

Abstract
Dormancy is a common strategy adopted by bacterial cells as a means of surviving adverse environmental conditions. For Streptomyces bacteria, this involves developing chains of dormant exospores that extend away from the colony surface. Both spore formation and subsequent spore germination are tightly controlled processes, and while significant progress has been made in understanding the underlying regulatory and enzymatic bases for these, there are still significant gaps in our understanding. One class of proteins with a potential role in spore-associated processes are the so-called resuscitation-promoting factors, or Rpfs, which in other actinobacteria are needed to restore active growth to dormant cell populations. The model species Streptomyces coelicolor encodes five Rpf proteins (RpfA to RfpE), and here we show that these proteins have overlapping functions during growth. Collectively, the S. coelicolor Rpfs promote spore germination and are critical for growth under nutrient-limiting conditions. Previous studies have revealed structural similarities between the Rpf domain and lysozyme, and our in vitro biochemical assays revealed various levels of peptidoglycan cleavage capabilities for each of these five Streptomyces enzymes. Peptidoglycan remodeling by enzymes such as these must be stringently governed so as to retain the structural integrity of the cell wall. Our results suggest that one of the Rpfs, RpfB, is subject to a unique mode of enzymatic autoregulation, mediated by a domain of previously unknown function (DUF348) located within the N terminus of the protein; removal of this domain led to significantly enhanced peptidoglycan cleavage.
INTRODUCTION
Streptomyces species are Gram-positive bacteria that abound in soil environments. They are renowned for their secondary metabolic capabilities; they produce a multitude of compounds that have found utility in both medicine and agriculture, including a vast array of antibiotics, chemotherapeutics, immunosuppressants, and antiparasitic agents (1). They are also well known for their complex, multicellular developmental cycle (2). The Streptomyces life cycle can be broadly divided into two stages: vegetative growth and reproductive development. Unlike most bacteria, Streptomyces organisms are filamentous, and during the vegetative phase of their life cycle, they grow by hyphal tip extension and branching, ultimately forming an interwoven network of cells known as the vegetative mycelium (3). Under certain as yet poorly defined stress conditions, vegetative growth ceases, and reproductive growth ensues. Here, unbranched filamentous cells, termed aerial hyphae, grow away from the vegetative mycelium and extend into the air. These cells then undergo a synchronous round of chromosome segregation, cell division, and cell wall modification to yield chains of dormant exospores (4). These spores are resistant to a wide variety of environmental insults and can be widely dispersed.
A dormant cell state is not unique to the streptomycetes, and indeed many well-studied bacteria (e.g., Bacillus, Clostridium, and Myxococcus) have adopted sporulation as a means of surviving adverse environmental conditions (5). Other bacteria have evolved less extreme dormant states and instead adopt cell states with low metabolic activity such that these cells are better able to tolerate stresses such as antibiotic exposure or immune system assault (6). In addition to low metabolic activity, dormant cells of all types often have a cell wall architecture that differs from that of their vegetative counterparts. For example, Bacillus spores possess thick cell walls comprising peptidoglycan with altered cross-linking compared with that of vegetative cells (7). Similarly, latent or dormant Mycobacterium tuberculosis cells display a different cell wall structure relative to that of actively growing cells (8).
While a thick wall constitutes an effective protective barrier, it can also be refractory to cell growth. Consequently, cell wall remodeling must be integral to restoring dormant cells to an active growth state. In the actinobacteria, a group that includes both the mycobacteria and the streptomycetes, a protein family dubbed the resuscitation-promoting factors (Rpfs) have been implicated in the cleavage of dormant cell walls and subsequent promotion of growth and metabolic reactivation (9). The first Rpf protein was identified in Micrococcus luteus, an actinobacterium that forms dormant cells following prolonged starvation. In an elegant set of experiments, Mukamolova and colleagues found that picomolar concentrations of this secreted protein were sufficient to convert dormant Micrococcus cells into actively growing cells (10, 11). Intriguingly, the Rpf protein in M. luteus is essential for viability (11), suggesting that its function may extend beyond dormant cell resuscitation. Subsequent studies have focused on the Rpf proteins in Mycobacterium; M. tuberculosis encodes five Rpf (RpfMTB) proteins (12). These proteins share some functional redundancy (13), and while it is possible to delete all five rpf genes without affecting viability, the corresponding mutant is unable to exit dormancy and is further compromised in its ability to initiate/establish infections in mouse models (14–16). Several Rpfs in the mycobacteria appear to act as part of a larger peptidoglycan cleavage complex, associating with RipA, an essential enzyme with endopeptidase activity (17, 18).
The mechanistic basis underlying Rpf-mediated emergence from dormancy has not been fully elucidated. An important step toward understanding Rpf activity came with the solution structure of the Rpf domain from RpfB of M. tuberculosis, which revealed a protein fold with similarity to both lysozyme and lytic transglycosidases (19). These results suggested that proteins with an Rpf domain may cleave within the polysaccharide backbone of the peptidoglycan. Given this predicted activity, two—not mutually exclusive—hypotheses have been put forward to explain how Rpf proteins could promote exit from dormancy: (i) Rpf activity relieves the physical constraints imposed by the altered cell wall structure that had otherwise inhibited growth of the dormant cell; (ii) Rpf activity serves as a signal (or generates a signal) that is, in turn, sensed by the cell, and this signal stimulates the resumption of growth (9).
Like M. tuberculosis, the best-studied streptomycete, Streptomyces coelicolor, encodes five Rpf (RpfSC) proteins (20). Generally speaking, these proteins are not orthologous to the M. tuberculosis proteins, with only one of the five (RpfB) sharing a similar domain organization. Here, we probe the role of these proteins in the growth and development of S. coelicolor, compare their peptidoglycan cleavage capabilities and interaction potential, and uncover an intriguing modulatory role for a domain of unknown function associated with RpfB.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
Bacterial strains used or created in this work are detailed in Table 1. S. coelicolor A3(2) strain M145 and its derivatives were grown at 30°C on R5, mannitol soya flour (MS), Difco nutrient agar (NA), or LB agar medium supplemented with antibiotics to maintain selection where appropriate, or in either a 50:50 mixture of tryptic soy broth–yeast extract-malt extract (TSB-YEME) liquid medium or liquid new minimal medium with phosphate (NMMP), as described by Kieser et al. (21). All Escherichia coli strains were grown at 37°C on LB or NA agar plates or in LB or super optimal broth (SOB) liquid medium, supplemented with antibiotics where appropriate, with the exception of BW25113, which was grown at 30°C. All Saccharomyces cerevisiae strains were grown at 30°C in yeast-peptone-dextrose-adenine (YPDA) liquid medium or on YPDA or synthetic defined (SD) amino acid drop-out agar plates (Clonetech) to maintain selection where appropriate.
TABLE 1.
