

ABSTRACT
Many of the small DNA tumor viruses encode transforming proteins that function by targeting critical cellular pathways involved in cell proliferation and survival. In this study, we have examined whether some of the functions of the polyomavirus small T antigens (ST) are shared by the E6 and E7 oncoproteins of two oncogenic papillomaviruses. Using three different assays, we have found that E7 can provide some simian virus 40 (SV40) or murine polyomavirus (PyV) ST functions. Both human papillomavirus 16 (HPV16) and bovine papillomavirus (BPV1) E7 proteins are capable of partially substituting for SV40 ST in a transformation assay that also includes SV40 large T antigen, the catalytic subunit of cellular telomerase, and oncogenic Ras. Like SV40 ST, HPV16 E7 has the ability to override a quiescence block induced by mitogen deprivation. Like PyV ST, it also has the ability to inhibit myoblast differentiation. At least two of these activities are dependent upon the interaction of HPV16 E7 with retinoblastoma protein family members. For small T antigens, interaction with PP2A is needed for each of these functions. Even though there is no strong evidence that E6 or E7 share the ability of small T to interact with PP2A, E7 provides these functions related to cellular transformation.
IMPORTANCE DNA tumor viruses have provided major insights into how cancers develop. Some viruses, like the human papillomaviruses, can cause cancer directly. Both the papillomaviruses and the polyomaviruses have served as tools for understanding pathways that are often perturbed in cancer. Here, we have compared the functions of transforming proteins from several DNA tumor viruses, including two papillomaviruses and two polyomaviruses. We tested the papillomavirus E6 and E7 oncoproteins in three functional assays and found that E7 can provide some or all of the functions of the SV40 small T antigen, another well-characterized oncoprotein, in two of these assays. In a third assay, papillomavirus E7 has the same effect as the murine polyomavirus small T protein. In summary, we report several new functions for the papillomavirus E7 proteins, which will contribute new insights into the roles of viruses in cancer and the cellular pathways they perturb in carcinogenesis.
INTRODUCTION
The small DNA tumor viruses (the polyomaviruses, the adenoviruses, and the papillomaviruses) have contributed to major advances in basic mammalian cell and molecular biology. For example, these viruses were used in the discovery of processes such as mammalian transcription, RNA splicing, DNA replication, and the delineation of pathways and genes often perturbed in human cancer. Simian virus 40 (SV40) and the murine polyomavirus (PyV) have been key viruses used in these experiments. Other small DNA tumor viruses play etiologic roles in certain human cancers. High-risk human papillomaviruses (HPV) of the alpha genus, such as HPV16 and HPV18, cause cervical cancer, other anogenital cancers, and approximately 20% of upper airway carcinomas (1). The Merkel cell polyomavirus (MCV) is the best candidate for a human polyomavirus causative agent in human cancer (2).
Comparative studies of the oncoproteins encoded by the DNA tumor viruses have revealed that there are remarkable similarities among their functions and cellular targets. One early example of a shared target is the retinoblastoma protein (RB1) and its related pocket proteins (RBL1/p107 and RBL2/p130), which are targeted and inactivated by adenovirus E1A, SV40 large T antigen (LT), and the high-risk HPV E7 proteins (3–5). In addition, the p53 tumor suppressor protein is targeted and inactivated by the adenovirus E1B 55-kDa protein, SV40 LT, and the high-risk HPV E6 proteins (6–9). HPV E6 hijacks the E3 ubiquitin ligase E6AP (also known as UBE3A) and directs it to p53, which then is ubiquitylated and degraded by the proteasome (10, 11).
The early region of many polyomaviruses encodes not only LT but also a small T antigen (ST) that contributes to oncogenesis. SV40 mutants defective for producing ST have long been known to have a diminished ability to transform rodent cells (12, 13). More recently, small T has been shown to have a critical role in the full transformation of human cells in combination with the catalytic subunit of telomerase (hTERT), oncogenic Ras, and SV40 LT (14).
A major host cell target of polyomavirus small T antigens is the serine- and threonine-specific phosphatase 2A (PP2A), an abundant cellular enzyme that is composed of a catalytic C subunit bound to structural and regulatory A and B subunits. The enzyme exists in many forms consisting of different combinations of A, B, and C subunits (reviewed in reference 15). PP2A has numerous protein substrates, and in interacting with PP2A, small T antigens are believed to alter the signaling pathways regulated by PP2A. Both SV40 ST and PyV ST interact with the PP2A complex (16), but they do so in different ways and with distinct downstream effects. Some of the functions of the two have been compared directly, illustrating both the similarities and the differences that exist among these proteins (17, 18). SV40 ST binds the A subunit of PP2A and displaces B subunits, which confer substrate specificity on PP2A. In particular, depletion of the B56γ subunit from cells can mimic the effects of introducing SV40 ST (19). PyV ST promotes progression through the cell cycle in a PP2A binding-dependent manner (20). Although it seems clear that either ST can redirect PP2A to dephosphorylate new targets, the differences between the targets of PP2A in the presence and absence of small T antigens remain to be fully defined. Overall, the small T-PP2A interactions have varied and significant effects on cell cycle progression, differentiation, and tumorigenesis (14, 17, 18, 21).
Therefore, the polyomavirus ST antigens target the same cellular complex, PP2A, to promote some similar and some differing changes in cells. Another feature of DNA tumor virus oncoproteins is that some of them may use different mechanisms to target a single cellular pathway. For example, the SV40 LT blocks the downstream functions of p53, not by targeting it for degradation (as does high-risk HPV E6) but rather by stabilizing and presumably sequestering p53 away from its cellular targets. Thus, it is possible that HPV oncoproteins activate the same mitogenic pathways as those that are triggered by small T, but perhaps not through binding PP2A. In this study, we have explored the possibility that the high-risk HPV E6 and/or E7 proteins share some functions and targets with polyomavirus small T antigens. We have tested whether E6 and/or E7 can (i) substitute for SV40 ST in the transformation of human cells, (ii) overcome an override of a G0 quiescence block, and (iii) modulate myoblast differentiation. We find that E7, but not E6, can partially or fully replace SV40 or PyV ST antigen functions in each of these assays. We further analyze the requirement for RB1 binding by E7 in these assays and discuss potential new RB binding-dependent and -independent functions of E7.
