

ABSTRACT
Human herpesvirus 6A (HHV-6A), a member of the betaherpesvirus family, is associated with several human diseases. Like all herpesviruses, HHV-6A establishes a lifelong, latent infection in its host. Reactivation of HHV-6A is frequent within the immunosuppressed and immunocompromised populations and results in lytic viral replication within multiple organs, often leading to severe disease. MicroRNAs (miRNAs) are key regulators of multiple cellular processes that regulate the translation of specific transcripts. miRNAs carried by herpesviruses play important roles in modulating the host cell, thereby facilitating a suitable environment for productive viral infection and/or latency. Currently, there are approximately 150 known human herpesvirus-encoded miRNAs, although an miRNA(s) encoded by HHV-6A has yet to be reported. We hypothesized that HHV-6A, like other members of the human herpesvirus family, encodes miRNAs, which function to promote viral infection. We utilized deep sequencing of small RNA species isolated from cells harboring HHV-6A to identify five novel small noncoding RNA species that originate from the viral genome, one of which has the characteristics of a viral miRNA. These RNAs are expressed during productive infection by either bacterial artificial chromosome (BAC)-derived virus in Jjhan cells or wild-type HHV-6A strain U1102 virus in HSB2 cells and are associated with the RNA induced silencing complex (RISC) machinery. Growth analyses of mutant viruses that lack each individual miRNA revealed that a viral miRNA candidate (miR-U86) targets the HHV-6A IE gene U86, thereby regulating lytic replication. The identification and biological characterization of this HHV-6A-specific miRNA is the first step to defining how the virus regulates its life cycle.
IMPORTANCE A majority of the human population is infected with human herpesvirus 6A (HHV-6A), a betaherpesvirus family member. Infections usually occur in young children, and upon resolution, the virus remains in a latent state within the host. Importantly, during times of weakened immune responses, the virus can reactivate and is correlated with significant disease states. Viruses encode many different types of factors that both undermine the host antiviral response and regulate viral replication, including small RNA species called microRNAs (miRNAs). Here we report that HHV-6A encodes at least one miRNA, which we named miR-U86. We have characterized the requirement of this viral miRNA and its impact on the viral life cycle and found that it functions to regulate a viral protein important for efficient viral replication. Our data suggest that viral miRNAs are important for HHV-6A and that they may serve as an important therapeutic target to inhibit the virus.
INTRODUCTION
Human herpesvirus 6 (HHV-6), a ubiquitous pathogen in the general population, is a member of the betaherpesvirus family. HHV-6 consists of two variants: HHV-6A and HHV-6B. Even though they are nearly 90% homologous at the genomic level and they have common cell tropism (e.g., T-lymphocytes), HHV-6A and HHV-6B were classified as different species in 2012 (1) based on their distinct cellular receptors, CD46 for HHV-6A (2) and CD134 for HHV-6B (3), and end-organ disease etiology (reviewed in reference 4). In industrial nations, roughly 90% of children by the age of 2 years will acquire a primary HHV-6 infection. Primary infections with HHV-6B often result in exanthem subitum (roseola infantum), which is the most common cause of infant emergency room visits (5). HHV-6A primary infection is correlated with several diseases, including encephalitis (6), chronic fatigue syndrome (7, 8), and multiple sclerosis (9); however, the underlying connections between the virus and these diseases remain to be elucidated.
Viruses utilize many factors in promoting efficient viral replication and homeostasis within host cells. Among these factors are virus-encoded microRNAs (miRNAs) (reviewed in reference 10). miRNAs are small noncoding RNAs, whose lengths range from ∼17 to 23 nucleotides and which function by binding to target mRNAs. Depending on the extent of complementarity, miRNAs either repress translation of their mRNA targets or destabilize target mRNA. The net result of either process is reduced levels of protein encoded by the target mRNA. miRNAs are involved in the regulation of multiple cellular processes such as apoptosis (for an example, see reference 11), cell fate determination (for an example, see reference 12), and tumorigenesis (for an example, see references 13 and 14). Over the past decade, more than 15,000 miRNA sequences in mammals, plants, and several DNA viruses were identified and listed in the miRNA registry, miRBase (15). Thus far, six human herpesviruses are known to encode miRNAs (reviewed in reference 16), which function to modulate the translation of both host and viral transcripts, resulting in immune evasion, undermining of host antiviral responses, inhibition of host cell apoptosis, and maintenance of latency.
Here, we report a novel miRNA encoded by HHV-6A that is distinct from the miRNAs encoded by HHV-6B (17). We further show that the expression of this miRNA is important for efficient viral replication, suggesting that like other human herpesviruses, HHV-6A encodes at least one miRNA to ensure a successful viral infection. Future work aimed at understanding the biological function of this HHV-6A-encoded miRNA will undoubtedly provide insight into gaining a better understanding of HHV-6A pathogenesis.
MATERIALS AND METHODS
Cell culture, virus production, and virus titration.
Jjhan and HSB2 cells were propagated in RPMI 1640 medium with 8% fetal bovine serum (Sigma-Aldrich, St. Louis, MO) and supplemented with 100 U/ml each of penicillin and streptomycin. HEK293 cells were propagated in Dulbecco's modified Eagle medium (DMEM) with 10% newborn calf serum (Sigma-Aldrich) and supplemented with 100 U/ml each of penicillin and streptomycin. Bacterial artificial chromosome (BAC)-derived HHV-6A (HHV-6A-BAC) strain U1102 was obtained from Yasuko Mori (Kobe University, Japan) and was previously engineered to express green fluorescent protein (GFP) (18). HHV-6A-BAC DNA was isolated as described previously (18). HHV-6A-BAC DNA (5 μg) and the human cytomegalovirus (HCMV) pp71-expressing plasmid pCGN1-pp71 (1 μg) (19) were transfected into 3.0 × 106 Jjhan cells with transfection reagent “V” utilizing a Nucleofector (Lonza AG, Basel, Switzerland) per the manufacturer's protocols. After 5 to 7 days, the medium was changed and supplemented with 20 ng/ml tissue plasminogen activator (TPA; Sigma) and 3 mM Na-butyrate (Sigma) for 24 h. Cells were washed 3 times with 1× phosphate-buffered saline (PBS) to remove the TPA and Na-butyrate and cocultured with an equal number of HSB2 cells that were prestimulated for 24 h with 2 ng/ml interleukin-2 (IL-2; Sigma) and 5 μg/ml phytohemagglutinin (PHA; Sigma). Fresh, prestimulated HSB2 cells were added every 4 to 6 days to allow accumulation of the virus by cell-to-cell spread. To isolate virus, the cultures were pelleted by low-speed centrifugation and the supernatant was reserved. Infected cells were disrupted by bath sonication to release virus from infected cells. The medium was then cleared of cellular debris, and the supernatant was added to the reserved medium. Virus was then purified by ultracentrifugation through a 20% sorbitol cushion for 90 min at 72,000 × g at 25°C. The resulting pellet was resuspended in medium supplemented with 1.5% bovine serum albumin (BSA), and aliquots were stored at −80°C following snap-freezing in liquid nitrogen.
Titers of HHV-6A were calculated using standard 50% tissue culture infective dose (TCID50) assays. Briefly, Jjhan cells were plated into a 96-well plate. Aliquots of HHV-6A were thawed, serially diluted in 10-fold increments, and used to inoculate Jjhan cells. Two weeks following infection, GFP-positive wells were scored to determine the titer of the stock.
Generation of viral recombinants.
The miRNA mutant viruses were generated by using galK selection and counterselection to modify the HHV-6A-BAC DNA, as described previously (20). In brief, the galK cassette was amplified from pGalK vector using the primers designed for each of the miRNA candidates (see Table S1 in the supplemental material). The resulting PCR products were purified using Illustra GFX PCR DNA and Gel Band Purification kits (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom) as per the manufacturer's instructions and then used to transform HHV-6A-BAC-containing SW102 cells for insertion by homologous recombination. The resulting recombinants were plated on minimal medium that contained galactose and chloramphenicol for positive selection. Colonies were then further screened on MacConkey's medium containing galactose and chloramphenicol, and successful recombinants that contained the galK cassette were used for reversion. The galK cassette was replaced by homologous recombination using double-stranded oligonucleotides (see Table S1 in the supplemental material), which contain the mutated sequence for each miRNA candidate by counterselection on minimal plates containing 2-deoxy-galactose (2-DOG) and glycerol as the carbon source. Each recombinant was confirmed by sequencing using miRNA-specific sequencing primers (see Table S1 in the supplemental material).