Plasmids and Streptomyces coelicolor, Escherichia coli, and Saccharomyces cerevisiae strains
| Strain or plasmid | Genotype, characteristic(s), and/or usea | Reference or source |
|---|---|---|
| Streptomyces coelicolor A3(2) strains | ||
| M145 | SCP1− SCP2− | 21 |
| E104 | M145 SCO3097::aac(3)IV | 25 |
| E104a (ΔrpfA) | M145 ΔSCO3097 | This study |
| E110 (ΔrpfC) | M145 SCO3098::aac(3)IV | This study |
| E111 (ΔrpfB) | M145 SCO3150::vph | This study |
| E112 (ΔrpfD) | M145 SCO0974::TOPO 2.1 | This study |
| E113 (ΔrpfE) | M145 SCO7458::aac(3)IV | This study |
| E114 (ΔrpfAB) | M145 SCO3097-SCO3098::aac(3)IV | This study |
| E114a | M145 ΔSCO3097-SCO3098 | This study |
| E115 (ΔrpfA DrpfB DrpfC) (3× mutant) | E114a SCO3150::vph | This study |
| E116 | E115 SCO7458::aac(3)IV | This study |
| E116a (ΔrpfA DrpfB DrpfC DrpfE) (4× mutant) | E115 ΔSCO7458 | This study |
| E117 (ΔrpfA ΔrpfB ΔrpfC ΔrpfD ΔrpfE) (5× mutant) | E116a SCO0974::TOPO 2.1 | This study |
| Escherichia coli strains | ||
| DH5α | Used for routine cloning | |
| ET12567(pUZ8002) | dam dcm; with transmobilizing plasmid pUZ8002 | 42, 43 |
| Rosetta 2 | Protein overexpression | Novagen |
| BW25113 | Construction of cosmid-based knockouts | 44 |
| BT340 | DH5α-carrying pCP20, used for FLP recombinase-mediated removal of disruption cassettes | 44 |
| Saccharomyces cerevisiae strains | ||
| Y2H Gold | MATa trp1-901 leu2-3,112 ura3-52 his3-200 gal4Δ gal80Δ LYS2::GAL1UAS-Gal1TATA-His3 GAL2UAS-Gal2TATA-Ade2URA3::MEL1UAS-Mel1TATA AUR1-C MEL1 | Clontech |
| Y187 | MATα ura3-52 his3-200 ade2-101 trp1-901 leu2-3,112 gal4Δ gal80Δ met− URA3::GAL1UAS-Gal1TATA-LacZ MEL1 | Clontech |
| Plasmids | ||
| StE41 | Cosmid used for knockout of rpfA and rpfC | 45 |
| StE87 | Cosmid used for knockout of rpfB | 45 |
| St5C11 | Cosmid used for knockout of rpfE | 45 |
| pIJ82 | pSET152 derivative, hyg replacing aac(3)IV | Gift from H. Kieser |
| pMC175 | pIJ82 carrying rpfD | This work |
| pMC176 | pIJ82 carrying rpfE | This work |
| pET15b | Overexpression of His6-tagged proteins | Novagen |
| pMC177 | pET15b carrying rpfA | 25 |
| pMC178 | pET15b carrying rpfB | This work |
| pMC179 | pET15b carrying rpfBΔDUF348 | This work |
| pMC180 | pET15b carrying rpfC | This work |
| pMC181 | pET15b carrying rpfD | This work |
| pMC182 | pET15b carrying rpfE | This work |
| pGAD T7 AD | Used to generate transcriptional fusions to the GAL4 transcriptional activation domain | Clontech |
| pMC183 | pGAD carrying rpfA | This work |
| pMC184 | pGAD carrying rpfB | This work |
| pMC185 | pGAD carrying rpfBΔDUF348 | This work |
| pMC186 | pGAD carrying rpfC | This work |
| pMC187 | pGAD carrying rpfD | This work |
| pMC188 | pGAD carrying rpfE | This work |
| pGBK T7 BD | Used to generate transcriptional fusions to the GAL4 DNA binding domain | Clontech |
| pMC189 | pGBK carrying rpfA | This work |
| pMC190 | pGBK carrying rpfB | This work |
| pMC191 | pGBK carrying rpfBΔDUF348 | This work |
| pMC192 | pGBK carrying rpfC | This work |
| pMC193 | pGBK carrying rpfD | This work |
| pMC194 | pGBK carrying rpfE | This work |
| pFLUX | Integrative transcriptional reporter vector; ori (pUC18) oriT (RK2) int ϕBT1 attP ϕBT1 luxCDABE (promoterless) aac(3)IV | 32 |
| pFLUX-Pos | pFLUX carrying the ermE* promoter | This work |
| pMC195 | pFLUX carrying the rpfA promoter | This work |
| pMC196 | pFLUX carrying the rpfB promoter (upstream of SCO3152) | This work |
| pMC197 | pFLUX carrying the rpfC promoter | This work |
| pMC198 | pFLUX carrying the rpfD promoter | This work |
| pMC199 | pFLUX carrying the rpfE promoter | This work |
UAS, upstream activation sequence.
Total RNA isolation.
S. coelicolor M145 was grown at 30°C on MS agar plates overlaid with cellophane discs. At different developmental stages (vegetative growth, aerial development, and early and late sporulation), biomass was harvested using a sterile spatula. M145 was also grown at 30°C in TSB-YEME medium, and cells were recovered from culture aliquots by centrifugation. Samples were frozen at −80°C for 3 to 6 days. Total RNA was extracted by mechanical disruption, and coextracted DNA was digested using Turbo DNase (Ambion), as previously described by Moody et al. (22). Total RNA was quantified using a NanoDrop ND-1000, and RNA integrity was confirmed by size fractionating 2 μg of total RNA on an agarose gel. RNA extract purity was verified by measuring the A260/A280 and A260/A230 ratios; these ranged from 1.88 to 2.03 and from 2.18 to 2.38, respectively.
Primer design for reverse transcription-quantitative PCR (RT-qPCR) analyses.
The predicted rpfA (SCO3097), rpfB (SCO3150), rpfC (SCO3098), rpfD (SCO0974), and rpfE (SCO7458) coding sequences were retrieved from the Streptomyces annotation server StrepDB (http://strepdb.streptomyces.org.uk/). Gene-specific PCR primers, generating products of 50 to 150 nucleotides (nt), were designed using Primer-BLAST (http://www.ncbi.nlm.nih.gov/tools/primer-blast/) (see Table S1 in the supplemental material). Standard desalted primers were purchased from Integrated DNA Technologies.
The specificity of each primer pair to its intended target was tested in silico by searching the S. coelicolor genome for potential product-generating annealing sites. Generation of a single amplicon was confirmed by PCR. Briefly, 20-μl reaction mixtures contained 1× Taq reaction buffer, 200 μM each deoxynucleoside triphosphate (dNTP), 5% (vol/vol) dimethyl sulfoxide, 300 nM each primer (see Table S1 in the supplemental material), 0.125 U/μl Taq DNA polymerase, and 1 ng/μl S. coelicolor M145 genomic DNA. Nuclease-free water was used as the template for the negative-control reactions. The following amplification conditions were used: initial denaturation at 95°C for 3 min; 35 amplification cycles consisting of denaturation at 95°C for 30 s, annealing for 30 s (temperatures were optimized for each primer pair) (see Table S1), and extension at 72°C for 45 s, followed by a final extension step at 72°C for 5 min.
Reverse transcription and real-time PCR.
rpfA, rpfB, rpfC, rpfD, and rpfE transcripts were reverse transcribed using SuperScript III reverse transcriptase (Invitrogen). An initial reaction mixture containing 167 ng/μl total RNA, 167 nM each reverse primer, and 833 μM each dNTP (Fermentas or Invitrogen) was heated at 95°C for 5 min and immediately cooled on ice. Each reaction mixture was then supplemented with a second solution containing 2.5× First-Strand buffer (Invitrogen), 12.5 mM dithiothreitol, 5 U/μl RNaseOUT (Invitrogen), and 25 U/μl SuperScript III reverse transcriptase (RT) (Invitrogen). Transcripts were reverse transcribed at 55°C for 60 min, prior to heat inactivation of the enzymes at 70°C for 15 min. To verify the absence of undigested genomic DNA, no-RT controls were conducted as described above, except that RNaseOUT and SuperScript III reverse transcriptase were omitted. The resulting cDNA samples were stored at −20°C for no more than 5 days.
cDNA was quantified using real-time PCR. Triplicate 20- or 25-μl singleplex reactions were carried out in 1× PerfeCTa SYBR green SuperMix (Quanta Biosciences), together with 300 nM each gene-specific primer and 8 ng/μl reverse-transcribed total RNA. No-template negative controls, in which cDNA was replaced with an equal volume of nuclease-free water, were included in each PCR run. Reactions were performed in clear 96-well PCR plates (Bio-Rad) with a CFX96 Touch Real-Time PCR detection system (Bio-Rad) under the following conditions: initial denaturation at 95°C for 3 min, followed by 50 amplification cycles consisting of denaturation at 95°C for 15 s and annealing/extension at a primer-specific temperature (see Table S1 in the supplemental material) for 45 s. Fluorescence was measured during each extension step. A melt curve analysis (65 to 95°C with 5-s fluorescence reads every 0.5°C increase) was also conducted immediately following product amplification.
Fluorescence data were baseline corrected using CFX Manager software, version 2.1 (Bio-Rad). Transcript levels were then calculated using the DART-PCR, version 1.0 workbook (23). Starting fluorescence values (R0), which are proportional to initial template quantities, were calculated for each sample using the average midpoint and amplification efficiency for a given primer pair, as described by Peirson et al. (23). Expression levels of each gene were normalized to total RNA mass and PCR product length.
Luciferase assays.
To monitor rpf expression during germination, the promoter regions for each rpf gene were cloned into the BamHI and KpnI sites of pFLUX (Table 1; see also Table S2 and Fig. S1 in the supplemental material), and these constructs, together with pFLUX alone (negative control) and pFLUX plus the ermE* promoter (positive control, where ermE* is a constitutive, highly active promoter) (Table 1), were conjugated into wild-type S. coelicolor M145. Equal numbers of spores were inoculated into 150 μl of TSB-YEME liquid medium in white 96-well plates (Thermo-Fisher). Plates were then incubated at 30°C with shaking, and luminescence was measured (integration time of 4,000 ms) every 10 min for 12 h using a Tecan Ultra Evolution plate reader. Luminescence levels of the negative control were subtracted from those of the rpf fusions and positive control. Eight biological replicates were prepared, and experiments were conducted at least twice.
rpf mutant strain construction.
