

Abstract
Small noncoding RNAs (sRNAs) are posttranscriptional regulators that have been identified in multiple species and shown to play essential roles in responsive mechanisms to environmental stresses. The natural ability of specific bacteria to resist high levels of radiation has been of high interest to mechanistic studies of DNA repair and biomolecular protection. Deinococcus radiodurans is a model extremophile for radiation studies that can survive doses of ionizing radiation of >12,000 Gy, 3,000 times higher than for most vertebrates. Few studies have investigated posttranscriptional regulatory mechanisms of this organism that could be relevant in its general gene regulatory patterns. In this study, we identified 199 potential sRNA candidates in D. radiodurans by whole-transcriptome deep sequencing analysis and confirmed the expression of 41 sRNAs by Northern blotting and reverse transcriptase PCR (RT-PCR). A total of 8 confirmed sRNAs showed differential expression during recovery after acute ionizing radiation (15 kGy). We have also found and confirmed 7 sRNAs in Deinococcus geothermalis, a closely related radioresistant species. The identification of several novel sRNAs in Deinococcus bacteria raises important questions about the evolution and nature of global gene regulation in radioresistance.
INTRODUCTION
Small RNAs (sRNAs), ranging from 21 to >400 nucleotides (nt), are an intriguing class of RNAs that typically do not encode functional proteins but have demonstrated intrinsic roles as cellular regulators of transcription and translation (1–4). A key property of sRNAs is their ability to simultaneously turn on and off a variety of metabolic pathways in response to environmental signals such as changes of temperature, pH, and other potentially lethal stressors (2, 5–7). To exert their function, sRNAs can either base pair with mRNAs to prevent or promote protein translation or sequester proteins into ribonucleic protein complexes to intervene in protein activity (8). Although a variety of mechanisms for sRNA function continue to be reported, it is well documented that sRNAs are highly dependent on their secondary structure and on their ability to undergo rapid conformational changes to exert their regulatory effects (2, 5–7). Noncoding RNAs can be broadly categorized into two classes based on where they are encoded relative to their targets (9, 10), but this classification is continuously evolving. For instance, a newly tRNA-derived sRNA from Escherichia coli and other organisms continues to challenge these classifications (11). While most of the cis-encoded sRNAs control one specific target, some trans-encoded sRNAs are capable of binding and regulating multiple targets (2, 5–7). Recent studies have also discovered more potential mechanisms of sRNA function, such as a coupled action with riboswitch elements that are regulated by different ligands (12). Moreover, the versatile role and the specificity of sRNA targeting are gaining increasing traction for engineering applications, particularly in the context of metabolic engineering (13–16).
Currently, with the development of advanced prediction and sequencing techniques, an increase number of sRNAs have been identified throughout bacteria (17). Hundreds of sRNA candidates have been computationally predicted with different algorithms, such as sRNApredict, QRNA, or NAPP (18). On the other hand, many sRNAs have been identified experimentally by deep sequencing techniques (e.g., transcriptome sequencing [RNA-seq]) and other experimental techniques (e.g., Northern blotting and microarray analyses) (19–22).
Deinococcus species represent an interesting group of bacteria given their ability to survive extraordinarily high doses of ionizing radiation. Deinococcus radiodurans can survive acute doses of up to 12 to 20 kGy, which cause massive DNA damage, and can grow under chronic irradiation at dose rates as high as 60 Gy/h without inducing mutations (23). Moreover, D. radiodurans is amenable to genetic engineering and has been subjected to whole-genome sequencing and functional genomic analyses (23–28). In comparison, vertebrates and Escherichia coli cannot typically survive doses higher than 5 Gy and 1 kGy, respectively. This makes D. radiodurans a leading model for studies of DNA repair and a top candidate for bioremediation of radioactive waste sites (29–31).
Various hypotheses have been tested to understand the extreme radioresistance of D. radiodurans (32–36). This phenotype is complex, relying on a set of DNA repair proteins that operate far more efficiently than in naturally radiation-sensitive organisms (32–36). The molecular basis for the high efficiency of DNA repair proteins in D. radiodurans appears to include the accumulation of manganese antioxidants, which prevent the inactivation of enzymes by reactive oxygen species (ROS) (31, 37). Over the last 15 years, a diverse set of genes (including some involved in metabolism, DNA repair, and ROS scavenging) have been shown to be differentially regulated following high-dose exposures (5 to 16 kGy) (32, 33); however, most of the upregulated genes were subsequently shown not to be essential to radioresistance (37). Since then, the main strategy for delineating a minimal set of genes involved in extreme resistance has been to compare the whole-genome sequences of phylogenetically related but distinct Deinococcus species, whereby unique genes have been ruled out but shared genes have been pooled as candidates for involvement in resistance. This bioinformatics approach eliminated almost all the novel genes first implicated in the extreme radiation resistance of D. radiodurans (23), and few unique genes in Deinococcus spp., such as recA, remain implicated in contributing to its remarkable DNA repair capacity (38, 39). Indeed, the conserved set of radiation resistance determinants of D. radiodurans consists mainly of genes present in many other organisms (37). For instance, a common palindromic DNA motif of a dedicated transcriptional regulator (HucR) within the set of conserved genes was predicted (40).
The question of how radioresistance in D. radiodurans is regulated remains unresolved, and we have hypothesized that sRNAs in D. radiodurans may be important based not only on their regulatory roles in other bacteria but also on their small size. The linear densities of DNA damage caused by ionizing radiation in D. radiodurans and other organisms are very similar (29, 30). Approximately 0.005 double-strand breaks (DSBs)/Gy/Mb of DNA DSBs are introduced to the genome, and this rate is ∼10 times higher for single-stranded breaks (SSBs). We hypothesize that the small size of sRNA genes would leave them largely undamaged at 15 kGy and could contribute to irradiation resistance. Previous work hinted at the existence of noncoding RNAs of potential importance to Deinococcus spp. A transcript that resembles a Y RNA has been identified in D. radiodurans (41). This particular noncoding RNA is able to bind Rsr, a Ro protein ortholog that contributes to radioresistance and is structurally similar to Hfq (42).
In this study, we merged computational and experimental techniques to identify novel potential sRNAs in D. radiodurans. We used computational tools to find hundreds of loci that were predicted to be sRNA candidates and applied a previously developed criterion to further filter most plausible sRNA candidates (43–45). We also used deep sequencing techniques using total RNA from D. radiodurans and discovered 199 sRNA candidates in intergenic regions (IGRs). Upon confirmation by Northern blotting and analysis, we uncovered the expression of 41 novel sRNAs in D. radiodurans, 8 of which showed differential expression following recovery from ionizing radiation. We also found and experimentally validated the presence of homologous sRNA candidates in a closely related radioresistant species, Deinococcus geothermalis. Our studies also identify other well-characterized noncoding RNAs that have not been previously annotated in current versions of the D. radiodurans genome (NCBI GenBank accession numbers NC_001263.1 and NC_001264.1). We suspect that validation of sRNA expression in D. radiodurans will contribute another dimension to ongoing studies of the mechanisms of radiation resistance.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Deinococcus radiodurans strain R1 (ATCC 13939) and Deinococcus geothermalis strain DSM 11300 were cultured according to methods reported previously (46–48). In brief, cells were grown overnight at 30°C (D. radiodurans) or 37°C (D. geothermalis) in TGY broth (1% tryptone–0.1% glucose–0.5% yeast extract) to exponential phase (optical density at 600 nm [OD600] = 1) or stationary phase (OD600 = 3).
