

Abstract
Yersinia pestis, the agent of plague, forms a biofilm in its flea vector to enhance transmission. Y. pestis biofilm development is positively regulated by hmsT and hmsD, encoding diguanylate cyclases (DGCs) involved in synthesis of the bacterial second messenger c-di-GMP. rcsA, encoding an auxiliary protein in Rcs phosphorelay, is nonfunctional in Y. pestis, while in Yersinia pseudotuberculosis, rcsA is functional and represses biofilms. Previously we showed that Rcs phosphorelay negatively regulates transcription of hmsT in Y. pestis and its ancestor Yersinia pseudotuberculosis. In this study, we show that Rcs positively regulates hmsCDE operon (encoding HmsD) in Y. pestis; while in the presence of functional rcsA, Rcs represses hmsCDE operon in Y. pseudotuberculosis. Loss of rcsA's function in Y. pestis not only causes derepression of hmsT but also causes activation of hmsD, which may account for the increased biofilm formation in Y. pestis. In addition, differential regulation of the two DGCs, HmsT and HmsD by Rcs may help Y. pestis to adapt to different environment.
Yersinia pestis, the causative agent of plague, is transmitted to mammals by infected flea bites. Transmission of Y. pestis is greatly enhanced after it forms a bacterial biofilm in the proventriculus of the flea1. Y. pestis biofilm is characterized by a dense aggregate of bacteria surrounded by a polysaccharide-rich extracellular matrix, which is synthesized and exported by the Y. pestis hmsHFRS genes2,3,4,5,6.
Y. pestis biofilms are positively regulated by cyclic-di-GMP (c-di-GMP), a second messenger existing in numerous gram negative bacteria7,8,9,10. C-di-GMP is synthesized by diguanylate cyclase (DGC) enzymes and is degraded by phosphodiesterase (PDE) enzymes11,12,13. The Y. pestis genome encodes two DGCs, HmsT and HmsD, related to biofilm formation7,8,9,10. The two DGCs of Y. pestis play differential roles in environment-dependent biofilm formation. HmsT plays a major role on in vitro biofilm formation, while HmsD plays a more prominent role in producing proventricular-blocking biofilm in the flea10.
The Rcs phosphorelay, a signal transduction system conserved in Enterobacteriacea14,15, was involved in regulation of biofilm formation, pathogenesis, motility, as well as the general stress response16,17,18,19,20,21. Rcs consists of the transmembrane sensor kinase RcsC, the response regulator RcsB and the transmembrane protein RcsD, which functions as the phosphorelay intermediate between RcsC and RcsB14,15. RcsB controls gene transcription initiation, acting in homodimers, or together with auxiliary proteins including RcsA, BglJ, GadE and others22,23,24,25,26. Interaction of RcsB with the auxiliary proteins extends its regulatory repertoire and represents a special mechanism of transcriptional control in bacteria27.
Y. pestis and its closely related ancestor Yersinia pseudotuberculosis biofilms are negatively regulated by the Rcs phosphorelay system in vitro24,28. rcsA is a nonfunctional pseudogene in Y. pestis, while in Y. pseudotuberculosis, rcsA is functional and represses biofilms24,29. We previously reported that Rcs repressed hmsT transcription in Y. pestis24. In this study, we identified another regulatory target of the Rcs phosphorelay in Yersinia. We show that in Y. pestis, RcsB activates hmsCDE transcription; while with the functional auxiliary protein RcsA in Y. pseudotuberculosis, RcsB represses hmsD transcription.
Results
Functional RcsA represses the hmsCDE operon
We previously reported that Y. pestis rcsA is mutated to a nonfunctional pseudogene and that introducing the functional Y. pseudotuberculosis rcsA into Y. pestis results in decreased biofilm formation28. Further study showed that Rcs phosphorelay repressed transcription of the diguanylate cyclase gene hmsT24. However, Y. pestis with functional rcsA forms almost no biofilm in the digestive tract of fleas, whereas the Y. pestis hmsT mutant forms intermediate levels of biofilm in the digestive tract of flea24, suggesting that rcsA may regulate other target genes to repress Yersinia spp. biofilms.
Since hmsD, another DGC encoding gene in Y. pestis, plays a more important role in the biofilm formation in vivo10, we hypothesized that hmsD may be regulated by Rcs. To verify this hypothesis, we constructed transcriptional fusions using E. coli lacZ as the reporter. As hmsD is located in the hmsCDE operon, hmsC::lacZ was constructed in the pGD926 vector30. hmsT::lacZ and lcrQ::lacZ were constructed in the pGD926 vector as positive and negative controls. The resulting plasmids were transformed into Y. pestis lacZ mutants and assayed for β-galactosidase activity. As shown in Figure 1a and Supplementary Fig. S1, functional RcsA represses the transcription of hmsC::lacZ and hmsT::lacZ but not that of lcrQ::lacZ. To confirm this result of lacZ reporter assays, we tested the mRNA level of hmsD using the quantitative reverse transcription polymerase chain reaction (qRT-PCR). Transcription of hmsD is strongly decreased when functional RcsA is present (Figure 1b). We also examined HmsD expression by western blotting. Addition of 2× Myc (Myc2) to the C-terminus of HmsD does not affect its function31. The Myc2 C-terminal tag was introduced into the chromosome of Y. pestis. Consistent with the lacZ reporter assay and qRT-PCR result, HmsD is reduced in Y. pestis with functional rcsA (11% of the levels of wild type, Figure 1c, lane 3). Taken together, these results suggest functional rcsA repress the transcription of hmsCDE in Y. pestis.
Figure 1. Regulation of hmsCDE by Rcs.