Single null mutations were made in rpfB, rpfC, and rpfE using ReDirect technology (24); the rpfA mutation had been created previously (25). rpfB was replaced with a viomycin resistance (vph) cassette on cosmid StE87, whereas rpfC and rpfE were each replaced with apramycin resistance [aac(3)IV] cassettes on cosmids StE41 and St5C11, respectively (Table 1 gives plasmid and strain information; see Table S2 in the supplemental material for primer information). For mutation of rpfD, the typical ReDirect gene replacement protocol was unsuccessful due to spurious cosmid recombination, and instead rpfD was disrupted in the chromosome. A 700-bp region encompassing the Rpf domain of rpfD was amplified and cloned into the TOPO vector (Invitrogen). The kanamycin resistance gene on the TOPO vector backbone was then replaced with an aac(3)IV-oriT-containing DNA fragment using the ReDirect protocol (24).
Mutant cosmids/disruption plasmids were introduced into the nonmethylating E. coli strain ET12567/pUZ8002 prior to conjugation into S. coelicolor M145. Resulting exconjugants were subsequently screened for double-crossover recombinants (or in the case of the rpfD disruption, selected for single-crossover recombinants). Correct replacement of each rpf gene with the appropriate antibiotic resistance cassette or, for rpfD, disruption of the coding sequence, was confirmed using diagnostic PCRs with mutant chromosomal DNA as the template, alongside wild-type chromosomal DNA and mutant cosmids (where appropriate) as negative and positive controls, respectively. These reactions were conducted to confirm that the cosmids/plasmids had recombined at the appropriate positions in the chromosome and that the wild-type gene was no longer present or intact (see Table S2 in the supplemental material for primer combinations).
To create markerless mutants and to permit recycling of the aac(3)IV resistance marker when multiple mutations were created, the apramycin resistance cassette was removed from the rpfA rpfC double mutant cosmid and from the rpfE single mutant cosmid using the temperature-sensitive FLP recombination plasmid in E. coli DH5α (strain BT340) (Table 1). Cosmids were introduced into these cells and grown nonselectively at 42°C to stimulate expression of the FLP recombinase. To identify colonies carrying cosmids in which the antibiotic resistance marker had been excised by FLP recombinase, colonies were tested for kanamycin resistance and apramycin sensitivity. Diagnostic PCR using primers specific for regions upstream and downstream of the Rpf coding sequences (see Table S2 in the supplemental material) was used to confirm appropriate removal of the apramycin cassette from the cosmid. These cosmids were then reintroduced into their corresponding marker-containing S. coelicolor mutant strain by protoplast transformation. The kanamycin-resistant transformants were then grown nonselectively and subsequently screened for double-crossover recombination and loss of both kanamycin and apramycin resistance markers; this was confirmed by PCR.
To create a multiple rpf mutant strain, we used the approach detailed in the schematic diagram in Fig. S2 in the supplemental material. The rpfA rpfC double mutant was created by replacing both genes (which are adjacent on the S. coelicolor chromosome) with the aac(3)IV resistance gene on cosmid StE41. This resistance gene was then removed, and the rpfB::vph mutant cosmid was next introduced, followed by screening and confirmation of the rpfB mutation as described above. Next, the rpfE::aac(3)IV mutant cosmid was introduced, and the rpfE mutation was confirmed before the apramycin resistance cassette was removed from rpfE, as outlined previously. Finally, the rpfD disruption construct was introduced into a mutant strain with deletions of rpfA, rpfB, rpfC, and rpfE (the 4× Δrpf mutant), selected for integration into rpfD in the chromosome, and confirmed by PCR.
rpf mutant strain complementation.
Within the single rpf mutations, the greatest phenotypic effects were seen for rpfA, rpfD, and rpfE mutant strains, with the rpfA mutant phenotype having been complemented previously (25). To ensure that the mutant phenotypes associated with the ΔrpfD and ΔrpfE strains were due to loss of their respective Rpf proteins, complementation clones were generated. The rpfD coding sequence was amplified, together with sufficient upstream and downstream sequence so as to encompass all necessary regulatory elements (325 nt upstream and 244 nt downstream of the coding sequence). This fragment was then cloned into the integrating plasmid pIJ82 (Table 1), and the integrity of the resulting construct was confirmed by sequencing. The plasmid was subsequently introduced into the ΔrpfD mutant strain, where hygromycin-resistant colonies were selected. Complementation of rpfE followed a similar scheme, only the complementing fragment extended 175 bp upstream of the translation start site and 237 bp downstream of the stop codon. Empty plasmid vectors were also introduced into each mutant strain as a control for any plasmid-specific effects. Complementation was confirmed by comparing germination profiles of plasmid alone-containing mutant and wild-type strains with that of the complemented mutant strain, as described below.
TEM.
Wild-type and mutant strains were grown on MS agar at 30°C for 7 days before being examined by transmission electron microscopy (TEM). TEM was performed as described by Haiser et al. (25). Images were processed using Adobe Photoshop Elements 11 Editor. Cell wall thickness was measured using ImageJ software (26) for a minimum of 25 spores per strain.
Spore germination assay.
Mutant and wild-type spores were plated on MS agar overlaid with cellophane discs and incubated at 30°C for up to 12 h. At intermediate time points, a 1-cm square was excised from the cellophane disc and was examined using phase-contrast microscopy to score germinated spores (those possessing at least one germ tube) versus nongerminated spores. A minimum of 200 spores were counted per strain per time point in at least three independent experiments.
Antibiotic sensitivity assays.
Two million wild-type and mutant spores were spread over 500 μl of MS agar containing bacitracin (Bioshop Canada) or d-cycloserine (Sigma-Aldrich) in 48-well microplates (Thermo-Fisher). Plates were incubated for 4 days at 30°C. For overlay experiments, spores were inoculated on 500 μl of MS agar in 48-well microplates and incubated at 30°C for 16 h (to allow for complete spore germination) before wells were overlaid with the indicated amounts of bacitracin or d-cycloserine. Plates were then incubated for an additional 3 days at 30°C. The experiment was repeated at least three times.
Protein overexpression and purification.
To assess the enzymatic activity of each Rpf, the sequence encoding the extracellular domain of each protein (excluding the SignalP-predicted signal peptide sequence [27]) was amplified from S. coelicolor M145 chromosomal DNA using the primers described in Table S2 in the supplemental material. The amplified products were sequentially digested with BamHI and NdeI and ligated with pET15b (Novagen), which had been digested with the same enzymes and dephosphorylated prior to ligation. All constructs were verified by sequencing using promoter and terminator primers (see Table S2) before being introduced into chemically competent E. coli Rosetta 2 cells (Novagen). The resulting colonies were grown overnight in 5-ml cultures supplemented with ampicillin and chloramphenicol, and these overnight cultures were then used to inoculate 500 ml of LB medium containing ampicillin and chloramphenicol. Cultures were grown at 37°C to an optical density at 600 nm (OD600) of 0.8, at which point isopropyl-β-d-thiogalactopyranoside (IPTG) was added in the amounts indicated in Table S3 in the supplemental material to induce protein overexpression. Cultures were incubated for the times/temperatures indicated in Table S3 before cells were collected by centrifugation.
Cell pellets were resuspended in 10 ml of lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, 1 mg/ml lysozyme, pH 8.0) containing one Complete Mini EDTA-free protease inhibitor pellet (Roche) and incubated on ice for 30 min. The cell suspension was sonicated on ice six times for 10 s each before being treated with 40 μg of RNase A and 20 U of Turbo DNase (Ambion) for 15 min on ice. The lysate was then centrifuged at 10,000 × g for 30 min at 4°C. The supernatant was removed and incubated with 1 ml of Ni-nitrilotriacetic acid (NTA) slurry (Qiagen) for 1.5 h at 4°C before being applied to a PolyPrep chromatography column (Bio-Rad). The column was washed with buffer (50 mM NaH2PO4, 300 mM NaCl, pH 8.0) containing increasing concentrations of imidazole (4 ml each of wash buffer containing 20 mM, 50 mM, and 70 mM imidazole) before His6-tagged proteins were eluted with imidazole-containing buffer at concentrations optimized for each protein (see Table S3 in the supplemental material). Purified proteins were applied to an Ultra Centricon centrifugal unit (Amicon), which allowed for exchange of the imidazole-containing buffer with protein storage buffer (50 mM NaH2PO4, 300 mM NaCl, pH 8.0). Protein purification was assessed following separation of purified proteins (and their accompanying washes) on a 12% SDS polyacrylamide gel and staining with Coomassie brilliant blue. Protein concentrations were determined using a Bradford assay (28), with bovine serum albumin as a standard.