The epitope-tagged recombinants for the U86 protein in either the wild-type (WT) or the ΔmiR-U86 background were generated using Kan-frt recombination, as previously described (21). The C-terminal end of the U86 open reading frame (ORF) in HHV-6A BAC (WT and ΔmiR-U86) was FLAG epitope tagged with pGTE-3×FLAG-Kan-FRT to amplify the 3×FLAG-Kan-FRT (21) cassette using the following primers: FOR, 5′-ATCCTATCAGCAAAGAGTTTAAATCGAAATTTAGTACACTTTCAAAATGTTAGATTATAAAGATGATGATGATAAA −3′, and REV, 5′ CAAGGATTTCCGTTATACTGTTTTATTTTTTTTTAAAAAAAAGTTGGGAGGGCCGCGGGAATTCGAAGTT-3′, where the underlined sequences bind to the 3×FLAG-Kan-FRT cassette and the italic sequences bind the BAC DNA for homologous recombination. The resulting product was used for homologous recombination using either HHV-6A-BAC-WT- or HHV-6A-ΔmiR-U86-containing bacteria. The Kan-FRT cassette was excised by the arabinose-inducible Flp, as described previously (22) to generate WT-U86-3xF and ΔmiR-U86:U86-3xF mutant.
Deep sequencing.
Total RNA was extracted from HHV-6A-BAC-transfected Jjhan cells and mock-transfected Jjhan cells using the mirVana miRNA isolation kit (Life Technologies, Carlsbad, CA) according to the manufacturer's protocol. Total RNA isolated from normal brain tissue was purchased (catalog number 636530; Clontech, Mountain View, CA) and used as a control for sequencing library generation. The sequencing library was size selected and generated using the Illumina Small RNA sample preparation kit version 1.5 (Illumina, San Diego, CA) per the manufacturer's protocols. Sequencing reads were performed on an Illumina Genome Analyzer IIx system. Sequencing reads were analyzed by custom Linux programs that first filter reads that do not align to the human genome. The remaining reads are then aligned to HHV-6A strain Uganda 1102 (GenBank accession number X83413.1). Results were then further parsed to identify sequencing reads that matched the HHV-6A reference strain with no less than 100% match for reads of >20 nucleotides (nt) and <23 nt to identify reads corresponding to the predicted size of miRNAs. The resulting pool of candidates were mapped to the HHV-6A genome using MacVector DNA software version 12.0 (MacVector, Cary, NC) to identify the location of the reads. Reads represented >10 times in the original sequence corresponding to genomic locations that were intergenic, intronic, or opposite known ORFs were further pursued. The appropriate sequencing output files were submitted to the NCBI repository (see below).
Detection of RNA and protein species.
To assess the expression levels of the miRNAs, we employed a modified TaqMan-based stem-loop reverse transcriptase quantitative PCR (RT-qPCR). For a positive control, the level of human small-nucleolar RNA (snoRNA) RNU44 was examined using the RNU44 TaqMan control assay (Applied BioSystems, Carlsbad, CA). cDNA of the RNU44 and each candidate viral RNA was synthesized using the TaqMan MicroRNA reverse transcription kit (Applied Biosystems) according to the manufacturer's protocol with viral RNA candidate-specific stem-loop oligonucleotides (see Table S1 in the supplemental material). TaqMan qPCR was performed using a 1:15 dilution of the product from the reverse transcriptase reaction along with 0.7 μM common reverse primer (5′-GTGCAGGGTCCGAGGT-3′), 1.5 μM candidate specific forward primer (see Table S1 in the supplemental material), 0.2 μM candidate specific TaqMan probe (see Table S1 in the supplemental material), and 1× TaqMan Universal PCR master mix, No AmpErase UNG (Applied Biosystems).
To detect mRNA, cells were infected as described in the text and harvested at time points indicated in the text. Total RNA was extracted using TRI reagent (Sigma-Aldrich) and amplified using TaqMan Universal PCR master mix, No AmpErase UNG (Life Technologies), to quantify viral transcripts (U90, U12, and U100), and cellular transcript (β-actin) was quantified using TaqMan β-actin detection reagents (Life Technologies). Primers and TaqMan probes used for U90, U12, and U100 are listed in Table S1 in the supplemental material. Each reaction was performed in triplicate with 0.3 μM final concentration of each primer and 0.2 μM final concentration of TaqMan probe. All values are expressed as fold changes using the delta-delta threshold cycle (ΔΔCT) method and were normalized to input, which corresponds to 3 h after the original inoculation.
To evaluate the expression of U86, Jjhan cells were infected with either WT-U86-3xF or ΔmiR-U86:U86-3xF (multiplicity of infection [MOI], 0.1) and cells were harvested at 6 days p.i. Total protein was extracted, and an equal concentration of protein (30 μg) was separated by SDS-PAGE followed by Western blotting with the following primary antibodies: anti-m2 FLAG (1:7,500; Sigma-Aldrich) and anti-α-tubulin (1:5,000; Sigma-Aldrich). Both were visualized using a secondary antibody conjugated to horseradish peroxidase (goat-anti-mouse HRP, 1:10,000; Jackson ImmunoResearch, West Grove, PA).
IP of viral RNA/human Argonaute 2 complexes.
Immunoprecipitation (IP) protocols were followed as previously described (17). Briefly, for each IP, 1 × 108 HHV-6A-infected HSB2 cells were used. Cells were washed twice with PBS before lysis in 2 ml lysis buffer, containing 25 mM Tris-HCl (pH 7.5), 150 mM KCl, 2 mM EDTA, 0.5% NP-40, 0.5 mM dithiothreitol (DTT), and complete protease inhibitor (Roche). Lysates were incubated for 30 min at 4°C and then centrifuged at 16,100 × g for 30 min to remove nuclei and clear the cytosolic fraction. Ten micrograms of hAgo2 antibody (11A9; Millipore) or negative-control rat IgG (Millipore) was incubated with 50 μl of Dynabeads protein G (Life Technologies) in 2 ml of RPMI medium with constant rotation at 4°C overnight. Antibody-coupled beads were washed once with lysis buffer and incubated with 2 ml of lysate for 2.5 h. Beads were then washed four times with IP wash buffer (50 mM Tris-HCl [pH 7.5], 300 mM NaCl, 5 mM MgCl2, 0.1% NP-40, 1 mM NaF) and once with cold PBS. RNA was extracted from the beads by adding 500 μl TRIzol reagent (Life Technologies) twice. Isolated RNA was used for TaqMan qPCR as described above.
Detection of viral RNA species by Northern blotting.
Total RNA was extracted from HHV-6A-infected or mock-infected HSB2 cells as well as HHV-6A small RNA-overexpressing cells using TRIzol reagent (Life Technologies) according to the manufacturer's instructions. Total RNA (30 μg) was mixed with an equal volume of Gel Loading Buffer II (Applied Biosystems), and the mixture was heated at 95°C for 5 min prior to separation in a 15% urea-acrylamide gel. RNA was transferred to a HybondNX membrane (Amersham Biosciences) in 0.5× Tris-borate-EDTA (TBE) buffer and UV cross-linked. Membranes were prehybridized for 30 min in ExpressHyb (Clontech) at 37°C. For each of the 5 candidate RNA species, antisense DNA oligonucleotides were 5′-end labeled with 30 μCi of [γ-32P]dATP by using T4 polynucleotide kinase (New England BioLabs). The labeled probes were added directly to the ExpressHyb and allowed to hybridize overnight at 37°C. The blot was then washed four times for 15 min at 25°C with wash solution 1 (2× SSC, where 1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate, 0.05% SDS) and then washed twice for 20 min at 25°C with wash solution 2 (0.1 × SSC, 0.1% SDS). The blots were exposed to X-ray film at −80°C for 4 days.
Production of viral small RNA-overexpressing Jjhan cells.
To generate individual cell lines that overexpress each candidate small viral RNAs, we designed specific hairpins (see Table S1 in the supplemental material) and cloned each into a lentiviral expression plasmid, pCMV-U6, which contains the dsRed gene. To confirm the insert, all clones were sequenced using the following primer: 5′-CCCGCTAGCATCCGACGCCGCCATCTCTA-3′. HEK293 cells were transfected with HHV-6A-sncRRNA-pCMV-U6, pΔ8.9, and pVSV-G (Clontech) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Medium was collected 2 days and 4 days posttransfection, from which virus was concentrated by filtration through a 0.4-μm filter, followed by ultracentrifugation at 112,000 × g for 90 min at 4°C. The resulting viral pellet was resuspended in Jjhan RPMI medium (see above). Jjhan cells were transduced with individual lentivirus constructs expressing each candidate small RNA. Successfully transfected cells were further enriched by fluorescence-activated cell sorter (FACS) using the dsRed marker on a BD FACSAria II cell sorter (Becton Dickinson; Franklin Lakes, NJ).
Viral growth analyses.
To assess viral growth, either Jjhan cells or HHV-6A small RNA-overexpressing cells were infected (MOI = 0.01) with HHV-6A WT or the mutant viruses that were isolated by sorbitol purification from dissociated cells. Infected cells were harvested at the time points indicated in the text. At each time point, infected cells were isolated and cell-associated, total DNA was extracted as described previously (21). To quantify viral DNA, the following primers directed at GFP, which is within the BAC-derived viral genome, were used: FOR, 5′-ACCACTACCTGAGCACCCAGTC-3′, and REV, 5′-GTCCATGCCGAGAGTGATCC-3′. Each sample was normalized to cellular DNA using primers directed at cellular MDM2 FOR (5′-CCCCTTCCATCACATTGCA-3′) and REV (5′-AGTTTGGCTTTCTCAGAGATTTCC-3′). Each sample was analyzed in triplicate. All values are expressed as a ratio of HHV-6A-BAC genome copies per cell (GFP/mouse double minute 2 homolog [MDM2]) and were normalized to input as described above.