MATERIALS AND METHODS
Plasmids and virus production.
Plasmid pMIG-SV40ST (9059; Addgene) was used as the starting material to generate all of the pMIG vectors used in the study. First, a BamHI site (lost in the original cloning) was introduced at the 5′ end of the SV40 ST open reading frame (ORF) by site-directed mutagenesis to create pMIG-SV40ST-BamHI. To create the pMIG-empty vector, pMIG-SV40ST was digested with BamHI and EcoRI, the fragment containing the vector backbone was gel purified, the ends were filled by Klenow fragment action, and the vector was recircularized with T4 DNA ligase. To create pMIG-E6 and -E7 plasmids, the appropriate papillomavirus ORFs were PCR amplified out of pL(E6)SH (04755; Howley laboratory), pL(E7)SH (04756; Howley laboratory), or bovine papillomavirus 1 (BPV1) in pML2D (142-06; Howley laboratory) with primers that added a 5′ BamHI site, a 3′EcoRI site, and an in-frame hemagglutinin (HA) tag. The HA tag was added to the N terminus of E6 proteins and the C terminus of E7 proteins. These DNA fragments then were digested with BamHI and EcoRI and subcloned into the modified pMIG vector in place of SV40 ST. HPV16 E7 mutants and BPV1 E7 mutants were subcloned from their original vectors (22, 23) into pMIG in the same way. pMIG SV40 ST110 has been described previously (14). pMIG SV40 ST C103S was used as described previously (21), and the mutation was characterized earlier 24. Retrovirus stocks were produced from the pMIG and pBabe vectors using previously published protocols (25).
Cell lines.
T98G cells obtained from the American Type Culture Collection (ATCC) and C2C12 cells were cultivated in high-glucose Dulbecco's modified Eagle medium (DMEM) plus 110 mg/ml sodium pyruvate, 10% fetal bovine serum (FBS), and 2 mM l-glutamine (all from Invitrogen, Life Technologies, Carlsbad, CA, USA) at 37°C and 5% CO2. C2C12 medium also contained 50 U/ml penicillin and 50 μg/ml streptomycin. HEK TER cells expressing SV40 LT, hTERT, and G12V H-Ras have been described previously (14) and were cultured in MEM plus 10% bovine serum. To create stable HEK TER cell lines, undiluted virus stocks produced as described above were mixed 1:1 with media containing 16 μg/ml Polybrene and used to infect HEK TER, T98G, or C2C12 cells. New HEK TER cell lines were expanded and sorted by fluorescence-activated cell sorting (FACS) to generate populations of green fluorescent protein (GFP)-positive cells. T98G cells were transduced in the same way, such that cells were 95 to 100% GFP positive 48 h postinfection with pMIG retroviruses and were used in cell cycle analysis experiments with growth in low serum beginning at the same 48-h postinfection time point. C2C12 cells were seeded at 20 to 30% confluence and similarly transduced with pMIG and/or pBabe retrovirus stocks overnight in the presence of Polybrene (8 μg/ml final concentration). Cells were allowed to recover for 8 h and then subjected to another round of infection.
Anchorage-independent growth assays.
Growth of cells in soft agar was performed as previously described (26). In each experiment, 5 × 103 or 5 × 104 cells were seeded into 0.4% Noble agar in MEM plus 10% bovine serum. Colonies were counted in each plate 3 weeks after cell seeding. Each cell line was analyzed in three independent soft-agar experiments. Each independent experiment included three replicate plates for each condition tested. Soft-agar plates were visualized under a dissection microscope, and colonies were counted manually. Western blot analyses of the HEK TER cell lines used in the anchorage-independent growth assays were performed as previously described (25).
Cell cycle analysis.
Twenty-four hours after retroviral transduction, T98G cells were replated in 6-well plates with 1.8 × 105 cells per well. An extra well of 2.5 ×104 empty vector cells also was plated and was maintained in 10% serum throughout the experiment. One day after replating, cells were switched to low-serum growth by washing twice with the low-serum media and incubating in DMEM containing 0.1% FBS for 72 h. Following growth in low serum, cells were incubated with bromodeoxyuridine (BrdU; RPN 201V; Amersham) for 4 h. Cells then were trypsinized, fixed in 70% ethanol, and stained with fluorescein isothiocyanate (FITC)-coupled anti-BrdU antibody (347583; BD Biosciences) and propidium iodide/RNase staining buffer (550825; BD Biosciences). Flow cytometry was performed on a BD LSR Fortessa cell analyzer.
C2C12 cell differentiation assay.
Control cells, HPV protein-expressing cells, and small T-expressing cells were seeded at 50% confluence 24 h after infection and allowed to reach confluence. The cells then were induced with differentiation medium (DMEM plus 2% horse serum) and refed with the same every 2 days thereafter. Samples were harvested after 7 days of cultivation. The cells were washed once, collected in extraction buffer (137 mM NaCl, 20 mM Tris, 0.92 mM Ca2+, 0.49 mM Mg2+, 1% NP-40, and 10% glycerol, adjusted to pH 7), and incubated for 30 min on ice in the presence of protease inhibitors (1 μM pepstatin, 1 μg/ml aprotinin, 1 mM phenylmethanesulfonyl fluoride [PMSF]). The cleared supernatant was boiled with SDS loading buffer. The extent of differentiation was measured by Western blotting for myosin heavy-chain levels (MF-20 antibody; Developmental Studies Hybridoma Bank, University of Iowa). Anti-GFP antibody (G1544; Sigma-Aldrich) was used to assess the relative level of infection, while expression of PyV ST (clone HA11, MMS-101P; Covance) and SV40 ST (DP01; Calbiochem) also were validated. Actin (A2668; Sigma-Aldrich) was used as a loading control.
RESULTS
E7 oncoproteins partially substitute for SV40 ST in HEK TER transformation.