Cell wall cleavage assays.
An EnzChek lysozyme assay kit (Molecular Probes) was used to assess the ability of Rpfs to cleave fluorescein-labeled Micrococcus lysodeikticus (more commonly known as M. luteus) cell wall material in vitro. One nanomole of purified Rpf was added to each reaction mixture, and the volume was brought to 50 μl with 1× reaction buffer (100 mM sodium phosphate, 100 mM NaCl, pH 7.5) before the addition of 50 μl of the fluorescein-labeledM. lysodeikticus peptidoglycan substrate to each reaction mixture. One nanomole of the DNA-binding protein sIHF (29) was used as a negative control, while lysozyme (60 pmol, as per the manufacturer's recommendation) served as a positive control. Assays were performed in black 96-well plates (Thermo-Fisher). The amount of fluorescein released was measured every hour for 8 h using a Synergy 4 Microplate Reader (BioTek), with an excitation wavelength of 494 nm and an emission wavelength of 521 nm. Each assay was conducted using at least three independently isolated protein samples, with each protein activity assessed in triplicate.
Rpf interactions.
To probe the potential for Rpf-Rpf protein interactions, the DNA corresponding to the mature Rpf sequences (lacking the signal peptide) were excised from their corresponding pET15b plasmid (Table 1) using NdeI and BamHI and cloned into both pGAD T7 activating domain (AD) and pGBK T7 binding domain (BD) vectors (Clontech) digested with the same enzymes. Conveniently, these vectors contain antibiotic selection markers to facilitate cloning in E. coli (ampicillin and kanamycin, respectively) and prototrophic markers for selection in yeast (Leu biosynthesis and Trp biosynthesis, respectively). pGAD T7 AD-Rpf and pGBK T7 BD-Rpf constructs were transformed into S. cerevisiae strains Y187 and Y2H Gold (Clontech), respectively, according to the TRAFO high-efficiency transformation protocol (30). Transformants were isolated on SD medium lacking Leu (Y187) or Trp (Y2H Gold) following incubation at 30°C for 3 days. To test for Rpf-Rpf interactions, single colonies (2 to 4 mm in diameter) of Rpf construct-containing Y2H Gold and Y187 strains were picked from 3-day-old plates and mixed together in 1 ml of 2× YPDA medium for mating. Cultures were incubated overnight at 30°C, after which cells were collected by centrifugation and plated on SD medium lacking both Trp and Leu. Single colonies were then patched onto SD plates lacking Trp and Leu to confirm the viability of the mated strains and on SD plates lacking Trp, Leu, adenine, and His to test for protein-protein interactions. Plates were examined for growth after 2 days of incubation at 30°C.
Gel filtration chromatography.
Purified RpfB and a mutant variant lacking the three DUF348 domains (RpfBΔDUF348) were separated on a Superdex 75 column (GE Healthcare) preequilibriated with storage buffer (300 mM NaCl, 50 mM NaH2PO4, pH 8) to remove protein aggregates. Pure protein was applied to the column and eluted with storage buffer at a flow rate of 0.4 ml/min at 4°C. The absorbance of the eluate at 230 nm was recorded to confirm protein presence. Standards for column calibration (and assessment of protein oligomeric status) included RNase A (13.7 kDa), chymotrypsinogen A (25 kDa), ovalbumin (43 kDa), and albumin (67 kDa). Peak fractions were pooled and separated on a 12% SDS polyacrylamide gel stained with Coomassie brilliant blue to confirm the presence of RpfB/RpfBΔDUF348.
RESULTS
Bioinformatic analysis of the Rpfs in S. coelicolor.
S. coelicolor encodes at least 60 proteins having domains with predicted peptidoglycan cleavage capabilities, and of these, seven possess an Rpf-like domain (20, 25). Of these seven, two (SCO2326 and SCO5029) appeared to be more distantly related to the well-characterized Rpf domains from Micrococcus and Mycobacterium (20) and lacked key catalytic residues (see Fig. S3 in the supplemental material); these were not considered further. The remaining five proteins (SCO0974, SCO3097, SCO3098, SCO3150, and SCO7458) bore domains that were highly similar to the crystallized M. tuberculosis RpfB protein (RpfBMTB) and contained both critical catalytic amino acids and important substrate binding residues (Fig. 1A). Of these five proteins, SCO3150 showed the greatest sequence divergence relative to RpfBMTB (Fig. 1A) but shared a similar domain architecture, with its Rpf domain positioned at the extreme C-terminal end of the protein, where it was preceded by a G5 domain having potential N-acetylglucosamine-binding activity (31) and three DUF348 domains whose functions are unknown (Fig. 1B). We have therefore designated this protein RpfBSC. For all other S. coelicolor Rpf proteins, the Rpf domain was immediately adjacent to the signal peptide at the N-terminal end. SCO3097 (RpfA), SCO3098 (RpfC), and SCO0974 (RpfD) each also carried a downstream LysM peptidoglycan-binding domain, like the first characterized Rpf from M. luteus (Fig. 1B), while RpfD possessed an additional domain with predicted peptidase activity at its C-terminal end. SCO7458, or RpfE, in its mature form comprised solely an Rpf domain (Fig. 1B).
FIG 1.

Sequence analysis of five Rpf homologues. (A) Multiple-sequence alignment of the Rpf domain from the five predicted Rpf proteins in S. coelicolor, together with RpfB from Mycobacterium tuberculosis (RpfBMTB). Identical residues are highlighted in black. Similar residues are indicated in gray background. Key catalytic (Glu) and substrate-binding residues (Ser/Thr and Trp) are indicated with asterisks. (B) Schematic representation of the functional domain organization of the predicted Rpf proteins, along with M. luteus Rpf and RpfBMTB. Sec, general secretion signal; Rpf, resuscitation-promoting factor domain; LysM, lysin motif; DUF348, domain of unknown function 348; G5, peptidoglycan-binding domain. Mtb, M. tuberculosis.
Within the streptomycetes, four of the five Rpf proteins were broadly conserved (RpfA, RpfB, RpfC, and RpfD), while the smallest protein, RpfE, was not encoded by any of the other well-characterized Streptomyces species (Table 2). Instead, Streptomyces venezuelae and Streptomyces avermitilis encoded additional LysM-containing Rpfs that lacked equivalent orthologues in S. coelicolor and Streptomyces griseus (Table 2).
TABLE 2.
Rpf domain-containing proteins in S. coelicolor, S. avermitilis, S. venezuelae, and S. griseusa
| S. coelicolor protein | Orthologue ina: |
||
|---|---|---|---|
| S. avermitilis | S. venezuelae | S. griseus | |
| RpfA (SCO3097) | SAV3534 | SVEN2900 | SGR4438 |
| RrfB (SCO3150) | SAV3588 | SVEN2970 | SGR4355 |
| RpfC (SCO3098) | SAV3535 | SVEN2901 | SGR4437 |
| RpfD (SCO0974) | SAV7250 | SVEN6803 | SGR745 |
| RpfE (SCO7458) | |||
Although the other species did not encode RpfE, additional LysM-containing Rpfs were present in S. avermitilis (SAV6528 and SAV6527) and S. venezuelae (SVEN1384) that lacked orthologues in S. coelicolor and S. griseus.
Temporal expression of rpf genes during solid and liquid culture growth.
As an initial characterization step, we monitored the transcription profiles of the five rpf genes to determine when these genes were expressed and whether their expression was coordinately regulated. RNA was harvested from plate-grown cultures during vegetative growth (24 h), aerial hyphae formation (48 h), and early and late sporulation (72 and 96 h). Transcript levels were then assessed using RT-qPCR. We could detect expression for only three of the rpf genes (rpfA, rpfC, and rpfD), with the levels of rpfB and rpfE being too low to obtain transcript profiles. For the detectable rpf genes, expression was highest during vegetative growth before levels dropped at the onset of aerial development (Fig. 2A).
FIG 2.

