

Abstract
The olfactory bulb (OB) is one of the first brain regions in Parkinson’s disease (PD) to contain alpha-synuclein (α-syn) inclusions, possibly associated with nonmotor symptoms. Mechanisms underlying olfactory synucleinopathy, its contribution to progressive aggregation pathology and nigrostriatal dopaminergic loss observed at later stages, remain unclear. A second hit, such as environmental toxins, is suggestive for α-syn aggregation in olfactory neurons, potentially triggering disease progression. To address the possible pathogenic role of olfactory α-syn accumulation in early PD, we exposed mice with site-specific and inducible overexpression of familial PD-linked mutant α-syn in OB neurons to a low dose of the herbicide paraquat. Here, we found that olfactory α-syn per se elicited structural and behavioral abnormalities, characteristic of an early time point in models with widespread α-syn expression, including hyperactivity and increased striatal dopaminergic marker. Suppression of α-syn reversed the dopaminergic phenotype. In contrast, paraquat treatment synergistically induced degeneration of olfactory dopaminergic cells and opposed the higher reactive phenotype. Neither neurodegeneration nor behavioral abnormalities were detected in paraquat-treated mice with suppressed α-syn expression. By increasing calpain activity, paraquat induced a pathological cascade leading to inhibition of autophagy clearance and accumulation of calpain-cleaved truncated and insoluble α-syn, recapitulating biochemical and structural changes in human PD. Thus our results underscore the primary role of proteolytic failure in aggregation pathology. In addition, we provide novel evidence that olfactory dopaminergic neurons display an increased vulnerability toward neurotoxins in dependence to presence of human α-syn, possibly mediating an olfactory-striatal dopaminergic network dysfunction in mouse models and early PD.
Introduction
Parkinson’s disease (PD) is characterized by progressive locomotor impairments, linked to loss of dopaminergic neurons in the nigrostriatal system. Prior to the onset of motor symptoms the majority of PD patients develop neuropsychiatric symptoms such as reduced stress tolerance, anxiety, olfactory deficits and depression [38, 39, 55, 65].
Particular relevance was attributed to the olfactory bulb (OB), being the first brain region to contain alpha-synuclein (α-syn)-positive aggregates [4] together with the presence of smell deficits in ~90 % of PD patients [25]. The olfactory epithelium is the first relay station for odor perception and conveyed to higher brain structures, such as the piriform and entorhinal cortex. Olfactory processing relies on the interaction of mitral cells with inhibitory dopaminergic (DAergic) periglomerular and granule cells, which are located in the glomerular layer (GL). Structural and imaging studies show atrophy of neurons [5, 35, 74] and α-syn inclusions in granule, mitral and periglomerular cells in OB of PD patients [84]. However, the contribution of olfactory periglomerular DAergic cells in PD is not yet clear, since numerous studies observed a negative role of α-syn on dopamine synthesis and release [13, 43, 68, 75, 108], but olfactory DAergic cell numbers might be increased [36, 66]. α-Syn-positive Lewy bodies and Lewy neurites, the hallmark of PD, are seen primarily in the OB and the dorsal motor nucleus in early PD [4]. These lesions may share prion-like characteristics by recruiting their endogenously expressed counterparts [95] and gradually propagate through the brainstem toward the midbrain; whereas, olfactory α-syn pathology only slowly evolves into related areas and does not advance to non-olfactory brain regions [4].
The accumulation of α-syn oligomers, rather than fibrils, has been proposed as key event for nigral DAergic cell vulnerability [17] and is possibly caused by either (1) its interaction with dopamine (DA) [10, 16, 73]; (2) single point mutations such as the A30P mutation [15], or (3) proteolytic cleavage leading to C-terminally truncation [26, 48, 62]. Several pan-neuronal α-syn transgenic animal models develop early symptoms that may precede nigral DAergic cell loss, including smell deficiency [29, 45, 70], and hyperactivity [43, 44, 81, 106]. The higher activity towards mild stress was associated with an increase of striatal DAergic marker, eventually normalized and lost at old age [43, 44, 81, 106]. In our previous study, we observed that mice with site-specific α-syn expression in OB neurons develop a similar phenotype and neurochemical changes [71], implying that olfactory circuits may play a role in regulation of the nigrostriatal DAergic tone. Epidemiological studies suggest exposure to environmental toxins as contributing factor to PD pathogenesis that may enter the brain via the olfactory neuroepithelium, laying out the concept of the olfactory vector hypothesis [24, 78]. Paraquat, is widely used herbicide and has been suggested as a risk factor for PD [23]. It was shown that paraquat increases α-syn aggregation pathology [28, 53] as a result of impaired protein clearance pathways [101]. Dysfunction of autophagy is a common pathogenic pathway in α-synucleinopathies [18, 50]. However, the sequence of events leading to impaired autophagy and accumulation of neurotoxic α-syn species remain to be elucidated.
Since the OB is likely to be entrance for environmental toxins and given its involvement in early PD, we generated conditional transgenic mice with site-specific expression in olfactory neurons. These mice exhibit hyperactivity, and a dysregulation of olfactory and midbrain DAergic circuits in absence of olfactory DAergic cell-death [71]. A similar phenotype was described in young transgenic mice with widespread expression of α-syn [43, 44, 81, 106]. With these conditional transgenic mice we tested whether an additional exposure to paraquat would increase the vulnerability of olfactory DAergic neurons and to further unravel a potential interplay between the olfactory and striatal DAergic circuit. Here, we describe a series of biochemical, structural, cell biological and human brain studies, that together implicate a synergistic effect of human α-syn and paraquat on activation of calpain that led to adverse protein clearance and thus accumulation of calpain-cleaved C-terminally truncated, insoluble and oligomeric α-syn, culminating in olfactory DAergic cell-death. Finally, suppression of the transgene expression reversed paraquat-induced olfactory pathology and dramatically improved olfactory and nigrostriatal DA dysregulation and associated neurobehavioral changes.
Materials and methods
Animals and treatment
Mice with doxycycline (dox, D)-responsive overexpression of human mutant A30P α-syn under conditional control of the prion protein promoter (PrP_A30P; in the present study designated as A30P) were created and previously described [71]. Transgenic A30P (n = 13) and dox-treated A30P (n = 13) aged 12 months at the beginning of the experiment were used (see also Fig. 1d). One mouse of each group died during test period and was excluded from the entire study. Of each group, randomized n = 6 animals were injected intraperitoneally with either saline or paraquat (5 mg/kg; Sigma) twice a week for 6 weeks. Animals were anesthetized, decapitated, brains were sagitally split upon removal and half of the brain either immediately frozen or fixed. All animals were handled in strict accordance with good animal practice and all procedures were completed under the specifications set forth by the UCSD Institutional Animal Care and Use Committee.
Fig. 1.

Hyperactivity is reversed by paraquat treatment in A30P mice with site-specific expression in the OB. a, b A30P α-synucleinopathy led to olfactory DA deficiency in absence of DAergic cell-death, elevated midbrain DA levels and an associated higher reactive phenotype in A30P mice [71]. Neurochemical changes and phenotype appear to be related to young age in several other mice with widespread α-syn expression [43, 44, 81] and may associate with higher reactivity to mild stress in early PD. Exposure to paraquat potentially exacerbates α-syn-induced toxicity in olfactory DAergic neurons and if leading to changes in phenotype and striatal DAergic tone, further supporting a potential regulative role of OB in the striatal DAergic activity. c Representative images of sagittal brain sections of A30P mice stained with anti-human α-syn antibody. Predominant expression was observed in the GL. Dox-treated mice show strong suppression of immunoreactivity. d Experimental design. The A30P+D and A30P+D+P received 2 mg/ml doxycycline (dox, D) during all the experimental period to suppress A30P expression. After 4 weeks, mice were randomized into two treatment groups receiving either paraquat (P) or saline. Groups were behaviorally tested prior (OF1) and after P treatment (OF2). e Horizontal and vertical (rearing) open field activity was recorded over 10 min. A30P mice are hyperactive compared to controls without expression (A30P+D and non-transgenic controls; Ntg). Horizontal activity and rearing was strongly reduced after paraquat treatment and when compared to dox-treated mice (A30P+P+D). Mice did not display differences in anxiety, as thigmotaxis (expressed in percent of time) was unchanged among groups. The data are mean ± SEM. *p < 0.05, **p < 0.01 A30P vs. A30P+D or A30P+P vs. A30P+D+P. §§§p < 0.001 A30P+P vs. A30P. Scale bar in overviews: 3 mm, left panel 60 μm
Behavioral tests
Briefly, as previously described [90], total activity of 12-month-old animals (A30P, n = 12; non-transgenic controls (Ntg), n = 9; A30P+D, n = 12) was performed at the beginning and the end of the paraquat experiment and calculated as total beam breaks in a 10-min period. Thigmotaxis was calculated as the ratio of time spent in the periphery:center and is expressed as % of total time.
Sequential extraction and Western blot analyses
Sequential extraction and immunoblotting was performed as previously described [70]. For methods see Supplemental Methods.
Calpain digestion
Calpain digestion of recombinant α-syn was performed as previously described [26]. Briefly, 15 μg of recombinant α-syn was incubated with calpain I (2 U) in buffer containing 40 mM HEPES and 1 mM calcium at 37 °C. In order to demonstrate fragment specificity to calpain cleavage, 4 mM of calpeptin was added to calpain buffer and coincubated for 60 min. To stop proteolysis samples were frozen at −20 °C for 10 min and subsequently used for Western blot analysis.
For detection of calpain-resistant α-syn, free-floating sections were incubated in calpain activation solution (20 mM tris/HCl pH 7.8, 3.6 mM CaCl2, 100 mM KCl2), containing 150 μg/ml calpain I (208712, Calbiochem) for 16 h at 37 °C.
Immunohistochemistry and cell culture
For methods of immunohistochemistry and cell culture see Supplemental Methods.