(a) β-galactosidase activities of hmsC::lacZ reporter in Y. pestis. Y. pestis KIM6+ (WT) transformed with empty vector (vector) and functional RcsA (p-rcsA), RcsB deletion mutant transformed with empty vector (vector), functional RcsA (p-rcsA), wild-type RcsB (p-rcsB), inactive RcsB (p-rcsB (D56Q)) and active RcsB (p-rcsB (D56E)). (b) mRNA levels of hmsD regulated by Rcs in Y. pestis. hmsD mRNA levels were determined by qRT- PCR (Methods), and normalized to wild type. The mean and standard deviation of three independent experiments with three replicates are indicated. *P < 0.05. (c) Expression of HmsD regulated by Rcs in Y. pestis. Western blots of total protein-matched lysates prepared from stationary phase LB cultures and probed with anti-Myc antibody. Levels of HmsD were quantitated by densitometry using ImageJ from at least two independent experiments: numbers below blots indicate the ratio of HmsD from the indicated strain compared to that from wild-type hmsD-Myc2 strain (WT). ns, none specific band. Strain designations (Supplementary Table S1) are: Control, KIM6+ without Myc tag; WT, hmsD-Myc2; rcsA+, functional rcsA hmsD-Myc2; ΔrcsB, ΔrcsB hmsD-Myc2; ΔrcsB Vector, ΔrcsB hmsD-Myc2/pUC19; ΔrcsB p-rcsB, ΔrcsB hmsD-Myc2/pYC332; ΔrcsD Vector, ΔrcsD-N-terminal hmsD- Myc2/pET-32a; ΔrcsD p-rcsD, ΔrcsD-N-terminal hmsD- Myc2/pYC225. Full-length blots are presented in Supplementary Fig. S5 online.
RcsB positively regulates the hmsCDE operon
We next used the lacZ reporters to determine the effects of RcsB. To our surprise, over-expression of RcsB produced a moderate increase while mutation of RcsB resulted in a slight decrease of the promoter activity of hmsC (Figure 1a). The positive control hmsT::lacZ was repressed while the negative control lcrQ::lacZ was not affected by over-expression of RcsB (Supplementary Fig. S1). qRT-PCR and western analysis confirmed that deletion of RcsB resulted in decreased expression of hmsD, while over-expression of RcsB activated the expression of hmsD (Figure 1b and c). Phosphorylation of aspartic acid 56 (D56) is usually required for RcsB's function. Replacement D56 with glutamic acid (E) can mimic the phosphorylated status of RcsB, while mutation of D56 with glutamine (Q) abolishes the phosphorylation of RcsB32,33. Over-expression of RcsB(D56E) but not RcsB(D56Q) results in induction of hmsC expression (Figure 1a), suggesting that phosphorylation of RcsB is necessary for the regulation of hmsC. Consistent with this and the previous finding that RcsD might dephosphorylate RcsB in Y. pestis24,28, deletion of rcsD resulted in slightly increased expression of HmsD (111% of the levels of wild type, Figure 1c), while over-expression of RcsD produced a moderate decrease in expression of HmsD (61% of the levels of wild type, Figure 1c). These results imply that RcsB positively regulates the hmsCDE operon.
Since hmsD is located in the hmsCDE operon and since hmsC negatively regulates the function of hmsD31, we wanted to know the effect of expression of the hmsCDE operon on biofilm formation. To detect this, we cloned the hmsCDE genes into the pVTRA vector, where the hmsCDE genes are driven by an IPTG-inducible promoter. The resulting plasmid was transformed into the Y. pestis hmsCDE mutant and analyzed for biofilm formation. Consistent with previous findings that deletion of hmsCDE operon resulted in decreased biofilm formation, over-expression of hmsCDE following addition of IPTG resulted in increased biofilm formation (Figure 2), suggesting that increased transcription of hmsCDE activates biofilm formation. Taken together, these results suggest that Rcs might control biofilm formation in Y. pestis by upregulating the hmsCDE operon.
Figure 2. hmsCDE positively regulates biofilm formation in Y. pestis.
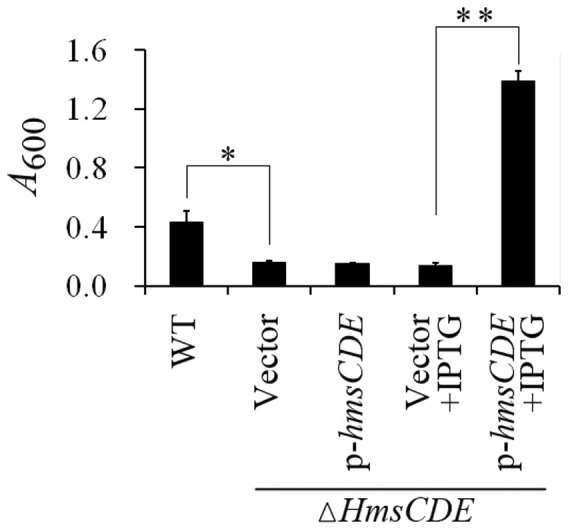
Y. pestis biofilms produced in polystyrene culture dishes and quantified by crystal violet staining (Methods). The mean and standard deviation of three independent experiments with three replicates are indicated. *P < 0.05, **P < 0.01.
An RcsAAB box in the hmsC promoter mediates the regulation of the hmsCDE operon by Rcs
Using two different 5′ RACE Kits (Methods), we determined the hmsCDE transcription start site to be 64-bp upstream of the initial ATG of hmsC. To verify this, we constructed a lacZ reporter with the mutated putative −10 box. The mutation of the −10 box resulted in nearly complete loss of activity of the hmsC promoter, indicating that the identified transcription start site is the sole or predominant one (Supplementary Fig. S2). To further rule out the possibility that hmsD has another promoter in the ORF of hmsC, we constructed a lacZ reporter that contained the hmsC ORF region but not its upstream sequence, and assayed for β-galactosidase activity. As shown in Supplementary Fig. S2, almost no promoter activity was detected in the analysis. Taken together, these results suggested that 64-bp upstream of ATG of hmsC is the sole or predominant transcription start site.