Assay for cell viability.
To assess cell viability, cell proliferation of Jjhan cells and viral small RNA-overexpressing cells was determined using the CellTiter 96 Non-Radioactive Cell Proliferation assay kit (Promega, Madison, WI) according to the company's protocol. Metabolic activity was measured using a “Victor2 Multilabel Counter” (PerkinElmer, Waltham, MA) as indicated in the text. Samples were measured in triplicate, and all values were normalized to input.
Nucleotide sequence accession number.
The sequencing output files were submitted to the NCBI repository and assigned the accession number GSE62233.
RESULTS
Identification of seven HHV-6A small noncoding RNAs.
To identify small RNAs originating from the HHV-6A genome, we utilized Illumina deep sequencing technology to profile small RNA transcripts isolated from Jjhan cells, a human CD4+ T-lymphocyte cell line (23), transfected with a BAC-derived HHV-6A (strain U1102; HHV-6A-BAC) (18). Importantly, this BAC construct was previously engineered to express GFP from a nonviral promoter (18), which allows us to monitor transfection as well as replication efficiency. One week following transfection, we observed ∼5% GFP-positive Jjhan cells. Total RNA isolated from a mock-transfected population of Jjhan cells and commercially available human brain total RNA were utilized as controls for sequencing. We extracted total RNA from each of the aforementioned cells, size selected for small (<30-bp) transcripts, and then used the resulting sequences to generate a cDNA library suitable for sequence analyses to identify viral RNAs encoded by HHV-6A. We obtained more than 3.4 × 106 sequence reads from each sample. Comparison of the sequenced libraries revealed that ∼100,000 reads were unique to the HHV-6A-transfected cells, as these were not present in the mock-transfected Jjhan cells or the library generated from the human brain total RNA. We further enriched for HHV-6A miRNA candidates by passing the sequence reads through several filters. First, the results were aligned to the human genome to remove any reads of human origin. Next, we aligned the remaining reads to an HHV-6A reference genome (GenBank accession number X83413.1). Notably, neither the mock-transfected Jjhan cells nor the human brain library contained a single transcript that mapped to the HHV-6A genome. Of the >100,000 reads, more than 10,000 transcripts from the HHV-6A-BAC-transfected cells mapped to the HHV-6A genome with 100% identity (Fig. 1A). Upon further parsing, ∼97% of those transcripts were longer than 24 nt and matched known open reading frames (ORFs) of HHV-6A, suggesting that they were of viral mRNA origin. We next mapped the remaining transcripts (∼300) to regions of the viral genome that represented intronic sequences, intergenic sequences, and those on opposite strands from known, published HHV-6A ORFs (Fig. 1A and B), as other herpesviral miRNAs originate from these genomic regions (24–26). We prioritized our efforts on these remaining candidate small viral RNAs that were 22 nt or 23 nt in length and were represented at least 10 times within our filtered reads. This resulted in a total of seven candidate viral RNAs, which we further pursued. The sequences and nucleotide coordinates are shown in Fig. 1B. Six of the identified viral RNAs failed to have characteristics conserved with known miRNAs, including the ability to form a stem-loop structure, and as such are designated small noncoding RNAs (sncRNAs). One identified candidate was found to have characteristics of an miRNA, and we have designated it miR-U86 since it is found on the opposite strand from the U86 ORF. Additionally, the predicted precursor of this candidate forms a hairpin as predicted by RNAfold analysis (Fig. 1B).
FIG 1.

Genomic localization of HHV-6A sncRNA candidates in strain U1102. Seven HHV-6A small viral transcripts map to the genome of HHV-6A strain U1102. (A) Three candidates (sncRNA-U2, sncRNA-U3-1, and sncRNA-U77) map to the intergenic region, and four candidates (sncRNA-U3-2, sncRNA-U14, sncRNA-U54, and miR-U86) map to the opposite strand of known ORFs (U3, U14, U54, and U86, respectively). Three candidates (sncRNA-U3-2, sncRNA-U54, and miR-U86) map to the plus (+) strand, and four candidates (sncRNA-U2, sncRNA-U3-1, sncRNA-U14, and sncRNA-U77) map to the minus (−) strand. (B) Candidate names, sequenced nucleotides, and genomic location within HHV6-A U1102 (GenBank accession number X83413.1). The predicted hairpin for miR-U86 is shown on the right. (C) To determine if candidate viral transcripts are associated with RISC components, lysates from HHV-6A-infected HSB-2 cells (MOI = 0.1) were immunoprecipitated with either an Ago2-specific antibody or nonspecific rat IgG antibody. Complexes were washed and purified, and RNA isolated from the immunoprecipitated complexes was analyzed by RT-qPCR using virus-specific primers. The data are shown as the fold increases of amplification from the Ago2-specific complexes compared to the nonspecific complexes. N.D., not detected.
In order to determine if the candidate viral RNAs were associated with the host RNA induced silencing complex (RISC) components, we infected HSB-2 cells with wild-type HHV-6A. We used lysates from these cells in an immunoprecipitation (IP) assay using Argonaute 2 (Ago2)-specific antibody or a nonspecific antibody, which served as a negative control. We eluted associated RNAs and quantified specific HHV-6A RNAs using RT-qPCR. We found a significant enrichment of five candidate viral RNAs bound to complexes immunoprecipitated by the Ago2-specific antibody compared to the nonspecific antibody (Fig. 1C), suggesting that four viral sncRNAs and the proposed viral miRNA are found within the host RISC machinery.
HHV-6A-infected cells express at least five small noncoding RNAs.
To validate the expression of the seven viral transcripts, we utilized stem-loop RT-qPCR methodology (27). We extracted small RNAs from Jjhan cells transfected with HHV-6A-BAC and HSB2 cells, a human T-cell lymphoblastoid cell line (28), infected with HHV-6A (strain U1102), as well as the respective parent cell lines lacking HHV-6A. We assessed the relative levels of each of the seven candidate transcripts, using the cellular small-nucleolar RNA (snoRNA) RNU44 as an internal control. Of the seven candidates, we were unable to detect sncRNA-U3-2 or sncRNA-U77 in either HHV-6A BAC-transfected Jjhan cells or HHV-6A-infected HSB2 cells (Table 1). Additionally, we were unable to detect any candidate miRNAs from the parent cell lines in the absence of HHV-6A infection. However, we did find the remaining five HHV-6A transcripts (sncRNA-U2, sncRNA-U3-1, sncRNA-U14, sncRNA-U54, and miR-U86) both in HHV-6A-BAC-transfected Jjhan cells and in HHV-6A-infected HSB-2 cells (Table 1). The viral transcripts were not amplified in our control experiments in which reverse transcriptase was not added, suggesting that the amplification observed was specific and not due to contaminating viral DNA. Importantly, we were unable to detect any of the seven candidates in mock-transfected Jjhan cells or mock-infected HSB2 cells (Table 1).
TABLE 1.
Expression of HHV-6A small RNAs in transfected Jjhan cells and infected HSB2 cells
| Cells and transcript name | Small RNA expressiona |
|||
|---|---|---|---|---|
| Mock CT ± SD | −RT CT ± SD | +RT CT ± SD | Level relative to RNU44 | |
| HHV6-transfected Jjhan cells | ||||
| RNU44 | 20.9 ± 0.1 | ND | 21.7 ± 0.3 | 1.0 |
| sncRNA-U2 | ND | ND | 29.6 ± 0.3 | 4.0 × 10−3 |
| sncRNA-U3-1 | ND | ND | 33.4 ± 0.3 | 2.9 × 10−4 |
| sncRNA-U3-2 | ND | ND | ND | NA |
| sncRNA-U14 | ND | ND | 35.8 ± 0.4 | 5.6 × 10−5 |
| sncRNA-U54 | ND | ND | 33.9 ± 0.3 | 2.0 × 10−4 |
| sncRNA-U77 | ND | ND | ND | NA |
| miR-U86 | ND | ND | 38.3 ± 0.3 | 9.7 × 10−6 |
| HHV-6A-infected HSB2 cells | ||||
| RNU44 | 21.4 ± 0.1 | ND | 21.8 ± 0.1 | 1.0 |
| sncRNA-U2 | ND | ND | 29.7 ± 0.1 | 4.3 × 10−3 |
| sncRNA-U3-1 | ND | ND | 35.0 ± 0.5 | 1.1 × 10−4 |
| sncRNA-U3-2 | ND | ND | ND | NA |
| sncRNA-U14 | ND | ND | 32.4 ± 0.2 | 6.9 × 10−4 |
| sncRNA-U54 | ND | ND | 30.0 ± 0.2 | 2.7 × 10−4 |
| sncRNA-U77 | ND | ND | ND | NA |
| miR-U86 | ND | ND | 32.4 ± 0.1 | 6.8 × 10−4 |
ND, not detected; NA, not applicable.
