Alzheimer’s disease. J Neurochem. 1999;73:1635–1640. doi: 10.1046/j.1471-4159.1999.0731635.x. [DOI] [PubMed] [Google Scholar]
- 80.Davies P, Feisullin S. Postmortem stability of alpha-bungarotoxin binding sites in mouse and human brain. Brain Res. 1981;216:449–454. doi: 10.1016/0006-8993(81)90148-7. [DOI] [PubMed] [Google Scholar]
- 81.Sugaya K, et al. Nicotinic acetylcholine receptor subtypes in human frontal cortex: changes in Alzheimer’s disease. Journal of neuroscience research. 1990;27:349–359. doi: 10.1002/jnr.490270314. [DOI] [PubMed] [Google Scholar]
- 82.Hyman BT, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2012;8:1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Davis KL, et al. Cholinergic markers in elderly patients with early signs of Alzheimer disease. JAMA. 1999;281:1401–1406. doi: 10.1001/jama.281.15.1401. [DOI] [PubMed] [Google Scholar]
- 84.DeKosky ST, et al. Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann Neurol. 2002;51:145–155. doi: 10.1002/ana.10069. [DOI] [PubMed] [Google Scholar]
- 85.Counts SE, et al. Alpha7 nicotinic receptor up-regulation in cholinergic basal forebrain neurons in Alzheimer disease. Arch Neurol. 2007;64:1771–1776. doi: 10.1001/archneur.64.12.1771. [DOI] [PubMed] [Google Scholar]
- 86.Albert MS, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:270–279. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McKhann GM, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2011;7:263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dineley KT, et al. beta-Amyloid peptide activates alpha 7 nicotinic acetylcholine receptors expressed in Xenopus oocytes. J Biol Chem. 2002;277:25056–25061. doi: 10.1074/jbc.M200066200. [DOI] [PubMed] [Google Scholar]
- 89.Puzzo D, et al. Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J Neurosci. 2008;28:14537–14545. doi: 10.1523/JNEUROSCI.2692-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bell KA, et al. MAPK recruitment by beta-amyloid in organotypic hippocampal slice cultures depends on physical state and exposure time. J Neurochem. 2004;91:349–361. doi: 10.1111/j.1471-4159.2004.02722.x. [DOI] [PubMed] [Google Scholar]
- 91.Dziewczapolski G, et al. Deletion of the alpha 7 nicotinic acetylcholine receptor gene improves cognitive deficits and synaptic pathology in a mouse model of Alzheimer’s disease. J Neurosci. 2009;29:8805–8815. doi: 10.1523/JNEUROSCI.6159-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Puzzo D, Arancio O. Amyloid-beta peptide: Dr. Jekyll or Mr. Hyde? Journal of Alzheimer’s disease : JAD. 2013;33(Suppl 1):S111–120. doi: 10.3233/JAD-2012-129033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Teaktong T, et al. Alzheimer’s disease is associated with a selective increase in alpha7 nicotinic acetylcholine receptor immunoreactivity in astrocytes. Glia. 2003;41:207–211. doi: 10.1002/glia.10132. [DOI] [PubMed] [Google Scholar]
- 94.Wang H, et al. Dissociating beta-amyloid from alpha7 nicotinic acetylcholine receptor by a novel therapeutic agent, S 24795, normalizes alpha 7 nicotinic acetylcholine and NMDA receptor function in Alzheimer’s diseasebrain. J Neurosci. 2009;29:10961–10971. doi: 10.1523/JNEUROSCI.6088-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chu LW, et al. Increased alpha 7 nicotinic acetylcholine receptor protein levels in Alzheimer’s disease patients. Dement Geriatr Cogn Disord. 2005;19:106–112. doi: 10.1159/000082661. [DOI] [PubMed] [Google Scholar]