An RcsAB box is located within 59-bp upstream of the hmsC transcription start site, which matches the consensus sequence at 9 of 14 nucleotides, including all five of the most conserved nucleotides(Figure 3). We noticed that the left half of the conserved RcsAB box is repeated immediately upstream of the RcsAB box. Since the right half of the RcsAB box is believed to be the RcsB binding site23,26,34, the left part might be the RcsA binding site. Thus, we designated this region as the RcsAAB box (Figure 3), which is found in all sequenced Y. pestis and Y. pseudotuberculosis (data not shown).
Figure 3. Organization of hmsCDE promoter-proximal region and the predicted or verified cis-acting elements.
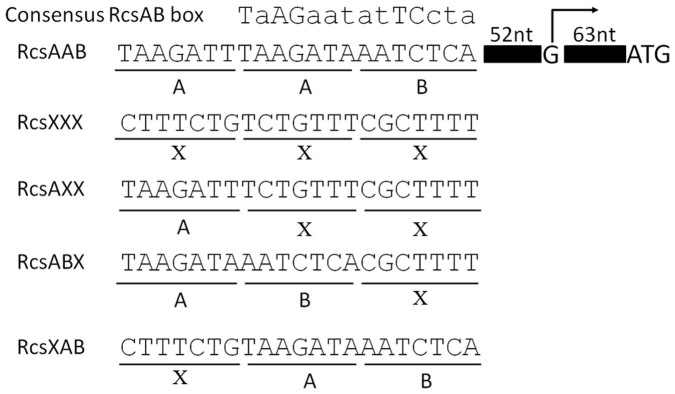
Top, the consensus RcsAB box TaAGaatatTCcta. The capital letters indicate conserved nucleotides. The right half is reported as the RcsB binding site, named B for short while the left part might be the RcsA binding site, named A for short. Middle, the organization of hmsCDE promoter-proximal region. Transcription start site G (+1) (under bent arrow) was mapped 64-bp upstream of the translation initiator codon ATG by 5′ RACE. By alignment with the consensus RcsAB box, the putative Rcs box of hmsC is located at 59-bp upstream of the transcription start site. A repeat of the left half of the conserved putative RcsA binding site is located immediately upstream of RcsAB box, thus it is designated as the RcsAAB box. Bottom, Rcs box mutations were introduced into the putative binding sites located at positions indicated. X indicates the binding site mutated.
To test the role of this region, we constructed a series of mutations of the RcsAAB box in the hmsC::lacZ fusion reporter (Figure 3) and analyzed the effects of Rcs on these mutated reporters in Y. pestis. Mutation of RcsAAB box to RcsXXX, RcsAXX or RcsABX but not RcsXAB, activated the hmsC::lacZ reporter. Functional rcsA strongly repressed the hmsC promoter with the RcsXXX, RcsAXX and RcsABX mutations in an rcsB-dependent manner (Figure 4a, b, c), but only slightly repressed the hmsC promoter with the RcsXAB mutation (Figure 4d), indicating that repression of hmsCDE by RcsA requires RcsB but is independent of the RcsAB binding site. To our surprise, RcsB also repressed hmsC::lacZ reporter when the RcsAAB box was mutated to RcsXXX, RcsAXX or RcsABX (Figure 4a, b, c), but slightly activated the hmsC::lacZ reporter in the RcsXAB mutant (Figure 4d). This result suggests that activation of hmsCDE by RcsB is dependent on the RcsB binding site in the RcsAAB box.
Figure 4. Role of the RcsAAB box on transcriptional regulation of hmsCDE by Rcs.

β-galactosidase activities of hmsC::lacZ reporters with the RcsAAB box mutated to (a) RcsXXX, (b) RcsAXX, (c) RcsABX or (d) RcsXAB were analyzed. The mean and standard deviation of three independent experiments with three replicates are indicated. The mean and standard deviation of three independent experiments with three replicates are indicated. *P < 0.05, **P < 0.01.
To confirm the results of the lacZ reporter assays, we further mutated the RcsAAB box on the chromosome and carried out western blotting analysis. Consistent with the lacZ analysis, HmsD expression was increased in Y. pestis with RcsXXX, RcsAXX and RcsABX (Figure 5a, b, c, lane 4) but not in Y. pestis with RcsXAB (Figure 5d, lane 4). Functional rcsA strongly repressed HmsD expression in Y. pestis with RcsAAB box mutation (Figure 5a, b, c, lane 4). RcsB repressed HmsD expression when the RcsAAB box was mutated to RcsXXX, RcsAXX or RcsABX (Figure 5a, b, c, lane 7), but slightly activated HmsD expression in Y. pestis with RcsXAB (Figure 5d, lane 7). The western blotting results further suggest that activation but not repression of HmsD expression by Rcs requires the RcsB binding site.
Figure 5. Role of the RcsAAB box on regulation of expression of HmsD by Rcs in Y. pestis.