Elucidation of the contributions of the SV40 early region to the transformation of human cells in cooperation with the catalytic subunit of telomerase (hTERT) and oncogenic Ras revealed an essential role for both SV40 small T and large T antigens (14, 26). This transformation assay included the entire SV40 early region (encoding both large T and small T), hTERT, and oncogenic Ras. SV40 large T antigen could not replace the entire SV40 early region in this assay, indicating the critical contribution of SV40 small T. Also, although E6 and E7 from HPV16 and BPV1 function as oncogenes in a variety of different assays, they were unable to replace the entire SV40 early region in this assay (14). That study did not examine whether E6 and/or E7 could replace SV40 ST in cells that expressed SV40 LT, hTERT, and activated Ras, so the question of whether the HPV oncoproteins shared functions with SV40 small T antigen had not been addressed. Therefore, we established an HEK TER-based transformation assay (Fig. 1A) and began our studies by testing whether E6 or E7 could substitute for SV40 ST in this assay. In our initial experiments, we tested the E6 and E7 genes from both HPV16 and BPV1.
FIG 1.

HEK TER anchorage independence transformation assay. (A) The HEK TER cell-based system was used to determine whether papillomavirus oncoproteins can replace SV40 ST antigen in cells expressing SV40 large T antigen, hTERT, and activated Ras (14). HEK TER cells were transduced with pMIG-based retroviruses, which consist of a test oncoprotein gene located upstream of the EMCV IRES and a GFP open reading frame. Test oncoproteins included wild-type SV40 ST, the SV40 ST110 mutant, HPV16 E6, HPV16 E7, BPV1 E6, and BPV1 E7. GFP-positive cell populations were sorted by FACS and tested for growth in soft agar. LTR, long terminal repeat. (B) HEK TER cells were transduced with pMIG retroviruses encoding SV40 small T antigen (SV40 ST), HPV16 E6, HPV16 E7, BPV1 E6, or BPV1 E7. The various panels depict phase-contrast and green fluorescence images of HEK TER cell lines after sorting and expansion. Mock-transduced HEK TER cells were included as a control but were not sorted. (C) In a separate set of transductions, HEK TER cells were transduced with a pMIG vector encoding the SV40 ST110 mutant or with an empty version of the pMIG vector. Images depict phase-contrast and green fluorescence images of HEK TER cell lines after sorting and expansion. HEK TER parental cells are included as a control but were not sorted.
HA-tagged E6 and E7 from HPV16 or BPV1 were cloned into a pMIG-based retrovirus vector upstream of the encephalomyocarditis virus (ECMV) internal ribosomal entry site (IRES) and the GFP open reading frame. As controls in this experiment, we also tested wild-type (wt) SV40 ST as well as an SV40 ST110 C-terminal truncation mutant that is unable to bind PP2A (27). HEK TER cells were transduced with the bicistronic pMIG retrovirus vectors, and the GFP-positive cell populations were selected by FACS (Fig. 1B and C). Mock-infected and parental HEK TER cells were not sorted and are included as controls to indicate the absence of GFP signal in the initial HEK TER populations. Since GFP is downstream of the IRES in the bicistronic retrovirus vector, its expression served as a surrogate for expression from the retroviral vectors in each of the HEK TER cell lines.
The HEK TER oncoprotein-expressing cell lines then were plated in soft agar at a density of either 5 × 103 or 5 × 104 cells per 6-cm dish, and colonies were counted at 3 weeks after plating. As shown in Fig. 2A, no colonies were observed for the HEK TER-HPV16 or -BPV1 E6 cell lines. However, both HPV16 and BPV1 E7 genes promoted the formation of some HEK TER cell colonies. In agreement with previous analysis, the SV40 ST truncation mutant ST110 was unable to transform HEK TER cells, reflecting a role for SV40 ST/PP2A complex formation in this assay (14). Based on this result, we continued the analysis of HPV16 and BPV1 E7 oncoprotein-expressing cell lines in three independent experiments. E7s reproducibly supported the growth of HEK TER cell colonies at approximately 10 to 20% of the efficiency of wild-type SV40 ST (Fig. 2B). SV40 ST110 never allowed colony formation. We did note that colonies under the E7 conditions sometimes were smaller than SV40 ST colonies, in addition to being less numerous. Each HEK TER cell line was analyzed in three independent experiments, with three replicate plates per condition each time, and both the reduction in colony number and size were reproducible across these experiments.
FIG 2.
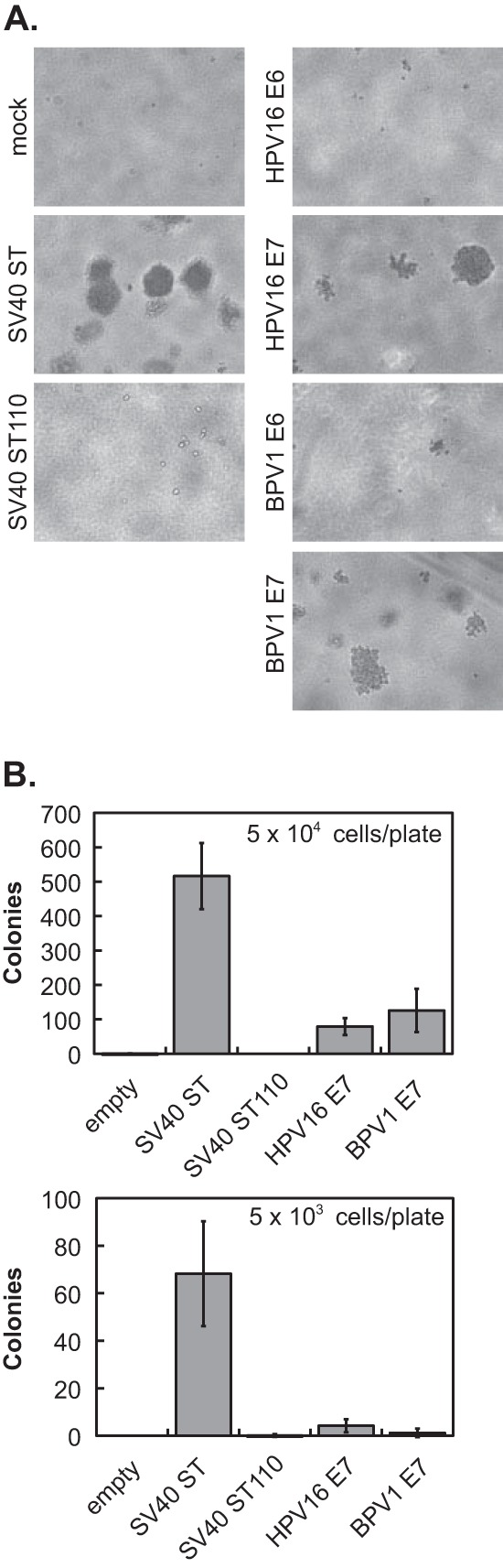
E7 oncoproteins partially replace SV40 ST in the HEK TER transformation assay. HEK TER-oncoprotein cell lines were plated in soft agar at a density of 5 × 103 or 5 × 104 cells per 6-cm dish. Cells were plated in triplicate for each condition and incubated at 37°C. Colonies were photographed after plating at the higher density (A) and counted after plating at both densities (B) 3 weeks later. Values shown in the graphs are the averages from three independent experiments ± standard deviations.