- 96.Yu WF, et al. High selective expression of alpha7 nicotinic receptors on astrocytes in the brains of patients with sporadic Alzheimer’s disease and patients carrying Swedish APP 670/671 mutation: a possible association with neuritic plaques. Exp Neurol. 2005;192:215–225. doi: 10.1016/j.expneurol.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 97.Dineley KT, et al. Accelerated plaque accumulation, associative learning deficits, and up-regulation of alpha 7 nicotinic receptor protein in transgenic mice co-expressing mutant human presenilin 1 and amyloid precursor proteins. J Biol Chem. 2002;277:22768–22780. doi: 10.1074/jbc.M200164200. [DOI] [PubMed] [Google Scholar]
- 98.Liu Q, et al. A novel nicotinic mechanism underlies beta-amyloid-induced neuronal hyperexcitation. J Neurosci. 2013;33:7253–7263. doi: 10.1523/JNEUROSCI.3235-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang HY, et al. S 24795 limits beta-amyloid-alpha7 nicotinic receptor interaction and reduces Alzheimer’s disease-like pathologies. Biol Psychiatry. 2010;67:522–530. doi: 10.1016/j.biopsych.2009.09.031. [DOI] [PubMed] [Google Scholar]
- 100.Wang HY, et al. Dissociating beta-amyloid from alpha 7 nicotinic acetylcholine receptor by a novel therapeutic agent, S 24795, normalizes alpha 7 nicotinic acetylcholine and NMDA receptor function in Alzheimer’s disease brain. J Neurosci. 2009;29:10961–10973. doi: 10.1523/JNEUROSCI.6088-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Perea G, Araque A. Astrocytes potentiate transmitter release at single hippocampal synapses. Science. 2007;317:1083–1086. doi: 10.1126/science.1144640. [DOI] [PubMed] [Google Scholar]
- 102.Fellin T, et al. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron. 2004;43:729–743. doi: 10.1016/j.neuron.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 103.Fiacco TA, McCarthy KD. Intracellular astrocyte calcium waves in situ increase the frequency of spontaneous AMPA receptor currents in CA1 pyramidal neurons. J Neurosci. 2004;24:722–732. doi: 10.1523/JNEUROSCI.2859-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Palop JJ, et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Vossel KA, et al. Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. 2013;70:1158–1166. doi: 10.1001/jamaneurol.2013.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Samochocki M, et al. Galantamine is an allosterically potentiating ligand of the human alpha4/beta2 nAChR. Acta neurologica Scandinavica Supplementum. 2000;176:68–73. doi: 10.1034/j.1600-0404.2000.00310.x. [DOI] [PubMed] [Google Scholar]
- 107.Maelicke A, et al. Allosteric sensitization of nicotinic receptors by galantamine, a new treatment strategy for Alzheimer’s disease. Biol Psychiatry. 2001;49:279–288. doi: 10.1016/s0006-3223(00)01109-4. [DOI] [PubMed] [Google Scholar]
- 108.Barnes CA, et al. Chronic treatment of old rats with donepezil or galantamine: effects on memory, hippocampal plasticity and nicotinic receptors. Neuroscience. 2000;99:17–23. doi: 10.1016/s0306-4522(00)00180-9. [DOI] [PubMed] [Google Scholar]
- 109.Rogers SL, et al. Long-term efficacy and safety of donepezil in the treatment of Alzheimer’s disease: final analysis of a US multicentre open-label study. Eur Neuropsychopharmacol. 2000;10:195–203. doi: 10.1016/s0924-977x(00)00067-5. [DOI] [PubMed] [Google Scholar]
- 110.Hurst R, et al. Nicotinic acetylcholine receptors: from basic science to therapeutics. Pharmacol Ther. 2013;137:22–54. doi: 10.1016/j.pharmthera.2012.08.012. [DOI] [PubMed] [Google Scholar]
- 111.Selkoe DJ. Preventing Alzheimer’s disease. Science. 2012;337:1488–1492. doi: 10.1126/science.1228541. [DOI] [PubMed] [Google Scholar]