We next attempted to characterize the candidate miRNAs by Northern blot analyses using specific probes for the five viral RNA species described above. While we did not observe strong hybridization to RNA species within the size range of mature miRNAs (20 to 30 bases), we did observe significant hybridization of the probes to RNA species within the 100-base range (Fig. 2A), suggesting that the probes were able to bind to the precursor. It is likely that the weak hybridization to mature species is due to the low abundance of the small viral transcripts in relation to the total amount of RNA isolated from the cells. This is evident given the relative levels of candidate miRNAs in relation to the cellular snoRNA RNU44 (Table 1) (27, 28). We next mapped the exact ends of the small viral RNAs using the specificity of primer binding and extension using an RT-qPCR protocol. We designed “unfit” looped RT primers, which are modified versions of “fit” looped RT primers used to generate cDNA versions of the miRNAs suitable for TaqMan PCR (Fig. 2BI). We exploited the required specificity of the primers used in the stem-loop RT-qPCR to allow for validation of the 3′ ends of our small viral RNAs. To properly prime the reaction for generating cDNA, the looped primer must bind within the last seven 3′ terminal nucleotides without gaps or additional nucleotides (27). We took advantage of this requirement to confirm the ends of the small viral RNAs by using looped primers that bind inside our predicted 3′ end (Fig. 2BII) or immediately outside our predicted end (Fig. 2BIII).
FIG 2.

RT-qPCR using looped RT primers confirms that HHV-6A carries at least five sncRNAs. (A) In order to characterize candidate transcripts and their precursors, total RNA isolated from either mock-infected (M) or HHV-6A-infected (I; MOI = 0.1) HSB-2 cells or lentiviral transduced cells (as designated) was separated by PAGE and immobilized on nitrocellulose membranes. Each membrane was probed with a radiolabeled oligonucleotide corresponding to the antisense sequence of the predicted viral RNA. A radiolabeled ladder is shown for size determination. (B) To confirm accurate mapping of the 3′ end of the five HHV-6A RNA candidates, stem-loop primers for cDNA synthesis were designed and labeled based on their binding of the 3′ end of the candidate miRNAs. The stem-loop primers are shown and labeled I for binding to the predicted 3′ end of the viral transcript, II for binding 3 nt upstream of the predicted end of the viral transcript, and III for binding 3 nt downstream of the predicted end of the viral transcript. (C) Representative RT-qPCR amplification profiles are shown for each of the candidate viral transcripts along with the designation of each stem-loop primer from panel B. (D) Summary table detailing the amplification by RT-qPCR that confirms the ends of the HHV-6A viral transcript candidates. Transcripts that were detected are scored as positive (+), whereas transcripts not detected are scored as negative (−).
In order to characterize our candidate small RNAs, we isolated RNA from either mock-transfected or HHV-6A BAC-transfected Jjhan cells for use in RT-qPCR assays. We can detect miRNA amplification using the “fit primers” (Fig. 2C). Using these modified primers, we did not detect amplification of our candidate viral RNAs by RT-qPCR where the primer bound inside our predicted end (primer II) (Fig. 2C). Four of the five viral RNAs tested (sncRNA-U2, sncRNA-U3-1, sncRNA-U14, and miR-U86) failed to amplify using a primer that resulted in a gap (primer III), suggesting that we accurately mapped the 3′ end of these candidate viral transcripts (Fig. 2C). We did, however, amplify candidate sncRNA-54 using a loop primer that should have resulted in a gap. This result suggests that various isoforms of this RNA exist, as they are indeed apparent in the sequencing data, and this may explain why sncRNA-54 demonstrated the weakest hybridization to probe in the Northern blot. We are currently characterizing these additional isoforms. Nonetheless, taken together, these data argue that HHV-6A encodes at least four sncRNAs and at least one miRNA. For the remainder of this study, we focused on the biological role of miR-U86.
HHV-6A miR-U86 inhibits viral lytic replication.
To determine the requirement of miR-U86 for viral replication, we took advantage of bacterial recombineering (20) to disrupt the expression of the candidate miRNA within our infectious HHV-6A-BAC construct. For miR-U86, which mapped to a region opposite a known HHV-6A ORF critical for viral replication, U86, we mutated the seed sequence as previously described (29, 30). Such mutations are designed to alter the miRNA sequence as much as possible while preserving the amino acid sequence encoded by the ORF on the opposite strand. This prevents the synthesis of a mature miRNA, as the mutations disrupt the ability to generate a pre-miRNA hairpin precursor. Following sequence confirmation of the generated recombinant, we transfected the mutant BAC into Jjhan cells. We monitored transfection efficiency by fluorescence microscopy for GFP expression and subsequent productive viral transmission in HSB2 cells. Within 72 h posttransfection (hpt), we observed GFP expression from the ΔmiR-U86 mutant construct in the Jjhan cells, suggesting that we had successfully transferred virus to these cells (data not shown). We generated purified virus for ΔmiR-U86 as well as wild-type (WT) virus for use in subsequent infectivity experiments aimed at understanding the function of this novel miRNA.
To first confirm that the mutant virus was unable to express miR-U86, we performed RT-qPCR using primers specific for either the mutated miRNA or the WT miRNA, as a positive control. Our results confirmed that the mutation that we generated in fact inhibited detectable expression of miR-U86 (Fig. 3A).
FIG 3.
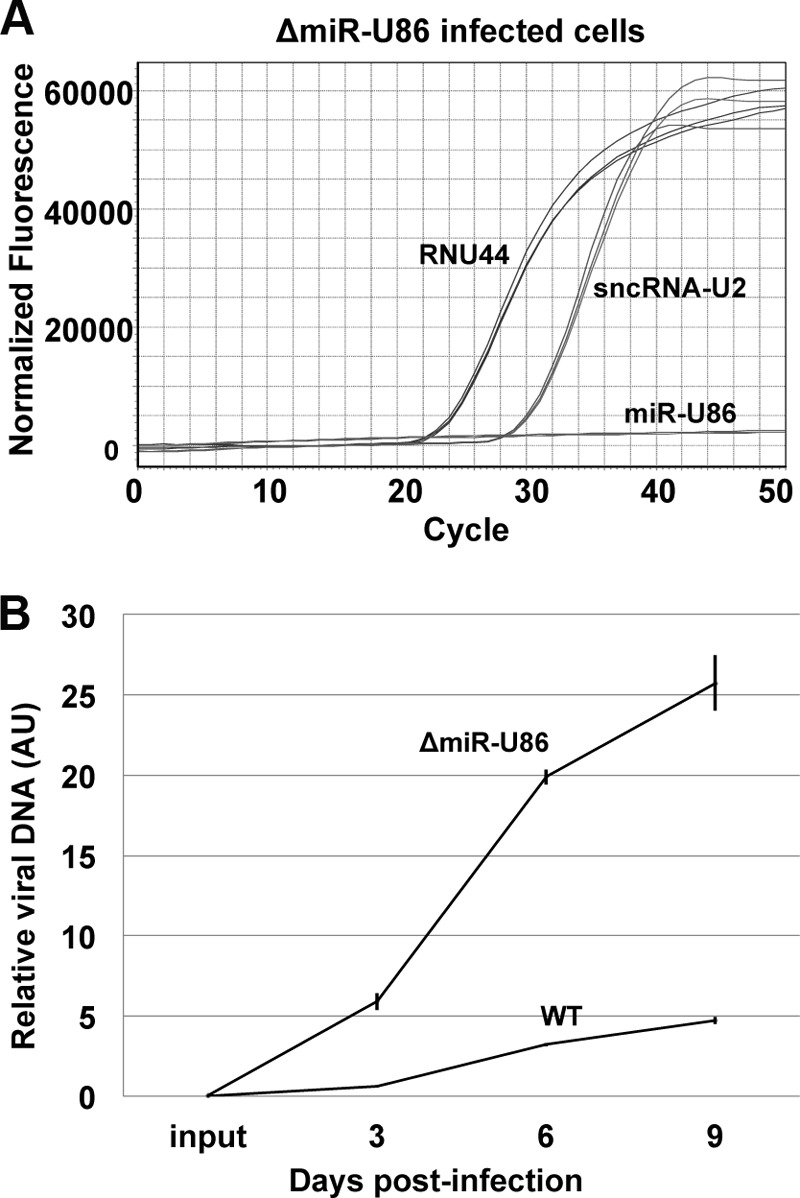
ΔmiR-U86 mutant infection displays increased viral growth in Jjhan cells. (A) To validate the disruption of mutated miR-U86 expression, RNA isolated from ΔmiR-U86 mutant-infected Jjhan cells (MOI = 0.01) was used to profile the expression of miR-U86 compared to the host snoRNA, RNU44, as well as the virally encoded sncRNA-U2. Representative real-time amplification profiles are shown. (B) Cell-free, sorbitol-purified virus (WT and ΔmiR-U86 mutant) was used to infect Jjhan cells (MOI = 0.01), which were collected at 0, 3, 6, and 9 days p.i. for analysis of DNA by qPCR. Values are expressed as the number of HHV-6A-BAC genome copies per cells (GFP/MDM2) and normalized to input. Each sample was analyzed in triplicate (n = 3). AU, arbitrary units.