E7 activity in the HEK TER assay does not require RB1 binding and does not map to a single domain.
Since E7s, but not E6s, exhibited some activity in the HEK TER assay, we next examined a series of well-characterized HPV16 E7 and BPV1 E7 mutants to understand which of the E7 functions might be contributing to HEK TER cell transformation. Schematic representations of both wild-type E7s and the location of the mutations are shown in Fig. 3A. These mutants have been characterized extensively. For HPV16 E7 mutants, the ability of each to cooperate with ras in BRK cell transformation, to transactivate the adenovirus E2 promoter, and to bind to cellular proteins, including RB1 and UBR4, has been defined (22, 28). One HPV16 E7 mutant that has been used frequently in the literature is the ΔDLYC mutant, which is the only mutant included in this study that lacks the ability to bind RB1. The BPV1 E7 mutants have been characterized for their ability to bind UBR4, to stimulate transformation by E6, and to inhibit anoikis (23). Many of the mutations present in the BPV1 E7 ORF were introduced as alanine substitutions rather than as amino acid deletions.
FIG 3.

Regions of HPV16 E7 that contribute to HEK TER anchorage independence. (A) Schematic of HPV16 E7 and BPV1 E7 amino acid deletion and substitution mutants used in the study. The HPV16 E7 schematic was adapted from reference 39. (B) Western blot of stable HPV16 E7 expression in HEK TER cell lines. HPV16 E7 proteins tagged with the hemagglutinin epitope at the C terminus were detected with anti-HA antibody. +MG132, cells were treated with 30 μM the proteasome inhibitor MG132 for 4 h prior to harvest. (C) HEK TER-HPV16 E7 mutant cell lines were plated in soft agar at a density of 5 × 104 cells per 6-cm dish. Cells were plated in triplicate for each condition and incubated at 37°C. Colonies were counted 3 weeks after plating, and values shown in the graphs represent the averages from three independent experiments ± standard deviations. (D) Western blot of stable BPV1 E7 expression in HEK TER cell lines. BPV1 E7 proteins tagged with the hemagglutinin epitope at the C terminus were detected with anti-HA antibody. (E) HEK TER-BPV1 E7 cell lines were plated in soft agar at a density of 5 × 104 cells per 6-cm dish. Cells were plated in triplicate for each condition and incubated at 37°C. Colonies were counted 3 weeks after plating, and values shown in the graphs represent the averages from three independent experiments ± standard deviations.
Each of the E7 proteins, tagged with a hemagglutinin (HA) epitope at the C terminus, was cloned into the pMIG bicistronic vectors and transduced into HEK TER cells. The GFP-positive cell populations that had been sorted by FACS were tested for HPV16 E7 expression by Western blotting (Fig. 3B) with or without MG132 to enhance the E7 protein expression levels and to validate expression. The ΔDLYC mutant expressed at a level comparable to that of the wt and the other six mutants expressed at lower levels, but they were comparable to one another. The cells were assayed for anchorage-independent growth in triplicate at a density of 5 × 104 cells per 6-cm dish (Fig. 3C). In this experiment, wild-type HPV16 E7 transformed at approximately 50% the level of SV40 ST. None of the HPV16 E7 mutants could fully replace the ability of wt HPV16 E7 to transform in this assay, but the three mutants with deletions in CR2 of HPV16 E7 (ΔDLYC, ΔSS, and ΔEDE) retained substantial transforming activity compared to the other mutants, even though the expression of the ΔSS and ΔEDE mutants appeared to be lower than that of the ΔDLYC mutant in the Western blot analysis (Fig. 3B). Statistical analysis gave P values of 0.08 or higher for comparisons of colony formation in the presence of any one of these mutants to colony formation by wt HPV16 E7. Although the ΔDLYC mutant cannot bind to RB1, the HPV16 E7 ΔSS and ΔEDE mutants bind RB1 similarly to wild-type HPV16 E7 (22) but are not phosphorylated by casein kinase II. In contrast, the mutants with deletions or substitutions in CR1 or CR3 were more impaired in colony formation in the HEK TER assay, and each comparison of colony formation by these mutants to that of the wild-type HPV16 E7 had a P value of 0.01 or less. These regions of E7 are not involved in RB1 binding, but the N-terminal CR1 region is known to contribute to an interaction with the putative N-end rule ubiquitin ligase UBR4 (28).
Similarly, analysis of the BPV1 E7 mutants did not assign the activity in the HEK TER transformation assay to a single small domain. Again, the HEK TER cells expressing the various BPV1 E7 mutant proteins were sorted by FACS, and the resulting GFP-positive cell populations were validated for BPV1 E7 expression by Western blotting (Fig. 3D). The cell lines were tested in the HEK TER colony formation assay (Fig. 3E). As in the analysis of HPV16 E7 mutants, some of the BPV1 alanine substitution mutants (RR and TSSTS) retained more activity than others in the transformation assay. Other mutants (Δ5-9, LLIL, and Δ90-94) were unable to support the formation of any HEK TER colonies. We note that the binding of UBR4 to BPV1 E7 also has been mapped to the Δ5-9 and LLIL regions, and that studies with BPV1 E7 have correlated its binding to UBR4 with the ability to inhibit anoikis and enhance anchorage independence (23, 29). Similar to HPV16 E7, the BPV1 E7 UBR4 binding mutants (Δ5-9 and LLIL) were impaired in colony formation, but several mutants still capable of engaging UBR4 also were impaired, indicating that other parts of E7 also contribute in this assay. It is possible that transforming activities for BPV1 E7 act through other binding partners that are not shared with HPV16 E7 and that have not yet been studied in detail.