Electron microscopy and immunogold analysis
For ultrastructural analyses, coronal vibratome sections of the main OB (40-μm thickness) were prepared and post-fixed with 2 % of glutaraldehyde/0.1 % osmium tetroxide in 0.1 M sodium cacodylate buffer, embedded in epoxy and analyzed with a Zeiss EM 10 electron microscope as described [58]. For immunogold labeling, sections were mounted in nickel grids, etched and incubated with 105 antibody followed by labeling with 10 nm Aurion ImmunoGold particles (1:50, Electron Microscopy Sciences, Fort Washington, PA) with silver enhancement. Autophagosomes were characterized by containment of partially degraded cytoplasmic components and double membranes and counted of 50 cell profiles per group.
Cell counting
The number of TH-positive cells was estimated with systematic random sampling of about every third section to yield 6–8 sections to encompass the full rostral-caudal extent of the olfactory bulb (OB) and the substantia nigra (SN). The respective region was outlined using a 4× objective attached to an Olympus BX51 microscope. TH-positive neurons of were estimated using 8.21.1 Stereo-Investigator Software (MicroBrightField, Biosciences) as previously described [70]. Briefly, total cell numbers were estimated according to the optical fractionator method according to West et al. [100]. Region definitions were based on the Paxinos and Watson mouse brain atlas and sections were analyzed using a 100 × 1.3 Plan Apo oil immersion objective. A guard height of 5 μm was used for sampling brick depth of around 20 μm. Counts were taken at predetermined intervals (x = 150, y = 150) and with a counting frame of 86 × 86 μm (=7.396 μm2). For assessment of TH and GFAP immunoreactivity levels in striatum of mice, sections were digitized to grayscale pictures and analyzed for optical densities using ImageQuant 1.43 software (NIH). Data were corrected for tissue background staining by subtracting optical density values from fiber tracts within identical sections and presented as corrected optical density. All analyses were performed on a blinded basis.
Human samples
Striatum and olfactory bulbs of PD human brain samples (Suppl. Table 1) were supplied by the Parkinson’s UK Tissue Bank, funded by Parkinson’s UK, a charity registered in England and Wales (258197) and Scotland (SC037554) and analyses done in accordance with the Ethics Committee guidelines. Tissue was sequentially extracted and 25 μg of TBS and urea extracts used for immunoblot studies as described above. Olfactory bulb postmortem brain tissues from humans either without neurological disease and dementia with Lewy bodies (DLB) were obtained from the tissue bank of the Alzheimer’s Disease Research Center (ADRC) at the University of California at San Diego. Patients are listed in Supplementary Table 1.
Production of lentiviral (LV) constructs
Vector plasmids were constructed for the production of third generation LV vectors that expressed wild-type human α-syn (LV-α-syn) or the empty plasmid backbone as a control. The human CMV promoter was used to drive expression of the transgenes. Lentiviral vectors were generated by transient transfection with the vector and packaging plasmids in 293T cells as described previously [88].
Statistical analyses
Statistical analyses were conducted with GraphPadPrism version 6.0. Differences between means were analyzed by two-tailed t test and one-way ANOVA with post hoc tests as appropriate; p < 0.05 was considered significant.
Results
Hyperactivity caused by expression of mutant A30P in OB neurons is reversed by paraquat treatment
Treatment with paraquat was shown to induce dose-dependent neurotoxicity [6] and accumulation of α-syn in nigrostriatal brain regions of mice overexpressing wild-type α-syn [28, 53]; however, its impact on olfactory DAergic neurons is unknown. We used conditional transgenic mice presenting site-specific and doxycycline-responsive expression of familial PD-linked A30P α-syn in OB neurons, with its strongest expression in olfactory glomerular layer (GL) [71]. In our previous study, overexpression of α-syn reduced olfactory DA levels in absence of olfactory DAergic cell but increased midbrain DA, functionally associating with higher reactivity to novelty stress and aversive odor stimuli in our mice (Fig. 1a). This hyperactive phenotype is also commonly observed at an early time point in different α-syn transgenic mouse lines [30, 34, 43, 44, 81, 92, 106]. Additionally, increased striatal TH and locomotor response is observed in olfactory bulbectomized animal models of depression, possibly owing to direct connections between the striatum and areas of the limbic system, including the olfactory bulb and amygdala [80]. The amygdala is essential for perception and valence of stress [49] and thus may be implicated in the hyperdopaminergic transmission (Fig. 1a) either by direct stimulation of DAerig neurons [79] or indirectly by increasing steroid release through its projections to the hypothalamus, thereby upregulating TH synthesis and potentially DA production by a complex network interaction (Fig. 1a; see also discussion, [52, 54, 72]).
We therefore asked, whether a subtoxic paraquat dose would elicit olfactory DAergic cell-death and alternate the behavioral phenotype towards novelty stress in our conditional mice (Fig. 1b). Doxycycline (dox) treatment suppressed transgene expression in the respective neuronal layers without affecting the phenotype of non-transgenic control (Ntg) mice (Fig. 1c), [71]. We therefore used dox-treated A30P mice (A30P+D) as stringent controls allowing a direct relation of the observed changes to the expression of the human mutant α-syn. Mice were subsequently treated with either i.p. paraquat (A30P+P, A30P+D+P) or saline (A30P, A30P+D) (for schematic treatment paradigm, see Fig. 1d). We analyzed mice in the open field to test for differences in behavioral motivation and locomotor activity. First, mice were tested prior paraquat treatment to confirm the previously observed phenotype (OF1, Fig. 1e). At 12 months of age untreated A30P mice were clearly hyperactive (90 % increase when compared to Ntg controls, set as a baseline) and to respective dox-treated controls (A30P+D) (Fig. 1e). Similarly, rearing activity was increased, when compared to Ntg and A30P+D mice (Fig. 1e). After treatment with paraquat for 6 weeks (Fig. 1b) animals were re-tested in the open field (OF2). Planned comparisons to study the paraquat effect in A30P transgenic mice (A30P+P) showed a dramatic reduction of horizontal and rearing activity (approx. −120 %, p < 0.001) when compared to untreated A30P mice and dox-treated controls (p = 0.01; Fig. 1e). Rearing showed a mild, however, non-significant reduction compared to A30P+D (p = 0.07), probably owing to repeated testing. Thus our data imply that paraquat treatment opposed the higher active phenotype of A30P mice. We did not observe differences in thigmotaxis, expressed as the ratio center:periphery activity, suggesting that the score of locomotor performance was not influenced by differences in anxiety among groups.
Paraquat treatment reverses the increase in nigral-striatal TH immunoreactivity caused by expression of mutant A30P in the OB and leads to OB TH-cell loss
Increased striatal DAergic markers were reported to cause higher activity to mild stress (novelty, footshock) in animal models of neuropsychiatric disorders, such as bulbectomized rats as model of depression [57], but also in mice with widespread expression of human α-syn [30, 34, 43, 44, 81, 92, 106]. To estimate whether alterations in tyrosine hydroxylase (TH) immunoreactivity associate with the phenotype, DAergic cell counts in the olfactory GL and substantia nigra (SN) were performed on transgenic and control animals with and without paraquat treatment using stereological estimations of TH-positive neurons. Additionally, density of TH-positive nerve fibers in the striatum was estimated (Fig. 2e, f). A significant loss of TH neurons was observed in the outer GL in OB sections of A30P+P mice (Fig. 2a, b). This was associated by an increase in astrogliosis (Fig. S1a, b). Olfactory TH was not significantly reduced in untreated A30P+P (Fig. 2a, b), which was in accordance to our previous observation [71]. We did not observe significant TH-cell loss in paraquat-treated controls (A30P+D+P), suggesting that expression of human α-syn selectively increases vulnerability of GL DAergic neurons towards the used low-paraquat dose.
Fig. 2.

Paraquat-induced degeneration of olfactory DAergic neurons reverses abnormalities in nigrostriatal TH immunore-activity in A30P mice. a, b The stereologically estimated total number of DAergic neurons (detected by TH immunohistochemistry) showed olfactory TH-cell loss in paraquat-treated tg mice (A30P+P) when compared to respective controls (A30P+D). c, d Reduction of the increased TH immunoreactivity of TH-cell counts and e, f striatal fibers was observed by paraquat treatment in tg mice when compared to untreated A30P mice. No effect of paraquat was detected in non-expressing controls (A30P+D; A30P+D+P). For visual clarity, images were converted into greyscale. The data are expressed as mean ± SEM. *p < 0.05 vs. A30P+D; §p < 0.05; §§p < 0.01 vs. A30P. Scale bar 25 μm in a and c, 15 μm in e
Although A30P mice had no expression in nigrostriatal neurons, they had significantly increased TH-positive neurons (40 %; Fig. 2c, d) and TH-positive fibers (140 %; Fig. 2e, f) than A30P+D controls. In contrast, TH levels were lower in A30P+P mice (Fig. 2c–f). However, paraquat treatment did not lead to significant differences in TH immunoreactivity compared to respective controls (A30P+D, A30P+D+P), suggesting that TH reduction is not related to cell-death. Taken together, the altered levels of TH in OB and nigrostriatal system strongly suggest that changes observed in both brain regions are directly related to olfactory A30P expression, despite its topographically restricted expression in OB neurons, and are opposed by paraquat treatment.