We then focused on evaluating viral replication of the mutant virus compared to WT infection. To this end, we monitored viral replication by quantifying the accumulation of viral DNA—a method valid for determining titers of cell-associated virus (21). We used sorbitol-purified ΔmiR-U86 and WT viruses to infect Jjhan cells at a low multiplicity of infection (MOI = 0.01). Infected cells were collected at 0, 3, 6, and 9 days postinfection for subsequent qPCR analyses. We observed that ΔmiR-U86 virus showed a significant increase in viral DNA replication compared to WT virus (Fig. 3B), suggesting that miR-U86 plays an important biological role during lytic replication in Jjhan cells.
HHV-6A miR-U86 impacts viral gene transcription and U86 protein expression.
Of the five novel HHV-6A small noncoding RNAs that we identified, miR-U86 is particularly interesting, as this miRNA is derived from sequences opposite the essential viral immediate early (IE) gene U86 (Fig. 1A and 4A). HHV-6A U86, along with U90, generates a spliced transcript encoding an IE protein that is the orthologue of the human cytomegalovirus (HCMV) IE2 protein (31). As other herpesviruses, including HCMV, utilize miRNAs to regulate their IE transcripts, we prioritized our efforts to better characterize the effect of miR-U86 on regulating viral lytic replication. To further investigate the increased replication that we observed following infection of Jjhan cells with this mutant virus (Fig. 3B), we first profiled the kinetics of viral RNA expression during infection of Jjhan cells with ΔmiR-U86. HHV-6A, like all herpesviruses, expresses its genes in a temporal fashion, whereby one observes the synthesis of the IE genes, followed by the early genes (E) and lastly the late genes (L), the last of which (L genes) are generated following viral DNA replication (32). We compared the accumulation levels of a representative viral mRNA from each of the three kinetic classes using RT-qPCR for U90 (IE gene), U12 (E gene), and U100 (L gene). We observed an increased level of RNA expression for each gene in the ΔmiR-U86-infected cells compared to the WT-infected cells at 3, 6, and 9 days p.i. (Fig. 4B), suggesting that ΔmiR-U86 might play a role in the viral life cycle as early as IE gene transcription. It is important to note that the primers directed at U90 amplify a region of the viral mRNA that is spliced to the U86 ORF (see Table S1 in the supplemental material), which we predict is a target of ΔmiR-U86.
FIG 4.

HHV-6A ΔmiR-U86 mutant infection upregulates viral gene transcription and U86 protein expression. (A) miR-U86 maps to the opposite strand of U86. The mutation of HHV-6A-miR-U86 was designed to alter the miRNA seed sequence while preserving the integrity of the U86 coding sequence. Black vertical bars designate mutated nucleotides. (B) Expression kinetics of HHV-6A mRNAs from the three kinetic classes of transcripts. Total RNA was extracted and analyzed by RT-qPCR. All values are expressed as fold change (ΔΔCT) and normalized to input. (C) Jjhan cells were infected with WT:U86-3xFlag or ΔmiR-U86:U86-3xFlag (MOI = 0.1), and total protein was harvested at 6 days p.i. U86 expression was assessed by immunoblot analysis using an antibody directed at the FLAG epitope tag. Tubulin is shown as a loading control.
We next sought to determine if deletion of miR-U86 resulted in an increase of U86 at the protein level. As an antibody that recognizes HHV-6A U86 is not available, we generated recombinant viruses in the WT and ΔmiR-U86 viral backgrounds using BAC recombineering protocols to generate three tandem FLAG epitope tags on the carboxy-terminal end of U86, termed WT-U86-3xF and ΔmiR-U86:U86-3xF, respectively. Neither of the tagged recombinant viruses demonstrated growth defects compared to their respective parental WT or ΔmiR-U86 viruses (data not shown), suggesting that the epitope tag does not alter the growth kinetics of either virus. We next utilized these epitope-tagged viral recombinants to monitor U86 expression in each background (i.e., WT-U86-3xF versus ΔmiR-U86:U86-3xF) during infection of Jjhan cells. We observed an increase in U86 protein expression by immunoblotting in the ΔmiR-U86:U86-3xF virus compared to the WT-U86-3xF virus (Fig. 4C), suggesting that miR-U86 expression regulates the protein expression levels of U86.
To confirm that the phenotypes that we observed in response to ΔmiR-U86 were due to the disturbance of the miRNA and not due to off-site mutations, we generated a revertant of the miR-U86 mutation, thus restoring expression of this miRNA. We infected Jjhan cells with sorbitol-purified virus reconstituted from BAC-derived WT, ΔmiR-U86, and the miR-U86 revertant (ΔmiR-U86REV) and monitored the viral growth properties across the three infections by qPCR for viral genomes. Our analyses of their growth properties revealed that consistent with our earlier results, the ΔmiR-U86 virus replicated to a higher level than the WT virus (Fig. 5). Importantly, the ΔmiR-U86REV virus replicated to levels similar to those of the WT virus, thereby confirming that the phenotype that we observed was due to the mutation of the miRNA. Taken together, these findings reveal that the deletion of miR-U86 directly impacts viral replication, perhaps by modulating the levels of U86 protein.
FIG 5.
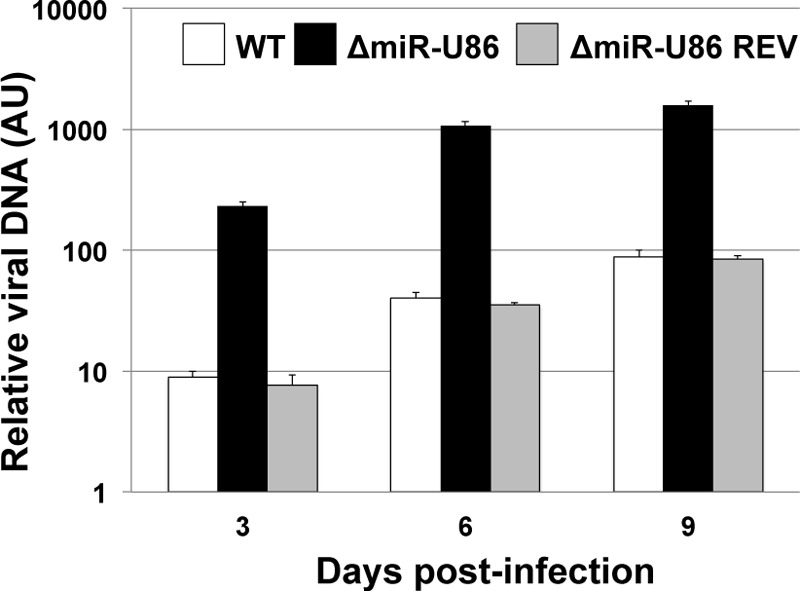
Increased viral replication results from mutating miR-U86. To assess viral replication, sorbitol-purified virus (WT, ΔmiR-U86, and ΔmiR-U86 REV) was used to infect Jjhan cells (MOI = 0.01). Infected cells were harvested for qPCR at 3, 6, and 9 days p.i., and viral genomes were detected using primers directed at GFP and normalized to cellular MDM2. All values are shown relative to input. Each sample was analyzed in triplicate (n = 3). AU, arbitrary units.
HHV-6A miR-U86 expression inhibits wild-type virus replication.
To further examine the effect of miR-U86 on viral replication, we generated a stable cell line that expresses the mature miR-U86 miRNA using a lentiviral expression construct (Jj-miR-U86). This construct also encodes a fluorescent marker, dsRed, thereby allowing us to enrich for the HHV-6A miRNA-expressing cells by FACS. We isolated total RNA to quantify the abundance of the specific viral small RNA species expressed by the transduced cells by RT-qPCR. We compared the level of the exogenous HHV-6A miRNA to that of endogenous levels expressed by HHV-6A-infected Jjhan cells. Our analyses revealed that the FACS-enriched Jj-miR-U86 cells express miR-U86 at higher levels than the HHV-6A-infected Jjhan cells (Fig. 6A), suggesting that these stable cell lines are a valid tool for assessing the function of this miRNA. Importantly, no miRNA was detected in mock-infected cells (Fig. 6A). We further characterized the expression of the miR-U86 mature miRNA from the Jj-miR-U86 cells by Northern blotting. Using probes specific for miR-U86, we were able to detect hybridization of the probe to RNA species corresponding to the appropriate miRNA size from the transduced cells (Fig. 2).
FIG 6.