Western blots of total protein-matched lysates prepared from cells with mutation of (a) RcsXXX, (b) RcsAXX, (c) RcsABX or (d) RcsXAB were analyzed by anti-Myc antibody. Levels of HmsD were quantitated by using ImageJ from at least two independent experiments: numbers below blots indicate the ratio of HmsD from the indicated strain compared to that from wild-type hmsD-Myc2 strain (WT). Strain designations (Supplementary Table S1) are: 1, KIM6+; 2, hmsD-Myc2; 3, Rcs box mutation, hmsD-Myc2; 4, functional rcsA, Rcs box mutation, hmsD-Myc2; 5, ΔrcsB, Rcs box mutation, hmsD-Myc2; 6, ΔrcsB, Rcs box mutation, hmsD-Myc2/pUC19; 7, ΔrcsB, Rcs box mutation, hmsD-Myc2/pYC332. pYC332: p-rcsB. Full-length blots are presented in Supplementary Fig. S6–S9 online.
hmsD promoter binding by RcsB
We directly assayed RcsB binding to the RcsAAB box using an electrophoretic mobility shift assay (EMSA). The DNA probe was a 112-bp promoter sequence comprising the RcsAAB box. The promoter DNA was electrophoresed alone or after incubation with purified RcsB (Methods). The promoter DNA of hmsT and lcrQ were analyzed by EMSA as positive and negative controls. Similar to hmsT promoter (Supplementary Fig. S3a), half of the hmsD probe was retarded by 800 ng of RcsB (Figure 6a, lane 3), and at 2000 ng the free probe was almost undetectable (Figure 6a, lane 6). In contrast, more than 2000 ng of RcsB was required to retard half of the lcrQ probe, and 4000 ng for essentially complete shifting (Supplementary Fig. S3b).To further examine the role of RcsAAB box, we also performed EMSA for RcsB with the mutated HmsD promoter. With the mutated RcsXXX or RcsAXX, more than 2000 ng of RcsB was required to retard half of the probe, and 4000 ng for essentially complete shifting (Figure 6b, c, lanes 6, 8), which is similar to that of the negative control lcrQ promoter (Supplementary Fig. S3b). However, with the mutated RcsXAB or RcsABX, about 1200 ng of RcsB was required to retard half of the probe, and 2400 ng for essentially complete shifting (Figure 6d, e, lane 5, 7).
Figure 6. RcsB binds to the hmsC promoter.
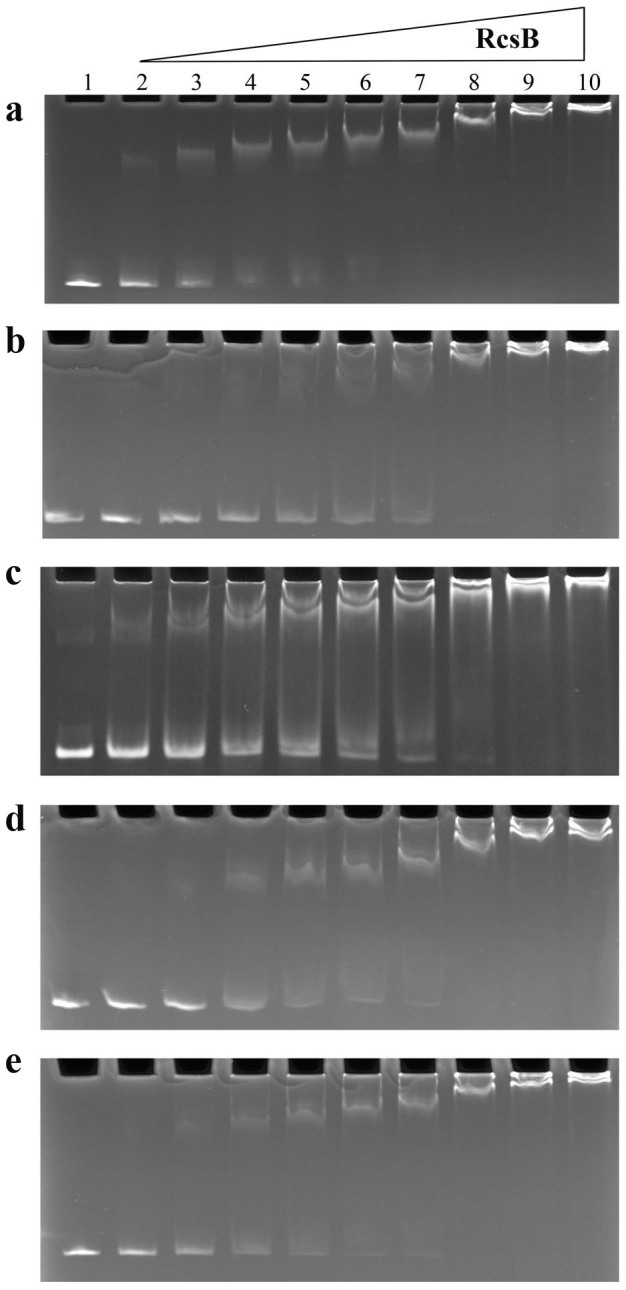
Electrophoretic mobility shift assays (EMSA) of hmsC promoter DNA incubated with increasing concentrations of RcsB. hmsD promoters with wild-type RcsAAB box (a) or with the mutated RcsAAB box RcsXXX (b), RcsAXX (c), RcsABX (d), or RcsXAB (e) were tested with identical protein combinations. Lane 1, hmsD probe alone; lanes 2–10, 100 ng hmsCDE probe with 400, 800, 1200, 1600, 2000, 2400, 4000, 6000 or 8000 ng of RcsB in the 16 μL reaction.
RcsB positively regulates but RcsAB negatively regulates hmsD in Y. pesuodtuberculosis
We next tested regulatory roles of RcsB and RcsA on hmsD in Y. pesudotuberculosis IP32953 using western blotting analysis. Consistent with the results in Y. pestis, RcsB mutation resulted in increased expression HmsD in Y. pseudotuberculosis (Supplementary Fig. S4a, lane 3). To our surprise, either mutation or over-expression of RcsB resulted in increased expression HmsD in Y. pseudotuberculosis (Supplementary Fig. S4a, lane 3, 7). To verify the role of the RcsB binding site, we also mutated the RcsAAB region to RcsAXX in Y. pseudotuberculosis. Consistent with the result in Y. pestis, repression of hmsD by Rcs was independent of the RcsB binding site (Supplementary Fig. S4b).