α-Syn insolubility and C-terminal truncation in paraquat-treated A30P mice and in PD
Structural alterations in GL TH-cell numbers of A30P+P mice could result from α-syn-induced aggregation pathology, which correlates well with accumulation of full-length and C-terminally truncated (CT) α-syn [11, 46, 48, 70]. To address this question, we next estimated levels of soluble and insoluble α-syn in OB and striatum of A30P+P mice, utilizing antibodies against full-length and truncated human (105 antibody; [31] and total α-syn (syn-1 antibody; [76]. As expected, human α-syn as well as increased levels of total α-syn was detected in OB only (Fig. 3a, b), confirming histological data showing site-specific human α-syn in A30P mice (Fig. 1a). A30P+P mice showed an increase in full-length and truncated α-syn species (Fig. 3c, d). Mouse brains were also subjected to sequential extractions to evaluate presence of insoluble α-syn aggregates as previously published [70, 82, 89]. After urea extraction, a strong accumulation of 14-kDa full-length α-syn and also to a lesser extent the 12-kDa CT α-syn was detected by immunoblotting in A30P+P mice compared to either paraquat or dox-treated controls (Fig. 3e). Insoluble α-syn was neither detected within the striatum (Fig. 3e) nor the nigral brain region (Fig. S2a). To assess the oligomeric state of α-syn without a potential breakdown of its assemblies by detergents, protein extracts were subjected to native gel electrophoresis. A30P and A30P+P mice revealed non-denaturated ~60 to 70 kDa α-syn as the main species in the buffer-soluble fraction (Fig. 3f). Relatively strong α-syn signals were also found at a higher molecular level in A30P+P mice, in addition to lower migrating signals with a size distribution pattern between 35 and 60 kDa; however, the latter were also detected at around equal levels in dox-treated controls suggesting an unspecific cross-reactivity of the 105 antibody (Fig. 3f). Since complex protein conformations may display a higher electrophoretic mobility compared to unbound structures in native gels [86], we performed calpain digestion of recombinant α-syn to analyze migration pattern of truncated and full-length α-syn. Under these conditions the full-length, monomeric α-syn migrated at ~60 kDa. Additional signals consisting of numerous calpain-specific fragments migrated below the monomeric form at around 10–40 kDa (Fig. 3g). These signals were not seen when recombinant α-syn was incubated with calpain-1 in the presence of calpeptin, an inhibitor (I, Fig. 3g). Although native gel techniques are not quantitative, it suggests that higher oligomeric α-syn forms appear to be more represented in OB of A30P+P mice. Together, these data suggest that paraquat treatment increased truncation and assembly of human α-syn selectively in OB neurons.
Fig. 3.

Biochemical characterization of α-syn pattern reveals paraquat-dependent increase in oligomeric and CT α-syn. a, b Immunoblots from western blot analysis of OB and striatal extracts from non-transgenic (WT), A30P and paraquat-treated A30P+P and dox-treated control (A30P+D+P, A30P+D) mice. Antibodies that either recognized full-length (FL) and C-terminal truncated (CT) human α-syn (105) or total (murine and human) α-syn (syn-1) showed that human α-syn was only present in OB tissue. c Quantification of immunoblots showed that A30P+P mice present an increase of TBS-soluble FL α-syn and d CT α-syn. A series of ultracentrifugation and extraction steps were used to obtain urea-insoluble fractions of FL α-syn and CT α-syn. A faint CT α-syn signal in urea extracts was solely present in A30P+P mice. e WT and dox-treated (A30P+D+P, A30P+D) control mice only showed minor levels of urea-insoluble α-syn. In contrast, quantification of urea-fractions of A30P+P mice revealed strong increase of insoluble α-syn when compared to untreated A30P mice. f Native PAGE. α-Syn at ~60 to 70 kDa and higher molecular species accumulate in A30P+P mice. Bands below the main species may present truncated α-syn or cross-reaction of 105 antibody. g Left SDS-PAGE/Western blot (antibody syn-1) analysis of recombinant α-syn digested with calpain-1 for 0, 10, 30 and 60 min, and with recombinant α-syn incubated with calpain-1 and calpeptin, an inhibitor (I). Right same samples were subjected to native PAGE/ Western blot (antibody syn-1) showing main ~55 to 60 kDa monomeric α-syn (FL-Syn) and below calpain-1 cleaved α-syn fragments (CT-Syn). Immunoblots are highlighted by separating boxes for clarity of display. The data are expressed as mean ± SEM. **p < 0.01, ***p < 0.001 vs. WT; §p < 0.05 vs. A30P; +p < 0.05, ++p < 0.01, +++p < 0.001 vs. A30P+P
Sequential extraction of human brains with LB pathology revealed accumulation of insoluble full-length and CT α-syn species [1, 46, 70, 82, 89] that may promote aggregation pathology by recruiting soluble α-syn into aggregates [48, 62]; however, less is known about pattern of α-syn in human OB. To determine whether this insolubility pattern, as also observed in A30P+P mice, consistently applies for OB in PD and DLB, we compared soluble and insoluble α-syn levels in human striatal and the OB tissues (see also Table S1). Immunoblots were probed with the antibody 105, which displays a high preference to CT α-syn, when compared with immunosignals gained from the syn1 antibody, recognizing an epitope at aa 91–96, thereby harboring specificity to the same target-antigen (Fig. S2b), [31].
In the soluble fraction, full-length and truncated α-syn was readily detected in both tissues (Fig. 4a, d) and co-migrated with calpain-cleaved recombinant α-syn (Fig. 4d). Significant accumulation of full-length soluble α-syn was only detected in striatal tissues of PD and DLB patients when compared to control brain (Fig. 4b, d) and may relate to its accumulation at striatal synapses; whereas, soluble CT α-syn accumulated in both tissues analyzed. The α-syn in PD and DLB patients was highly insoluble in the OB and striatum when compared with that in control brains (Fig. 4a, d). In addition to the accumulation of full-length insoluble α-syn, a similar strong accumulation of CT α-syn was observed in the OB of PD patients (Fig. 4a, c) compared to the striatum of PD and DLB patients (Fig. 4d, f). Thus, α-syn insolubility pattern in OB of PD shares hallmark features detected in striatal PD and DLB tissues allowing us to suggest that its presence correlates with pathologic aggregates.
Fig. 4.

α-Syn pattern in PD OB tissue resembles biochemical changes within striatum of PD and DLB brain. a Western blots show α-syn in the TBS-soluble and urea-insoluble fraction in OBs of PD and DLB brain. b, c Antibody specific for full-length and (calpain-cleaved) CT α-syn was used and revealed relative strong signals of insoluble α-syn in PD and DLB brain. Quantification of immunoblots showed a significant increase of insoluble Fl and CT α-syn in OB of PD patients. d Western blots show striatal protein extracts of α-syn in the TBS-soluble and urea-insoluble fraction of PD and DLB brain. d, e Quantification revealed an increase of soluble and insoluble FL and CT α-syn in striatum of PD and DLB patients. Immunoblots are highlighted by separating boxes for clarity of display. The data are expressed as mean ± SEM *p < 0.05, **p < 0.01
Paraquat-related increase of calpain-resistant α-syn in A30P mice
Accumulation of CT α-syn could result from elevated proteolytic cleavage by enzymes, such as calpain [26, 62], or from nonspecific breakdown due to detergents used in the SDS-PAGE. Since A30P+P mice displayed an increased amount of CT α-syn, co-migrating with calpain-cleaved recombinant α-syn, we hypothesized that an additional calpain treatment would accentuate its immunodominance. For this, we incubated sections of A30P+P mice and controls in presence of calpain. Postmortem olfactory brain sections from a human control and a PD patient were included to compare immunoreactivity in human disease condition and the animal model. The presence of calpain-resistant fragments was then analyzed with an antibody with high preference against CT α-syn (105 antibody [31]) and compared with the 15G7 antibody, exhibiting decreased C-terminal affinity [11, 70]. The 15G7 antibody recognizes a broader epitope ranging from aa 116–131. Therefore, calpain digestion at aa 122 not only cleaves the α-syn molecule, but also disrupts the antibody epitope (schemes in Fig. 2b, d). In A30P and A30P+P mice, significant reduction of immunoreactivity was observed in the GL of the OB when calpain-treated sections were stained with the 15G7 antibody compared to sections without treatment (Fig. 5a, b). In contrast, A30P+P had significantly higher levels of α-syn immunoreactivity after calpain treatment when compared to A30P mice (Fig. 5c, d). A similar pattern was detected in human olfactory sections, displaying a selective calpain-resistant 105 immunoreactivity in the PD patient. This α-syn pattern strongly suggests accumulation of calpain-cleaved α-syn in A30P+P mice and olfactory PD tissue as opposed to simple breakdown artifacts in the SDS-PAGE.
Fig. 5.

A30P+P mice show elevated levels of calpain-resistant α-syn in the GL. a, c Representative sections of GL from A30P and A30P+P mice with and without treatment of calpain-1 and, b, d quantitative results. a After calpain1 digestion, brain sections were immunostained with either 15G7 antibody, displaying a calpain-cleavage site within its main epitope or c 105 antibody, with high preference to calpain-cleaved CT α-syn and immunoreactivity quantified by densitometry. Schematic diagrams (b, d) indicate antibody epitopes and location of a major calpain 1 cleavage site at aa 122. ANOVA revealed a 105 antibody-specific and significant accumulation of calpain-resistant α-syn in A30P+P mice, indicating accumulation of CT α-syn. The data are expressed as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 vs. Ntg; §p < 0.05, §§p < 0.01 vs. A30P; +p < 0.05 A30P+calp vs. A30P+P+calp. Scale bar 25 μm
Formation of CT α-syn depends on calpain activity
We have previously demonstrated that accumulation of CT α-syn is associated with neuropathological alterations in transgenic animal brains and possibly generated by elevated proteolytic cleavage via calpain [31, 70]. In order to provide additional experimental evidence in support of calpain implicated in the detected OB synucleinopathy, we measured protein expression levels of calpain and calpastatin. Calpastatin is a selective inhibitor and substrate of calpain, and its formation into complexes is indicative for increased calpain activity [22, 86]. When compared to A30P+D mice, A30P and paraquat-treated mice showed increased expression levels of calpain (Fig. 6a) but not a decrease in calpastatin (data not shown); however, expression levels may not directly correlate with enzymatic activity. We therefore utilized native PAGE, enabling the detection of free and calpain-associated complex calpastatin (CAST) formations by its shift in electrophoretic mobility. CAST complex formations are thought to correlate with calpain activation [86]. The calculated ratio between free CAST and complex CAST revealed an increase in presence of the complex formation in A30P+P mice compared to A30P mice and Ntg controls (Fig. 6c, d), thus further suggesting calpain activation implicated in the formation of CT α-syn.
Fig. 6.