- 112.Becker RE, Greig NH. Fire in the ashes: can failed Alzheimer’s disease drugs succeed with second chances? Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2013;9:50–57. doi: 10.1016/j.jalz.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sperling RA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schneider LS, et al. Clinical trials and late-stage drug development for Alzheimer’s disease: an appraisal from 1984 to 2014. Journal of internal medicine. 2014;275:251–283. doi: 10.1111/joim.12191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Resende RR, Adhikari A. Cholinergic receptor pathways involved in apoptosis, cell proliferation and neuronal differentiation. Cell communication and signaling : CCS. 2009;7:20. doi: 10.1186/1478-811X-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Narla S, et al. alpha7 nicotinic receptor agonist reactivates neurogenesis in adult brain. Biochem Pharmacol. 2013;86:1099–1104. doi: 10.1016/j.bcp.2013.07.028. [DOI] [PubMed] [Google Scholar]
- 117.State MW, Levitt P. The conundrums of understanding genetic risks for autism spectrum disorders. Nat Neurosci. 2011;14:1499–1506. doi: 10.1038/nn.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ebert DH, Greenberg ME. Activity-dependent neuronal signalling and autism spectrum disorder. Nature. 2013;493:327–337. doi: 10.1038/nature11860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Perry EK, et al. Cholinergic activity in autism: Abnormalities in the cerebral cortex and basal forebrain. Am J Psychiat. 2001;158:1058–1066. doi: 10.1176/appi.ajp.158.7.1058. [DOI] [PubMed] [Google Scholar]
- 120.Martin-Ruiz CM, et al. Molecular analysis of nicotinic receptor expression in autism. Brain Res Mol Brain Res. 2004;123:81–90. doi: 10.1016/j.molbrainres.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 121.Deutsch SI, et al. Cholinergic abnormalities in autism: is there a rationale for selective nicotinic agonist interventions? Clin Neuropharmacol. 2010;33:114–120. doi: 10.1097/WNF.0b013e3181d6f7ad. [DOI] [PubMed] [Google Scholar]
- 122.Pandya A, Yakel JL. Allosteric modulators of the alpha4beta2 subtype of neuronal nicotinic acetylcholine receptors. Biochem Pharmacol. 2011;82:952–958. doi: 10.1016/j.bcp.2011.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lippiello PM. Nicotinic cholinergic antagonists: A novel approach for the treatment of autism. Medical Hypotheses. 2006;66:985–990. doi: 10.1016/j.mehy.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 124.Poirier MF, et al. Prevalence of smoking in psychiatric patients. Progress in neuro-psychopharmacology & biological psychiatry. 2002;26:529–537. doi: 10.1016/s0278-5846(01)00304-9. [DOI] [PubMed] [Google Scholar]
- 125.Bejerot S, Nylander L. Low prevalence of smoking in patients with autism spectrum disorders. Psychiatry research. 2003;119:177–182. doi: 10.1016/s0165-1781(03)00123-9. [DOI] [PubMed] [Google Scholar]
- 126.Dani JA, Heinemann S. Molecular and cellular aspects of nicotine abuse. Neuron. 1996;16:905–908. doi: 10.1016/s0896-6273(00)80112-9. [DOI] [PubMed] [Google Scholar]
- 127.Fenster CP, et al. Upregulation of surface alpha4beta2 nicotinic receptors is initiated by receptor desensitization after chronic exposure to nicotine. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1999;19:4804–4814. doi: 10.1523/JNEUROSCI.19-12-04804.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Arnold LE, et al. Placebo-Controlled Pilot Trial of Mecamylamine for Treatment of Autism Spectrum Disorders. Journal of Child and Adolescent Psychopharmacology. 2012;22:198–205. doi: 10.1089/cap.2011.0056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lopez MG, et al. Can cholinesterase inhibitors provide additional effects to cholinergic neurotransmission enhancement? Journal of molecular neuroscience : MN. 2006;30:141–144. doi: 10.1385/JMN:30:1:141. [DOI] [PubMed] [Google Scholar]
- 130.Srivastava RK, et al. Role of donepezil in autism: its conduciveness in psychopharmacotherapy. Case reports in psychiatry. 2011;2011:563204. doi: 10.1155/2011/563204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ghaleiha A, et al. Galantamine efficacy and tolerability as an augmentative therapy in autistic children: A randomized, double-blind, placebo-controlled trial. J Psychopharmacol. 2013 doi: 10.1177/0269881113508830. [DOI] [PubMed] [Google Scholar]