Discussion
Y. pestis diverged from Y. pseudotuberculosis only 1,500-6,400 years ago35,36,37. We previously obtained evidence suggesting that, during the evolution of Y. pestis, mutation of rcsA was the product of natural selection and not genetic drift28. The mutation was required for Y. pestis to colonize its flea vector with a biofilm28,29. We also previously found that a target of Rcs regulation is hmsT24, encoding a DGC that regulates biofilms. Repressing hmsT apparently is not the only mechanism by which Rcs negatively regulates biofilms, because Y. pestis hmsT mutation and Y. pestis rcsA-pstb strains have similar biofilm phenotype in vitro, but different biofilm phenotypes in vivo10,24.
In this study, we showed that hmsCDE, encoding another DGC (hmsD) that regulates biofilms in Yersinia28,31, is also a target of Rcs regulation. Most importantly, we found a dual effect of the Rcs phosporelay on expression of hmsD: RcsB alone activates hmsD transcription, but RcsAB represses hmsD transcription. Several lines of evidence support this conclusion. First, hmsD transcription is reduced when functional RcsA is present (Figure 1a, b), and HmsD protein levels are also reduced (Figure 1c and Supplementary Fig. S4a, lane 2). Secondly, hmsD transcription is reduced when RcsB is mutated and stimulated when RcsB is over-expressed (Figure 1a), and HmsD protein levels are consistent with the transcriptional levels (Figure 1c). Thirdly, deletion of hmsCDE operon results in less biofilm formation but over-expression of hmsCDE activates biofilm formation (Figure 2). Finally, we showed that RcsB binds to the hmsD promoter in an RcsB binding site-dependent manner (Figure 6). Contrary to the finding that rcsB positively regulates hmsCDE, our previously result revealed that mutation of rcsB resulted in increased biofilm formation in vitro24,28. It can be explained by the fact that hmsT but not hmsD plays a dorminant role on in vitro biofilm formation10. Mutation of rcsB causes increased expression of hmsT24, which may account for the increased biofilm formation in vitro.
The Rcs phosphorelay signal transduction system is an important signaling pathway in the Enterobacteriaceae. It has been reported that RcsB functioned as an activator and also a repressor in the regulation of gadA in E. coli23. RcsB/GadE heterodimer binds to the GAD box, while RcsB alone binds to the RcsB box. Compared with the affinity of the RcsB/GadE for the GAD box, the affinity of RcsB for RcsB box is very low23. Thus, RcsB functions as an activator at lower concentration and as a repressor at high concentration in the regulation of gadA. Apparently it is not the same case for the regulation hmsCDE. An Rcs box is present at 59-bp upstream of the transcription start site of hmsCDE. This box is necessary for the activation but not the repression of hmsCDE transcription (Figure 4 and Figure 5). RcsB might bind to this box and then directly activate the transcription of hmsCDE.
The regulatory mechanism by which RcsAB represses hmsCDE transcription is still unknown. One hypothesis is that RcsAB acts indirectly by activating or repressing another regulator, which subsequently regulates hmsCDE. This hypothesis is supported by the facts that both RcsB and RcsAB negatively regulate hmsCDE expression when RcsAAB box is mutated to RcsXXX and RcsAXX (Figure 4 and Figure 5). In addition, there is still another question: why Y. pestis and Y. pseudotuberculosis have an RcsAAB box rather than an RcsAB box? One explanation is that the presence of left RcsA binding site affects the activation role of Rcs on hmsCDE. This hypothesis is supported by the result that Rcs regulation of hmsCDE is almost gone in the RcsXAB background (Figure 4d and Figure 5d).
Our data support a model of multi-level control of hmsCDE by Rcs in Yersinia (Figure 7). In the presence of rcsB and functional rcsA, as is found in Y. pseudotuberculosis, RcsB together with RcsA repress the transcription of hmsCDE indirectly, which is independent of RcsAB binding site. At the same time, RcsB activates the transcription of hmsCDE in an RcsB binding site-dependent manner. As a whole, Rcs negatively regulates hmsCDE expression in Y. pseudotuberculosis. In the presence of rcsB alone, as found in Y. pestis, rcsB directly activates and indirectly represses hmsCDE expression. As a whole, Rcs negatively regulates hmsCDE expression in Y. pestis. In Y. pseudotuberculosis, hmsT and hmsD transcriptions are tightly repressed. This tight repression was relieved during Y. pestis evolution by the mutation of rcsA to a pseudogene28. There remains a residual repression of hmsT mediated by RcsB24, but expression of hmsD is activated by RcsB. We know that HmsT plays a dominant role on in vitro biofilm formation, while HmsD plays a major role on biofilm formation in the flea7,10. Thus RcsB negatively regulates Y. pestis biofilm formation in vitro, but might positively regulate biofilms in vivo. In summary, it appears that Y. pestis evolved to occupy the flea niche not only by derepressing hmsT but also by activating hmsD, thereby activating biofilm development.
Figure 7. Regulation of hmsCDE transcription by Rcs in Y. pseudotuberculosis and Y. pestis.