Effect of paraquat on calpain activation and accumulation of CT α-syn. a, b OB protein extracts from non-transgenic (WT), A30P and paraquat-treated A30P+P and dox-treated control (A30P+D+P, A30P+D) mice were loaded on a SDS-PAGE or c native PAGE. a, c Antibodies specific for calpain 1 and calpastatin (CAST), its specific inhibitor, were used. b Quantification of immunoblots shows that A30P+P mice present increased level of calpain-1 and d complex formation of CAST. The data are expressed as mean ± SEM. **p < 0.01 vs. Ntg, §p < 0.05 vs. A30P, +p < 0.05 vs. A30P+D. e Cell biological analysis of SH-SY5Y human neuroblastoma cells treated with increasing concentration of paraquat. Cell viability was assessed at 48 and 72 h after treatment with paraquat, with and without 10 μM of the calpain inhibitor calpeptin, using LDH assay. f Non-cytotoxic concentration (150 μM) was used to analyze paraquat-induced activation of calpain and g, h a correlating formation of CT α-syn. f Increase in calpain activity in SH-SY5Y cells exposed to 50, 150 μM of paraquat was reduced by parallel treatment with calpeptin (10 μM) for 72 h. g Representative immunoblot of SH-SY5Y with 105 antibody against human full-length and CT α-syn shows increase of endogenous full-length and CT α-syn. Infecting cells with lentiviral human α-syn (+LV Syn) was used as a positive control and showed expected relative strong reactivity of 105 against full-length and CT α-syn. Calpeptin, a specific inhibitor (Inh) of calpain treatment reduced level of FL α-syn and CT α-syn. h Correlation between (fold) increase of CT α-syn and (fold) increase of calpain activity dependent on concentration of paraquat. Immunoblots are highlighted by separating boxes for clarity of display. The data are expressed as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 vs. WT; §p < 0.05, §§p < 0.01 vs. A30P; +p < 0.05, A30P+calp vs. A30P+P+calp
We next defined, whether exposure to various amounts of paraquat concentrations, correlated with calpain-induced accumulation of CT α-syn. We used SH-SY5Y neuroblastoma cells as an in vitro model of PD DAergic neurons. The cells were first exposed to increasing concentrations of paraquat (0–300 μM) for 72 h to evaluate cytotoxicity by measurement of LDH activity in the cell culture medium, showing that paraquat induced cell death at 300 μM (Fig. 6e). Additional treatment with calpeptin, an inhibitor of calpain, increased viability at 150 and 300 μM after 48 h of exposure. However, inhibition of calpain was not sufficient to reduce the strong cytotoxic effect at a dosage of 300 μM at 72 h; indicating a dose- and time-dependent response. We therefore used non-toxic concentrations ≤150 μM to further investigate the effects on calpain activity and α-syn pattern. We identified that 150 μM of paraquat led to a significant increase in calpain activity (Fig. 6e) and that co-treatment of cells with calpeptin blocked the observed effect (Fig. 6f). Moreover, exposure to increasing concentrations of paraquat led to increased levels of endogenous full-length and CT α-syn. This pattern was substantiated when cells were additionally treated with lentivirus-overexpressing human α-syn, which was used as a positive control (Fig. 6g). Analysis of calpain and CT α-syn formation showed a significant correlation dependent on the paraquat concentration (Fig. 6h), further indicating impact of calpain cleavage in truncation of α-syn in OB of A30P+P mice.
Paraquat-induced calpain activation impairs autophagic pathways
Calpain regulates basal activity of autophagy via degradation of ATG12-ATG5 conjugate, required for proper formation of autophagosomal membranes [105]. We therefore analyzed, whether paraquat-induced elevation of calpain activity impaired autophagy, thereby contributing to the detected high α-syn levels via reduced clearance of aggregated α-syn. Electron microscopical images of the OB from A30P+P mice demonstrated an abnormal accumulation of autophagosomes (Fig. 7a). This accumulation may reflect either an increased formation of autophagosomes or reduction of lysosomal degradation. To differentiate between these two possibilities, we additionally performed double-labeling immunofluorescence and Western blot analyses of autophagolysosome marker in OB and striatum of A30P+P mice and respective controls. A30P+P mice displayed a relatively strong accumulation of α-syn with LC3 in OB periglomerular cells (Fig. 7b). On biochemical level, paraquat treatment substantially increased free ATG12, LC3-II and Lamp2A (Fig. 7c, d). LC3-II and Lamp2A are constituents of the autolysosome membrane and eventually degraded by lysosomal hydrolases [19, 37]. Thus, the detected increased levels further suggest reduced autolysosomal clearance. Thus, it appears that impaired autophagic clearance mechanisms lead to higher α-syn levels in OB neurons of A30P+P mice.
Fig. 7.

Reduced autophagolysosome clearance in OB of A30P+P mice. a Representative electron micrographs of GL from A30P+P mice. Clustered autophagosomal structures with higher electron density were increased in A30P+P mice. Right number of autophagosomes per cell profile (50 profiles per group). b Double-fluorescence confocal photomicrographs showed a relative strong accumulation of LC3 immunoreactive puncta colocalizing with human α-syn in A30P+P mice (see also inset) when compared to A30P or dox-treated controls. c Protein extracts from OB and striatum increase detected with antibodies against membrane proteins of the autolysosme. d Quantification revealed increase in free ATG12, and autolysosome membrane marker Lamp2A and reduced clearance of autolysosome substrate LC3-II in OB neurons of A30P+P mice. Immunoblots are highlighted by separating boxes for clarity of display. The data are expressed as mean ± SEM. **p < 0.01 vs. Ntg, §p < 0.05 vs. A30P, +p < 0.05, ++p < 0.05, vs. A30P+D. Scale bar 1 μm in a, 10 μm in b and 5 μm in inset
Ultrastructural analysis of the effects of paraquat in OB periglomerular cells in A30P mice
In the light of the effects of paraquat on TH neuronal cell numbers along with increased activation of calpain and inhibition of autophagy in the olfactory bulb, we further analyzed the ultrastructure of periglomerular cell profiles of A30P+P mice. Following paraquat treatment, ultrastructural analysis revealed accumulation of autophagic vacuoles and multivesicular bodies, which may further be indicative for impaired autophagy pathways (Fig. 8a–c). Other forms of pathological changes in dendritic terminals included the evidence of mitochondrial condensation and damage (Fig. 8e, f), whereas this morphological alteration was not observed in dox-treated controls (Fig. 8d). Occasionally, synaptic and cytoplasmic mitochondria showed drastic morphological alterations, appearing swollen, with condensed inner and outer membranes and disrupted cristae (Fig. 8e, f). Immunogold electron microscopy was used to define the subcellular localization of human 105 α-syn, displaying preference for CT α-syn [31]. Immunolabeled α-syn was detected accumulating at dendritic terminals and often translocalized within damaged mitochondria, showing either subtle or strong signs of dark degeneration in A30P+P mice (Fig. 8g–i).
Fig. 8.

Ultrastructural characterization of the effects of paraquat treatment in A30P mice a Exposure to paraquat leads to dendrite abnormalities, such as presence of numerous multilaminar bodies (indicated by triangles), multivesicular bodies (mvb) and damaged mitochondria (mit). b, c In some cases signs of synaptic dendrite degeneration are more pronounced, displaying dark-condensed structures, reminiscent of mitochondria that are undergoing autophagocytosis (indicated by asterisk). d In A30P+D controls mitochondria with regular shape and size are observed. e, f Dendritic terminal showing abnormal mitochondria with severe disruption of mitochondrial cristae and central vacuolization (arrows), sometimes associated with multilaminar bodies (f). g Using immunogold labeling technique, scattered immunoparticles localizing human α-syn 105 (arrowhead) are abundant in the cytoplasm of periglomerular cells, intermixed with numerous electrodense structures (asterisk). h Human α-syn 105 immunoparticles are localized to normal mitochondria and i mitochondria with prominent disruption of mitochondrial cristae. Scale bar 100 nm in i, 250 nm in d–f, 400 nm in a–c, g, h
Discussion
It is not known whether olfactory α-syn pathology may contribute to the development of widespread α-syn aggregation and progressive degeneration of the DAergic nigrostriatal system in PD brains and whether a selective vulnerability of olfactory neurons initiates the disease process. To address these questions, we studied transgenic mice with spatially restricted overexpression of mutant A30P primarily in OB neurons exposed to a low dose of the herbicide paraquat. Here we show, that selective death of OB DAergic neurons responsible for a stress-related hyperactive phenotype can result from a combination of multiple hits resulting from the elevated activity of calpain that adversely affects protein clearance mechanisms, inducing the accumulation of full-length insoluble and calpain-cleaved CT α-syn and its conversion into oligomeric structures. The pathological activation of calpain may originate from a paraquat-induced oxidative stress response, leading to lipid peroxidation and elevation of Ca2+ levels [20, 33] (Fig. 9).
Fig. 9.
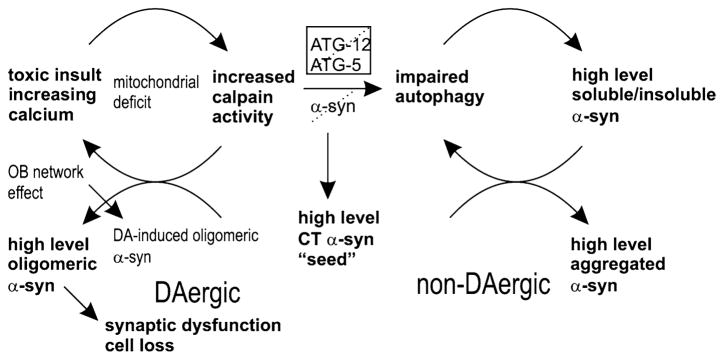
Schematic representation of the proposed hypothetical cascade for DAergic vulnerability in PD. Given that environmental toxins, such as paraquat, interfere with calcium homeostasis via mitochondrial damage, the resulting increase in calpain activity may lead to an inhibition of autophagy and increased generation of CT α-syn, “seeding” α-syn co-assembly. In nigrostriatal DAergic neurons, an early increase of TH and associated DA production, which is triggered by OB α-synucleinopathy, may incite α-syn oligomerization at synaptic membranes. Additional accumulation of CT α-syn may further promote its assembly, culminating in synaptic dysfunction and cell loss
We also found that the degeneration of OB DAergic neurons opposed the higher striatal DAergic tone and higher reactive phenotype. These findings support a new model of progressive α-synucleinopathy, based on the functional interplay between the olfactory and striatal DAergic circuits, that may render striatal synapses vulnerable to DA-induced α-syn oligomerization in early PD. Therefore, low doses of paraquat may first mediate α-syn accumulation in OB neurons, triggering upregulation of striatal TH and a consecutive DA-induced striatal synaptic deficit that may further contribute to the progressive phenotype (Fig. 9).