Preparation of the protein lysate and Western blotting.
Cell pellets were harvested by centrifugation, washed with sterilized water, and resuspended in lysis buffer (1 mM Tris-HCl [pH 8.0] containing 1 mM phenylmethylsulfonyl fluoride [PMSF]). The cells were frozen with liquid nitrogen, thawed on ice, and lysed by sonication. The cell extracts were collected by centrifugation, and the concentrations were measured with Direct Detect (EMD Millipore). Proteins were resolved by SDS-PAGE using 12% stacking and 5% resolution polyacrylamide gels and immunoblotted according to standard protocols (49). Anti-RecA Escherichia coli rabbit (catalog number BAM-61-003-EX; Cosmo Bio Co.) and a goat anti-rabbit IgG(H+L) horseradish peroxidase (HRP) conjugate (catalog number PR-W4011; Fisher) were used as primary and secondary antibodies, respectively.
Total RNA extraction.
Whole RNA was extracted as previously reported, with minor changes (50). Briefly, cells were pelleted and resuspended in TRIzol reagent (catalog number 15596-026; Invitrogen) and lysed by using a bead beater (catalog number 3110BX; Bio Spec Products Inc.) with four 100-s pulses. The top aqueous phase containing RNA was extracted with chloroform-isoamyl alcohol (24:1) and precipitated with isopropanol. The resulting pellet was dissolved in RNase-free water. The RNA concentration was measured by using a spectrophotometer, and RNA was stored at −20°C for short-term use. The integrity and purity of total RNAs were verified by using a spectrometer (OD260/OD280) and by RNA gel staining.
Whole-transcriptome deep sequencing and data analyses.
cDNA libraries were prepared from total RNAs that were extracted from irradiated or nonirradiated cells by using standard methods (51). We used a NEBNext Small RNA Library Prep Set for Illumina (catalog number E7330S; New England BioLabs Inc.) to prepare cDNA for all samples according to the protocol provided by the manufacturer. The cDNA library was then sequenced with Illumina HiSeq 2000 in one run with 200 cycles. All sequenced reads were trimmed to remove the adapter sequence for mapping to the D. radiodurans R1 genome (GenBank accession numbers NC_001263.1 and NC_001264.1) with Bowtie2 Aligner (52). For alignment visualization, Integrated Genome Viewer (IGV) was used to identify expression in noncoding regions that could indicate potential sRNA candidates.
Selection of computationally predicted candidates.
Computationally predicted sRNA candidates were selected from both previous reports and predictions made with SIPHT (43, 53). In a previous study, 265 potential sRNAs were predicted by QRNA with a comparative algorithm, and 127 sRNA candidates were predicted by SIPHT with default parameters (43, 53). The genome coordinates of all computationally predicted candidates are included in Table S1 in the supplemental material. We used criteria from our previous study to narrow the list of highly potential sRNA candidates for confirmation by Northern blotting (45). In short, sRNA candidates found in longer and generally conserved intergenic regions (among all bacteria) were selected for Northern blotting. In total, 35 candidates from QRNA prediction and 24 from SIPHT prediction were selected and added to the list compiled from transcriptomic analysis.
Ionizing irradiation.
D. radiodurans cells were cultured to exponential (OD600 = 1) and stationary (OD600 = 3) phases, packed, frozen with dry ice, and transported in sterilized plastic bags for irradiation. Exponential and stationary phases were determined by growth curve analysis and tested via a spectrophotometer (OD600), as previously described (34). Samples were thawed at room temperature at the radiation facility before irradiation. These samples were kept cold on wet ice (0°C) while being irradiated with a 10-MeV, 18-kW linear accelerator (LINAC) β-ray source at the National Center for Electron Beam Research, Texas A&M University. Cell samples were subjected to sham and 15-kGy (250-Gy/s) exposures. This initial high-radiation exposure was designed to elicit a response strong enough to allow differential expression of sRNAs that is detectable by Northern blotting (37). Cells were diluted 4- to 5-fold to an OD600 of 1, recovered in fresh culture (TGY) medium for 2 h at 30°C immediately following irradiation, and processed for RNA extraction or stored at −80°C for future analysis. Cell survival rates were measured by plating recovered sham and irradiated cells on TGY plates for CFU comparison.
Northern blotting.
Total RNA was prepared from exponential-phase and stationary-phase D. radiodurans cells exposed to 0 kGy and 15 kGy. Northern blotting (performed as previously described [49]) was used to confirm sRNA expression and to evaluate differential expression as a result of irradiation. We used a 10% polyacrylamide gel for total RNA electrophoresis (under denaturing conditions), and a total of 10 μg of RNA was loaded onto each lane for sampling. A radioactive labeled phiX174 DNA/HinfI ladder (catalog number E3511; Promega) was used as a size marker. The separated RNAs were transferred onto a positively charged membrane (Hybond N+, catalog number RPN119B; GE Life Sciences) and cross-linked with 254 nm UV light. PerfectHyb Plus hybridization buffer (catalog number H7033; Sigma-Aldrich) was used for probe hybridizations over 3 h of incubation at 42°C. Radioactivity was recorded by phosphor storage imaging (Typhoon; GE). The probes were designed to have a complementary sequence toward the target sRNA and radiolabeled with γ-32P by T4 polynucleotide kinase (catalog number M0236S; New England BioLabs). A full list of the probes is included in Table S2 in the supplemental material. Each candidate was tested with probes in the forward and reverse orientations. Each sRNA was experimentally verified in only one direction, and at least two sets of probes were used to test each candidate.
RT-PCR and cotranscription.
cDNAs were prepared with a Super Script III kit (Invitrogen, USA) according to the manufacturer's protocol. Forward and reverse primers,18 to 22 nt long, were designed for each sRNA candidate based on RNA-seq data. Primers were also designed to amplify the upstream or downstream open reading frame (ORF) by itself or by including the candidate sRNA coding region to test the possibility of sRNA cotranscription with the flanking gene. The PCRs were run with cDNA and a minus-reverse transcriptase (RT) control. The annealing temperature was optimized depending on the melting temperature of each primer (see Table S3 in the supplemental material for PCR primer sequences).
Deep 5′ RACE.
Deep 5′ rapid amplification of cDNA ends (RACE) was performed as previously described, with minor modifications (50). Deep 5′-RACE libraries were pooled and sequenced by using an Ion Torrent 316 chip (Wadsworth Center Applied Genomic Technologies Core Facility) (50). For deep 5′ RACE, sequence reads were identified by the presence of the expected adapter sequence at the 5′ end of the transcript. Adapter sequences were removed, and reads of >20 nt were mapped to the reference genomes (GenBank accession numbers NC_001263.1 and NC_001264.1) by using Bowtie2 (54). The 5′ ends were identified as the farthest position with sequenced reads mapped from the 5′ end of the probe. Sequences of all primers used for deep RACE are listed in Table S4 in the supplemental material.
ELISA.
The total RNA samples from irradiated cells were analyzed by an enzyme-linked immunosorbent assay (ELISA) as another way to confirm the oxidative stress induced by irradiation. The test was done with an OxiSelect Oxidative RNA Damage ELISA kit (catalog number STA-325; Cell Biolabs Inc.) according to the protocol provided by the manufacturer.
Nucleotide sequence accession number.
Newly determined sequence data were deposited in the GEO database under accession number GSE64952.
RESULTS
Deep sequencing reveals hundreds of potential transcripts from noncoding regions.