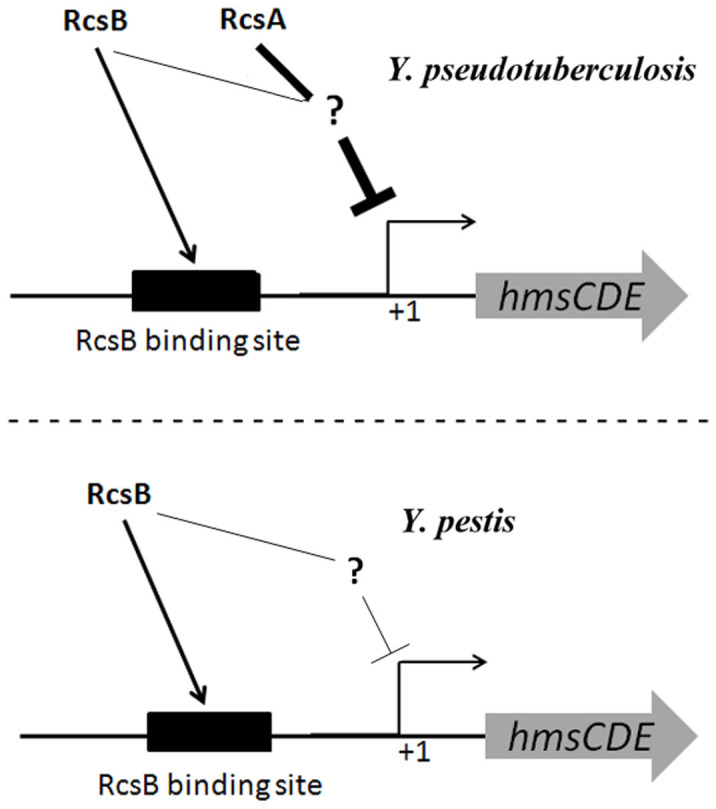
In Y. pseudotuberculosis (top), RcsB together with RcsA indirectly repress the transcription of hmsCDE independent of the RcsAB binding site, while RcsB activates transcription of hmsCDE dependent on the RcsB binding site. As a consequence, Rcs negatively regulates the hmsCDE operon in Y. pseudotuberculosis. In Y. pestis (bottom), as rcsA is mutated to nonfunctional pseudogene, RcsB alone directly positively but indirectly negatively regulates hmsCDE operon. As a consequence, Rcs positively regulates the hmsCDE operon in Y. pestis.
The Rcs system might respond to different environmental signals, resulting in precise regulation of biofilm formation through control of hmsT and hmsD. In Y. pseudotuberculosis, RcsA and RcsB could respond to different environmental signals respectively and then precisely regulate hmsCDE transcription, which in turn control biofilm formation to adapt different environment. In Y. pestis, HmsT and HmsD are differentially regulated by Rcs, which may be partially responsible for the two DGCs played differential roles on environment-dependent biofilm formation10.
Methods
Bacterial strains and plasmids
The strains and plasmids used are shown in Supplementary Table S1. For construction of hmsC::lacZ and hmsD::lacZ reporter, the 353-bp upstream region together with the first 7 condons of the hmsC or the 624-bp upstream region together with the first 7 condons of the hmsD was amplified by PCR using KIM6+ chromosome DNA as template, respectively. The DNA fragments were digested with HindIII and BamHI, and cloned into pGD92630, resulting in plasmid pYC287 and pYC487. For construction of hmsT::lacZ and lcrQ::lacZ reporter, the 353-bp upstream region together with the first 7 condons of the hmsT or the 353-bp upstream region together with the first 7 condons of the lcrQ was amplified, digested, and cloned into pGD926, resulting in plasmid pYC593 and pYC597. RcsB(D56Q) and RcsB(D56E) were generated by overlapping PCR as described previously33.
The Rcs box mutation strains were made by two-step allelic replacement38, the mutation sequences are listed in Supplementary Table S2. Briefly, a 2-kb PCR product containing the promoter of hmsCDE was amplified from Y. pestis KIM6+ and cloned into pUC19. Using the resulting plasmid as template, the Rcs box was mutated using a mutagenic PCR primer, and the product was substituted into the Y. pestis chromosome by allelic replacement38. Oligonucleotide primers used are shown in Supplementary Table S2. All strains were verified by PCR, DNA sequencing or plasmid complementation, as appropriate.
β-galactosidase assays
β-galactosidase activities were measured as previously described24. Overnight cultures of Y. pestis harboring lacZ reporters were diluted to OD600 0.05 and grown in LB broth at room temperature to OD600 about 1.5. ONPG (o-nitrophenyl-β-D-galactopyranoside) was cleaved by lysates of cells at 37°C and expressed in Miller units39. Results from three independent experiments done in triplicate were analyzed.
Quantitative real time PCR
qRT-PCR was carried out as previously described10. Briefly, cells were grown in LB broth overnight and diluted to OD600 0.05 and grown in LB broth at room temperature to OD600 about 0.8. Total RNA was isolated from collected cells using the RNeasy Mini Kit (Qiagen). Residual DNA was removed by treatment with rDNase I (Ambion) and confirmed by PCR. cDNA was synthesized from the RNA and used for quantitative PCR on an ABI Prism 7900 Sequence Detection System (Taqman, Applied Biosystems). The quantity of mRNA was normalized relative to the quantity of the reference gene crr (y1485)40. The ratio of the normalized quantity of hmsD mRNA in different strains to the normalized quantity in the wild-type samples was calculated. Primers and probe sets used are listed in Supplementary Table S2. Results from three independent experiments done in triplicate were analyzed by one-way ANOVA with Bonferroni's test.
Western blotting
Western blotting was carried out as previously described24. Samples were extracted from same amount of stationary phase cells cultured in 26°C, separated on 10% SDS-PAGE gels, transferred to PVDF membrane (Millipore), analyzed by immunoblot with antibodies to the Flag (Invitrogen) or Myc (Invitrogen), and detected with Immobilon Western HRP Substrate (Millipore). Results were quantitated by densitometry using NIH ImageJ.
Transcription start site of hmsCDE operon
The transcription start site of hmsCDE gene was determined by using the 5′ RACE System for Rapid Amplification of cDNA Ends Kit (Invitrogen) and Smart®RACE cDNA Amplification Kit (Clontech) according to the manufacturer's instructions. Primer sequences are listed in Supplementary Table S2.