Potential involvement of an olfactory-striatal interplay in early PD
In PD the OB is one of the first brain regions to contain α-syn aggregates [4] and a strong correlation of olfactory synucleinopathy with advanced stages in LB disease [84] favor the hypothesis, that olfactory synucleinopathy may contribute to disease progression. Additionally, the OB can serve as a suitable entry point for environmental toxins, such as paraquat or rotenone, contributing to PD pathogenesis. Environmental toxins possibly enter the brain via olfactory sensory neurons that converge onto postsynaptic targets within specialized structures, so-called glomeruli (GL). The GL are distributed as a layer and encircled by periglomerular DAergic neurons near the olfactory surface. The GL has reciprocal connections to the mitral cell layer and mediates a lateral inhibition on mitral cell activity [77]. A higher activation of mitral cells was hypothesized to influence the capacity to discriminate between odors, being impaired at a low DAergic tone [3, 102]. Consistently, DA and TH reduction via olfactory deprivation was related to higher activation of mitral cells [103], which is in turn associated with an increased alertness in rodents [12]. Our previous and present results confirmed a relationship between a physiological reduction of olfactory DAergic marker with increased reactivity of mice towards novelty stress, caused by site-specific overexpression of α-syn [(Fig. 1), [71]]. Although olfactory TH-positive cell numbers in PD are still a matter of debate [36, 40, 84], these cells may reflect a compensatory response to striatal DA dysfunction [2, 104]. We previously demonstrated that overexpression of human mutant α-syn reduced DA level and TH immunoreactivity [71]. Additionally, its constant high expression levels due to its overexpression via an artificial promoter may hinder a compensatory genesis of new DAergic olfactory neurons [56, 59]. This is in line with previous studies, suggesting an inverse correlation of α-syn overexpression on DAergic function [14, 68, 75, 99]. Here, we found that in untreated A30P mice, striatal and nigral TH immunoreactivity was increased (Fig. 2), suggesting a relationship between the hyperactive phenotype and increased TH expression levels. Consistently, several transgenic mice with widespread α-syn expression display a higher reactive phenotype and an associated increase of striatal DAergic marker at early ages, which is eventually normalized and opposed at old age [43, 44, 81, 106]. The similarities in striatal DAergic tone suggest two possibilities. First, the elevated DA level emerges in relation to the overexpression of α-syn. However, in particular its normalization in absence of cellular pathology albeit permanent transgenic overexpression in mid-aged mice argues against a possible physiological role of human α-syn. Second, higher DA levels may be caused indirectly by α-syn-induced pathology in other brain regions, suggesting that the striatal DAergic tone may be part of a broader network regulation. In support of this hypothesis, elevated DA levels and higher reactivity to novelty stress are also present in animals with olfactory bulbectomy [57]. Indeed, glucocorticoids, the primary humoral effector of the physiological response to stress, can upregulate TH synthesis and thereby potentially also regulate DA production [52, 54, 72]. In addition, perinatal exposure of glucocorticoids increased the DAergic cell numbers [60] and responsiveness of the adult hypothalamo-pituitary-adrenal (HPA) axis [98].
It has long been suggested that DA itself might be the culprit of PD, by inciting striatal α-syn oligomerization that may disrupt membrane integrity [10, 16, 73, 94]. Therefore, olfactory α-synucleinopathy could contribute to deficits in the striatal DA function in early PD without α-syn spreading from the olfactory bulb into the nigrostriatal system (see also Fig. 9), which however deserves future studies.
Treatment with a low dose of paraquat did not alter TH immunoreactivity in animals without transgenic expression in the present study, supporting the need for α-syn over-expression as an additional stressor of DAergic cell-death (Fig. 2); this is consistent with previous data showing that α-syn and exposure to paraquat can synergistically lead to DAergic degeneration [27, 28, 69]. Additionally, TH immunoreactivity and hyperactivity was reduced in dependence of the paraquat-induced olfactory cell-death in A30P mice, thus further indicating a potential regulative role of the OB in the higher reactive phenotype.
Dependence of neurotoxicity on CT α-syn
Mutations in the α-syn gene have been identified to cause familial PD; in particular, triplication of the SNCA locus [85] was correlated with an early-onset and a rapid progressive DAergic phenotype [83], suggesting a toxic gain-of-function related to high protein levels. Consistently, α-syn-deficient mice display reduced susceptibility to environmental neurotoxins [21]. A common finding of paraquat treatment in vivo and in vitro is its ability to increase α-syn level and fibrillization in DAergic neurons [28, 53, 93]. Recently, Wills and colleagues (2011) demonstrated that paraquat-induced accumulation of α-syn was accompanied with its decreased degradation via proteasomal and autophagic pathways. The increase in insoluble α-syn probably reflects altered protein clearance mechanisms, which is in accord to our and other previous studies, that α-syn fibrils are preferentially degraded via autophagy [11, 97]. Interestingly, calpain, a calcium-dependent cysteine protease involved in PD [64] and other neurodegenerative diseases [96] is activated by a series of neurotoxins, including paraquat [107]. Calpain regulates basal autophagy by cleavage of the ATG12-ATG5 conjugate [105]. This conjugate is of particular interest, owing to its participation in protein quality control after exposure to oxidative stress in cellular models [51] and its role in incorporation of α-syn into the autophagosome [97]. We show that a subtoxic dose of paraquat readily induces calpain activity in olfactory DAergic neurons, which was associated with an increase of free ATG12, accumulation of autophagosomal membrane proteins and autophagosomal structures. Consistently, autophagic vacuoles, mitochondrial autophagy and damage were observed at olfactory dendritic terminals in A30P+P mice. Therefore, paraquat toxicity, which is known to induce lipid peroxidation of mitochondrial membranes [9], is potentially mediated via defective autophagy/mitophagy in A30P+P mice. Notably, paraquat toxicity is neither mediated via the DA transporter nor does it inhibit mitochondrial complex I [9], thus the selective vulnerability of DAergic mitochondria towards paraquat toxicity [87] is not clear. However, recent studies suggest localization of α-syn to mitochondria under normal and stress conditions, inhibiting complex I [8, 61, 62]. Therefore, it may be speculated that mitochondrial α-syn as detected in olfactory nerve terminals (Fig. 8) aggravates mitochondrial dysfunction and therefore dopaminergic cytotoxicity.
Additionally, α-syn is a substrate of calpain 1 and its proteolytic cleavage alters α-syn fibrillogenesis: whereas, calpain fragments of soluble α-syn did not increase fibrillization, cleavage of fibrillized α-syn resulted in generation of ~10 to 12 kDa CT α-syn fragments, inducing co-assembly of soluble full-length α-syn [62]. We demonstrated a significant accumulation of CT α-syn by biochemistry and immunostaining in paraquat-treated A30P mice, using the 105 antibody with high preference to CT α-syn [31]. The CT α-syn fragment was shown to co-migrate with calpain 1 cleaved recombinant α-syn peptide in our present (Fig. 4, Fig. S2) and previous studies [31, 70], thus adding further evidence for a pathophysiological role of calpain 1 in PD [32, 41]. Its accumulation is in accord with the detected accumulation in the OB and the striatum of PD patients (Fig. 4). In addition, calpain-specific and therefore resistant α-syn structures accumulated in calpain 1-pretreated tissue sections of A30P+P mice and PD patients. Importantly, the increased immunoreactivity was detected using the 105 antibody, but not with the 15G7 α-syn antibody; the latter is known for its decreased binding capacity towards CT α-syn [7, 11, 70]. Taken together, this strongly suggests accumulation of calpain 1-cleaved α-syn in A30P+P mice as opposed to breakdown artifacts in the SDS-PAGE. Indeed, owing to the presence of CT α-syn in LB [1, 26] and its capacity to aggravate aggregation pathology in vitro [46] and in vivo [91] it is widely believed that CT of α-syn plays a central role in α-syn aggregation pathology.
Using native gels, we detected an increase in higher oligomeric forms in paraquat-treated A30P mice (Fig. 3). Notably, recombinant α-syn fragments generated by cal-pain 1-cleavage, thus, likely disrupting secondary and tertiary structures, migrated at a relative higher molecular mass in native gels, arguing against folded multimers. Interestingly, Chinta and co-workers (2010) found that decreased clearance of α-syn correlated with its presence as monomer and oligomers in defective mitochondria.
Therefore, we propose a model for activation of calpain may contribute to the observed significant neurotoxicity in a sequential manner by inhibition of autophagic clearance pathways, leading to an increase in insoluble α-syn, which in turn results in CT α-syn, promoting additional oligomerization, mitochondrial translocalization and damage, culminating in cell-death (Fig. 9).
Conclusion
Previous studies have highlighted the adverse effect of calpain on α-syn aggregation and its strong presence in core of LB formations [67], suggest a possible role as “seed” of protein aggregation. Our findings provide a direct link between high calpain activation, accumulation of insoluble full-length and CT α-syn and its conversion into oligomeric structures as part of a synergistic effect between α-syn and environmental toxins that may underlie the selective vulnerability of DAergic neurons.