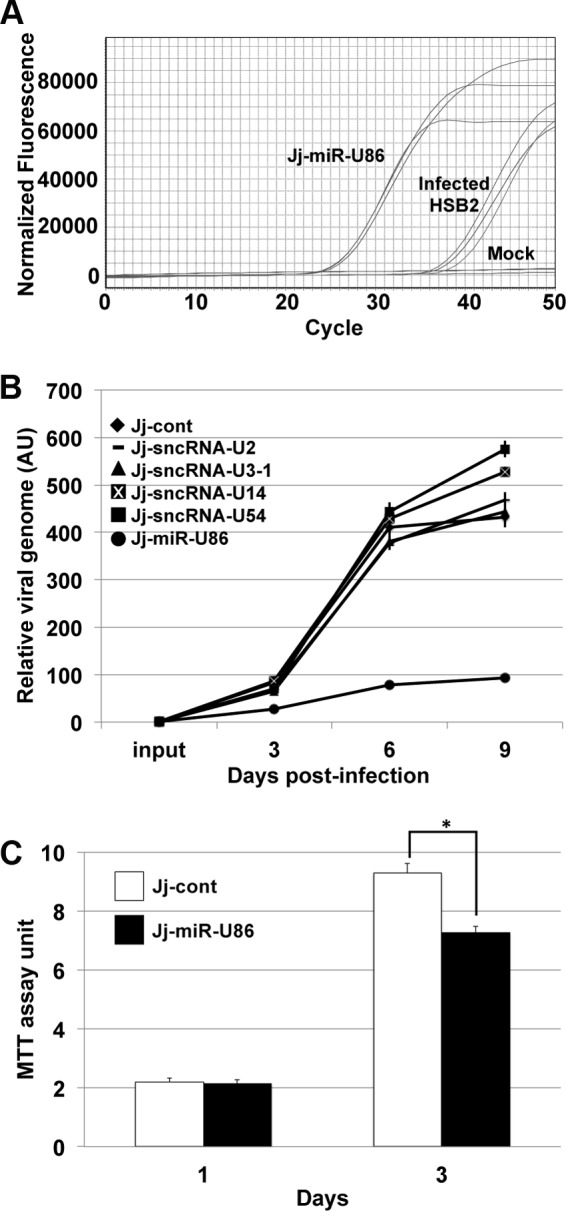
Overexpression of miR-U86 results in a reduction of WT viral replication. (A) To determine the relative expression of miR-U86 from transduced cells compared to viral infection, transduced miR-U86-expressing Jjhan cells (Jj-miR-U86), HHV-6A-infected HSB-2 cells (MOI = 0.01), and mock-infected cells were compared. Representative RT-qPCR amplification profiles are shown for miR-U86 levels in each sample. (B) HHV-6A WT virus was used to infect control and viral-transcript-overexpressing Jjhan cells (MOI = 0.01). Cells were harvested at 0, 3, 6, and 9 days p.i. for qPCR analysis of viral genomes, using primers directed at GFP and then normalized to cellular MDM2. All values are shown relative to input, and each sample was analyzed in triplicate (n = 3). AU, arbitrary units. (C) Cell viability was assessed by MTT assay at 1 and 3 days for each cell type indicated. Values were normalized to input cells. *, P < 0.01.
To determine the effect of exogenous viral sncRNA and miRNA expression on the growth properties of WT HHV-6A virus, we infected transduced Jjhan cell lines to evaluate lytic replication by multistep growth analyses. We assessed cell-associated viral DNA at 0, 3, 6, and 9 days p.i. by qPCR. HHV-6A WT infection of sncRNA-U2-, -U3-1-, -U14-, and -U54-expressing cells had no significant growth defect compared to infected control-transduced cells (Jj-cont) (Fig. 6B). In contrast, miR-U86-expressing cells (Jj-miR-U86) infected with WT virus displayed a significant growth defect compared to all other transduced Jjhan cells tested, including the control-transduced Jjhan cells (Fig. 6B), a finding that is consistent with our data using the ΔmiR-U86 viral BAC mutant. To determine if the reduced viral load in the Jjhan-miR-U86 cells is a consequence of cellular changes irrespective of viral replication, we monitored cellular viability. Ectopic expression of miR-U86 resulted in a slight but statistically significant reduction in cell viability, as Jj-miR-U86 cells grew slower than control-transduced Jjhan cells in a 3-day 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay (Fig. 6C). While miR-U86 expression did demonstrate marginal effects on cell growth, we speculate that the significant impact on viral replication is possibly due to miRNA targeting of either viral or cellular factors that are required for efficient viral replication, as discussed below.
To better understand the effects of miR-U86 expression on HHV-6A replication, we quantified both viral mRNAs and proteins during WT infection of Jj-cont cells and Jj-miR-U86 cells. To this end, we infected the transduced cells with sorbitol-purified WT virus and monitored the production of viral mRNA by RT-qPCR over the course of infection. At each time point, we assessed a representative transcript from each of the kinetic classes of HHV-6A genes: U90 (IE gene), U12 (E gene), and U100 (L gene). We found an inhibition of viral IE, E, and L transcripts (Fig. 7A) that was consistent with the observed reduction of viral DNA replication (Fig. 6B), suggesting that the mechanism of action occurs during the initial stages of the replication cycle. Additionally, Western blot analyses revealed that the U86 protein, which we hypothesize is the target of miR-U86, was expressed to higher levels in WT-infected Jj-cont cells than in WT-infected Jj-miR-U86 cells (Fig. 7B). Taken together, these findings suggest that miR-U86 plays an important role in viral replication, likely during the initial events of infection.
FIG 7.

Overexpression of miR-U86 results in decreased viral transcripts and IE protein during WT infection. (A) Jjhan control transduced cells (JJ-cont) and Jjhan cells overexpressing miR-U86 (Jj-miR-UL86) were infected with WT HHV-6A, and total RNA was extracted at the indicated times. Values are expressed as fold changes (ΔΔCT) normalized to cellular β-actin and further normalized to input. Each sample was analyzed in triplicate (n = 3). AU, arbitrary units. (B) Control-transduced (Jj-cont) and miR-U86-transduced (Jj-miR-U86) Jjhan cells were infected with WT:U86-3xFlag or mock infected (MOI = 0.1) for 6 days. Cell lysates were then analyzed by immunoblotting for the expression of U86 using the FLAG epitope tag. Human α-tubulin is shown as a loading control.
HHV-6A miR-U86, in part, regulates viral lytic replication.
Viral miRNAs are reported to have multiple biological functions in which individual miRNAs can target both host antiviral responses and viral transcripts (30, 33). Thus, in order to define the role of miR-U86 on the viral life cycle versus impacting the host cell, we analyzed the lytic replication of the ΔmiR-U86 recombinant HHV-6A that has mutations within the miRNA site within the cells ectopically expressing miR-U86. As the ΔmiR-U86 virus lacks the sequences required to generate the miRNA, it also lacks the sequences where the ectopically expressed miRNA would bind. Thus, the ORF on the opposite strand of the genome, U86, will not be targeted by exogenous expression of miR-U86. We infected the Jj-miR-U86 cells with either WT or ΔmiR-U86 virus and quantified genomic amplification over a defined time course. We found that the ΔmiR-U86 virus was able to replicate to higher levels in the Jj-miR-U86 cells than in the WT cells as monitored by microscopy for enhanced GFP (eGFP) expression (Fig. 8A) as well as by qPCR for genome copy (Fig. 8B). These observations, coupled with the earlier results, suggest that the main function of miR-U86 is to regulate viral transcripts, specifically U86, as opposed to cellular transcripts, as the growth of ΔmiR-U86 virus was not significantly impacted in cells expressing miR-U86.
FIG 8.
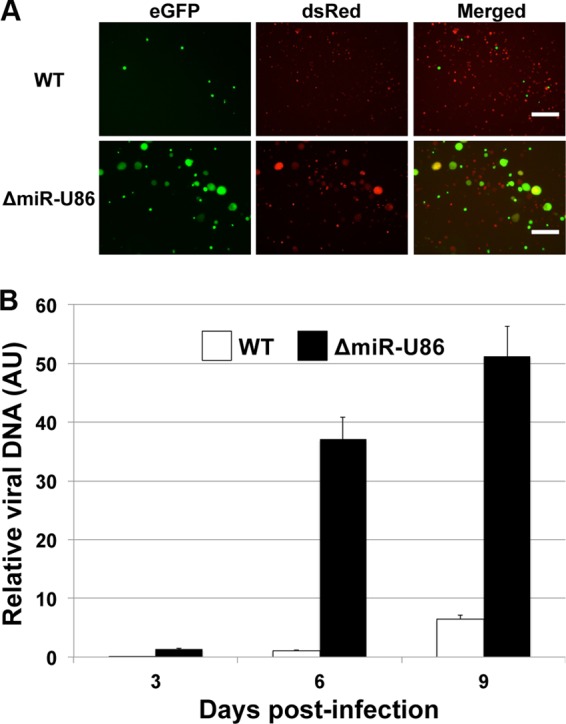
ΔmiR-U86-infected miR-U86-overexpressing cells grow more efficiently than do WT-infected cells. miR-U86-overexpressing Jjhan cells were infected with either WT or ΔmiR-U86 virus. (A) ΔmiR-U86 virus replicates more efficiently than WT virus in Jjhan-miR-U86 cells at 6 days p.i. as assessed by eGFP expression. Images were collected at a magnification of ×125. Bar, 200 μm. (B) Jjhan-miR-U86-infected cells were collected at 0, 3, 6, and 9 days p.i., and viral genomes were detected using primers that recognize GFP. Values are expressed as the number of HHV-6A-BAC genome copies per cell (GFP/MDM2) normalized to input. Samples were analyzed in triplicate (n = 3). AU, arbitrary units.