Transcript levels of Rpf-encoding genes in S. coelicolor throughout growth and development. (A) The S. coelicolor wild-type strain M145 was grown at 30°C on MS agar medium and TSB-YEME liquid medium. At the times indicated, total RNA was extracted from cells, and transcripts bearing the rpfA, rpfC, and rpfD coding regions were quantified using RT-qPCRs. Transcript levels were normalized to total RNA mass and PCR product length. The quantification cycle value of each no-RT control was greater than that of the +RT samples by at least 10 cycles. Quantification cycle values of all no-template controls were consistently greater than 40 cycles. All data are presented as means ± standard errors (n = 1 to 3). au, arbitrary units. (B) rpf promoter activities were monitored in TSB-YEME liquid medium for 12 h postinoculation using a luminescence-based reporter. The inset panel (positive control) represents the activity of the strong constitutive ermE* promoter fused to the lux genes over the same 12-h time course. Data are presented as means ± standard errors (n = 8). RLU, relative light units.
Similar profiles were observed for rpfA and rpfC when transcripts extracted from liquid-grown cultures were examined. Note that S. coelicolor does not differentiate in liquid culture, and thus these profiles reflect expression in vegetatively growing cells (Fig. 2A). A different pattern was detected for rpfD, however, where initial transcript levels were relatively low before rising during entry into stationary phase (∼40 h) (Fig. 2A). As was the case for our solid culture samples, rpfB and rpfE transcripts were below the level of reliable detection.
Given the importance of rpf gene products in resuscitating dormant cells in Mycobacterium and Micrococcus (10, 11), we were interested in monitoring rpf transcription during spore germination. We were unable to do this using RT-qPCR (we could not isolate sufficient levels of RNA), so instead we constructed rpf promoter fusions to the lux reporter genes (32) and followed luminescence at 10-min intervals for the first 12 h of growth in liquid culture (spore germination was first seen at ∼4 h under these growth conditions). We found that rpfB transcription began almost immediately and continued through the first 5 h of growth/germination (a similar expression pattern was occasionally seen for rpfC as well although it was not consistently observed), while rpfA expression was first detected at ∼3 h, increased to maximal levels from ∼5 to 6 h, and then dropped back to a low steady-state level (Fig. 2B). Expression from the other rpf promoters was below the detectable limit of the luminometer during this time course.
Rpfs are important for initiating germination.
Given the early transcription of rpfA and rpfB (and sometimes rpfC) and the resuscitation-associated function of the Rpfs as a whole, we sought to determine whether the Rpfs played a role in spore germination, i.e., the “dormant cell resuscitation” equivalent in Streptomyces. We first constructed strains bearing individual rpf deletions (rpfA, rpfB, rpfC, and rpfE) or disruptions (rpfD). As peptidoglycan lytic enzymes are notoriously redundant in their activities (33), we also generated strains with deletions in increasing numbers of rpf genes (deletions of two, three, four, and five rpf genes) with the goal of gaining a clearer picture as to the role of Rpf domain-containing proteins in S. coelicolor germination, growth, and development.
Spore germination for single and multiple rpf mutant strains was monitored over a 12-h time course, with spores being scored for the presence of one or more germ tubes using light microscopy (Fig. 3A). We found that, generally, loss of individual rpf genes was correlated with a delay in the onset of germination. The most significant delays were observed for rpfA and rpfE single mutants, and subtle but reproducible germination delays were seen for the rpfD mutant; loss of rpfB had no effect on spore germination, whereas rpfC mutants occasionally took longer than the wild type to initiate germination. Somewhat surprisingly, we found that the initial delay in germination did not translate into a longer time frame needed for complete germination to occur: all spores (mutant and wild type) were fully germinated by 12 h. The germination defect for the rpfA mutant has been complemented previously (25), and here we were able to partially complement the defect observed for the rpfD and rpfE mutants with a wild-type copy of their respective genes (see Fig. S4 in the supplemental material).
FIG 3.

Rpfs are involved in initiating germination of S. coelicolor spores. (A) Wild-type (WT), individual rpf mutants, and 5× Δrpf mutant spores were monitored for germination using light microscopy over 12 h. The proportion of germinated spores was determined for each strain at the indicated time points. Data are representative of three independent trials (n > 100 spores for every time point, for every strain). (B) Transmission electron micrographs of wild-type, individual rpf mutants, and 5× Δrpf mutant strains. Scale bar, 250 nm. (C) Wall widths of spores (n > 25) from the samples shown in panel B were determined from transmission electron micrographs. Box plots indicate the median (center band), the 25th and 75th percentiles (lower and upper limits of the box, respectively), and the 5th and 95th percentiles (lower and upper whiskers, respectively). Asterisks indicate outliers.
Of the multiple rpf mutant strains, the strain with deletions of rpfA through rpfE (5× Δrpf mutant) showed the greatest impairment in germination: germ tubes had emerged from fewer than 40% of spores after 9 h although, as was seen for the single mutants, germination was complete by 12 h (Fig. 3A; data not shown). Collectively, the delay in initiating germination seen for the rpf mutants indicates that these proteins play an important (although not essential) role in promoting spore germination.
Delayed germination in these strains could stem directly from the lack of Rpfs, which may be needed to remodel the spore peptidoglycan during germination and/or release a germination-promoting signal. Alternatively, the germination defects could stem from Rpf-dependent changes in spore peptidoglycan architecture such that the spore wall is less amenable to germ tube emergence. Indeed, deleting peptidoglycan hydrolases has been associated with increased cell wall thickness in Bacillus subtilis (34, 35). To determine whether this was a possibility, we probed spore wall thickness for those rpf mutants exhibiting germination delays and found that there was no correlation between spore wall thickness and germination rate. Of the single mutants, the rpfA strain exhibited the greatest germination defect, yet rpfA mutant spores had the thinnest walls of all strains tested (25) (Fig. 3B and C). In contrast, the spore walls of the 5× mutant were at least as thick as those of the wild type, and this mutant showed the greatest delay of all strains (Fig. 3B and C). While these experiments do not preclude the possibility that altered cell wall structure contributes to delayed germination, they do indicate that wall thickness alone cannot be used as a surrogate for germination competence.
Germinating spores lacking Rpfs are more sensitive to cell wall-specific antibiotics.
To determine whether the germination defects and heterogeneous spore wall thickness of the different rpf mutants might reflect altered cell wall properties, we tested the different mutants for their sensitivity to antibiotics targeting the cell wall. Mutant and wild-type spores were plated on MS agar containing increasing amounts of d-cycloserine (inhibitor of d-Ala–d-Ala ligase) (Fig. 4A) and bacitracin (inhibitor of C55-bactoprenol pyrophosphate dephosphorylation) (Fig. 4B). As a control, we also tested all strains for their sensitivity to hygromycin, an antibiotic targeting protein synthesis. Relative to the wild type, all rpf mutants showed increased sensitivity to d-cycloserine and bacitracin, with the ΔrpfA, ΔrpfD, and the 5× Δrpf strains being the most sensitive; all strains (wild type and mutant) were equally sensitive to hygromycin (data not shown). To determine if the increased rpf mutant sensitivity reflected a germination-specific defect, mutant and wild-type spores were spread on MS agar and grown for 16 h to allow for complete germination. The vegetatively growing cells were then overlaid with increasing amounts of d-cycloserine (Fig. 4C) or bacitracin (Fig. 4D). In contrast to our previous results, the rpf mutant strains were resistant to all concentrations of both antibiotics. This suggested that Rpf activity alters the spore wall—or that of the initial germ tubes—such that Rpf-deficient strains display enhanced sensitivity to antibiotics that block cell wall synthesis.
FIG 4.

Rpf deletion enhances antibiotic sensitivity during germination. Equal numbers of mutant and wild-type (WT) spores were spread on MS agar containing the indicated amounts of d-cycloserine (A) or bacitracin (B) or were inoculated onto MS agar and allowed to germinate for 16 h before being overlaid with the indicated amounts of d-cycloserine (C) or bacitracin (D). Plates were incubated for 4 days at 30°C. Images are representative of three independent replicates.
Deleting rpf genes impacts vegetative growth in liquid culture.
To further probe the biological role of the Rpf proteins in S. coelicolor, we followed the growth and development of the individual and multiple rpf mutant strains on solid agar plates (sporulation-specific [MS] and rich [R5] media). After 2 days, the majority of rpf mutants appeared largely wild type (Fig. 5A) and remained so after additional incubation (data not shown).
FIG 5.