- 132.Schaaf CP. Nicotinic acetylcholine receptors in human genetic disease. Genetics in medicine : official journal of the American College of Medical Genetics. 2014 doi: 10.1038/gim.2014.9. [DOI] [PubMed] [Google Scholar]
- 133.Young JW, Geyer MA. Evaluating the role of the alpha-7 nicotinic acetylcholine receptor in the pathophysiology and treatment of schizophrenia. Biochem Pharmacol. 2013;86:1122–1132. doi: 10.1016/j.bcp.2013.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Leonard S, et al. Smoking, Genetics and Schizophrenia: Evidence for Self Medication. J Dual Diagn. 2007;3:43–59. doi: 10.1300/J374v03n03_05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hong LE, et al. Effects of moderate-dose treatment with varenicline on neurobiological and cognitive biomarkers in smokers and nonsmokers with schizophrenia or schizoaffective disorder. Arch Gen Psychiatry. 2011;68:1195–1206. doi: 10.1001/archgenpsychiatry.2011.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wing VC, et al. Varenicline modulates spatial working memory deficits in smokers with schizophrenia. Schizophrenia research. 2013;149:190–191. doi: 10.1016/j.schres.2013.06.032. [DOI] [PubMed] [Google Scholar]
- 137.Shim JC, et al. Adjunctive varenicline treatment with antipsychotic medications for cognitive impairments in people with schizophrenia: a randomized double-blind placebo-controlled trial. Neuropsychopharmacology. 2012;37:660–668. doi: 10.1038/npp.2011.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mihalak KB, et al. Varenicline is a partial agonist at alpha4beta2 and a full agonist at alpha7 neuronal nicotinic receptors. Mol Pharmacol. 2006;70:801–805. doi: 10.1124/mol.106.025130. [DOI] [PubMed] [Google Scholar]
- 139.Freedman R, et al. Linkage of a neurophysiological deficit in schizophrenia to a chromosome 15 locus. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:587–592. doi: 10.1073/pnas.94.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Freedman R. alpha7-nicotinic acetylcholine receptor agonists for cognitive enhancement in schizophrenia. Annual review of medicine. 2014;65:245–261. doi: 10.1146/annurev-med-092112-142937. [DOI] [PubMed] [Google Scholar]
- 141.Hurst RS, et al. A novel positive allosteric modulator of the alpha7 neuronal nicotinic acetylcholine receptor: in vitro and in vivo characterization. J Neurosci. 2005;25:4396–4405. doi: 10.1523/JNEUROSCI.5269-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Dunlop J, et al. Old and new pharmacology: positive allosteric modulation of the alpha7 nicotinic acetylcholine receptor by the 5-hydroxytryptamine(2B/C) receptor antagonist SB-206553 (3,5-dihydro-5-methyl-N-3-pyridinylbenzo[1,2-b:4,5-b’]di pyrrole-1(2H)-carboxamide) J Pharmacol Exp Ther. 2009;328:766–776. doi: 10.1124/jpet.108.146514. [DOI] [PubMed] [Google Scholar]
- 143.Dinklo T, et al. Characterization of 2-[[4-fluoro-3-(trifluoromethyl)phenyl]amino]-4-(4-pyridinyl)-5-thiazolemethanol (JNJ-1930942), a novel positive allosteric modulator of the {alpha}7 nicotinic acetylcholine receptor. J Pharmacol Exp Ther. 2011;336:560–574. doi: 10.1124/jpet.110.173245. [DOI] [PubMed] [Google Scholar]
- 144.Olincy A, et al. Proof-of-concept trial of an alpha7 nicotinic agonist in schizophrenia. Archives of general psychiatry. 2006;63:630–638. doi: 10.1001/archpsyc.63.6.630. [DOI] [PubMed] [Google Scholar]