In light that there is yet no effective drug therapy for PD, alternative approaches, such as stem cell or trophic factor gene therapy, are of increasing interest [42, 47]. The effectiveness of these therapies, however, depends on whether of PD is initiated by a cellular dysfunction of multiple brain regions in parallel or in sequence. DA itself is one of the most potent inducers of α-syn oligomerization [10], and nigral cell-death [63]. Thus, if olfactory α-synucleinopathy is one of the brain regions that regulate striatal DAergic activity throughout a functional network, interfering with its expression level may halt the progressive nature of the disease. Since α-syn was shown to form pathological aggregates that spread perhaps through dysfunctional synapses [95], it will be interesting to assess the effects of nigral human α-syn overexpression under condition of endogenously elevated DA level.
Supplementary Material
Acknowledgments
PD human and control brain samples were supplied by the Parkinson’s UK Tissue Bank, funded by Parkinson’s UK, a charity registered in England and Wales (258197) and Scotland (SC037554), from the tissue bank of the Alzheimer’s Disease Research Center (ADRC) at the University of California at San Diego and analyses done in accordance with the Ethics Committee guidelines. PrP-A30P mice were generated at the University of Tuebingen, Germany. S.N. was awarded with the fellowship of the German Parkinson’s Society. The work was supported by National Institutes of Health grants AG 18440, AG022704, NS057096 and AG5131to E.M, and by NGFNplus 01GS08134 to O.R/S.N.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00401-014-1255-5) contains supplementary material, which is available to authorized users.
Contributor Information
Silke Nuber, Email: snuber@ucsd.edu, Department of Neurosciences, University of California San Diego, 9500 Gilman Dr., MTF 344, La Jolla, CA 92093-0624, USA.
Daniel Tadros, Department of Neurosciences, University of California San Diego, 9500 Gilman Dr., MTF 344, La Jolla, CA 92093-0624, USA.
Jerel Fields, Department of Pathology, University of California San Diego, 9500 Gilman Dr., La Jolla, CA 92093-0624, USA.
Cassia Rose Overk, Department of Neurosciences, University of California San Diego, 9500 Gilman Dr., MTF 344, La Jolla, CA 92093-0624, USA.
Benjamin Ettle, Department of Molecular Neurology, Friedrich-Alexander-University, Erlangen-Nuremberg, Germany.
Kori Kosberg, Department of Neurosciences, University of California San Diego, 9500 Gilman Dr., MTF 344, La Jolla, CA 92093-0624, USA.
Michael Mante, Department of Neurosciences, University of California San Diego, 9500 Gilman Dr., MTF 344, La Jolla, CA 92093-0624, USA.
Edward Rockenstein, Department of Neurosciences, University of California San Diego, 9500 Gilman Dr., MTF 344, La Jolla, CA 92093-0624, USA.
Margarita Trejo, Department of Neurosciences, University of California San Diego, 9500 Gilman Dr., MTF 344, La Jolla, CA 92093-0624, USA.
Eliezer Masliah, Department of Neurosciences, University of California San Diego, 9500 Gilman Dr., MTF 344, La Jolla, CA 92093-0624, USA. Department of Pathology, University of California San Diego, 9500 Gilman Dr., La Jolla, CA 92093-0624, USA.
References
- 1.Anderson JP, Walker DE, Goldstein JM, et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem. 2006;281:29739–29752. doi: 10.1074/jbc.M600933200. [DOI] [PubMed] [Google Scholar]
- 2.Bedard A, Parent A. Evidence of newly generated neurons in the human olfactory bulb. Brain Res Dev Brain Res. 2004;151:159–168. doi: 10.1016/j.devbrainres.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 3.Berkowicz DA, Trombley PQ. Dopaminergic modulation at the olfactory nerve synapse. Brain Res. 2000;855:90–99. doi: 10.1016/s0006-8993(99)02342-2. [DOI] [PubMed] [Google Scholar]
- 4.Braak H, Del Tredici K, Rub U, et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 5.Brodoehl S, Klingner C, Volk GF, et al. Decreased olfactory bulb volume in idiopathic Parkinson’s disease detected by 3.0-tesla magnetic resonance imaging. Mov Disord. 2012;27:1019–1025. doi: 10.1002/mds.25087. [DOI] [PubMed] [Google Scholar]
- 6.Brooks AI, Chadwick CA, Gelbard HA, et al. Paraquat elicited neurobehavioral syndrome caused by dopaminergic neuron loss. Brain Res. 1999;823:1–10. doi: 10.1016/s0006-8993(98)01192-5. [DOI] [PubMed] [Google Scholar]
- 7.Buchman VL, Ninkina N. Modulation of alpha-synuclein expression in transgenic animals for modelling synucleinopathies–is the juice worth the squeeze? Neurotox Res. 2008;14:329–341. doi: 10.1007/BF03033857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cali T, Ottolini D, Brini M. Mitochondria, calcium, and endoplasmic reticulum stress in Parkinson’s disease. Biofactors. 2011;37:228–240. doi: 10.1002/biof.159. [DOI] [PubMed] [Google Scholar]
- 9.Cali T, Ottolini D, Negro A, et al. alpha-Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J Biol Chem. 2012;287:17914–17929. doi: 10.1074/jbc.M111.302794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cappai R, Leck SL, Tew DJ, et al. Dopamine promotes alpha-synuclein aggregation into SDS-resistant soluble oligomers via a distinct folding pathway. FASEB J. 2005;19:1377–1379. doi: 10.1096/fj.04-3437fje. [DOI] [PubMed] [Google Scholar]
- 11.Casadei N, Pohler AM, Tomas-Zapico C, et al. Overexpression of synphilin-1 promotes clearance of soluble and misfolded alpha-synuclein without restoring the motor phenotype in aged A30P transgenic mice. Hum Mol Genet. 2014;23:767–781. doi: 10.1093/hmg/ddt467. [DOI] [PubMed] [Google Scholar]
- 12.Cattarelli M. The role of the medial olfactory pathways in olfaction: behavioral and electrophysiological data. Behav Brain Res. 1982;6:339–364. doi: 10.1016/0166-4328(82)90017-1. [DOI] [PubMed] [Google Scholar]
- 13.Choi BK, Choi MG, Kim JY, et al. Large alpha-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc Natl Acad Sci USA. 2013;110:4087–4092. doi: 10.1073/pnas.1218424110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chu Y, Kordower JH. Age-associated increases of alpha-synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: is this the target for Parkinson’s disease? Neurobiol Dis. 2007;25:134–149. doi: 10.1016/j.nbd.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 15.Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med. 1998;4:1318–1320. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- 16.Conway KA, Rochet JC, Bieganski RM, et al. Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science. 2001;294:1346–1349. doi: 10.1126/science.1063522. [DOI] [PubMed] [Google Scholar]
- 17.Cookson MR, van der Brug M. Cell systems and the toxic mechanism(s) of alpha-synuclein. Exp Neurol. 2008;209:5–11. doi: 10.1016/j.expneurol.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crews L, Spencer B, Desplats P, et al. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS One. 2010;5:e9313. doi: 10.1371/journal.pone.0009313. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19.Cuervo AM, Dice JF. Regulation of lamp2a levels in the lysosomal membrane. Traffic. 2000;1:570–583. doi: 10.1034/j.1600-0854.2000.010707.x. [DOI] [PubMed] [Google Scholar]
- 20.Daher JP, Ying M, Banerjee R, et al. Conditional transgenic mice expressing C-terminally truncated human alpha-synuclein (alphaSyn119) exhibit reduced striatal dopamine without loss of nigrostriatal pathway dopaminergic neurons. Mol Neurodegener. 2009;4:34. doi: 10.1186/1750-1326-4-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dauer W, Kholodilov N, Vila M, et al. Resistance of alpha-synuclein null mice to the parkinsonian neurotoxin MPTP. Proc Natl Acad Sci USA. 2002;99:14524–14529. doi: 10.1073/pnas.172514599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Tullio R, Cantoni C, Broggio C, et al. Involvement of exon 6-mediated calpastatin intracellular movements in the modulation of calpain activation. Biochim Biophys Acta. 2009;1790:182–187. doi: 10.1016/j.bbagen.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 23.Di Monte D, Sandy MS, Ekstrom G, et al. Comparative studies on the mechanisms of paraquat and 1-methyl-4-phenylpyridine (MPP+) cytotoxicity. Biochem Biophys Res Commun. 1986;137:303–309. doi: 10.1016/0006-291x(86)91210-6. [DOI] [PubMed] [Google Scholar]
- 24.Doty RL. The olfactory vector hypothesis of neurodegenerative disease: is it viable? Ann Neurol. 2008;63:7–15. doi: 10.1002/ana.21327. [DOI] [PubMed] [Google Scholar]
- 25.Doty RL. Olfaction in Parkinson’s disease and related disorders. Neurobiol Dis. 2012;46:527–552. doi: 10.1016/j.nbd.2011.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dufty BM, Warner LR, Hou ST, et al. Calpain-cleavage of alpha-synuclein: connecting proteolytic processing to disease-linked aggregation. Am J Pathol. 2007;170:1725–1738. doi: 10.2353/ajpath.2007.061232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feng LR, Maguire-Zeiss KA. Dopamine and paraquat enhance alpha-synuclein-induced alterations in membrane conductance. Neurotox Res. 2011;20:387–401. doi: 10.1007/s12640-011-9255-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fernagut PO, Hutson CB, Fleming SM, et al. Behavioral and histopathological consequences of paraquat intoxication in mice: effects of alpha-synuclein over-expression. Synapse. 2007;61:991–1001. doi: 10.1002/syn.20456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fleming SM, Salcedo J, Fernagut PO, et al. Early and progressive sensorimotor anomalies in mice overexpressing wild-type human alpha-synuclein. J Neurosci. 2004;24:9434–9440. doi: 10.1523/JNEUROSCI.3080-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Freichel C, Neumann M, Ballard T, et al. Age-dependent cognitive decline and amygdala pathology in alpha-synuclein transgenic mice. Neurobiol Aging. 2007;28:1421–1435. doi: 10.1016/j.neurobiolaging.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 31.Games D, Seubert P, Rockenstein E, et al. Axonopathy in an alpha-synuclein transgenic model of Lewy body disease is associated with extensive accumulation of C-terminal-truncated alpha-synuclein. Am J Pathol. 2013;182:940–953. doi: 10.1016/j.ajpath.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gasser T. Molecular pathogenesis of Parkinson disease: insights from genetic studies. Expert Rev Mol Med. 2009;11:e22. doi: 10.1017/S1462399409001148. [DOI] [PubMed] [Google Scholar]