Protein purification
RcsB was purified as previously described24. Briefly, Y. pestis strain containing pCBD209 was grown at 26°C to OD600 0.6 and 0.2% arabinose was added to induce protein expression for 4 h. Cells were harvested by centrifugation and disrupted by sonication. The protein was then purified by using a Ni-nitrilotriacetic acid (NTA) His Bind Purification Kit (Novagen), as recommended by the manufacturer.
Electrophoretic mobility shift assay (EMSA)
EMSA was performed as previously described. Briefly, a 112-bp PCR product containing the Rcs box or mutated Rcs box of the hmsCDE operon promoter region was amplified using the primers shown in Supplementary Table S2. Purified recombinant protein was added to DNA binding reaction mixtures containing 50 mmol Tris-HCl pH 7.5, 100 mmol NaCl, 10 mmol DTT, 500 μg/mL bovine serum albumin (BSA) and 100 ng PCR products. The binding assays were performed in a volume of 16 μL at room temperature for 30 min. After incubation samples were electrophoresed at 70 V for 1.5 hour in 6% DNA retardation gels. The gels were stained with ethidium bromide.
In vitro biofilms
Microtiter plate biofilm assays were carried out as previously described24. Briefly, bacteria were cultured overnight in LB broth supplemented with 4 mmol CaCl2 and 4 mmol MgCl2 overnight and diluted to 96-well polystyrene plates, shaking for 24 hour at 26°C. The wells were washed and the adherent biofilm was stained with crystal violet, solubilized with 80% ethanol-20% acetone and measured by A600. Results from three independent experiments with five replicates per experiment were analyzed by one-way ANOVA with Dunnet's posttest to compare the wild type to the other strains.
Author Contributions
Y.S. and X.G. wrote the main manuscript text. X.G. and G.R. prepared figures 1–6. H.Z. and X.M. prepared for some strains constructions. All authors reviewed the manuscript.
Supplementary Material
Supplementary information
Acknowledgments
We thank B. Joseph Hinnebusch for critical reading of the manuscript. This work was supported by National Basic Research Program (2015CB554200), National Science and Technology Major Project of China (No. 2013ZX10004-601), Intramural Research Program of the Institute of Pathogen Biology, CAMS, the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry, Rising Star of PUMC, the Research Fund for the Doctoral Program of Higher Education of China (20131106120052), the Youth Fund of PUMC, the Program for Changjiang Scholars and Innovative Research Team in University (IRT13007).
References
- Chouikha I. & Hinnebusch B. J. Yersinia--flea interactions and the evolution of the arthropod-borne transmission route of plague. Curr. Opin. Microbiol. 15, 239–246 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darby C., Hsu J. W., Ghori N. & Falkow S. Plague bacteria biofilm blocks food intake. Nature 417, 243–244 (2002). [DOI] [PubMed] [Google Scholar]
- Hinnebusch B. J., Perry R. D. & Schwan T. G. Role of the Yersinia pestis hemin storage (hms) locus in the transmission of plague by fleas. Science 273, 367–370 (1996). [DOI] [PubMed] [Google Scholar]
- Jarrett C. O. et al. Transmission of Yersinia pestis from an infectious biofilm in the flea vector. J. Infect. Dis. 190, 783–792 (2004). [DOI] [PubMed] [Google Scholar]
- Perry R. D., Pendrak M. L. & Schuetze P. Identification and cloning of a hemin storage locus involved in the pigmentation phenotype of Yersinia pestis. J. Bacteriol. 172, 5929–5937 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D. & Yang R. Formation and regulation of Yersinia biofilms. Protein & Cell 2, 173–179 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobrov A. G. et al. Systematic analysis of cyclic di-GMP signalling enzymes and their role in biofilm formation and virulence in Yersinia pestis. Mol. Microbiol. 79, 533–551 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones H. A., Lillard J. W. & Perry R. D. HmsT, a protein essential for expression of the haemin storage (Hms+) phenotype of Yersinia pestis. Microbiology 145, 2117–2128 (1999). [DOI] [PubMed] [Google Scholar]
- Kirillina O., Fetherston J. D., Bobrov A. G., Abney J. & Perry R. D. HmsP, a putative phosphodiesterase, and HmsT, a putative diguanylate cyclase, control Hms-dependent biofilm formation in Yersinia pestis. Mol. Microbiol. 54, 75–88 (2004). [DOI] [PubMed] [Google Scholar]
- Sun Y. C. et al. Differential control of Yersinia pestis biofilm formation in vitro and in the flea vector by two c-di-GMP diguanylate cyclases. PLoS. One 6, e19267 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan R. P. et al. Cell-cell signaling in Xanthomonas campestris involves an HD-GYP domain protein that functions in cyclic di-GMP turnover. Proc. Natl. Acad. Sci. U S A 103, 6712–6717 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ryjenkov D. A., Tarutina M., Moskvin O. V. & Gomelsky M. Cyclic diguanylate is a ubiquitous signaling molecule in bacteria: insights into biochemistry of the GGDEF protein domain. J. Bacteriol. 187, 1792–1798 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A. J., Ryjenkov D. A. & Gomelsky M. The ubiquitous protein domain EAL is a cyclic diguanylate-specific phosphodiesterase: enzymatically active and inactive EAL domains. J. Bacteriol. 187, 4774–4781 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke D. J. The Rcs phosphorelay: more than just a two-component pathway. Future Microbiol. 5, 1173–1184 (2010). [DOI] [PubMed] [Google Scholar]
- Majdalani N. & Gottesman S. The Rcs phosphorelay: a complex signal transduction system. Annu. Rev. Microbiol. 59, 379–405 (2006). [DOI] [PubMed] [Google Scholar]