- 145.Tregellas JR, et al. Effects of an alpha 7-nicotinic agonist on default network activity in schizophrenia. Biological psychiatry. 2011;69:7–11. doi: 10.1016/j.biopsych.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Preskorn SH, et al. Normalizing effects of EVP-6124, an alpha-7 nicotinic partial agonist, on event-related potentials and cognition: a proof of concept, randomized trial in patients with schizophrenia. Journal of psychiatric practice. 2014;20:12–24. doi: 10.1097/01.pra.0000442935.15833.c5. [DOI] [PubMed] [Google Scholar]
- 147.Cheng Q, Yakel JL. Presynaptic alpha7 nicotinic acetylcholine receptors enhance hippocampal mossy fiber glutamatergic transmission via PKA activation. J Neurosci. 2014;34:124–133. doi: 10.1523/JNEUROSCI.2973-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kolomeets NS, et al. Decreased numerical density of CA3 hippocampal mossy fiber synapses in schizophrenia. Synapse. 2007;61:615–621. doi: 10.1002/syn.20405. [DOI] [PubMed] [Google Scholar]
- 149.Backonja MM. Defining neuropathic pain. Anesthesia and analgesia. 2003;97:785–790. doi: 10.1213/01.ANE.0000062826.70846.8D. [DOI] [PubMed] [Google Scholar]
- 150.Grichnik KP, Ferrante FM. The difference between acute and chronic pain. The Mount Sinai journal of medicine, New York. 1991;58:217–220. [PubMed] [Google Scholar]
- 151.Woolf CJ, Mannion RJ. Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet. 1999;353:1959–1964. doi: 10.1016/S0140-6736(99)01307-0. [DOI] [PubMed] [Google Scholar]
- 152.(!!! INVALID CITATION !!!)
- 153.Umana IC, et al. Neuronal nicotinic receptors as analgesic targets: It’s a winding road. Biochemical Pharmacology. 2013;86:1208–1214. doi: 10.1016/j.bcp.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hamann SR, Martin WR. Opioid and nicotinic analgesic and hyperalgesic loci in the rat brain stem. The Journal of pharmacology and experimental therapeutics. 1992;261:707–715. [PubMed] [Google Scholar]
- 155.Sahley TL, Berntson GG. Antinociceptive effects of central and systemic administrations of nicotine in the rat. Psychopharmacology. 1979;65:279–283. doi: 10.1007/BF00492216. [DOI] [PubMed] [Google Scholar]
- 156.Sullivan JP, et al. (+/−)-Epibatidine elicits a diversity of in vitro and in vivo effects mediated by nicotinic acetylcholine receptors. The Journal of pharmacology and experimental therapeutics. 1994;271:624–631. [PubMed] [Google Scholar]
- 157.Qian C, et al. Epibatidine is a nicotinic analgesic. European journal of pharmacology. 1993;250:R13–14. doi: 10.1016/0014-2999(93)90043-h. [DOI] [PubMed] [Google Scholar]
- 158.Garraffo HM, et al. Epibatidine: From Frog Alkaloid to Analgesic Clinical Candidates. A Testimonial to “True Grit”! Heterocycles. 2009;79:207–217. [Google Scholar]
- 159.Daly JW, et al. Alkaloids from frog skin: the discovery of epibatidine and the potential for developing novel non-opioid analgesics. Nat Prod Rep. 2000;17:131–135. doi: 10.1039/a900728h. [DOI] [PubMed] [Google Scholar]
- 160.Spande TF, et al. Epibatidine – a Novel (Chloropyridyl)Azabicycloheptane with Potent Analgesic Activity from an Ecuadorian Poison Frog. Journal of the American Chemical Society. 1992;114:3475–3478. [Google Scholar]
- 161.Gerzanich V, et al. Comparative Pharmacology of Epibatidine – a Potent Agonist for Neuronal Nicotinic Acetylcholine-Receptors. Molecular Pharmacology. 1995;48:774–782. [PubMed] [Google Scholar]
- 162.Bannon AW, et al. Broad-spectrum, non-opioid analgesic activity by selective modulation of neuronal nicotinic acetylcholine receptors. Science. 1998;279:77–81. doi: 10.1126/science.279.5347.77. [DOI] [PubMed] [Google Scholar]
- 163.Kesingland AC, et al. Analgesic profile of the nicotinic acetylcholine receptor agonists, (+)-epibatidine and ABT-594 in models of persistent inflammatory and neuropathic pain. Pain. 2000;86:113–118. doi: 10.1016/s0304-3959(00)00233-5. [DOI] [PubMed] [Google Scholar]