Finally, it is important to note that RB1 binding to E7 likely is not a critical contributor to transformation in this assay and that this is illustrated in two ways. First, the HEK TER cells express SV40 LT that can inactivate retinoblastoma family proteins, so it is not surprising that the HPV16 E7 transformation complementation function does not require RB binding. Second, BPV1 E7 does not bind to RB family members (30), so its activity in this assay is independent of RB1 binding. The implications of these results for RB-independent functions of E7 in transformation are highlighted in Discussion. SV40 LT does not bind to UBR4 (31 and J. DeMasi and P. M. Howley, unpublished data).
HPV16 E7 can overcome G0 arrest.
Quiescence (G0) allows cycling cells to reversibly exit the cell cycle. In a collaborative study involving several of our laboratories, we found that inhibition of PP2A during G2 enables cells to override quiescence (21). These data pointed to the modulation of PP2A-B56γ-driven Ras signaling during G2 as the key to permitting normal G0. This study also established that SV40 ST can override quiescence (G0), and that this activity requires the binding and inactivation of PP2A (21). Specifically, wt SV40 ST allowed cells to bypass quiescence, whereas the C103S mutant form of SV40 ST did not. SV40 ST C103S is a mutant that is defective in PP2A inhibition, but unlike the SV40 ST110 mutant used in the HEK TER assay, it retains some ability to bind PP2A (21, 24). Since an override of G0 may be a key element of a transformed phenotype, we tested whether the HPV16 E6 or E7 oncogenes could, like SV40 ST, override a G0 block.
As in the previous studies, we used the T98G human glioblastoma cell line for these experiments. T98G cells enter quiescence when deprived of serum mitogens. The cells were transduced with retroviruses encoding wild-type SV40 ST antigen, the C103S mutant of SV40 ST, HPV16 E6, HPV16 E7, the ΔDLYC mutant of HPV16 E7, or appropriate control vectors. To test the ability of these oncoproteins to override quiescence, cells were arrested in G0 by growth in low serum, treated with BrdU, and stained with anti-BrdU antibodies and propidium iodide, and then BrdU uptake and DNA content were analyzed by flow cytometry. Cells were considered to override G0 when they continued to incorporate BrdU, indicating cells in S phase, following 72 h in 0.1% serum. A representative experiment is shown in Fig. 4A. Consistent with the previous work, 16.4% of T98G cells expressing wt SV40 ST incorporated BrdU during the experiment, compared to 4.5% of empty vector and 3.8% of SV40 ST C103S cells in S phase in the same time period. HPV16 E7 was able to promote the progression of 12.2% of the cell population into S phase, but HPV16 E6 did not allow cell cycle progression above the level of the negative controls. We repeated the experiment and summarized three independent replicates (Fig. 4B) with the same result. Wild-type SV40 ST and HPV16 E7 are able to promote the bypass of G0 arrest in T98G cells, but C103S ST and HPV16 E7 are not.
FIG 4.

HPV16 E7 promotes the bypass of G0 arrest in T98G cells. (A) T98G cells expressing wild-type SV40 small T (ST), SV40 ST C103S, HPV16 E6, HPV16 E7, HPV16 E7 ΔDLYC, or an empty vector (Empty) were grown in DMEM containing 0.1% FBS for 72 h, followed by BrdU labeling and propidium iodide (PI) staining for DNA content. An additional population of empty vector cells was maintained in 10% FBS and is included as a control. The percentage of cells in S phase was determined by flow cytometry. (B) Percentage of cells in S phase for T98G cells expressing HPV16 E6 or HPV16 E7 or containing the empty vector. The graph represents averages with standard deviations from three independent experiments, one of which is the experiment shown in panel A. A two-tailed t test was used to determine statistically significant differences between the groups, and selected P values are indicated on the graph.
Since HPV16 E7, unlike SV40 ST, is able to complex with and inactivate the retinoblastoma family of pocket proteins, we tested whether the E7 ΔDLYC mutant that is defective in binding the RB family of pocket proteins (30) also could override a G0 block. Such an activity would imply that E7 harbored a pocket protein-independent mechanism for overcoming quiescence. However, cells expressing HPV16 E7 ΔDLYC could not promote progression into S phase, as measured by BrdU incorporation, as well as the wild-type HPV16 E7 control could (Fig. 4A and B). Therefore, we conclude that the ability of HPV16 E7 to bypass quiescence is dependent at least in part upon the integrity of the domain involved in binding the retinoblastoma family proteins.
HPV16 E7 blocks myoblast differentiation.
We next tested the effects of HPV16 oncoproteins on myoblast differentiation in the C2C12 model. There is often an inverse relationship between cellular differentiation and transformation, and the C2C12 assay has been used to assess how viral proteins and other factors affect myoblast differentiation. The effects of both SV40 ST and PyV ST have been tested extensively in the C2C12 differentiation assay (17, 18). Briefly, PyV ST inhibits the differentiation of C2C12 myoblasts into myotubes, while SV40 ST does not. Here, we wanted to determine the effect of HPV16 oncoproteins in the same assay. C2C12 cells were transduced with pMIG retroviruses encoding SV40 ST, HPV16 E6, HPV16 E7, or the HPV E7 ΔPTLHE or ΔDLYC mutant, with a pBabe-puro retrovirus encoding murine polyomavirus small T, or with appropriate vector controls and were stimulated to differentiate into myotubes using standard culture conditions. The extent of differentiation was measured visually (Fig. 5A) and by Western blotting for myosin heavy chain (MHC), a marker of myoblast differentiation (Fig. 5B). As previously reported, the presence of SV40 ST allowed the formation of myotubes and induced MHC expression to levels higher than that of the control cells. In contrast, PyV ST inhibited both the formation of myotubes and MHC levels. We also observed that HPV16 E7 inhibited differentiation by both measures, while HPV16 E6 did not.
FIG 5.