- 33.Gomez-Isla T, Irizarry MC, Mariash A, et al. Motor dysfunction and gliosis with preserved dopaminergic markers in human alpha-synuclein A30P transgenic mice. Neurobiol Aging. 2003;24:245–258. doi: 10.1016/s0197-4580(02)00091-x. [DOI] [PubMed] [Google Scholar]
- 34.Graham DR, Sidhu A. Mice expressing the A53T mutant form of human alpha-synuclein exhibit hyperactivity and reduced anxiety-like behavior. J Neurosci Res. 2010;88:1777–1783. doi: 10.1002/jnr.22331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hawkes CH, Shephard BC, Daniel SE. Olfactory dysfunction in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1997;62:436–446. doi: 10.1136/jnnp.62.5.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huisman E, Uylings HB, Hoogland PV. Gender-related changes in increase of dopaminergic neurons in the olfactory bulb of Parkinson’s disease patients. Mov Disord. 2008;23:1407–1413. doi: 10.1002/mds.22009. [DOI] [PubMed] [Google Scholar]
- 37.Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Katzenschlager R, Lees AJ. Olfaction and Parkinson’s syndromes: its role in differential diagnosis. Curr Opin Neurol. 2004;17:417–423. doi: 10.1097/01.wco.0000137531.76491.c2. [DOI] [PubMed] [Google Scholar]
- 39.Kertelge L, Bruggemann N, Schmidt A, et al. Impaired sense of smell and color discrimination in monogenic and idiopathic Parkinson’s disease. Mov Disord. 2010;25:2665–2669. doi: 10.1002/mds.23272. [DOI] [PubMed] [Google Scholar]
- 40.Kirik D, Rosenblad C, Burger C, et al. Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. J Neurosci. 2002;22:2780–2791. doi: 10.1523/JNEUROSCI.22-07-02780.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koprich JB, Johnston TH, Huot P, et al. Progressive neurodegeneration or endogenous compensation in an animal model of Parkinson’s disease produced by decreasing doses of alpha-synuclein. PLoS One. 2011;6:e17698. doi: 10.1371/journal.pone.0017698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kordower JH, Bjorklund A. Trophic factor gene therapy for Parkinson’s disease. Mov Disord. 2013;28:96–109. doi: 10.1002/mds.25344. [DOI] [PubMed] [Google Scholar]
- 43.Kurz A, Double KL, Lastres-Becker I, et al. A53T-alpha-synuclein overexpression impairs dopamine signaling and striatal synaptic plasticity in old mice. PLoS One. 2010;5:e11464. doi: 10.1371/journal.pone.0011464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lam HA, Wu N, Cely I, et al. Elevated tonic extracellular dopamine concentration and altered dopamine modulation of synaptic activity precede dopamine loss in the striatum of mice overexpressing human alpha-synuclein. J Neurosci Res. 2011;89:1091–1102. doi: 10.1002/jnr.22611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lelan F, Boyer C, Thinard R, et al. Effects of human alpha-synuclein A53T-A30P mutations on SVZ and local olfactory bulb cell proliferation in a transgenic rat model of Parkinson disease. Parkinsons Dis. 2011;2011:987084. doi: 10.4061/2011/987084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li W, West N, Colla E, et al. Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations. Proc Natl Acad Sci USA. 2005;102:2162–2167. doi: 10.1073/pnas.0406976102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lindvall O. Developing dopaminergic cell therapy for Parkinson’s disease—give up or move forward? Mov Disord. 2013;28:268–273. doi: 10.1002/mds.25378. [DOI] [PubMed] [Google Scholar]
- 48.Liu CW, Giasson BI, Lewis KA, et al. A precipitating role for truncated alpha-synuclein and the proteasome in alpha-synuclein aggregation: implications for pathogenesis of Parkinson disease. J Biol Chem. 2005;280:22670–22678. doi: 10.1074/jbc.M501508200. [DOI] [PubMed] [Google Scholar]
- 49.Lo Bianco C, Schneider BL, Bauer M, et al. Lentiviral vector delivery of parkin prevents dopaminergic degeneration in an alpha-synuclein rat model of Parkinson’s disease. Proc Natl Acad Sci USA. 2004;101:17510–17515. doi: 10.1073/pnas.0405313101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lonskaya I, Hebron ML, Algarzae NK, et al. Decreased parkin solubility is associated with impairment of autophagy in the nigrostriatum of sporadic Parkinson’s disease. Neuroscience. 2012;232C:90. doi: 10.1016/j.neuroscience.2012.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mai S, Muster B, Bereiter-Hahn J, et al. Autophagy proteins LC3B, ATG5 and ATG12 participate in quality control after mitochondrial damage and influence lifespan. Autophagy. 2012;8:47–62. doi: 10.4161/auto.8.1.18174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Makino S, Smith MA, Gold PW. Regulatory role of glucocorticoids and glucocorticoid receptor mRNA levels on tyrosine hydroxylase gene expression in the locus coeruleus during repeated immobilization stress. Brain Res. 2002;943:216–223. doi: 10.1016/s0006-8993(02)02647-1. [DOI] [PubMed] [Google Scholar]
- 53.Manning-Bog AB, McCormack AL, Li J, et al. The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: paraquat and alpha-synuclein. J Biol Chem. 2002;277:1641–1644. doi: 10.1074/jbc.C100560200. [DOI] [PubMed] [Google Scholar]
- 54.Markey KA, Towle AC, Sze PY. Glucocorticoid influence on tyrosine hydroxylase activity in mouse locus coeruleus during postnatal development. Endocrinology. 1982;111:1519–1523. doi: 10.1210/endo-111-5-1519. [DOI] [PubMed] [Google Scholar]
- 55.Markopoulou K, Larsen KW, Wszolek EK, et al. Olfactory dysfunction in familial Parkinsonism. Neurology. 1997;49:1262–1267. doi: 10.1212/wnl.49.5.1262. [DOI] [PubMed] [Google Scholar]
- 56.Marxreiter F, Nuber S, Kandasamy M, et al. Changes in adult olfactory bulb neurogenesis in mice expressing the A30P mutant form of alpha-synuclein. Eur J Neurosci. 2009;29:879–890. doi: 10.1111/j.1460-9568.2009.06641.x. [DOI] [PubMed] [Google Scholar]
- 57.Masini CV, Holmes PV, Freeman KG, et al. Dopamine overflow is increased in olfactory bulbectomized rats: an in vivo microdialysis study. Physiol Behav. 2004;81:111–119. doi: 10.1016/j.physbeh.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 58.Masliah E, Rockenstein E, Veinbergs I, et al. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- 59.May VE, Nuber S, Marxreiter F, et al. Impaired olfactory bulb neurogenesis depends on the presence of human wild-type alpha-synuclein. Neuroscience. 2012;222:343–355. doi: 10.1016/j.neuroscience.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 60.McArthur S, McHale E, Gillies GE. The size and distribution of midbrain dopaminergic populations are permanently altered by perinatal glucocorticoid exposure in a sex- region- and time-specific manner. Neuropsychopharmacology. 2007;32:1462–1476. doi: 10.1038/sj.npp.1301277. [DOI] [PubMed] [Google Scholar]
- 61.Melachroinou K, Xilouri M, Emmanouilidou E, et al. Deregulation of calcium homeostasis mediates secreted alpha-synuclein-induced neurotoxicity. Neurobiol Aging. 2013;34:2853–2865. doi: 10.1016/j.neurobiolaging.2013.06.006. [DOI] [PubMed] [Google Scholar]
- 62.Mishizen-Eberz AJ, Norris EH, Giasson BI, et al. Cleavage of alpha-synuclein by calpain: potential role in degradation of fibrillized and nitrated species of alpha-synuclein. Biochemistry. 2005;44:7818–7829. doi: 10.1021/bi047846q. [DOI] [PubMed] [Google Scholar]
- 63.Mosharov EV, Larsen KE, Kanter E, et al. Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron. 2009;62:218–229. doi: 10.1016/j.neuron.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mouatt-Prigent A, Karlsson JO, Agid Y, et al. Increased M-calpain expression in the mesencephalon of patients with Parkinson’s disease but not in other neurodegenerative disorders involving the mesencephalon: a role in nerve cell death? Neuroscience. 1996;73:979–987. doi: 10.1016/0306-4522(96)00100-5. [DOI] [PubMed] [Google Scholar]
- 65.Muller T, Fuchs G, Hahne M, et al. Diagnostic aspects of early Parkinson’s disease. J Neurol. 2006;253(Suppl 4):IV29–IV31. doi: 10.1007/s00415-006-4006-2. [DOI] [PubMed] [Google Scholar]
- 66.Mundinano IC, Caballero MC, Ordonez C, et al. Increased dopaminergic cells and protein aggregates in the olfactory bulb of patients with neurodegenerative disorders. Acta Neuropathol. 2011;122:61–74. doi: 10.1007/s00401-011-0830-2. [DOI] [PubMed] [Google Scholar]
- 67.Muntane G, Ferrer I, Martinez-Vicente M. alpha-Synuclein phosphorylation and truncation are normal events in the adult human brain. Neuroscience. 2012;200:106–119. doi: 10.1016/j.neuroscience.2011.10.042. [DOI] [PubMed] [Google Scholar]