- 164.Donnelly-Roberts DL, et al. ABT-594 [(R)-5-(2-azetidinylmethoxy)-2-chloropyridine]: A novel, orally effective analgesic acting via neuronal nicotinic acetylcholine receptors: I. In vitro characterization. Journal of Pharmacology and Experimental Therapeutics. 1998;285:777–786. [PubMed] [Google Scholar]
- 165.Lynch JJ, et al. ABT-594 (a nicotinic acetylcholine agonist): anti-allodynia in a rat chemotherapy-induced pain model. European Journal of Pharmacology. 2005;509:43–48. doi: 10.1016/j.ejphar.2004.12.034. [DOI] [PubMed] [Google Scholar]
- 166.Rowbotham MC, et al. A randomized, double-blind, placebo-controlled trial evaluating the efficacy and safety of ABT-594 in patients with diabetic peripheral neuropathic pain. Pain. 2009;146:245–252. doi: 10.1016/j.pain.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 167.Arneric SP, et al. Neuronal nicotinic receptors: A perspective on two decades of drug discovery research. Biochemical Pharmacology. 2007;74:1092–1101. doi: 10.1016/j.bcp.2007.06.033. [DOI] [PubMed] [Google Scholar]
- 168.Gao BX, et al. Pharmacological effects of nonselective and subtype-selective nicotinic acetylcholine receptor agonists in animal models of persistent pain. Pain. 2010;149:33–49. doi: 10.1016/j.pain.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 169.Nirogi R, et al. Antinociceptive activity of alpha 4 beta 2*neuronal nicotinic receptor agonist A-366833 in experimental models of neuropathic and inflammatory pain. European Journal of Pharmacology. 2011;668:155–162. doi: 10.1016/j.ejphar.2011.06.032. [DOI] [PubMed] [Google Scholar]
- 170.Ji J, et al. A-366833: A novel nicotinonitrile-substituted 3,6-diazabicyclo[3.2.0]-heptane alpha 4 beta 2 nicotinic acetylcholine receptor selective agonist: Synthesis, analgesic efficacy and tolerability profile in animal models. Biochemical Pharmacology. 2007;74:1253–1262. doi: 10.1016/j.bcp.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 171.Cordero-Erausquin M, et al. Nicotine differentially activates inhibitory and excitatory neurons in the dorsal spinal cord. Pain. 2004;109:308–318. doi: 10.1016/j.pain.2004.01.034. [DOI] [PubMed] [Google Scholar]
- 172.Genzen JR, McGehee DS. Nicotinic modulation of GABAergic synaptic transmission in the spinal cord dorsal horn. Brain Res. 2005;1031:229–237. doi: 10.1016/j.brainres.2004.10.042. [DOI] [PubMed] [Google Scholar]
- 173.Rashid MH, et al. Tonic inhibitory role of alpha 4 beta 2 subtype of nicotinic acetylcholine receptors on nociceptive transmission in the spinal cord in mice. Pain. 2006;125:125–135. doi: 10.1016/j.pain.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 174.Yalcin I, et al. Nociceptive thresholds are controlled through spinal beta(2)-subunit-containing nicotinic acetylcholine receptors. Pain. 2011;152:2131–2137. doi: 10.1016/j.pain.2011.05.022. [DOI] [PubMed] [Google Scholar]
- 175.Kiyosawa A, et al. Nicotine facilitates glycine release in the rat spinal dorsal horn. J Physiol-London. 2001;536:101–110. doi: 10.1111/j.1469-7793.2001.t01-1-00101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Hurst R, et al. Nicotinic acetylcholine receptors: From basic science to therapeutics. Pharmacology & Therapeutics. 2013;137:22–54. doi: 10.1016/j.pharmthera.2012.08.012. [DOI] [PubMed] [Google Scholar]
- 177.Damaj MI, et al. Antinociceptive responses to nicotinic acetylcholine receptor ligands after systemic and intrathecal administration in mice. Journal of Pharmacology and Experimental Therapeutics. 1998;284:1058–1065. [PubMed] [Google Scholar]
- 178.Wang Y, et al. Antinociceptive effects of choline against acute and inflammatory pain. Neuroscience. 2005;132:49–56. doi: 10.1016/j.neuroscience.2004.12.026. [DOI] [PubMed] [Google Scholar]