HPV E7 inhibits myoblast differentiation. C2C12 cells were transduced with retroviruses encoding SV40 ST, PyV ST, HPV16 E6, HPV16 E7, and HPV16 E7 ΔPTHLE and ΔDLYC mutants or with an empty vector retrovirus, or they were mock transduced. The extent of differentiation was measured visually (A) or by blotting for MHC (B) 7 days after the initiation of differentiation.
In order to assess which function of HPV16 E7 was responsible for the block in differentiation, we used E7 mutants defective in binding certain interacting proteins. Specifically, we tested the HPV16 E7 ΔPTLHE mutant, which is defective in binding UBR4 (28), and the E7 ΔDLYC mutant, which is defective in binding RB1 (30). Each of these mutants was defective in blocking C2C12 myoblast differentiation. These data suggest a role for both RB and UBR4 in blocking myoblast differentiation; consistent with this, it should be noted that SV40 LT inhibition of RB1 previously has been shown to inhibit myoblast differentiation (32).
DISCUSSION
In this study, we investigated whether several of the activities of polyomavirus small T antigens are shared by the papillomavirus E6 and E7 oncoproteins. All members of the polyomaviruses encode a small T antigen, and there has been considerable long-term interest in SV40 and murine PyV small T antigens because of their transforming and cell cycle control functions. The more recent discovery that the small T encoded by the Merkel cell polyomavirus (MCV) is also an oncogene and appears to be the main oncogenic driver in Merkel cell cancers has brought renewed attention to the polyomavirus small T proteins as transforming elements (33). There are many similarities among the various polyomavirus small Ts. For instance, many, if not all, of the small Ts bind PP2A and, in doing so, perturb some of its downstream signaling pathways (16, 34). However, there are important functional differences. SV40 ST functions in transformation of human mammary epithelial cells (HMEC) expressing TERT, SV40 LT, and H-Ras (14), but murine PyV ST does not (17). PyV ST inhibits C2C12 differentiation, while SV40 ST does not (17).
In our study, we assayed the ability of the papillomavirus oncoproteins to substitute for three different polyomavirus small T functions that have been well established in the literature. We found that E7 encoded by either BPV1 or HPV16 was capable of partially substituting for SV40 small T in promoting anchorage-independent growth of HEK TER cells, which are human embryonic kidney cells that also express SV40 LT, hTERT, and oncogenic Ras. We found that HPV16 E7, but not HPV16 E6, could override a G0 block induced by mitogen deprivation in human T98G cells. Finally, we found that HPV16 E7 could function similarly to PyV ST in inhibiting myoblast differentiation.
Some of these newly characterized activities of E7 make it similar to SV40 ST antigen, while others make it similar to PyV ST. Like SV40 ST, HPV16 and BPV1 E7s can contribute to anchorage-independent growth in the HEK TER soft-agar assay (Fig. 2). Also similar to SV40 ST is the ability of HPV16 E7 to bypass G0 arrest in the T98G cell assay (Fig. 4). This is a function likely to be shared with PyV ST, since in a different assay system both PyV ST and SV40 small T stimulated BrdU incorporation in serum-deprived cells (17). For SV40 ST, this G0 bypass function is mediated through the inhibition of PP2A during G2 (21). Finally, in the C2C12 differentiation assay, HPV16 E7 is more similar to PyV ST, in that both proteins inhibit the differentiation of C2C12 myoblasts into myotubes (Fig. 5). The observation that E7 can inhibit differentiation in the C2C12 assay is consistent with previous studies that have shown that HPV16 E7 can inhibit cellular differentiation of keratinocytes (35).
Defining these new activities of E7 raises the question of the molecular mechanism behind each function. Each of the small T activities that we tested has been linked to the ability of small T to bind PP2A (14, 17). There is a report that HPV16 E7 also binds PP2A (36); however, our laboratories have been unable to confirm such an interaction. In a comprehensive mass spectrometry proteomic study that identified the cellular interacting partners of E7 proteins from 17 different HPVs (including HPV16), none of the subunits of PP2A bound to E7 (25). A similar analysis of BPV1 E7 also failed to identify any PP2A components as interacting partners (37). Furthermore, an independent analysis used mass spectrometry as well as yeast two-hybrid technologies to identify interaction networks for a variety of viral oncoproteins (31). That study identified components of PP2A as interaction partners of several polyomavirus small T antigens but did not identify components of PP2A in complex with any HPV E7 protein. Our studies suggest that E7 and polyomavirus small Ts affect some common or complementing downstream pathways, but that E7 must do so by perturbing one or more signaling pathways though interactions that do not directly involve PP2A. In addition, it is clear that SV40 ST and PyV ST themselves have different effects on and interactions with PP2A (17). Together, these observations suggest that the mechanisms involved are complex. Our study provides additional assays to examine E7's transforming activities that may help to understand these mechanisms.
Perhaps the best-recognized and most frequently documented activity of HPV16 E7 is its ability to bind and inactivate RB family proteins, so we also tested the ability of the HPV16 E7 ΔDLYC protein, which is unable to bind RB proteins, in each of these assays. In the HEK TER cell assay, although E7's transformation capacity in this assay was not as robust as that of SV40 ST, it was not dependent upon the ability of E7 to bind RB1 or the related retinoblastoma family of pocket proteins (Fig. 3). BPV1 E7 does not contain an RB1 LXCXE binding domain, and HPV16 E7 mutants unable to bind RB1 still were transformed in this assay. As previously noted, this could be due to the presence of SV40 LT antigen in the cells, which would negate any requirement for RB inactivation by E7. Nonetheless, including LT in this assay has the benefit of allowing the analysis of E7 functions in a context where RB activity has been eliminated. This allows us to show that other regions of E7 do contribute to transformation in the assay and suggests that further study of the RB-independent transforming functions of E7 are warranted. In the second assay, E7 also allowed cells to bypass the G0 senescence block and did so in an RB binding motif-dependent way (Fig. 4). This highlights the important contribution of the E7-RB interaction to cell cycle control. It will require further investigation to determine what RB-independent functions of E7, if any, also contribute here. Finally, PyV ST is well documented to promote progression through the cell cycle. Perhaps it is not surprising, then, that the E7-RB interaction, which also promotes cell cycle progression, was shown by our experiment to be important for E7 to inhibit C2C12 differentiation (Fig. 5). Other mutational analyses also were informative and highlighted possible new avenues of investigation. In the anchorage-independent growth assay, we noted a partial defect with the HPV16 E7 ΔPTLHE and BPV1 E7 Δ5-9 and LLIL mutants that are defective in binding UBR4, suggesting an additional activity of E7 in mediating this phenotype. An area of longtime interest in our laboratories has been the mechanisms by which BPV1 E7 transforms in the absence of RB binding, and this study highlights that there is more to learn in this regard. In particular, we plan to conduct proteomic studies that will allow a comparison of HPV16 E7 and BPV1 E7 binding partners, with the goal of validating and understanding the protein interactions unique to BPV1 E7 that contribute to transformation.