A range of 8,610,676 to 9,473,672 reads were generated per library in our Illumina RNA-seq analysis (GSE64952) of total RNA extracted from wild-type D. radiodurans that was cultured to exponential phase (OD600 = 1); of these, more than 80% were mapped to the genome of D. radiodurans by Bowtie2 (52). Reads that mapped to annotated rRNA and tRNA (∼30% of the total reads [see Fig. S1 in the supplemental material]) were excluded from our analysis. Intergenic regions that potentially encode noncoding RNAs were mapped and visualized by Integrated Genome Viewer (IGV). As a result, the sRNA candidates were manually annotated by inspecting the intergenic regions (IGRs). All intergenic loci mapped with >100 reads per base and longer than 30 nt were annotated as potential sRNA candidates. As a point of reference, the number of reads per base for annotated tRNAs ranged from 3,000 to 20,000. Depending on the genome location, the candidate sRNA was categorized as an overlapping coding region or an IGR transcript.
Given the continual evolution of genome annotation for D. radiodurans, we arbitrarily considered any sRNA candidates that overlapped the annotated coding region by <10 nt as being intergenic. As a result, 199 sRNA candidates were identified. These candidates included a transfer-messenger RNA (tmRNA) that was previously computationally identified (55) and the Y RNA-like sRNAs that were identified by deep sequencing (see Fig. S2 in the supplemental material) (42). The full list of 199 potential sRNA candidates is included in Table S5 in the supplemental material. Among all the small RNA candidates, 46% of them are encoded entirely in intergenic regions, and 54% overlap the 5′ or 3′ end of their adjacent coding region. These overlapping candidates, if on the same strand of the coding region, could be potential functional untranslated regions (UTRs). A functional UTR can form secondary structure and interact with a coding region to regulate translation. We found 56 overlapping candidates that are longer than 100 nt, which could support functional structures. On the other hand, these overlapping candidates can also act as cis-encoded sRNAs if they are on the opposite strand. Upon further bioinformatics analysis with the Rfam database (56), we also found two other candidates to be homologous with the T-box leader sequence (see Table S6 in the supplemental material) (55). The high number of reads observed for these candidates was consistent with the high level of expression expected from this class of RNAs.
sRNA transcripts verified by Northern blotting from deep sequencing results.
We selected 54 (Dsr1 to Dsr54) out of all 199 sRNA candidates identified from our deep sequencing analysis for further experimental verification. We hypothesized that these particular transcripts could be more easily detectable since they showed high expression levels relative to the average intergenic region (e.g., mapped with >500 reads); these candidates also exhibited higher-level conservation in closely related species (the conservation level analysis is discussed below). Unique probes were designed for each candidate to target the most expressed loci (see Table S2 in the supplemental material). Northern blotting was performed by using total RNA prepared from exponential-phase cultures, and 27 sRNAs were confirmed to be expressed (Fig. 1 and 2). To our knowledge, these sRNA candidates had not been identified previously. While most of the identified sRNA candidates are intergenic, eight candidates (Dsr1, Dsr7, Dsr12, Dsr17, Dsr35, Dsr40, Dsr46, and Dsr48) were categorized as 5′- or 3′-overlapping sRNAs. The expression of an RNA homologous to tmRNA was also validated by Northern blotting (Fig. 1 and 2). All confirmed sRNAs are renamed according to a recently proposed nomenclature which uses Mycobacterium as an example but can be applied to all bacterial species (57).
FIG 1.

Novel small RNA candidates in D. radiodurans confirmed by Northern blotting and/or RT-PCR deep sequencing. The angled arrows show the transcription start site confirmed by deep 5′ RACE. The middle arrows show where the Northern probes bind or where the PCR primers amplified. The gray middle arrows represent sRNAs that were identified by Northern blotting, and the black middle arrows represent sRNAs that identified with RT-PCR but not Northern blotting. The arrows on the sides are the annotated flanking protein-encoding regions. The black dashed lines indicate the estimated sRNA coding loci. The size of the RNAs was estimated by comparison to a phiX174 ladder. The 5′ ends were either identified by 5′ RACE or estimated by using deep sequencing data (indicated by asterisks). The 3′ ends are estimated with 5′-end coordinates and RNA size determined by Northern analysis or deep sequencing. The phylogenetic distribution shows the species for which homologous small RNAs were found: 1, Deinococcus gobiensis; 2, Deinococcus proteolyticus; 3, Deinococcus deserti; 4, Deinococcus peraridilitoris; 5, Deinococcus geothermalis; 6, Deinococcus maricopensis; 7, Deinococcus swuensis. The cotranscribed sRNAs that were identified by RT-PCR are marked in the far-right column.
FIG 2.
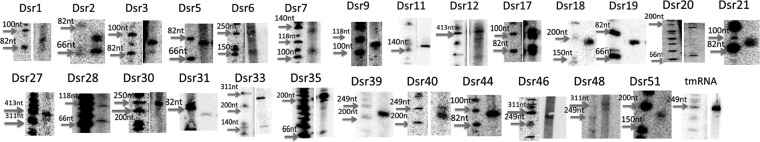
Images of Northern blots for confirmed sRNA candidates from deep sequencing analysis. Cells were cultured to exponential phase (OD600 = 1). Analysis was performed with 8 μg of total RNA sample. The expressions of predicted sRNAs were confirmed, and the sizes of the transcripts were estimated relative to the phiX174/HinfI marker. Some of the images of the ladder and the sRNA lanes are cut from the same gel in different parts, with the contrast being adjusted to show a clearer image.
While it is known that sRNAs can be independently transcribed from the genome or processed from an mRNA, most of the sRNAs identified in this study showed only one band by Northern blotting. For the remaining candidates, multiple or larger bands may suggest posttranscriptional processing of the sRNA or potential riboswitches.
Identification of more sRNA candidates by RT-PCR and cotranscription experiments.
To further confirm the expression of all sRNA candidates that were verified by Northern analysis, we conducted an RT-PCR analysis. These experiments were particularly beneficial for candidates that were expressed at very low levels and that yielded ambiguous results by Northern analysis. All sRNA candidates that were verified by Northern blotting were also confirmed by RT-PCR, except for Dsr5 (see Fig. S3 in the supplemental material). We also tested 6 additional sRNA candidates that were originally not detected by Northern probing (presumably because of their lower expression levels, as seen in our transcriptome data) but that were conserved in D. geothermalis or had predicted relevance to the radioresistance phenotype. This resulted in 4 more sRNAs candidates being identified: Dsr8, Dsr10, Dsr50, and Dsr52 (Fig. 1).
Since many identified sRNAs overlap or are in proximal distance to the upstream or downstream open reading frame, we hypothesized that some of these sRNAs could be cotranscribed with flanking genes. After testing for cotranscription with primers that amplify the sRNA and the corresponding flanking gene, we found Dsr8, Dsr10, Dsr12, Dsr19, Dsr33, and Dsr40 to be cotranscribed with their flanking coding regions (see Fig. S4 in the supplemental material). These data also suggest that sRNAs could be posttranscriptionally processed from larger transcripts.
5′ ends of the sRNA candidates mapped by deep RACE.
To verify the exact coordinates of the transcription start site (TSS) for each sRNA candidate, a deep 5′-RACE analysis was conducted on the confirmed sRNA candidates (58). After construction of a cDNA library for RNA samples prepared from exponential-phase cells, reverse primers (the same ones used as probes for Northern blotting) were used to amplify the 5′ end of each sRNA candidate (see Table S3 in the supplemental material). The amplicons were collected and sequenced. The sequenced reads were mapped to the genome of D. radiodurans as described in Materials and Methods. Annotations of the 5′ ends were done by manual inspection with IGV (see Fig. S5 in the supplemental material), and sequenced reads were successfully mapped to the 5′ ends of 14 sRNA candidates. Other sRNAs were not possible to map, potentially due to their lower expression levels. The identified coordinates of TSSs are shown in Fig. 1.