- 68.Nemani VM, Lu W, Berge V, et al. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65:66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Norris EH, Uryu K, Leight S, et al. Pesticide exposure exacerbates alpha-synucleinopathy in an A53T transgenic mouse model. Am J Pathol. 2007;170:658–666. doi: 10.2353/ajpath.2007.060359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nuber S, Harmuth F, Kohl Z, et al. A progressive dopaminergic phenotype associated with neurotoxic conversion of alpha-synuclein in BAC-transgenic rats. Brain. 2013;136:412–432. doi: 10.1093/brain/aws358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nuber S, Petrasch-Parwez E, Arias-Carrion O, et al. Olfactory neuron-specific expression of A30P alpha-synuclein exacerbates dopamine deficiency and hyperactivity in a novel conditional model of early Parkinson’s disease stages. Neurobiol Dis. 2011;44:192–204. doi: 10.1016/j.nbd.2011.06.017. [DOI] [PubMed] [Google Scholar]
- 72.Ortiz J, Fitzgerald LW, Lane S, et al. Biochemical adaptations in the mesolimbic dopamine system in response to repeated stress. Neuropsychopharmacology. 1996;14:443–452. doi: 10.1016/0893-133X(95)00152-4. [DOI] [PubMed] [Google Scholar]
- 73.Outeiro TF, Klucken J, Bercury K, et al. Dopamine-induced conformational changes in alpha-synuclein. PLoS One. 2009;4:e6906. doi: 10.1371/journal.pone.0006906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pearce RK, Hawkes CH, Daniel SE. The anterior olfactory nucleus in Parkinson’s disease. Mov Disord. 1995;10:283–287. doi: 10.1002/mds.870100309. [DOI] [PubMed] [Google Scholar]
- 75.Perez RG, Waymire JC, Lin E, et al. A role for alpha-synuclein in the regulation of dopamine biosynthesis. J Neurosci. 2002;22:3090–3099. doi: 10.1523/JNEUROSCI.22-08-03090.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Perrin RJ, Payton JE, Barnett DH, et al. Epitope mapping and specificity of the anti-alpha-synuclein monoclonal antibody Syn-1 in mouse brain and cultured cell lines. Neurosci Lett. 2003;349:133–135. doi: 10.1016/s0304-3940(03)00781-x. [DOI] [PubMed] [Google Scholar]
- 77.Pinching AJ, Powell TP. The neuron types of the glomerular layer of the olfactory bulb. J Cell Sci. 1971;9:305–345. doi: 10.1242/jcs.9.2.305. [DOI] [PubMed] [Google Scholar]
- 78.Prediger RD, Aguiar AS, Jr, Matheus FC, et al. Intranasal administration of neurotoxicants in animals: support for the olfactory vector hypothesis of Parkinson’s disease. Neurotox Res. 2012;21:90–116. doi: 10.1007/s12640-011-9281-8. [DOI] [PubMed] [Google Scholar]
- 79.Recchia A, Rota D, Debetto P, et al. Generation of a alpha-synuclein-based rat model of Parkinson’s disease. Neurobiol Dis. 2008;30:8–18. doi: 10.1016/j.nbd.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 80.Ricaurte GA, Guillery RW, Seiden LS, et al. Dopamine nerve terminal degeneration produced by high doses of methyl-amphetamine in the rat brain. Brain Res. 1982;235:93–103. doi: 10.1016/0006-8993(82)90198-6. [DOI] [PubMed] [Google Scholar]
- 81.Rothman SM, Griffioen KJ, Vranis N, et al. Neuronal expression of familial Parkinson’s disease A53T alpha-synuclein causes early motor impairment, reduced anxiety and potential sleep disturbances in mice. J Parkinsons Dis. 2013;3:215–229. doi: 10.3233/JPD-120130. [DOI] [PubMed] [Google Scholar]
- 82.Seidel K, Schols L, Nuber S, et al. First appraisal of brain pathology owing to A30P mutant alpha-synuclein. Ann Neurol. 2010;67:684–689. doi: 10.1002/ana.21966. [DOI] [PubMed] [Google Scholar]
- 83.Sekine T, Kagaya H, Funayama M, et al. Clinical course of the first Asian family with Parkinsonism related to SNCA triplication. Mov Disord. 2010;25:2871–2875. doi: 10.1002/mds.23313. [DOI] [PubMed] [Google Scholar]
- 84.Sengoku R, Saito Y, Ikemura M, et al. Incidence and extent of Lewy body-related alpha-synucleinopathy in aging human olfactory bulb. J Neuropathol Exp Neurol. 2008;67:1072–1083. doi: 10.1097/NEN.0b013e31818b4126. [DOI] [PubMed] [Google Scholar]
- 85.Singleton AB, Farrer M, Johnson J, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 86.Stifanese R, Averna M, De Tullio R, et al. Adaptive modifications in the calpain/calpastatin system in brain cells after persistent alteration in Ca2+ homeostasis. J Biol Chem. 2010;285:631–643. doi: 10.1074/jbc.M109.031674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tamamizu-Kato S, Kosaraju MG, Kato H, et al. Calcium-triggered membrane interaction of the alpha-synuclein acidic tail. Biochemistry. 2006;45:10947–10956. doi: 10.1021/bi060939i. [DOI] [PubMed] [Google Scholar]
- 88.Tiscornia G, Singer O, Verma IM. Production and purification of lentiviral vectors. Nat Protoc. 2006;1:241–245. doi: 10.1038/nprot.2006.37. [DOI] [PubMed] [Google Scholar]
- 89.Tofaris GK, Razzaq A, Ghetti B, et al. Ubiquitination of alpha-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function. J Biol Chem. 2003;278:44405–44411. doi: 10.1074/jbc.M308041200. [DOI] [PubMed] [Google Scholar]
- 90.Ubhi K, Rockenstein E, Mante M, et al. Neurodegeneration in a transgenic mouse model of multiple system atrophy is associated with altered expression of oligodendroglial-derived neurotrophic factors. J Neurosci. 2010;30:6236–6246. doi: 10.1523/JNEUROSCI.0567-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ulusoy A, Febbraro F, Jensen PH, et al. Co-expression of C-terminal truncated alpha-synuclein enhances full-length alpha-synuclein-induced pathology. Eur J Neurosci. 2010;32:409–422. doi: 10.1111/j.1460-9568.2010.07284.x. [DOI] [PubMed] [Google Scholar]
- 92.Unger EL, Eve DJ, Perez XA, et al. Locomotor hyperactivity and alterations in dopamine neurotransmission are associated with overexpression of A53T mutant human alpha-synuclein in mice. Neurobiol Dis. 2006;21:431–443. doi: 10.1016/j.nbd.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 93.Uversky VN, Li J, Fink AL. Pesticides directly accelerate the rate of alpha-synuclein fibril formation: a possible factor in Parkinson’s disease. FEBS Lett. 2001;500:105–108. doi: 10.1016/s0014-5793(01)02597-2. [DOI] [PubMed] [Google Scholar]
- 94.Volles MJ, Lee SJ, Rochet JC, et al. Vesicle permeabilization by protofibrillar alpha-synuclein: implications for the pathogenesis and treatment of Parkinson’s disease. Biochemistry. 2001;40:7812–7819. doi: 10.1021/bi0102398. [DOI] [PubMed] [Google Scholar]
- 95.Volpicelli-Daley LA, Luk KC, Patel TP, et al. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Vosler PS, Brennan CS, Chen J. Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Mol Neurobiol. 2008;38:78–100. doi: 10.1007/s12035-008-8036-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Watanabe Y, Tatebe H, Taguchi K, et al. p62/SQSTM1-dependent autophagy of Lewy body-like alpha-synuclein inclusions. PLoS One. 2012;7:e52868. doi: 10.1371/journal.pone.0052868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Welberg LA, Seckl JR, Holmes MC. Prenatal glucocorticoid programming of brain corticosteroid receptors and corticotrophin-releasing hormone: possible implications for behaviour. Neuroscience. 2001;104:71–79. doi: 10.1016/s0306-4522(01)00065-3. [DOI] [PubMed] [Google Scholar]
- 99.Wersinger C, Sidhu A. Disruption of the interaction of alpha-synuclein with microtubules enhances cell surface recruitment of the dopamine transporter. Biochemistry. 2005;44:13612–13624. doi: 10.1021/bi050402p. [DOI] [PubMed] [Google Scholar]
- 100.West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- 101.Wills J, Credle J, Oaks AW, et al. Paraquat, but not maneb, induces synucleinopathy and tauopathy in striata of mice through inhibition of proteasomal and autophagic pathways. PLoS One. 2012;7:e30745. doi: 10.1371/journal.pone.0030745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wilson DA, Sullivan RM. The D2 antagonist spiperone mimics the effects of olfactory deprivation on mitral/tufted cell odor response patterns. J Neurosci. 1995;15:5574–5581. doi: 10.1523/JNEUROSCI.15-08-05574.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wilson DA, Wood JG. Functional consequences of unilateral olfactory deprivation: time-course and age sensitivity. Neuroscience. 1992;49:183–192. doi: 10.1016/0306-4522(92)90086-h. [DOI] [PubMed] [Google Scholar]
- 104.Winner B, Geyer M, Couillard-Despres S, et al. Striatal deafferentation increases dopaminergic neurogenesis in the adult olfactory bulb. Exp Neurol. 2006;197:113–121. doi: 10.1016/j.expneurol.2005.08.028. [DOI] [PubMed] [Google Scholar]
- 105.Xia HG, Zhang L, Chen G, et al. Control of basal autophagy by calpain1 mediated cleavage of ATG5. Autophagy. 2010;6:61–66. doi: 10.4161/auto.6.1.10326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yamakado H, Moriwaki Y, Yamasaki N, et al. alpha-Synuclein BAC transgenic mice as a model for Parkinson’s disease manifested decreased anxiety-like behavior and hyperlocomotion. Neurosci Res. 2012;73:173–177. doi: 10.1016/j.neures.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 107.Yang W, Tiffany-Castiglioni E. Paraquat-induced apoptosis in human neuroblastoma SH-SY5Y cells: involvement of p53 and mitochondria. J Toxicol Environ Health A. 2008;71:289–299. doi: 10.1080/15287390701738467. [DOI] [PubMed] [Google Scholar]
- 108.Yu S, Zuo X, Li Y, et al. Inhibition of tyrosine hydroxylase expression in alpha-synuclein-transfected dopaminergic neuronal cells. Neurosci Lett. 2004;367:34–39. doi: 10.1016/j.neulet.2004.05.118. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.