Phenotypic analyses of rpf mutant strains. (A) Colony morphology of the wild type, individual rpf mutants, and 5× Δrpf deletion strains after 2 days growth on MS agar medium. (B) A total of 2 × 105 mutant and wild-type spores were plated on LB agar medium (a low-Mg2+ medium used to exacerbate any cell wall defects). Pictures were taken at the indicated time points and are representative of three independent trials. (C and D) Growth profile of the 5× Δrpf mutant compared to the wild type during liquid culture in TSB-YEME medium (C) and NMMP medium (D). Values are presented as means ± standard errors (n = 3). WT, wild type.
For the rpf genes whose expression was detectable, transcript levels were generally highest during germination and vegetative growth, with the exception of the level of rpfD in liquid culture. To determine whether any of the rpf mutations had an effect on vegetative growth, we followed colony growth over a 24-h time course on solid medium (Fig. 5B). We did not observe any delays in the growth of the rpfA or rpfB mutant, but the growth of rpfC, rpfD, and rpfE mutants was slightly retarded at 12 h. Intriguingly, the 5× Δrpf mutant failed to form a detectable colony after 12 h, and its growth was detectable only by 18 h (Fig. 5C). It is conceivable that these growth delays simply reflected slower germination rates; however, this cannot be the only explanation as the rpfC strain had largely normal germination but showed delayed vegetative growth, while the rpfA mutant had a significant germination delay but exhibited robust vegetative growth.
As defects in vegetative growth may be more pronounced in liquid culture, we followed the growth profile of the 5× Δrpf mutant relative to its wild-type parent strain in rich and minimal media. In rich medium, the growth curve of the 5× Δrpf mutant was virtually indistinguishable from that of the wild-type strain prior to stationary phase, at which point the 5× Δrpf mutant showed better growth than the wild type (Fig. 5D). In contrast, in minimal medium, the 5× Δrpf mutant grew far less robustly than the wild type at all time points, suggesting that the Rpfs confer a distinct competitive advantage during growth under nutrient-limiting conditions.
In vitro activity assays of each Rpf protein reveal widely various levels of peptidoglycan cleavage capabilities.
To begin probing the biochemical basis for Rpf function, we set out to assess the ability of each of these enzymes to cleave purified peptidoglycan. As a substrate, we opted to use commercially available and fluorescently labeled peptidoglycan from the close Streptomyces relative M. luteus. The fluorescein labeling density was sufficiently high that fluorescence was quenched; this quenching could be alleviated through cleavage of the labeled peptidoglycan, with increased fluorescence being directly proportional to cleavage activity. We overexpressed and purified each of the five Rpf enzymes and monitored their activity over 8 h. While all Rpfs were significantly less active than lysozyme (positive control), there were considerable differences in their activity levels. RpfE was the most active enzyme, followed by RpfA and RpfD. RpfB and RpfC had the lowest activities, with their levels always being less than half that of any other enzyme (Fig. 6). sIHF, a cytoplasmic DNA-binding protein from S. coelicolor, was also overexpressed, purified, and subjected to the cleavage assay to ensure that any activity observed was not due to contaminating E. coli proteins. These results indicate that all of the S. coelicolor Rpfs have some level of peptidoglycan cleavage capability and thus would be able to remodel Streptomyces cell walls.
FIG 6.

Rpf proteins cleave peptidoglycan in vitro. One nanomole of pure protein (as indicated) was mixed with fluorescein-labeled M. luteus cell walls. Data represent the average emission at 521 nm after 8 h of incubation of three trials ± 1 standard error. Negative control, cytoplasmic DNA binding protein sIHF; positive control, lysozyme (Lys). AU, arbitrary units.
Rpf interactions: RpfB forms a dimer.
Previous work in Mycobacterium has suggested that several Rpfs interact with other cell wall-lytic enzymes (17). In particular, the RipA endopeptidase interacts with RpfBMTB, and this association results in synergistic activity. RipA further interacts with RpfEMTB, an enzyme like RpfE in S. coelicolor that lacks an obvious means of associating with the peptidoglycan (17, 18). There is no RipA homologue encoded by the streptomycetes, and thus we wondered whether RpfD, with its endopeptidase domain, might interact with RpfB and whether any of the LysM domain-containing Rpfs (RpfA, RpfB, or RpfD) might associate with RpfE, helping to anchor it to the cell wall. To test this hypothesis we examined interactions between each Rpf protein (excluding their signal peptides) using a yeast two-hybrid system. Rpf interactions were tested in a pairwise fashion, with each mating pair spread on diagnostic medium such that only the strains with an interacting Rpf pair would be capable of growing. We found that RpfB associated with itself, based on the robust growth observed for this mating pair on selective medium, in contrast to all other Rpf pairs, which were unable to interact in this system (Fig. 7A).
FIG 7.
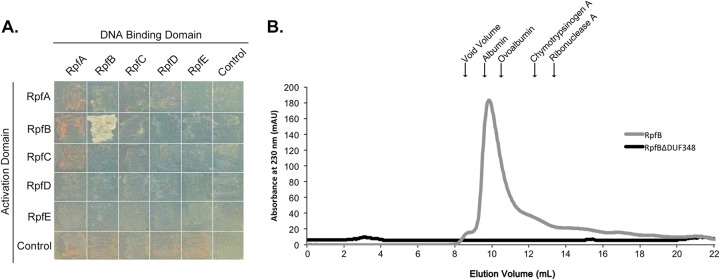
RpfB forms dimers. (A) Yeast two-hybrid analysis of interactions between Rpf proteins. Pictures were taken after 2 days of incubation at 30°C. Growth on selective medium indicates an interaction between bait (DNA-binding domain)- and prey (activation domain)-associated proteins. (B) Purified RpfB and RpfBΔDUF348 were separated on a Sephadex 75 gel filtration column. The peak at 9.4 ml represents likely RpfB dimers. RpfBΔDUF348 dimers were expected to elute at 12 ml. The molecular mass and elution volume, respectively, of each of the standards indicated above the figure are as follows: albumin, 67 kDa and 9.6 ml; ovalbumin, 43 kDa and 10.4 ml; chymotrypsinogen A, 25 kDa and 12.4 ml; and RNase A, 13.7 kDa and 13.3 ml.
To further test the oligomerization capabilities of RpfB, we overexpressed and purified mature RpfB and followed its oligomeric status using gel filtration chromatography. We found that RpfB eluted at a volume that was most consistent with a dimer form (molecular mass of the His-tagged fusion protein was 36.6 kDa), with no detectable monomer species observed (Fig. 7B), supporting our yeast two-hybrid observations.
Removal of the DUF348 domains from RpfB impacts dimerization and enhances enzyme activity.
RpfB does not possess any obvious protein interaction domains (Fig. 1B): its G5 domain is predicted to promote peptidoglycan binding (31), while the Rpf domain failed to promote dimerization in any of the other Rpf protein combinations tested here. DUF348 domains are frequently associated with proteins functioning at the cell wall (20, 36), but, as their name implies, their function is unknown; these domains therefore seemed the most likely candidates for mediating dimerization. To test the role of the DUF348 sequences in RpfB dimerization, we constructed a truncated version of RpfB in which the three DUF348 domains were removed (RpfBΔDUF348). We found that RpfBΔDUF348 failed to interact as robustly with itself as it did with the full-length mature RpfB protein, and its ability to interact with RpfB differed, depending on the vector system (Fig. 8A). To further probe the role of DUF348 domains in dimerization, we again conducted gel filtration chromatography. While RpfB appeared to elute predominantly as a dimer, no dimers were observed for RpfBΔDUF348 (molecular mass of the His-tagged fusion was 21.5 kDa) (Fig. 7B). Instead, it did not elute as a discrete peak at all and appeared to interact with the column, eluting only after 24 ml (data not shown), suggesting that it had a very different conformation than full-length RpfB.
FIG 8.

The DUF348 domains in RpfB are required for dimerization and inhibit RpfB activity. (A) Yeast two-hybrid analysis of interactions between full-length and truncated RpfB proteins, as described in the legend to Fig. 6. Pictures were taken after 2 days of incubation at 30°C on selective medium. The level of growth is correlated with the strength of the protein interaction. (B) One nanomole of pure protein was incubated with fluorescein-labeled M. luteus cell wall for 8 h. Fluorescence emitted at 521 nm was quantified. Data are presented as means ± standard errors (n = 3).
Given this observation, we sought to determine whether the DUF348 domains influenced RpfB activity. We tested the peptidoglycan cleavage activity of RpfBΔDUF348 in our fluorescent substrate assay and found its activity to be far greater than that of the full-length RpfB, suggesting that these domains may function to modulate enzyme activity (Fig. 8B).
DISCUSSION
Rpfs and their role in resuscitation.