Confirmation of additional sRNAs from computational predictions.
A total of 391 computationally predicted sRNA candidates in D. radiodurans were collected (see Table S1 in the supplemental material): 256 were generated by using QRNA, and 126 were predicted by using SIPHT (previously known as sRNApredict3) (43) (53). It is worth noting that among these two sets of predictions, 17 candidates overlapped (see Fig. S6 in the supplemental material). Of the 17 sRNAs predicted by both QRNA and SIPHT, 7 of them were also identified by deep sequencing as being potential sRNAs.
Based on a filter used successfully in previous works in our laboratory (45), we narrowed these predictions to include a smaller set of candidates that we rationalized would be more likely to be true sRNAs. Briefly, sRNA candidates that were encoded in larger (top 20% longest) and more isolated intergenic regions were selected for Northern blotting. Based on these results, we tested 60 additional candidates (36 from published QRNA predictions and 24 candidates from our SIPHT predictions). Importantly, 10 more sRNA candidates were validated by Northern blotting: 6 from QRNA predictions (Qpr1 to Qpr6) and 4 from SIPHT analysis (Spr1 to Spr4) (Fig. 3 and 4). The poor overlap between the two prediction methods or the high number of false positives is not surprising given that these algorithms are based on two different criteria (sequence homology and secondary structure conservation) and not on any functional information; similar observations have been reported in other bacterial studies (50). Interestingly, only two confirmed sRNAs (Spr1 and Qpr1) were detected by deep sequencing. This is not surprising, since some of the sRNAs might be degraded or not present in the cDNA pool due to biases in reverse transcription. These results confirm our above-described observation that better efficiency of sRNA experimental identification could be achieved by incorporating both computational and sequencing methods.
FIG 3.

Novel small RNA candidates predicted computationally and confirmed by Northern blotting. Spr1 to Spr4 are predicted by SIPHT, and Qpr1 to Qpr6 are predicted by QRNA. The middle arrows show where the Northern probes bind. The arrows on the sides are the annotated flanking protein-encoding regions. The size of the RNAs was estimated by comparison to a phiX174/HinfI ladder. The 5′ and 3′ ends were predicted computationally (SIPHT or QRNA). The phylogenetic distribution shows the species for which homologous small RNAs were found: 1, Deinococcus gobiensis; 2, Deinococcus proteolyticus; 3, Deinococcus deserti; 4, Deinococcus peraridilitoris; 5, Deinococcus geothermalis; 6, Deinococcus maricopensis; 7, Deinococcus swuensis.
FIG 4.

Images of Northern blots for confirmed sRNA candidates from computational prediction. The total RNA samples were extracted from D. radiodurans cell cultures at exponential phase (OD600 = 1). The expressions of predicted sRNAs were confirmed, and the sizes of the transcripts were estimated relative to the phiX174/HinfI ladder. Some of the images of the ladder and the sRNA lanes are cut from the same gel in different parts, with the contrast being adjusted to show a clearer image.
Differential expression of sRNAs during genome recovery after ionizing irradiation.
Following the discovery and validation of sRNAs in D. radiodurans, we investigated the possibility that these sRNAs were differentially expressed during recovery from high-dose irradiation, as an early indicator of their potential functional importance. For this analysis, we assayed the differential expression of all confirmed novel sRNAs following ionizing radiation (15 kGy), relative to sham irradiation controls. A scheme of the ionizing radiation procedure used is shown in Fig. 5A. To confirm the consistency of the overall biological trends observed after ionizing radiation (59, 60), we first verified that cells were viable after recovery postirradiation by making growth curves and counting CFU with plated cells (see Fig. S7 in the supplemental material). We found that the dose used in this study is equivalent to 15 to 18 kGy of gamma radiation (61). It is worth noting that this initial high-radiation exposure was designed to elicit a response strong enough to allow detectable potential differential expression of sRNAs (36).
FIG 5.

Scheme of the experimental procedure. (A) D. radiodurans R1 (ATCC 13939) cells were cultured in TGY medium to exponential (OD600 = 1) or stationary (OD600 = 3) phase. Cells were then irradiated while cold and then recovered in fresh medium for 120 min. RNA and protein total lysates were prepared for analysis. RT, room temperature. (B) Western blotting for RecA expression under 15 kGy of ionizing radiation. (C) ELISA (1 μg of RNA was used for each test) quantifying the oxidation damage of the DNA in irradiated samples and the control. 8OG, 8-oxoguanine.
Next, to quantitatively verify efficient irradiation, we also probed expression levels of RecA in irradiated samples (relative to nonirradiated samples) via Western blotting (Fig. 5B). An increase of the RecA protein expression level has been known to be an indirect indication of irradiation stresses, and this trend was confirmed in our study (Fig. 5B). Lastly, the expected higher levels of nucleic acid damage were confirmed by using an enzyme-linked immunosorbent assay (ELISA) to detect 8-oxoguanine levels (Fig. 5C). Higher levels of 8-oxoguanine were previously reported to accumulate in nucleic acids under oxidative stress (33, 62). Collectively, the above-described analysis confirmed radiation-induced damage while also confirming viability and the stress response.
To test differential sRNA expression during irradiation recovery, we prepared total RNA from sham- and 15-kGy-irradiated samples from both the exponential (OD600 = 1) and stationary (OD600 = 3) growth phases. These RNA samples were probed with radiolabeled oligonucleotides with complementarity to all identified novel sRNAs. The intensity changes were normalized by the average change of tRNA expression levels, as we assumed that the expression level changes of tRNAs was due only to loading or degradation effects. We found 8 sRNAs that showed differential expression following a 2-h genome recovery from 15-kGy irradiation (relative to sham irradiation). Figure 6 shows sRNAs that exhibited at least a 2-fold decrease or increase in band intensity after tRNA normalization (as quantified by GelQuant). Most of these sRNAs showed the same trend in both exponential and stationary phases, while some sRNAs exhibited these trends more obviously in one growth phase than in the other. sRNA blots that do not suggest differential expression are included in Fig. S8 in the supplemental material.
FIG 6.

Differential expression of selected sRNAs. In each Northern blot image, the left two lanes are RNA samples from exponential-phase (OD600 = 1) cells, and the right two lanes are from stationary-phase (OD600 = 3) cells. The first and third lanes are the control (sham irradiation) RNA samples, and the second and fourth lanes are the 15-kGy-irradiated RNA samples. The images of first two lanes and the last two lanes are cut from the same gel in different parts. The band intensity change of each candidate is normalized to the tRNA levels shown at the bottom of each blot. All the blots shown here have either a 2-fold decrease in activity after irradiation (Dsr2, Dsr5, Dsr7, Dsr18, Dsr27, Dsr30, and Dsr39) or a 2-fold increase in activity after irradiation (Dsr12).
Identification of conserved sRNAs in D. geothermalis.