In summary, our studies have identified shared functions between the papillomavirus E7 proteins and polyomavirus small T antigens. Defining the activity of E7 in these assays contributes to the already decades-long efforts to understand the mechanisms by which E7 and other oncoproteins transform, and our experiments also highlight the complexity of E7 effects. Although for the polyomavirus proteins the functions we assessed are dependent on binding to different subunits of PP2A, E7 does not appear to interact directly with this phosphatase complex. Like E7, another viral oncoprotein that has been shown to have transforming functions that are not dependent on PP2A binding is the ST antigen of MCV (38). Although MCV ST can bind to PP2A, the function of a PP2A binding-deficient mutant in transformation assays was not impaired in that study. It will be interesting to further analyze the PP2A-independent functions of these oncoproteins to understand how they replace the functions provided by SV40 and/or PyV ST binding to PP2A. It will also be important to continue to analyze the RB binding-independent functions of E7, especially those related to the E7-UBR4 interaction. Continuing to define and analyze the transforming properties of the DNA tumor virus oncoproteins will advance our understanding of cellular transformation and the mechanisms by which pathways are altered during cancer development.
ACKNOWLEDGMENTS
This work was supported by NIH grant P01CA050661 (P.M.H., T.M.R., B.S.S., D.M.L., and W.C.H.) and individual NRSA grant F32AI080075 to E.A.W.
REFERENCES
- 1.zur Hausen H. 2009. Papillomaviruses in the causation of human cancers–a brief historical account. Virology 384:260–265. doi: 10.1016/j.virol.2008.11.046. [DOI] [PubMed] [Google Scholar]
- 2.Chang Y, Moore PS. 2012. Merkel cell carcinoma: a virus-induced human cancer. Annu Rev Pathol 7:123–144. doi: 10.1146/annurev-pathol-011110-130227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeCaprio JA, Ludlow JW, Figge J, Shew JY, Huang CM, Lee WH, Marsillo E, Paucha E, Livingston DM. 1988. SV40 large tumor antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell 54:275–283. doi: 10.1016/0092-8674(88)90559-4. [DOI] [PubMed] [Google Scholar]
- 4.Dyson N, Howley PM, Munger K, Harlow E. 1989. The human papillomavirus-16 E7 oncoprotein is able to bind the retinoblastoma gene product. Science 243:934–937. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- 5.Whyte P, Buchkovich KJ, Horowitz JM, Friend SH, Raybuck M, Weinberg RA, Harlow E. 1988. Association between an oncogene and an anti-oncogene: the adenovirus E1A proteins bind to the retinoblastoma gene product. Nature 334:124–129. doi: 10.1038/334124a0. [DOI] [PubMed] [Google Scholar]
- 6.Lane DP, Crawford LV. 1979. T antigen is bound to a host protein in SV40-transformed cells. Nature 278:261–263. doi: 10.1038/278261a0. [DOI] [PubMed] [Google Scholar]
- 7.Linzer D, Levine AJ. 1979. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 17:43–52. doi: 10.1016/0092-8674(79)90293-9. [DOI] [PubMed] [Google Scholar]
- 8.Sarnow P, Ho YS, Williams J, Levine AJ. 1982. Adenovirus E1b-58kd tumor antigen and SV40 large tumor antigen are physically associated with the same 54 kd cellular protein in transformed cells. Cell 28:387–394. doi: 10.1016/0092-8674(82)90356-7. [DOI] [PubMed] [Google Scholar]
- 9.Werness BA, Levine AJ, Howley PM. 1990. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science 248:76–79. doi: 10.1126/science.2157286. [DOI] [PubMed] [Google Scholar]
- 10.Huibregtse JM, Scheffner M, Howley PM. 1991. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J 10:4129–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. 1993. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquination of p53. Cell 75:495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- 12.Rubin H, Figge J, Bladon MT, Chen LB, Ellman M, Bikel I, Farrell M, Livingston DM. 1982. Role of small t antigen in the acute transforming activity of SV40. Cell 30:469–480. doi: 10.1016/0092-8674(82)90244-6. [DOI] [PubMed] [Google Scholar]
- 13.Sleigh MJ, Topp WC, Hanich R, Sambrook JF. 1978. Mutants of SV40 with an altered small t protein are reduced in their ability to transform cells. Cell 14:79–88. doi: 10.1016/0092-8674(78)90303-3. [DOI] [PubMed] [Google Scholar]
- 14.Hahn WC, Dessain SK, Brooks MW, King JE, Elenbaas B, Sabatini DM, DeCaprio JA, Weinberg RA. 2002. Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol Cell Biol 22:2111–2123. doi: 10.1128/MCB.22.7.2111-2123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sablina AA, Hahn WC. 2008. SV40 small T antigen and PP2A phosphatase in cell transformation. Cancer Metastasis Rev 27:137–146. doi: 10.1007/s10555-008-9116-0. [DOI] [PubMed] [Google Scholar]
- 16.Pallas DC, Shahrik LK, Martin BL, Jaspers S, Miller TB, Brautigan DL, Roberts TM. 1990. Polyoma small and middle T antigens and SV40 small t antigen form stable complexes with protein phosphatase 2A. Cell 60:167–176. doi: 10.1016/0092-8674(90)90726-U. [DOI] [PubMed] [Google Scholar]
- 17.Andrabi S, Hwang JH, Choe JK, Roberts TM, Schaffhausen BS. 2011. Comparisons between murine polyomavirus and Simian virus 40 show significant differences in small T antigen function. J Virol 85:10649–10658. doi: 10.1128/JVI.05034-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hwang JH, Jiang T, Kulkarni S, Faure N, Schaffhausen BS. 2013. Protein phosphatase 2A isoforms utilizing Abeta scaffolds regulate differentiation through control of Akt protein. J Biol Chem 288:32064–32073. doi: 10.1074/jbc.M113.497644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen W, Possemato R, Campbell KT, Plattner CA, Pallas DC, Hahn WC. 2004. Identification of specific PP2A complexes involved in human cell transformation. Cancer Cell 5:127–136. doi: 10.1016/S1535-6108(04)00026-1. [DOI] [PubMed] [Google Scholar]