Rpf enzymes were originally defined on the basis of their resuscitation-promoting activities in Micrococcus and, later, in Mycobacterium. Our results support a role for the Rpfs in spore germination in S. coelicolor; however, they do not all contribute equivalently, and the fact that germination can proceed (albeit without wild-type kinetics) in the complete absence of Rpfs suggests that they must function in conjunction with other factors. Of the individual Rpfs, rpfA mutants had the most severe germination defect, with rpfB and rpfC mutants behaving most like the wild type. For the rpfB mutant, this could be explained in part by the fact that RpfB reproducibly exhibited the lowest in vitro cleavage activity of all the Rpfs. In contrast, rpfC was not routinely transcribed during germination, and we expected that it may have little to no enzymatic activity because, unlike the other Rpfs, it lacks a key substrate binding residue, having an Ala in place of a Ser/Thr residue (19) (Fig. 1A). Surprisingly, we found it to have intermediate levels of activity, suggesting that this residue is not critical for substrate recognition and binding. In examining the sequence of the RpfC orthologues in other streptomycetes, this particular residue varied considerably, with everything from Leu (Streptomyces sp. strain S4) and Asn (S. avermitilis and Streptomyces scabies) through to the expected Ser (S. venezuelae) and Thr (S. griseus and Streptomyces clavuligerus) residues found at this position (see Fig. S5 in the supplemental material).
The germination defect observed for the rpfE mutant was surprising, given that rpfE transcripts were undetectable. It is possible that low levels of expression are all that are required for its function; this would be consistent with previous observations in M. luteus, where the lone Rpf could restore active growth at extremely low (picomolar) concentrations (10).
The 5× Δrpf mutant exhibited the most severe defect in germination and subsequent vegetative outgrowth. These defects did not dramatically impact the growth of this strain under nutrient-rich conditions; however, in minimal medium, growth of this strain was severely attenuated compared with that of the wild type. This suggests that for Streptomyces species growing in a nutrient-poor soil environment, the lack of Rpf enzymes may confer a profound fitness cost, analogous to that seen for other bacteria (37).
Rpf redundancy in the Streptomycetes.
Within the Actinobacteria, M. luteus encodes a single Rpf enzyme that is essential for viability. In contrast, Streptomyces and Mycobacterium species encode four or more Rpf domain-containing enzymes, and at least in a laboratory environment, these Rpf-encoding genes can all be deleted without compromising viability. In M. tuberculosis, studies have suggested that deleting rpfBMTB (homologous to rpfB in S. coelicolor) delays M. tuberculosis resuscitation in a chronic tuberculosis model (14) although single deletions of any other rpf gene had no obvious effect. Combining mutations led to more severe phenotypic consequences, with double and triple rpf mutants attenuated in their ability both to resuscitate dormant cells in vitro and to establish chronic infections in vivo (16, 38, 39).
In S. coelicolor, we found that individual rpf deletions conferred modest germination defects, with differing levels of severity. This suggested that these enzymes make distinct contributions to germination and vegetative outgrowth, a possibility supported by the fact that individual Rpfs possessed distinct functional domains, and were expressed at various levels and, in some cases, at different times. We did not, however, observe directly additive phenotypic effects when multiple rpf genes were deleted, suggesting that there is some level of functional redundancy shared by these enzymes. Notably, in strains lacking all Rpfs, spore germination was still complete by 12 h, despite the marked delay in germination initiation. This suggests that the resuscitation process in Streptomyces is more complex than that in either Mycobacterium or Micrococcus and that the Rpfs may function as part of a larger peptidoglycan-remodeling network.
Rpf interactions.
In considering the Rpf proteins encoded by Streptomyces and Mycobacterium, only RpfB shared full-length similarity. Previous work in Mycobacterium had shown that while the rpfB mutation had phenotypic consequences, in vitro assays revealed RpfB to have little activity on its own (19), a phenomenon we also observed in our in vitro assays here. Subsequent investigations revealed a key interaction between RpfB and the endopeptidase RipA and synergistic peptidoglycan cleavage by the two enzymes (17, 18).
The dimerization capability of RpfB had not been previously recognized as the screen that led to RipA identification included only the Rpf domain of RpfB (17), and the Rpf domain does not appear to promote dimerization, based on the lack of interaction seen for all other Rpfs. Our results suggest that, instead, the DUF348 domains found at the N terminus of RpfB facilitate dimerization and inhibit RpfB enzyme activity. It is tantalizing to speculate that the RipA-RpfB association in Mycobacterium may lead to a conformational change in RpfB, alleviating the DUF348-mediated inhibition of enzyme activity. DUF348 domains are found in proteins throughout the Actinobacteria and Firmicutes and in most cases are found together with both G5 and peptidoglycan cleavage-associated domains (in S. coelicolor, these domains are not present in any protein but RpfB). Our results indicate that DUF348 domains may serve as a means of controlling the activity of peptidoglycan-cleaving enzymes.
Given the potentially destructive nature of cell wall-lytic enzymes like the Rpfs, it is critically important that their activity be tightly controlled. Increasingly, protein-protein interactions are being found to contribute to this regulation. In M. tuberculosis, RpfB-RipA activity is negatively influenced by interactions with penicillin binding protein 1 (PBP1) (40), while in E. coli, two amidases responsible for cell separation are controlled by peptidoglycan-binding enzymes that confer spatial and temporal regulation (41). Notably, the two regulatory proteins in E. coli were initially classified as having peptidoglycan cleavage activity themselves, specifically, endopeptidase activity. It will be interesting to see whether equivalent proteins control any of the Rpf enzymes in S. coelicolor.
Supplementary Material
ACKNOWLEDGMENTS
We thank Tamiza Nanji and Alba Guarné for their assistance with the gel filtration chromatography experiments and Matt Moody and Rachel Young for their assistance with figure construction.
This work was supported by a Vanier Canada Graduate Scholarship to R.J.S.-O., by an Ontario Graduate Scholarship to H.J.H., by the Canada Research Chairs program (to M.A.E.), by a grant from the Canadian Institutes for Health Research (to M.A.E.; grant no. MOP-93635), and by the Natural Sciences and Engineering Research Council of Canada (to M.A.E.; Discovery grant no. 312495).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02464-14.