To test the conservation level of the sRNA candidates in related radioresistant species, we used BLAST (Basic Logic Alignment Search Tool) (NCBI) to bioinformatically identify homologous sRNAs (E value of <0.01) in a sample of 6 representative Deinococcaceae species (Deinococcus gobiensis, D. proteolyticus, D. deserti, D. peraridilitoris, D. geothermalis, and D. maricopensis). All these species have been shown to be highly resistant to ionizing and UV radiation (63). We also performed a BLAST comparison of all sRNA candidates to other bacterial species to test if these sRNAs (particularly those differentially expressed) were conserved only in Deinococcus spp. Only the sRNAs that are conserved in Deinococcus spp. but not in other bacteria are included in Fig. 1, 3, and 7. Most of these sRNAs did not show sequence conservation in other species beyond Deinococcus spp.; this does not, however, rule out the possibility that the function of the identified sRNAs is conserved in other bacteria.
FIG 7.

Novel small RNA candidates in D. geothermalis confirmed by Northern blotting and homologous counterparts in D. radiodurans. The middle arrows show where the Northern probes bind. The gray middle arrows are the annotated flanking protein-encoding regions. The size of the RNAs was estimated by comparison to a phiX174 ladder. The 5′ end and 3′ end of D. geothermalis RNA are not applicable; instead, the coordinates of the 5′ end and 3′ end of the probe are shown. The phylogenetic distribution shows the species for which homologous small RNAs were found: 1, Deinococcus gobiensis; 2, Deinococcus proteolyticus; 3, Deinococcus deserti; 4, Deinococcus peraridilitoris; 5, Deinococcus geothermalis; 6, Deinococcus maricopensis; 7, Deinococcus swuensis.
We selected 10 sRNA candidates from the deep sequencing data that were highly conserved in Deinococcus spp. (in at least two species) from the above-described analysis and used Northern blotting to confirm sRNA expressions in D. geothermalis and D. radiodurans. We used BLAST to locate the homologous loci in the genome of D. geothermalis and designed complementary probes that were specific to D. geothermalis for Northern blotting. Seven sRNAs were identified in D. geothermalis (Fig. 7 and 8). Two of the D. geothermalis sRNAs were found in the antisense strand of a coding region (Gsr5 and Gsr7), while others were intergenic. All of these candidates were also confirmed in D. radiodurans by Northern blotting or RT-PCR, and four of the sRNAs reside in intergenic regions conserved between D. radiodurans and D. geothermalis (Fig. 8). We also found that Gsr1/Dsr50 and Gsr3/Dsr52 have downstream and upstream genes with similar functions. It is important to note that the observed difference in the sizes of the conserved sRNAs between D. geothermalis and D. radiodurans is not surprising since it is likely that they are processed differently in the two organisms; these trends have been reported previously for homologous sRNAs across species (49).
FIG 8.

Images of Northern blots for sRNAs candidates with homology to D. geothermalis. The RNA samples were extracted from nonirradiated D. geothermalis exponential-phase (OD600 = 1) cell cultures, and the size was estimated relative to the phiX174/HinfI ladder. Some of the images of the ladder and the sRNA lanes are cut from the same gel in different parts, with the contrast being adjusted to show a clearer image.
DISCUSSION
Genomic expression determined by whole-transcriptome analysis of D. radiodurans recovering from acute exposures to 15 kGy was previously reported (64, 65). During the early phase and midphase of recovery, D. radiodurans fails to grow, but within this interval, hundreds of genes within diverse functional groups are differentially regulated (64). After an exposure to 15 kGy, ∼150 DSBs are inflicted randomly over the four genomic partitions of D. radiodurans (66), followed by extensive exonucleolytic DNA degradation (67). In acutely irradiated cells, this causes a substantial lowering of the copy number of the more heavily damaged, larger genomic partitions compared to the smaller ones in the first hours of recovery, with global levels of RNA expression postirradiation being inversely related to partition size. Within the broader context of partition-specific expression, some of the genes were predicted to encode sRNAs. For example, DRA0234 is 171 bp in length, shows no similarity to any protein sequences, and has a transcript that was predicted to form a stable stem-loop structure (64); previous studies showed that DRA0234 was upregulated very early in recovery, displaying a 12-fold increase in irradiated D. radiodurans within the first 1.5 h of recovery (64). The expression of DRA0234 was also identified by our deep sequencing analysis (see Fig. S9 in the supplemental material). This gene, and perhaps other similar ones, might encode uncharacterized regulatory sRNAs.
In this study, we found 199 potential sRNA transcripts using a combined approach that involved deep sequencing analysis and computational predictions. We tested 125 candidates by Northern blotting and RT-PCR and confirmed the expression of 41 sRNAs in D. radiodurans and 7 sRNAs in D. geothermalis. We confirmed that a variety of potential sRNAs could be encoded by this organism, as observed in data acquired by deep sequencing analysis. One interesting example is Dsr12. It overlaps the 5′ end of the rpsF gene, which encodes the 30S ribosomal protein S6. Under sham irradiation, the size of the Dsr12 transcript approximately equaled the length of the identified TSS to the 3′ end of rpsF, indicating cotranscription of Dsr12 and rpsF. However, the signal of the larger transcript became weaker following acute irradiation, and a smaller transcript appeared. One possibility is that Dsr12 can serve mechanistically as a functional UTR that can change structure under radiation stress and induce downstream RNA degradation or posttranscriptional processing.
Besides Dsr12, most of the sRNAs that show differential expression during recovery from 15 kGy of irradiation are downregulated following irradiation. For example, Dsr2 is downregulated during irradiation recovery. Dsr2 is also predicted to bind the 5′ UTR of the recA mRNA, which is critical for homologous recombination. Therefore, one potential mechanism of Dsr2 is that under no radiation stress, Dsr2 could bind with recA and block translation; on the contrary, under irradiation stress, the downregulation of Dsr2 leads to an increased expression of recA. It is worth noting that, since most sRNAs do not exhibit expression changes during irradiation recovery, we suspect that the observed effects of sRNA downregulation are specific to these transcripts and not just a general by-product of irradiation.
In this work, we have also found by BLAST analysis that many sRNA candidates in D. radiodurans are conserved in other Deinococcus spp. We have experimentally confirmed that 7 of them are expressed in D. geothermalis (Fig. 7 and 8) (48, 68). While generally conserved sRNAs could act as housekeeping regulatory components, exclusively conserved sRNAs in radioresistant Deinococcus spp. (indicated in Fig. 1, 3, and 7) could have unique functionality and play critical roles in radioresistant species. The lack of experimental validation of all sRNAs predicted to be conserved in both organisms could be attributed to the possibility that these sRNAs are found at much lower levels in one of the two organisms (making them difficult to detect by Northern blotting).
Upon using TargetRNA2 to predict potential sRNA binding targets in D. radiodurans, we found predicted mRNA targets that could be functionally related to radioresistance mechanisms (those with an E value of <0.05 are listed in Table S7 in the supplemental material) (69). Many predicted mRNA targets found in this study also encode proteins that contribute to radiation survival, such as RecA, RuvA, and RadA (33, 39, 70). Other mRNAs do not encode proteins directly associated with DNA repair but are associated with stress response mechanisms or global gene regulation, such as the TetR family transcriptional regulator DR_0074, the cyclic AMP receptor protein (CRP)/ferredoxin-NADP+ reductase (FNR) family transcriptional regulator DR_0097, and the MerR family transcriptional regulator (71–73).