- 20.Mullane KP, Ratnofsky M, Cullere X, Schaffhausen B. 1998. Signaling from polyomavirus middle T and small T defines different roles for protein phosphatase 2A. Mol Cell Biol 18:7556–7564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Naetar N, Soundarapandian V, Litovchick L, Goguen KL, Sablina AA, Bowman-Colin C, Sicinski P, Hahn WC, DeCaprio JA, Livingston DM. 2014. PP2A-mediated regulation of Ras signaling in G2 is essential for stable quiescence and normal G1 length. Mol Cell 54:932–945. doi: 10.1016/j.molcel.2014.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Phelps WC, Münger K, Yee CL, Barnes JA, Howley PM. 1992. Structure-function analysis of the human papillomavirus type 16 E7 oncoprotein. J Virol 66:2418–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeMasi J, Chao MC, Kumar AS, Howley PM. 2007. Bovine papillomavirus E7 oncoprotein inhibits anoikis. J Virol 81:9419–9425. doi: 10.1128/JVI.00422-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mungre S, Enderle K, Turk B, Porras A, Wu YQ, Mumby MC, Rundell K. 1994. Mutations which affect the inhibition of protein phosphatase 2A by simian virus 40 small-T antigen in vitro decrease viral transformation. J Virol 68:1675–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.White EA, Sowa ME, Tan MJ, Jeudy S, Hayes SD, Santha S, Munger K, Harper JW, Howley PM. 2012. Systematic identification of interactions between host cell proteins and E7 oncoproteins from diverse human papillomaviruses. Proc Natl Acad Sci U S A 109:E260–E267. doi: 10.1073/pnas.1116776109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. 1999. Creation of human tumour cells with defined genetic elements. Nature 400:464–468. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 27.Mateer SC, Fedorov SA, Mumby MC. 1998. Identification of structural elements involved in the interaction of simian virus 40 small tumor antigen with protein phosphatase 2A. J Biol Chem 273:35339–35346. doi: 10.1074/jbc.273.52.35339. [DOI] [PubMed] [Google Scholar]
- 28.Huh KW, DeMasi J, Ogawa H, Nakatani Y, Howley PM, Munger K. 2005. Association of the human papillomavirus type 16 E7 oncoprotein with the 600-kDa retinoblastoma protein-associated factor, p600. Proc Natl Acad Sci U S A 102:11492–11497. doi: 10.1073/pnas.0505337102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DeMasi J, Huh KW, Nakatani Y, Munger K, Howley PM. 2005. Bovine papillomavirus E7 transformation function correlates with cellular p600 protein binding. Proc Natl Acad Sci U S A 102:11486–11491. doi: 10.1073/pnas.0505322102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Münger K, Werness BA, Dyson N, Phelps WC, Howley PM. 1989. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J 8:4099–4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rozenblatt-Rosen O, Deo RC, Padi M, Adelmant G, Calderwood MA, Rolland T, Grace M, Dricot A, Askenazi M, Tavares M, Pevzner SJ, Abderazzaq F, Byrdsong D, Carvunis AR, Chen AA, Cheng J, Correll M, Duarte M, Fan C, Feltkamp MC, Ficarro SB, Franchi R, Garg BK, Gulbahce N, Hao T, Holthaus AM, James R, Korkhin A, Litovchick L, Mar JC, Pak TR, Rabello S, Rubio R, Shen Y, Singh S, Spangle JM, Tasan M, Wanamaker S, Webber JT, Roecklein-Canfield J, Johannsen E, Barabasi AL, Beroukhim R, Kieff E, Cusick ME, Hill DE, Munger K, Marto JA, Quackenbush J, Roth FP, DeCaprio JA, Vidal M. 2012. Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature 487:491–495. doi: 10.1038/nature11288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maione R, Fimia GM, Holman P, Schaffhausen B, Amati P. 1994. Retinoblastoma antioncogene is involved in the inhibition of myogenesis by polyomavirus large T antigen. Cell Growth Differ 5:231–237. [PubMed] [Google Scholar]
- 33.DeCaprio JA, Imperiale MJ, Major EO. 2013. Polyomaviruses, p 1633–1661 InKnipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 34.Janssens V, Goris J, Van Hoof C. 2005. PP2A: the expected tumor suppressor. Curr Opin Genet Dev 15:34–41. doi: 10.1016/j.gde.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 35.Jones DL, Münger K. 1997. Analysis of the p53-mediated G1 growth arrest pathway in cells expressing the human papillomavirus type 16 E7 oncoprotein. J Virol 71:2905–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pim D, Massimi P, Dilworth SM, Banks L. 2005. Activation of the protein kinase B pathway by the HPV-16 E7 oncoprotein occurs through a mechanism involving interaction with PP2A. Oncogene 24:7830–7838. doi: 10.1038/sj.onc.1208935. [DOI] [PubMed] [Google Scholar]
- 37.Tan MJ. 2013. Identification of the cellular proteins and pathways engaged by the bovine papillomavirus type 1 E6 and E7 proteins. Ph.D. dissertation Harvard University, Cambridge, MA. [Google Scholar]
- 38.Shuda M, Kwun HJ, Feng H, Chang Y, Moore PS. 2011. Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J Clin Investig 121:3623–3634. doi: 10.1172/JCI46323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wise-Draper TM, Wells SI. 2008. Papillomavirus E6 and E7 proteins and their cellular targets. Front Biosci 13:1003–1017. doi: 10.2741/2739. [DOI] [PubMed] [Google Scholar]