REFERENCES
- 1.Hopwood DA. 2007. Streptomyces in nature and medicine: the antibiotic makers Oxford University Press; New York, NY. [Google Scholar]
- 2.Flärdh K, Buttner MJ. 2009. Streptomyces morphogenetics: dissecting differentiation in a filamentous bacterium. Nat Rev Microbiol 7:36–49. doi: 10.1038/nrmicro1968. [DOI] [PubMed] [Google Scholar]
- 3.Flärdh K, Richards DM, Hempel AM, Howard M, Buttner MJ. 2012. Regulation of apical growth and hyphal branching in Streptomyces. Curr Opin Microbiol 15:737–743. doi: 10.1016/j.mib.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 4.Elliot MA, Flärdh K. 2012. Streptomyces spores, encyclopedia of life sciences. John Wiley & Sons, Ltd., Chichester, United Kingdom. [Google Scholar]
- 5.Rittershaus ES, Baek SH, Sassetti CM. 2013. The normalcy of dormancy: common themes in microbial quiescence. Cell Host Microbe 13:643–651. doi: 10.1016/j.chom.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim JH, O'Brien KM, Sharma R, Boshoff HI, Rehren G, Chakraborty S, Wallach JB, Monteleone M, Wilson DJ, Aldrich CC, Barry CE III, Rhee KY, Ehrt S, Schnappinger D. 2013. A genetic strategy to identify targets for the development of drugs that prevent bacterial persistence. Proc Natl Acad Sci U S A 110:19095–19100. doi: 10.1073/pnas.1315860110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meador-Parton J, Popham DL. 2000. Structural Analysis of Bacillus subtilis spore peptidoglycan during sporulation. J Bacteriol 182:4491–4499. doi: 10.1128/JB.182.16.4491-4499.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seiler P, Ulrichs T, Bandermann S, Pradl L, Jorg S, Krenn V, Morawietz L, Kaufmann SH, Aichele P. 2003. Cell-wall alterations as an attribute of Mycobacterium tuberculosis in latent infection. J Infect Dis 188:1326–1331. doi: 10.1086/378563. [DOI] [PubMed] [Google Scholar]
- 9.Keep NH, Ward JM, Robertson G, Cohen-Gonsaud M, Henderson B. 2006. Bacterial resuscitation factors: revival of viable but non-culturable bacteria. Cell Mol Life Sci 63:2555–2559. doi: 10.1007/s00018-006-6188-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mukamolova GV, Kaprelyants AS, Young DI, Young M, Kell DB. 1998. A bacterial cytokine. Proc Natl Acad Sci U S A 95:8916–8921. doi: 10.1073/pnas.95.15.8916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mukamolova GV, Turapov OA, Kazarlan K, Telkov M, Kaprelyants AS, Kell DB, Young M. 2002. The rpf gene of Micrococcus luteus encodes an essential secreted growth factor. Mol Microbiol 46:611–621. doi: 10.1046/j.1365-2958.2002.03183.x. [DOI] [PubMed] [Google Scholar]
- 12.Mukamolova GV, Turapov OA, Young DI, Kaprelyants AS, Kell DB, Young M. 2002. A family of autocrine growth factors in Mycobacterium tuberculosis. Mol Microbiol 46:623–635. doi: 10.1046/j.1365-2958.2002.03184.x. [DOI] [PubMed] [Google Scholar]
- 13.Tufariello JM, Jacobs WR, Chan J. 2004. Individual Mycobacterium tuberculosis resuscitation-promoting factor homologues are dispensable for growth in vitro and in vivo. Infect Immun 72:515–526. doi: 10.1128/IAI.72.1.515-526.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tufariello JM, Mi K, Xu J, Manabe YC, Kesavan AK, Drumm J, Tanaka K, Jacobs WR Jr, Chan J. 2006. Deletion of the Mycobacterium tuberculosis resuscitation-promoting factor Rv1009 gene results in delayed reactivation from chronic tuberculosis. Infect Immun 74:2985–2995. doi: 10.1128/IAI.74.5.2985-2995.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kana BD, Gordhan BG, Downing KJ, Sung N, Vostroktunova G, Machowski EE, Tsenova L, Young M, Kaprelyants A, Kaplan G, Mizrahi V. 2008. The resuscitation-promoting factors of Mycobacterium tuberculosis are required for virulence and resuscitation from dormancy but are collectively dispensable for growth in vitro. Mol Microbiol 67:672–684. doi: 10.1111/j.1365-2958.2007.06078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Russell-Goldman E, Xu J, Wang X, Chan J, Tufariello JM. 2008. A Mycobacterium tuberculosis Rpf double-knockout strain exhibits profound defects in reactivation from chronic tuberculosis and innate immunity phenotypes. Infect Immun 76:4269–4281. doi: 10.1128/IAI.01735-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hett EC, Chao MC, Steyn AJ, Fortune SM, Deng LL, Rubin EJ. 2007. A partner for the resuscitation-promoting factors of Mycobacterium tuberculosis. Mol Microbiol 66:658–668. doi: 10.1111/j.1365-2958.2007.05945.x. [DOI] [PubMed] [Google Scholar]
- 18.Hett EC, Chao MC, Deng LL, Rubin EJ. 2008. A mycobacterial enzyme essential for cell division synergizes with resuscitation-promoting factor. PLoS Pathog 4:e1000001. doi: 10.1371/journal.ppat.1000001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cohen-Gonsaud M, Barthe P, Bagneris C, Henderson B, Ward J, Roumestand C, Keep NH. 2005. The structure of a resuscitation-promoting factor domain from Mycobacterium tuberculosis shows homology to lysozymes. Nat Struct Mol Biol 12:270–273. doi: 10.1038/nsmb905. [DOI] [PubMed] [Google Scholar]
- 20.Ravagnani A, Finan CL, Young M. 2005. A novel firmicute protein family related to the actinobacterial resuscitation-promoting factors by non-orthologous domain displacement. BMC Genomics 6:39. doi: 10.1186/1471-2164-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. 2000. Practical Streptomyces genetics. John Innes Foundation, Norwich, United Kingdom. [Google Scholar]
- 22.Moody MJ, Young RA, Jones SE, Elliot MA. 2013. Comparative analysis of non-coding RNAs in the antibiotic-producing Streptomyces bacteria. BMC Genomics 14:558. doi: 10.1186/1471-2164-14-558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peirson SN, Butler JN, Foster RG. 2003. Experimental validation of novel and conventional approaches to quantitative real-time PCR data analysis. Nucleic Acids Res 31:e73. doi: 10.1093/nar/gng073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gust B, Challis GL, Fowler K, Kieser T, Chater KF. 2003. PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc Natl Acad Sci U S A 100:1541–1546. doi: 10.1073/pnas.0337542100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haiser HJ, Yousef MR, Elliot MA. 2009. Cell wall hydrolases affect germination, vegetative growth, and sporulation in Streptomyces coelicolor. J Bacteriol 191:6501–6512. doi: 10.1128/JB.00767-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 28.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 29.Swiercz JP, Nanji T, Gloyd M, Guarne A, Elliot MA. 2013. A novel nucleoid-associated protein specific to the actinobacteria. Nucleic Acids Res 41:4171–4184. doi: 10.1093/nar/gkt095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gietz RD, Schiestl RH. 2007. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2:31–34. doi: 10.1038/nprot.2007.13. [DOI] [PubMed] [Google Scholar]
- 31.Bateman A, Holden MT, Yeats C. 2005. The G5 domain: a potential N-acetylglucosamine recognition domain involved in biofilm formation. Bioinformatics 21:1301–1303. doi: 10.1093/bioinformatics/bti206. [DOI] [PubMed] [Google Scholar]
- 32.Craney A, Hohenauer T, Xu Y, Navani NK, Li Y, Nodwell J. 2007. A synthetic luxCDABE gene cluster optimized for expression in high-GC bacteria. Nucleic Acids Res 35:e46. doi: 10.1093/nar/gkm086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Typas A, Banzhaf M, Gross CA, Vollmer W. 2012. From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat Rev Microbiol 10:123–136. doi: 10.1038/nrmicro2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fan DP, Beckman MM. 1971. Mutant of Bacillus subtilis demonstrating the requirement of lysis for growth. J Bacteriol 105:629–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fan DP, Pelvit MC, Cunningham WP. 1972. Structural difference between walls from ends and sides of the rod-shaped bacterium Bacillus subtilis. J Bacteriol 109:1266–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yeats CB, Bentley S, Bateman A. 2003. New knowledge from old: in silico discovery of novel protein domains in Streptomyces coelicolor. BMC Microbiol 3:3. doi: 10.1186/1471-2180-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Segev E, Rosengerg A, Mamou G, Sinai L, Ben-Yehuda S. 2013. Molecular kinetics of reviving bacterial spores. J Bacteriol 195:1875–1882. doi: 10.1128/JB.00093-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Downing KJ, Mischenko VV, Shleeva MO, Young DI, Young M, Kaprelyants AS, Apt AS, Mizrahi V. 2005. Mutants of Mycobacterium tuberculosis lacking three of the five rpf-like genes are defective for growth in vivo and for resuscitation in vitro. Infect Immun 73:3038–3043. doi: 10.1128/IAI.73.5.3038-3043.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Biketov S, Potapov V, Ganina E, Downing K, Kana BD, Kaprelyants A. 2007. The role of resuscitation promoting factors in pathogenesis and reactivation of Mycobacterium tuberculosis during intra-peritoneal infection in mice. BMC Infect Dis 7:146. doi: 10.1186/1471-2334-7-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hett EC, Chao MC, Rubin EJ. 2010. Interaction and modulation of two antagonistic cell wall enzymes of mycobacteria. PLoS Pathog 6:e1001020. doi: 10.1371/journal.ppat.1001020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Uehara T, Bernhardt TG. 2011. More than just lysins: peptidoglycan hydrolases tailor the cell wall. Curr Opin Microbiol 14:698–703. doi: 10.1016/j.mib.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.MacNeil DJ, Gewain KM, Ruby CL, Dezeny G, Gibbons PH, MacNeil T. 1992. Analysis of Streptomyces avermitilis genes required for avermectin biosynthesis utilizing a novel integration vector. Gene 111:61–68. doi: 10.1016/0378-1119(92)90603-M. [DOI] [PubMed] [Google Scholar]
- 43.Paget MS, Chamberlin L, Atrih A, Foster SJ, Buttner MJ. 1999. Evidence that the extracytoplasmic function sigma factor SigE is required for normal cell wall structure in Streptomyces coelicolor A3(2). J Bacteriol 181:204–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Redenbach M, Kieser HM, Denapaite D, Elchner A, Cullum J, Kinashi H, Hopwood DA. 1996. A set of ordered cosmids and a detailed genetic and physical map for the 8 Mb Streptomyces coelicolor A3(2) chromosome. Mol Microbiol 21:77–96. doi: 10.1046/j.1365-2958.1996.6191336.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.