The extraordinary survival of D. radiodurans cells exposed to ionizing radiation has been explicated with the hypothesis that the critical proteins for DNA repair and replication are protected by a mechanism of small-molecule Mn2+ antioxidants during irradiation (36, 74, 75). Proteins in D. radiodurans grown in TGY medium and exposed to massive acute doses of gamma rays (>10 kGy) are shielded from oxidation, preserving the functions of cytoplasmic enzymes as well as the transcriptional and translational potential of the cell (37). In contrast, naturally radiation-sensitive prokaryotes, such as E. coli and Pseudomonas putida, exposed to doses of >1 kGy display extensive global protein oxidation, which inactivates their repair and replication systems, rendering even minor DNA damage lethal. Evidently, the survival of cells exposed to high doses of radiation rests on a functional proteome (36). Within this context, the amount of genome damage caused per unit length is directly proportional to the dose of radiation. We argue that if the regulation of DNA repair genes in D. radiodurans is mediated by sRNAs, the small size of sRNA genes (<400 bp) would leave them largely intact compared to protein-encoding genes (∼1,000 to 2,000 bp) at the outer limits of D. radiodurans survival (15 kGy). This might yield functional sRNAs transcribed from genomic DSB fragments before the onset of DNA repair, acting on the pool of existing repair proteins (e.g., RecA) present in the cells prior to irradiation (35, 38). Our future studies will focus on better understanding the targets of the newly discovered sRNA candidates in this study. Other environmental stresses that cause DNA damage could also be tested, such as desiccation and UV-C light, yielding a more comprehensive view of sRNA regulatory pathways in oxidative stress responses.
Supplementary Material
ACKNOWLEDGMENTS
We thank Kevin Baldridge for performing and analyzing the ELISA.
We thank the University of Texas at Austin for an undergraduate research fellowship to R.L and B.C. This work was supported by the Welch Foundation (F-1756), the Defense Threat Reduction Agency Young Investigator Program (HDTRA1-12-0016), and the Air Force Office of Scientific Research Young Investigator Program (FA9550-13-1-0160).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03709-14.
REFERENCES
- 1.Gottesman S. 2004. The small RNA regulators of Escherichia coli: roles and mechanisms. Annu Rev Microbiol 58:303–328. doi: 10.1146/annurev.micro.58.030603.123841. [DOI] [PubMed] [Google Scholar]
- 2.Gottesman S. 2005. Micros for microbes: non-coding regulatory RNAs in bacteria. Trends Genet 21:399–404. doi: 10.1016/j.tig.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 3.Desnoyers G, Bouchard M-P, Massé E. 2013. New insights into small RNA-dependent translational regulation in prokaryotes. Trends Genet 29:92–98. doi: 10.1016/j.tig.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 4.Wiedenheft B, Sternberg SH, Doudna JA. 2012. RNA-guided genetic silencing systems in bacteria and archaea. Nature 482:331–338. doi: 10.1038/nature10886. [DOI] [PubMed] [Google Scholar]
- 5.Wassarman KM. 2002. Small RNAs in bacteria: diverse regulators of gene expression in response to environmental changes. Cell 109:141–144. doi: 10.1016/S0092-8674(02)00717-1. [DOI] [PubMed] [Google Scholar]
- 6.Guillier M, Gottesman S. 2006. Remodelling of the Escherichia coli outer membrane by two small regulatory RNAs. Mol Microbiol 59:231–247. doi: 10.1111/j.1365-2958.2005.04929.x. [DOI] [PubMed] [Google Scholar]
- 7.Majdalani N, Vanderpool CK, Gottesman S. 2005. Bacterial small RNA regulators. Crit Rev Biochem Mol Biol 40:93–113. doi: 10.1080/10409230590918702. [DOI] [PubMed] [Google Scholar]
- 8.Vazquez-Anderson J, Contreras LM. 2013. Regulatory RNAs: charming gene management styles for synthetic biology applications. RNA Biol 10:1778–1797. doi: 10.4161/rna.27102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arnvig KB, Young DB. 2009. Identification of small RNAs in Mycobacterium tuberculosis. Mol Microbiol 73:397–408. doi: 10.1111/j.1365-2958.2009.06777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Papenfort K, Vogel J. 2010. Regulatory RNA in bacterial pathogens. Cell Host Microbe 8:116–127. doi: 10.1016/j.chom.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 11.Garcia-Silva MR, Cabrera-Cabrera F, Güida MC, Cayota A. 2013. Novel aspects of tRNA-derived small RNAs with potential impact in infectious diseases. Adv Biosci Biotechnol 4:17–25. doi: 10.4236/abb.2013.45A002. [DOI] [Google Scholar]
- 12.DebRoy S, Gebbie M, Ramesh A, Goodson JR, Cruz MR, van Hoof A, Winkler WC, Garsin DA. 2014. A riboswitch-containing sRNA controls gene expression by sequestration of a response regulator. Science 345:937–940. doi: 10.1126/science.1255091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kang Z, Wang X, Li Y, Qian W, Qingsheng Q. 2012. Small RNA RyhB as a potential tool used for metabolic engineering in Escherichia coli. Biotechnol Lett 34:527–531. doi: 10.1007/s10529-011-0794-2. [DOI] [PubMed] [Google Scholar]
- 14.Li F, Wang Y, Gong K, Wang Q, Liang Q, Qi Q. 2014. Constitutive expression of RyhB regulates the heme biosynthesis pathway and increases the 5-aminolevulinic acid accumulation in Escherichia coli. FEMS Microbiol Lett 350:209–215. doi: 10.1111/1574-6968.12322. [DOI] [PubMed] [Google Scholar]
- 15.Sharma V, Yamamura A, Yokobayashi Y. 2012. Engineering artificial small RNAs for conditional gene silencing in Escherichia coli. ACS Synth Biol 1:6–13. doi: 10.1021/sb200001q. [DOI] [PubMed] [Google Scholar]
- 16.Na D, Yoo SM, Chung H, Park H, Park JH, Lee SY. 2013. Metabolic engineering of Escherichia coli using synthetic small regulatory RNAs. Nat Biotechnol 31:170–174. doi: 10.1038/nbt.2461. [DOI] [PubMed] [Google Scholar]
- 17.Waters LS, Storz G. 2009. Regulatory RNAs in bacteria. Cell 136:615–628. doi: 10.1016/j.cell.2009.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu X, Goodrich-Blair H, Tjaden B. 2011. Assessing computational tools for the discovery of small RNA genes in bacteria. RNA 17:1635–1647. doi: 10.1261/rna.2689811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miotto P, Forti F, Ambrosi A, Pellin D, Veiga DF, Balazsi G, Gennaro ML, Di Serio C, Ghisotti D, Cirillo DM. 2012. Genome-wide discovery of small RNAs in Mycobacterium tuberculosis. PLoS One 7:e51950. doi: 10.1371/journal.pone.0051950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raghavan R, Groisman EA, Ochman H. 2011. Genome-wide detection of novel regulatory RNAs in E. coli. Genome Res 21:1487–1497. doi: 10.1101/gr.119370.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saito S, Kakeshita H, Nakamura K. 2009. Novel small RNA-encoding genes in the intergenic regions of Bacillus subtilis. Gene 428:2–8. doi: 10.1016/j.gene.2008.09.024. [DOI] [PubMed] [Google Scholar]