- Francez-Charlot A., Castanie-Cornet M. P., Gutierrez C. & Cam K. Osmotic regulation of the Escherichia coli bdm (biofilm-dependent modulation) gene by the RcsCDB His-Asp phosphorelay. J. Bacteriol. 187, 3873–3877 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francez-Charlot A. et al. RcsCDB His-Asp phosphorelay system negatively regulates the flhDC operon in Escherichia coli. Mol. Microbiol. 49, 823–832 (2004). [DOI] [PubMed] [Google Scholar]
- Fredericks C. E., Shibata S., Aizawa S., Reimann S. A. & Wolfe A. J. Acetyl phosphate-sensitive regulation of flagellar biogenesis and capsular biosynthesis depends on the Rcs phosphorelay. Mol. Microbiol. 61, 734–747 (2006). [DOI] [PubMed] [Google Scholar]
- Pontel L. B., Pezza A. & Soncini F. C. Copper stress targets the rcs system to induce multiaggregative behavior in a copper-sensitive Salmonella strain. J. Bacteriol. 192, 6287–6290 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjit D. K. & Young K. D. The Rcs stress response and accessory envelope proteins are required for de novo generation of cell shape in Escherichia coli. J. Bacteriol. 195, 2452–2462 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta P., Clark E. R., Knutson K., Horne S. M. & Pruss B. M. OmpR and RcsB abolish temporal and spatial changes in expression of flhD in Escherichia coli Biofilm. BMC. Microbiol. 13, 182 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanie-Cornet M. P., Treffandier H., Francez-Charlot A., Gutierrez C. & Cam K. The glutamate-dependent acid resistance system in Escherichia coli: essential and dual role of the His-Asp phosphorelay RcsCDB/AF. Microbiology 153, 238–246 (2007). [DOI] [PubMed] [Google Scholar]
- Castanié-Cornet M. P. et al. Acid stress response in Escherichia coli: mechanism of regulation of gadA transcription by RcsB and GadE. Nucleic Acids Res. 38, 3546–3554 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y. C., Guo X. P., Hinnebusch B. J. & Darby C. The Yersinia pestis Rcs phosphorelay inhibits biofilm formation by repressing transcription of the diguanylate cyclase gene hmsT. J. Bacteriol. 194, 2020–2026 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh G. R. et al. BglJ-RcsB heterodimers relieve repression of the Escherichia coli bgl operon by H-NS. J. Bacteriol. 192, 6456–6464 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehland M. & Bernhard F. The RcsAB Box: characterization of a new operator essential for the regulation of exopolysaccharide biosynthesis in enteric bacteria. J. Biol. Chem. 275, 7013–7020 (2000). [DOI] [PubMed] [Google Scholar]
- Salscheider S. L., Jahn A. & Schnetz K. Transcriptional regulation by BglJ-RcsB, a pleiotropic heteromeric activator in Escherichia coli. Nucleic Acids Res. 42, 2999–3008 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y. C., Hinnebusch B. J. & Darby C. Experimental evidence for negative selection in the evolution of a Yersinia pestis pseudogene. Proc. Natl. Acad. Sci. U S A 105, 8097–8101 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y. C., Jarrett C. O., Bosio C. F. & Hinnebusch B. J. Retracing the evolutionary path that led to flea-borne transmission of Yersinia pestis. Cell Host Microbe 15, 578–586 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditta G. et al. Plasmids related to the broad host range vector, pRK290, useful for gene cloning and for monitoring gene expression. Plasmid 13, 149–153 (1985). [DOI] [PubMed] [Google Scholar]
- Ren G. X., Yan H. Q., Zhu H., Guo X. P. & Sun Y. C. HmsC, a periplasmic protein, controls biofilm formation via repression of HmsD, a diguanylate cyclase in Yersinia pestis. Environ. Microbiol. 16, 1202–1216 (2014). [DOI] [PubMed] [Google Scholar]
- Gupte G., Woodward C. & Stout V. Isolation and characterization of rcsB mutations that affect colanic acid capsule synthesis in Escherichia coli K-12. J. Bacteriol. 179, 4328–4335 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Hu Y., Francis M. S. & Chen S. RcsB positively regulates the Yersinia Ysc-Yop type III secretion system by activating expression of the master transcriptional regulator LcrF. Environ. Microbiol. 10.1111/1462-2920.12556 (2014). [DOI] [PubMed] [Google Scholar]
- Stratmann T., Pul U., Wurm R., Wagner R. & Schnetz K. RcsB-BglJ activates the Escherichia coli leuO gene, encoding an H-NS antagonist and pleiotropic regulator of virulence determinants. Mol. Microbiol. 83, 1109–1123 (2012). [DOI] [PubMed] [Google Scholar]
- Achtman M. et al. Microevolution and history of the plague bacillus, Yersinia pestis. Proc. Natl. Acad. Sci. U S A 101, 17837–17842 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chain P. S. et al. Insights into the evolution of Yersinia pestis through whole-genome comparison with Yersinia pseudotuberculosis. Proc. Natl. Acad. Sci. U S A 101, 13826–13831 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y. et al. Historical variations in mutation rate in an epidemic pathogen, Yersinia pestis. Proc. Natl. Acad. Sci. U S A 110, 577–582 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnenberg M. S. & Kaper J. B. Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect. Immun. 59, 4310–4317 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J. H. Experiments in Molecular Genetics. [352–355] (Cold Spring Harbor Laboratory Press, New York, 1972). [Google Scholar]
- Sebbane F., Jarrett C. O., Gardner D., Long D. & Hinnebusch B. J. Role of the Yersinia pestis plasminogen activator in the incidence of distinct septicemic and bubonic forms of flea-borne plague. Proc. Natl. Acad. Sci. U S A 103, 5526–5530 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information