DISCUSSION
It is well accepted that cellular encoded miRNAs are key regulators of both cellular and viral transcripts. Herpesviruses, as a family, encode their own viral miRNAs, although the functions of a majority of these viral miRNAs remain unknown (16). Recently, investigators have shown that HHV-6B encodes four miRNAs that are derived from the terminal repeats, as determined by sequencing small RNA species from HHV-6B-infected cells (17). In this work, we used a similar approach to identify virally encoded small noncoding RNAs from the related, yet distinct herpesvirus HHV-6A. Our deep sequencing analysis identified seven candidate viral transcripts that we further investigated, among which we were able to validate the expression for five upon subsequent analyses, one of which has the biological characteristics of a viral miRNA. This virally encoded miRNA, miR-U86, lacks any appreciable homology to host miRNAs, suggesting that it is unique to the virus. Additionally, miR-U86 is distinct from the four HHV-6B miRNAs, thereby underscoring the classification of these two pathogens as distinct viruses. However, it is also possible that the HHV-6A miRNA that we found is distinct from those of HHV-6B due to the types of cells used for the infection (Jjhan versus Sup-T-1s, respectively) or the depth of deep sequencing (17, 24–26).
To uncover the biological requirement of the four novel HHV-6A sncRNAs and one miRNA, we employed site-specific BAC mutagenesis in which we deleted or mutated each individual miRNA. Although we successfully generated mutant viruses lacking the expression of sncRNA-U2, sncRNA-U3-1, sncRNA-U54, and miR-U86, we were unable to produce a mutant virus lacking sncRNA-U14 expression in two independent attempts, suggesting that expression of this viral transcript may be absolutely essential for lytic replication within Jjhan cells. We attempted to complement the growth defect of the ΔsncRNA-U14 virus by growing the recombinant construct in sncRNA-U14-expressing Jjhan cells. While the viral infection within sncRNA-U14-expressing Jjhan cells did progress farther than that seen in mock-transduced cells, as monitored by the increased proportion of eGFP-positive cells, we were unable to generate sufficient virus for subsequent experimentation, suggesting that the levels of sncRNA-U14 expression are critical for efficient HHV-6A replication (data not shown). HHV-6A sncRNA-U14 is expressed on the opposite strand of the U14 gene, a structural tegument protein (34) that is the homolog of HCMV UL25 (35). It is possible that inhibition of viral replication from the Δ sncRNA-U14 virus may be indicative of a change independent from expression of the small noncoding RNA (e.g., changes in codon bias of U14 or modification of other cis binding factors required for efficient viral transcription). HHV-6 U14 binds the tumor suppressor p53, suggesting that the p53 interaction with the U14 tegument protein may be important for HHV-6 infection (36). It is attractive to speculate that sncRNA-U14 regulates the expression of U14 gene product, a prospect that we are currently pursuing.
The HHV-6A orthologue of the HCMV IE2 protein is encoded by a spliced transcript originating from ORFs U90 and U86 (31). HHV-6A miR-U86 is expressed from the opposite strand of the U86 ORF, and as a result miR-U86 has perfect complementarity with the HHV-6A IE2 mRNA. Therefore, we hypothesized that HHV-6A miR-U86 inhibits the expression of HHV-6A IE2 protein, thereby controlling viral lytic replication. We show that mutation of miR-U86 in HHV-6A increases viral mRNA expression of a representative gene from each of the three kinetic classes of transcripts. We also examined the effect of miR-U86 on expression of HHV-6A IE2 by using WT-U86-3xF and ΔmiR-U86:U86-3xF recombinant viruses. Our results indicate that HHV-6A miR-U86 expression is inversely correlated to the expression level of HHV-6A IE2 protein, suggesting that the U90/U86 transcript, encoding HHV-6A IE2, is indeed a target of this viral miRNA. Finally, deletion of the miRNA promotes viral DNA replication, and its ectopic expression inhibits viral DNA accumulation. Importantly, replication of a recombinant virus incapable of U86 regulation by miR-U86 grows efficiently in cells ectopically expressing miR-U86, suggesting that the main target of the viral miRNA is the HHV6A IE2 gene product, similar to that seen in other herpesviruses. Taken together, our results argue a role for HHV-6A miR-U86 in regulating viral lytic replication, likely by regulating the translation of the HHV-6A IE2 encoded by the U90/U86 transcript.
It has become evident that regulation of key herpesvirus IE transcripts via miRNA targeting is more common than it is the exception. miRNAs of cellular origin and/or viral origin target the IE transcripts in Epstein-Barr virus (EBV) (37), Kaposi's sarcoma-associated herpesvirus (KSHV) (38), herpes simplex virus 1 (HSV-1) (39, 40), HSV-2 (41), and the closely related betaherpesvirus HCMV (30, 42, 43). That the HHV-6A IE transcript(s) is also targeted by miRNAs underscores the commonality of herpesviruses. Whether our identified HHV-6A miRNA is involved in maintaining latency, as is suggested for other herpesvirus family members, remains to be elucidated. However, the identification of HHV-6A-encoded miRNAs offers a novel therapeutic target that may control viral infections and subsequent pathogenesis.
Supplementary Material
ACKNOWLEDGMENTS
We thank Christine O'Connor (University at Buffalo-SUNY) for helpful discussions, critical review of the manuscript, and technical support.
This study was supported by grants from the National Institute of Allergy and Infectious Diseases (grant no. RAI101080A) and the Midwest Regional Center of Excellence for Biodefense and Emerging Infectious Disease Research (grant no. U54AI057160) awarded to E.A.M.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02007-14.
REFERENCES
- 1.Adams MJ, Carstens EB. 2012. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2012). Arch Virol 157:1411–1422. doi: 10.1007/s00705-012-1299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Santoro F, Kennedy PE, Locatelli G, Malnati MS, Berger EA, Lusso P. 1999. CD46 is a cellular receptor for human herpesvirus 6. Cell 99:817–827. doi: 10.1016/S0092-8674(00)81678-5. [DOI] [PubMed] [Google Scholar]
- 3.Tang H, Serada S, Kawabata A, Ota M, Hayashi E, Naka T, Yamanishi K, Mori Y. 2013. CD134 is a cellular receptor specific for human herpesvirus-6B entry. Proc Natl Acad Sci U S A 110:9096–9099. doi: 10.1073/pnas.1305187110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ablashi D, Agut H, Alvarez-Lafuente R, Clark DA, Dewhurst S, DiLuca D, Flamand L, Frenkel N, Gallo R, Gompels UA, Hollsberg P, Jacobson S, Luppi M, Lusso P, Malnati M, Medveczky P, Mori Y, Pellett PE, Pritchett JC, Yamanishi K, Yoshikawa T. 2014. Classification of HHV-6A and HHV-6B as distinct viruses. Arch Virol 159:863–870. doi: 10.1007/s00705-013-1902-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamanishi K, Okuno T, Shiraki K, Takahashi M, Kondo T, Asano Y, Kurata T. 1988. Identification of human herpesvirus-6 as a causal agent for exanthem subitum. Lancet i:1065–1067. [DOI] [PubMed] [Google Scholar]
- 6.Ronmark E, Jonsson E, Platts-Mills T, Lundback B. 1999. Different pattern of risk factors for atopic and nonatopic asthma among children–report from the Obstructive Lung Disease in Northern Sweden Study. Allergy 54:926–935. doi: 10.1034/j.1398-9995.1999.00044.x. [DOI] [PubMed] [Google Scholar]
- 7.Ablashi DV, Eastman HB, Owen CB, Roman MM, Friedman J, Zabriskie JB, Peterson DL, Pearson GR, Whitman JE. 2000. Frequent HHV-6 reactivation in multiple sclerosis (MS) and chronic fatigue syndrome (CFS) patients. J Clin Virol 16:179–191. doi: 10.1016/S1386-6532(99)00079-7. [DOI] [PubMed] [Google Scholar]
- 8.Reeves WC, Stamey FR, Black JB, Mawle AC, Stewart JA, Pellett PE. 2000. Human herpesviruses 6 and 7 in chronic fatigue syndrome: a case-control study. Clin Infect Dis 31:48–52. doi: 10.1086/313908. [DOI] [PubMed] [Google Scholar]
- 9.Akhyani N, Berti R, Brennan MB, Soldan SS, Eaton JM, McFarland HF, Jacobson S. 2000. Tissue distribution and variant characterization of human herpesvirus (HHV)-6: increased prevalence of HHV-6A in patients with multiple sclerosis. J Infect Dis 182:1321–1325. doi: 10.1086/315893. [DOI] [PubMed] [Google Scholar]
- 10.Gottwein E. 2013. Roles of microRNAs in the life cycles of mammalian viruses. Curr Top Microbiol Immunol 371:201–227. doi: 10.1007/978-3-642-37765-5_8. [DOI] [PubMed] [Google Scholar]