- 22.Albrecht M, Sharma CM, Reinhardt R, Vogel J, Rudel T. 2010. Deep sequencing-based discovery of the Chlamydia trachomatis transcriptome. Nucleic Acids Res 38:868–877. doi: 10.1093/nar/gkp1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Makarova KS, Aravind L, Wolf YI, Roman L, Minton KW, Koonin EV, Daly J, Tatusov RL. 2001. Genome of the extremely radiation-resistant bacterium Deinococcus radiodurans viewed from the perspective of comparative genomics. Microbiol Mol Biol Rev 65:44–79. doi: 10.1128/MMBR.65.1.44-79.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lecointe F, Coste G, Sommer S, Bailone A. 2004. Vectors for regulated gene expression in the radioresistant bacterium Deinococcus radiodurans. Gene 336:25–35. doi: 10.1016/j.gene.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 25.Brim H, McFarlan SC, Fredrickson JK, Minton KW, Zhai M, Wackett LP, Daly MJ. 2000. Engineering Deinococcus radiodurans for metal remediation in radioactive mixed waste environments. Nat Biotechnol 18:85–90. doi: 10.1038/71986. [DOI] [PubMed] [Google Scholar]
- 26.Appukuttan D, Rao AS, Apte SK. 2006. Engineering of Deinococcus radiodurans R1 for bioprecipitation of uranium from dilute nuclear waste. Appl Environ Microbiol 72:7873–7878. doi: 10.1128/AEM.01362-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang JC, Bloom RJ, Smolke CD. 2011. Engineering biological systems with synthetic RNA molecules. Mol Cell 43:915–926. doi: 10.1016/j.molcel.2011.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lange CC, Wackett LP, Minton KW, Daly MJ. 1998. Engineering a recombinant Deinococcus radiodurans for organopollutant degradation in radioactive mixed waste environments. Nat Biotechnol 16:929–933. doi: 10.1038/nbt1098-929. [DOI] [PubMed] [Google Scholar]
- 29.Minton KW. 1994. DNA repair in the extremely radioresistant bacterium Deinococcus radiodurans. Mol Microbiol 13:9–15. doi: 10.1111/j.1365-2958.1994.tb00397.x. [DOI] [PubMed] [Google Scholar]
- 30.Cox MM, Battista JR. 2005. Deinococcus radiodurans—the consummate survivor. Nat Rev Microbiol 3:882–892. doi: 10.1038/nrmicro1264. [DOI] [PubMed] [Google Scholar]
- 31.Slade D, Radman M. 2011. Oxidative stress resistance in Deinococcus radiodurans. Microbiol Mol Biol Rev 75:133–191. doi: 10.1128/MMBR.00015-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tanaka M, Earl AM, Howell HA, Park M-J, Eisen JA, Peterson SN, Battista JR. 2004. Analysis of Deinococcus radiodurans's transcriptional response to ionizing radiation and desiccation reveals novel proteins that contribute to extreme radioresistance. Genetics 168:21–33. doi: 10.1534/genetics.104.029249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Basu B, Apte SK. 2012. Gamma radiation-induced proteome of Deinococcus radiodurans primarily targets DNA repair and oxidative stress alleviation. Mol Cell Proteomics 11:M111.011734. doi: 10.1074/mcp.M111.011734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu G, Wang L, Chen H, Lu H, Ying N, Tian B, Hua Y. 2008. RecO is essential for DNA damage repair in Deinococcus radiodurans. J Bacteriol 190:2624–2628. doi: 10.1128/JB.01851-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bonacossa de Almeida C, Coste G, Sommer S, Bailone A. 2002. Quantification of RecA protein in Deinococcus radiodurans reveals involvement of RecA, but not LexA, in its regulation. Mol Genet Genomics 268:28–41. doi: 10.1007/s00438-002-0718-x. [DOI] [PubMed] [Google Scholar]
- 36.Daly MJ, Gaidamakova EK, Matrosova VY, Vasilenko A, Zhai M, Venkateswaran A, Hess M, Omelchenko MV, Kostandarithes HM, Makarova KS, Wackett LP, Frederickson JK, Ghosal D. 2004. Accumulation of Mn(II) in Deinococcus radiodurans facilitates gamma-radiation resistance. Science 306:1025–1028. doi: 10.1126/science.1103185. [DOI] [PubMed] [Google Scholar]
- 37.Daly MJ. 2009. A new perspective on radiation resistance based on Deinococcus radiodurans. Nat Rev Microbiol 7:237–245. doi: 10.1038/nrmicro2073. [DOI] [PubMed] [Google Scholar]
- 38.Repar J, Cvjetan S, Slade D, Radman M, Zahradka D, Zahradka K. 2010. RecA protein assures fidelity of DNA repair and genome stability in Deinococcus radiodurans. DNA Repair (Amst) 9:1151–1161. doi: 10.1016/j.dnarep.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 39.Slade D, Lindner AB, Paul G, Radman M. 2009. Recombination and replication in DNA repair of heavily irradiated Deinococcus radiodurans. Cell 136:1044–1055. doi: 10.1016/j.cell.2009.01.018. [DOI] [PubMed] [Google Scholar]
- 40.Bordelon T, Wilkinson SP, Grove A, Newcomer ME. 2006. The crystal structure of the transcriptional regulator HucR from Deinococcus radiodurans reveals a repressor preconfigured for DNA binding. J Mol Biol 360:168–177. doi: 10.1016/j.jmb.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 41.Chen X, Taylor DW, Fowler CC, Galan JE, Wang H-W, Wolin SL. 2013. An RNA degradation machine sculpted by Ro autoantigen and noncoding RNA. Cell 153:166–177. doi: 10.1016/j.cell.2013.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen X, Quinn AM, Wolin SL. 2000. Ro ribonucleoproteins contribute to the resistance of Deinococcus radiodurans to ultraviolet irradiation. Genes Dev 14:777–782. doi: 10.1101/gad.14.7.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Livny J. 2012. Bioinformatic discovery of bacterial regulatory RNAs using SIPHT, p 3–14. In Keiler KC. (ed), Bacterial regulatory RNA: methods and protocols. Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- 44.Rivas E, Eddy SR. 2001. Noncoding RNA gene detection using comparative sequence analysis. BMC Bioinformatics 2:8. doi: 10.1186/1471-2105-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsai C, Liao R, Chou B, Palumbo M, Contreras M. 2015. Genome-wide analyses in bacteria show small-RNA enrichment for long and conserved intergenic regions. J Bacteriol 197:40–50. doi: 10.1128/JB.02359-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sukhi SS, Shashidhar R, Kumar SA, Bandekar JR. 2009. Radiation resistance of Deinococcus radiodurans R1 with respect to growth phase. FEMS Microbiol Lett 297:49–53. doi: 10.1111/j.1574-6968.2009.01652.x. [DOI] [PubMed] [Google Scholar]
- 47.Litthauer S, Gargiulo S, van Heerden E, Hollmann F, Opperman DJ. 2014. Heterologous expression and characterization of the ene-reductases from Deinococcus radiodurans and Ralstonia metallidurans. J Mol Catal B Enzym 99:89–95. doi: 10.1016/j.molcatb.2013.10.020. [DOI] [Google Scholar]