- 179.Pandya AA, Yakel JL. Effects of neuronal nicotinic acetylcholine receptor allosteric modulators in animal behavior studies. Biochem Pharmacol. 2013;86:1054–1062. doi: 10.1016/j.bcp.2013.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Pandya AA, Yakel JL. Activation of the alpha7 nicotinic ACh receptor induces anxiogenic effects in rats which is blocked by a 5-HT(1a) receptor antagonist. Neuropharmacology. 2013;70C:35–42. doi: 10.1016/j.neuropharm.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Pandya A, Yakel JL. Allosteric modulator Desformylflustrabromine relieves the inhibition of alpha2beta2 and alpha4beta2 nicotinic acetylcholine receptors by beta-amyloid(1–42) peptide. J Mol Neurosci. 2011;45:42–47. doi: 10.1007/s12031-011-9509-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Munro G, et al. The alpha7 nicotinic ACh receptor agonist compound B and positive allosteric modulator PNU-120596 both alleviate inflammatory hyperalgesia and cytokine release in the rat. Br J Pharmacol. 2012;167:421–435. doi: 10.1111/j.1476-5381.2012.02003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Freitas K, et al. In vivo pharmacological interactions between a type ii positive allosteric modulator of alpha7 nicotinic acetylcholine receptors and nicotinic agonists in a murine tonic pain model. Br J Pharmacol. 2012 doi: 10.1111/j.1476-5381.2012.02226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Freitas K, et al. Effects of alpha 7 positive allosteric modulators in murine inflammatory and chronic neuropathic pain models. Neuropharmacology. 2013;65:156–164. doi: 10.1016/j.neuropharm.2012.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Lee CH, et al. alpha4beta2 neuronal nicotinic receptor positive allosteric modulation: an approach for improving the therapeutic index of alpha4beta2 nAChR agonists in pain. Biochem Pharmacol. 2011;82:959–966. doi: 10.1016/j.bcp.2011.06.044. [DOI] [PubMed] [Google Scholar]
- 186.Zhu CZ, et al. Potentiation of analgesic efficacy but not side effects: co-administration of an alpha4beta2 neuronal nicotinic acetylcholine receptor agonist and its positive allosteric modulator in experimental models of pain in rats. Biochem Pharmacol. 2011;82:967–976. doi: 10.1016/j.bcp.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 187.Rode F, et al. Positive allosteric modulation of alpha4beta2 nAChR agonist induced behaviour. Brain Res. 2012;1458:67–75. doi: 10.1016/j.brainres.2012.03.064. [DOI] [PubMed] [Google Scholar]
- 188.Jackson KJ, et al. Role of alpha5 nicotinic acetylcholine receptors in pharmacological and behavioral effects of nicotine in mice. The Journal of pharmacology and experimental therapeutics. 2010;334:137–146. doi: 10.1124/jpet.110.165738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Lips KS, et al. Coexpression of alpha 9 and alpha 10 nicotinic acetylcholine receptors in rat dorsal root ganglion neurons. Neuroscience. 2002;115:1–5. doi: 10.1016/s0306-4522(02)00274-9. [DOI] [PubMed] [Google Scholar]
- 190.Billen B, et al. Molecular actions of smoking cessation drugs at alpha4beta2 nicotinic receptors defined in crystal structures of a homologous binding protein. Proc Natl Acad Sci U S A. 2012;109:9173–9178. doi: 10.1073/pnas.1116397109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Miller PS, Aricescu AR. Crystal structure of a human GABAA receptor. Nature. 2014;512:270–275. doi: 10.1038/nature13293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Hassaine G, et al. X-ray structure of the mouse serotonin 5-HT3 receptor. Nature. 2014;512:276–281. doi: 10.1038/nature13552. [DOI] [PubMed] [Google Scholar]
- 193.Li SX, et al. Ligand-binding domain of an alpha7-nicotinic receptor chimera and its complex with agonist. Nat Neurosci. 2011;14:1253–1259. doi: 10.1038/nn.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]