- 11.Moody R, Zhu Y, Huang Y, Cui X, Jones T, Bedolla R, Lei X, Bai Z, Gao SJ. 2013. KSHV microRNAs mediate cellular transformation and tumorigenesis by redundantly targeting cell growth and survival pathways. PLoS Pathog 9:e1003857. doi: 10.1371/journal.ppat.1003857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trifari S, Pipkin ME, Bandukwala HS, Aijo T, Bassein J, Chen R, Martinez GJ, Rao A. 2013. MicroRNA-directed program of cytotoxic CD8+ T-cell differentiation. Proc Natl Acad Sci U S A 110:18608–18613. doi: 10.1073/pnas.1317191110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Michael MZ, O'Connor SM, van Holst Pellekaan NG, Young GP, James RJ. 2003. Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol Cancer Res 1:882–891. [PubMed] [Google Scholar]
- 14.Zeng Z, Huang H, Huang L, Sun M, Yan Q, Song Y, Wei F, Bo H, Gong Z, Zeng Y, Li Q, Zhang W, Li X, Xiang B, Li X, Li Y, Xiong W, Li G. 2014. Regulation network and expression profiles of Epstein-Barr virus-encoded microRNAs and their potential target host genes in nasopharyngeal carcinomas. Sci China Life Sci 57:315–326. doi: 10.1007/s11427-013-4577-y. [DOI] [PubMed] [Google Scholar]
- 15.Kozomara A, Griffiths-Jones S. 2014. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42:D68–D73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cullen BR. 2011. Herpesvirus microRNAs: phenotypes and functions. Curr Opin Virol 1:211–215. doi: 10.1016/j.coviro.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tuddenham L, Jung JS, Chane-Woon-Ming B, Dolken L, Pfeffer S. 2012. Small RNA deep sequencing identifies microRNAs and other small noncoding RNAs from human herpesvirus 6B. J Virol 86:1638–1649. doi: 10.1128/JVI.05911-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang H, Kawabata A, Yoshida M, Oyaizu H, Maeki T, Yamanishi K, Mori Y. 2010. Human herpesvirus 6 encoded glycoprotein Q1 gene is essential for virus growth. Virology 407:360–367. doi: 10.1016/j.virol.2010.08.018. [DOI] [PubMed] [Google Scholar]
- 19.Kalejta RF, Bechtel JT, Shenk T. 2003. Human cytomegalovirus pp71 stimulates cell cycle progression by inducing the proteasome-dependent degradation of the retinoblastoma family of tumor suppressors. Mol Cell Biol 23:1885–1895. doi: 10.1128/MCB.23.6.1885-1895.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 33:e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O'Connor CM, Shenk T. 2011. Human cytomegalovirus pUS27 G protein-coupled receptor homologue is required for efficient spread by the extracellular route but not for direct cell-to-cell spread. J Virol 85:3700–3707. doi: 10.1128/JVI.02442-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lilja AE, Chang WL, Barry PA, Becerra SP, Shenk TE. 2008. Functional genetic analysis of rhesus cytomegalovirus: Rh01 is an epithelial cell tropism factor. J Virol 82:2170–2181. doi: 10.1128/JVI.02316-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Downing RG, Sewankambo N, Serwadda D, Honess R, Crawford D, Jarrett R, Griffin BE. 1987. Isolation of human lymphotropic herpesviruses from Uganda. Lancet ii:390. [DOI] [PubMed] [Google Scholar]
- 24.Pfeffer S, Sewer A, Lagos-Quintana M, Sheridan R, Sander C, Grasser FA, van Dyk LF, Ho CK, Shuman S, Chien M, Russo JJ, Ju J, Randall G, Lindenbach BD, Rice CM, Simon V, Ho DD, Zavolan M, Tuschl T. 2005. Identification of microRNAs of the herpesvirus family. Nat Methods 2:269–276. doi: 10.1038/nmeth746. [DOI] [PubMed] [Google Scholar]
- 25.Grey F, Nelson J. 2008. Identification and function of human cytomegalovirus microRNAs. J Clin Virol 41:186–191. doi: 10.1016/j.jcv.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dunn W, Trang P, Zhong Q, Yang E, van Belle C, Liu F. 2005. Human cytomegalovirus expresses novel microRNAs during productive viral infection. Cell Microbiol 7:1684–1695. doi: 10.1111/j.1462-5822.2005.00598.x. [DOI] [PubMed] [Google Scholar]
- 27.Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, Lao KQ, Livak KJ, Guegler KJ. 2005. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res 33:e179. doi: 10.1093/nar/gni178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salahuddin SZ, Ablashi DV, Markham PD, Josephs SF, Sturzenegger S, Kaplan M, Halligan G, Biberfeld P, Wong-Staal F, Kramarsky B, Gallo RC. 1986. Isolation of a new virus, HBLV, in patients with lymphoproliferative disorders. Science 234:596–601. doi: 10.1126/science.2876520. [DOI] [PubMed] [Google Scholar]
- 29.Sullivan CS, Grundhoff AT, Tevethia S, Pipas JM, Ganem D. 2005. SV40-encoded microRNAs regulate viral gene expression and reduce susceptibility to cytotoxic T cells. Nature 435:682–686. doi: 10.1038/nature03576. [DOI] [PubMed] [Google Scholar]
- 30.Murphy E, Vanicek J, Robins H, Shenk T, Levine AJ. 2008. Suppression of immediate-early viral gene expression by herpesvirus-coded microRNAs: implications for latency. Proc Natl Acad Sci U S A 105:5453–5458. doi: 10.1073/pnas.0711910105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gravel A, Tomoiu A, Cloutier N, Gosselin J, Flamand L. 2003. Characterization of the immediate-early 2 protein of human herpesvirus 6, a promiscuous transcriptional activator. Virology 308:340–353. doi: 10.1016/S0042-6822(03)00007-2. [DOI] [PubMed] [Google Scholar]
- 32.Tsao EH, Kellam P, Sin CS, Rasaiyaah J, Griffiths PD, Clark DA. 2009. Microarray-based determination of the lytic cascade of human herpesvirus 6B. J Gen Virol 90:2581–2591. doi: 10.1099/vir.0.012815-0. [DOI] [PubMed] [Google Scholar]
- 33.Lee SH, Kalejta RF, Kerry J, Semmes OJ, O'Connor CM, Khan Z, Garcia BA, Shenk T, Murphy E. 2012. BclAF1 restriction factor is neutralized by proteasomal degradation and microRNA repression during human cytomegalovirus infection. Proc Natl Acad Sci U S A 109:9575–9580. doi: 10.1073/pnas.1207496109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stefan A, Secchiero P, Baechi T, Kempf W, Campadelli-Fiume G. 1997. The 85-kilodalton phosphoprotein (pp85) of human herpesvirus 7 is encoded by open reading frame U14 and localizes to a tegument substructure in virion particles. J Virol 71:5758–5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baldick CJ Jr, Shenk T. 1996. Proteins associated with purified human cytomegalovirus particles. J Virol 70:6097–6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takemoto M, Koike M, Mori Y, Yonemoto S, Sasamoto Y, Kondo K, Uchiyama Y, Yamanishi K. 2005. Human herpesvirus 6 open reading frame U14 protein and cellular p53 interact with each other and are contained in the virion. J Virol 79:13037–13046. doi: 10.1128/JVI.79.20.13037-13046.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jung YJ, Choi H, Kim H, Lee SK. 2014. miR-BART20-5p stabilizes Epstein-Barr virus latency by directly targeting BZLF1 and BRLF1. J Virol 88:9027–9037. doi: 10.1128/JVI.00721-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu F, Stedman W, Yousef M, Renne R, Lieberman PM. 2010. Epigenetic regulation of Kaposi's sarcoma-associated herpesvirus latency by virus-encoded microRNAs that target Rta and the cellular Rbl2-DNMT pathway. J Virol 84:2697–2706. doi: 10.1128/JVI.01997-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Flores O, Nakayama S, Whisnant AW, Javanbakht H, Cullen BR, Bloom DC. 2013. Mutational inactivation of herpes simplex virus 1 microRNAs identifies viral mRNA targets and reveals phenotypic effects in culture. J Virol 87:6589–6603. doi: 10.1128/JVI.00504-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu W, Guo Z, Zhang X, Guo L, Liu L, Liao Y, Wang J, Wang L, Li Q. 2013. A microRNA encoded by HSV-1 inhibits a cellular transcriptional repressor of viral immediate early and early genes. Sci China Life Sci 56:373–383. doi: 10.1007/s11427-013-4458-4. [DOI] [PubMed] [Google Scholar]
- 41.Tang S, Patel A, Krause PR. 2009. Novel less-abundant viral microRNAs encoded by herpes simplex virus 2 latency-associated transcript and their roles in regulating ICP34.5 and ICP0 mRNAs. J Virol 83:1433–1442. doi: 10.1128/JVI.01723-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grey F, Meyers H, White EA, Spector DH, Nelson J. 2007. A human cytomegalovirus-encoded microRNA regulates expression of multiple viral genes involved in replication. PLoS Pathog 3:e163. doi: 10.1371/journal.ppat.0030163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O'Connor CM, Vanicek J, Murphy EA. 2014. Host microRNA regulation of human cytomegalovirus immediate early protein translation promotes viral latency. J Virol 88:5524–5532. doi: 10.1128/JVI.00481-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.