- 48.Makarova KS, Omelchenko MV, Gaidamakova EK, Matrosova VY, Vasilenko A, Zhai M, Lapidus A, Copeland A, Kim E, Land M, Mavrommatis K, Pitluck S, Richardson PM, Detter C, Brettin T, Saunders E, Lai B, Ravel B, Kemner KM, Wolf YI, Sorokin A, Gerasimova AV, Gelfand MS, Fredrickson JK, Koonin EV, Daly MJ. 2007. Deinococcus geothermalis: the pool of extreme radiation resistance genes shrinks. PLoS One 2:e955. doi: 10.1371/journal.pone.0000955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DiChiara JM, Contreras-Martinez LM, Livny J, Smith D, McDonough KA, Belfort M. 2010. Multiple small RNAs identified in Mycobacterium bovis BCG are also expressed in Mycobacterium tuberculosis and Mycobacterium smegmatis. Nucleic Acids Res 38:4067–4078. doi: 10.1093/nar/gkq101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsai C-H, Baranowski C, Livny J, McDonough KA, Wade JT, Contreras LM. 2013. Identification of novel sRNAs in mycobacterial species. PLoS One 8:e79411. doi: 10.1371/journal.pone.0079411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gelderman G, Contreras LM. 2013. Discovery of posttranscriptional regulatory RNAs using next generation sequencing technologies, p 269–295. In Alper HS. (ed), Systems metabolic engineering: methods and protocols. Methods in molecular biology. Humana Press, New York, NY. [DOI] [PubMed] [Google Scholar]
- 52.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Luban S, Kihara D. 2007. Comparative genomics of small RNAs in bacterial genomes. OMICS 11:58–73. doi: 10.1089/omi.2006.0005. [DOI] [PubMed] [Google Scholar]
- 54.Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gueneau de Novoa P, Williams KP. 2004. The tmRNA website: reductive evolution of tmRNA in plastids and other endosymbionts. Nucleic Acids Res 32:D104–D108. doi: 10.1093/nar/gkh102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burge SW, Daub J, Eberhardt R, Tate J, Barquist L, Nawrocki EP, Eddy SR, Gardner PP, Bateman A. 2013. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res 41:D226–D232. doi: 10.1093/nar/gks1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lamichhane G, Arnvig KB, McDonough KA. 2013. Definition and annotation of (myco)bacterial non-coding RNA. Tuberculosis (Edinb) 93:26–29. doi: 10.1016/j.tube.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 58.Olivarius S, Plessy C, Carninci P. 2009. High-throughput verification of transcriptional starting sites by deep-RACE. Biotechniques 46:130–132. doi: 10.2144/000113066. [DOI] [PubMed] [Google Scholar]
- 59.Kim J, Sharma AK, Abbott SN, Wood A, Dwyer DW, Jambura A, Kenneth W, Inman RB, Daly MJ, Cox MM, Wood EA, Minton KW. 2002. RecA protein from the extremely radioresistant bacterium Deinococcus radiodurans: expression, purification, and characterization. J Bacteriol 184:1649–1660. doi: 10.1128/JB.184.6.1649-1660.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Carroll JD, Daly MJ, Minton KW. 1996. Expression of recA in Deinococcus radiodurans. J Bacteriol 178:130–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Omelchenko MV, Wolf YI, Gaidamakova EK, Matrosova VY, Vasilenko A, Zhai M, Daly MJ, Koonin EV, Makarova KS. 2005. Comparative genomics of Thermus thermophilus and Deinococcus radiodurans: divergent routes of adaptation to thermophily and radiation resistance. BMC Evol Biol 5:57. doi: 10.1186/1471-2148-5-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bauche C, Laval J. 1999. Repair of oxidized bases in the extremely radiation-resistant bacterium Deinococcus radiodurans. J Bacteriol 181:262–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Murray RGE. 1992. The family Deinococcaceae, p 3732–3744. In Balows A, Tr̈uper HG, Dworkin M, Harder W, Schleifer K-H (ed), The prokaryotes. Springer-Verlag, New York, NY. [Google Scholar]
- 64.Liu Y, Zhou J, Omelchenko MV, Beliaev AS, Venkateswaran A, Stair J, Wu L, Thompson DK, Xu D, Rogozin IB, Gaidamakova EK, Zhai M, Makarova KS, Koonin EV, Daly MJ. 2003. Transcriptome dynamics of Deinococcus radiodurans recovering from ionizing radiation. Proc Natl Acad Sci U S A 100:4191–4196. doi: 10.1073/pnas.0630387100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Luan H, Meng N, Fu J, Chen X, Xu X, Feng Q, Jiang H, Dai J, Yuan X, Lu Y, Roberts AA, Luo X, Chen M, Xu S, Li J, Hamilton CJ, Fang C, Wang J. 2014. Genome-wide transcriptome and antioxidant analyses on gamma-irradiated phases of Deinococcus radiodurans R1. PLoS One 9:e85649. doi: 10.1371/journal.pone.0085649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.White O, Eisen JA, Heidelberg JF, Hickey EK, Peterson JD, Dodson RJ, Haft DH, Gwinn ML, Nelson WC, Richardson DL, Moffat KS, Qin H, Jiang L, Pamphile W, Crosby M, Shen M, Vamathevan JJ, Lam P, McDonald L, Utterback T, Zalewski C, Makarova KS, Aravind L, Daly MJ, Minton KW, Fleischmann RD, Ketchum KA, Nelson KE, Salzberg S, Smith HO, Venter JC, Fraser CM. 1999. Genome sequence of the radioresistant bacterium Deinococcus radiodurans R1. Science 286:1571–1577. doi: 10.1126/science.286.5444.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moseley BE, Evans DM. 1983. Isolation and properties of strains of Micrococcus (Deinococcus) radiodurans unable to excise ultraviolet light-induced pyrimidine dimers from DNA: evidence for two excision pathways. J Gen Microbiol 129:2437–2445. [DOI] [PubMed] [Google Scholar]
- 68.Ferreira AC, Nobre MF, Rainey FA, Silva MT, Wait R, Burghardt J, Chung AP, Costa MS. 1997. Deinococcus geothermalis sp. nov. and Deinococcus murrayi sp. nov., two extremely radiation-resistant and slightly thermophilic species from hot springs. Int J Syst Bacteriol 47:939–947. doi: 10.1099/00207713-47-4-939. [DOI] [PubMed] [Google Scholar]
- 69.Tjaden B. 2008. TargetRNA: a tool for predicting targets of small RNA action in bacteria. Nucleic Acids Res 36:W109–W113. doi: 10.1093/nar/gkn264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rath D, Mangoli SH, Pagedar AR, Jawali N. 2012. Involvement of pnp in survival of UV radiation in Escherichia coli K-12. Microbiology 158:1196–1205. doi: 10.1099/mic.0.056309-0. [DOI] [PubMed] [Google Scholar]
- 71.Potter AJ, Kidd SP, McEwan AG, Paton JC. 2010. The MerR/NmlR family transcription factor of Streptococcus pneumoniae responds to carbonyl stress and modulates hydrogen peroxide production. J Bacteriol 192:4063–4066. doi: 10.1128/JB.00383-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ramos JL, Martínez-bueno M, Antonio J, Terán W, Watanabe K, Gallegos MT, Brennan R, Martı M, Molina-Henares AJ, Tera W. 2005. The TetR family of transcriptional repressors. Microbiol Mol Biol Rev 69:326–356. doi: 10.1128/MMBR.69.2.326-356.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Khankal R, Chin JW, Ghosh D, Cirino PC. 2009. Transcriptional effects of CRP* expression in Escherichia coli. J Biol Eng 3:13. doi: 10.1186/1754-1611-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Daly MJ, Gaidamakova EK, Matrosova VY, Kiang JG, Fukumoto R, Lee D-Y, Wehr NB, Viteri GA, Berlett BS, Levine RL. 2010. Small-molecule antioxidant proteome-shields in Deinococcus radiodurans. PLoS One 5:e12570. doi: 10.1371/journal.pone.0012570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Daly MJ. 2012. Death by protein damage in irradiated cells. DNA Repair (Amst) 11:12–21. doi: 10.1016/j.dnarep.2011.10.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.