

DIF-1, a polyketide, has been primarily characterized as a transcriptional regulator, but proteome-wide identification of peptides that change phosphorylation level in DIF-treated cell reveals many novel targets. Thus a small-molecule regulator of gene transcription plays additional roles as a multifunctional regulator of protein modification.
Abstract
Differentiation-inducing factor-1 (DIF-1) is a polyketide that induces Dictyostelium amoebae to differentiate as prestalk cells. We performed a global quantitative screen for phosphorylation changes that occur within the first minutes after addition of DIF-1, using a triple-label SILAC approach. This revealed a new world of DIF-1–controlled signaling, with changes in components of the MAPK and protein kinase B signaling pathways, components of the actinomyosin cytoskeletal signaling networks, and a broad range of small GTPases and their regulators. The results also provide evidence that the Ca2+/calmodulin–dependent phosphatase calcineurin plays a role in DIF-1 signaling to the DimB prestalk transcription factor. At the global level, DIF-1 causes a major shift in the phosphorylation/dephosphorylation equilibrium toward net dephosphorylation. Of interest, many of the sites that are dephosphorylated in response to DIF-1 are phosphorylated in response to extracellular cAMP signaling. This accords with studies that suggest an antagonism between the two inducers and also with the rapid dephosphorylation of the cAMP receptor that we observe in response to DIF-1 and with the known inhibitory effect of DIF-1 on chemotaxis to cAMP. All MS data are available via ProteomeXchange with identifier PXD001555.
INTRODUCTION
Dictyostelium discoideum is an amoebozoan that lives as a unicellular organism in soil. It feeds on other microbes, and when the local food source is exhausted, a developmental cycle is initiated that produces a fruiting body consisting of a stalk holding up a spore mass that is dispersed to find a new food supply. Cells aggregate together in response to pulsatile emissions of cAMP emanating from a signaling center. The aggregated cells round up to form a mound that elongates to form a slug-shaped structure. During slug formation, cells adopt one of two presumptive fates. About 80% differentiate as prespore cells, precursors to the environmentally resistant spore cells. The other 20% differentiate as prestalk cells that act to lift the bolus of spore cells up off the substratum on a stalk that is embedded into a supporting basal disk. The basal disk derives from a prestalk subtype, the pstB cells, whereas the other major subtypes—the pstA and pstO cells—occupy the anterior, prestalk region of the slug, which enters the stalk at culmination.
At the slug stage, cells are uncommitted. This can be easily demonstrated by dividing the slug between the prestalk and prespore regions, whence, ultimately, both parts form a fruiting body. This implies the existence of extracellular signals that determine the proportions of different cell types and of guidance cues to direct their movement to the correct location. cAMP plays a key role in both processes. It is the chemoattractant that directs cellular aggregation to form a signaling center, and it is required to maintain prespore differentiation.
Prestalk differentiation is induced by differentiation-inducing factor-1 (DIF-1), a chlorinated polyketide, produced by the prespore cells, which directs cells to differentiate as prestalk cells. There is also regulated prestalk-to-stalk differentiation that is induced by di-c-GMP (Chen and Schaap, 2012). Several discrete signaling pathways mediate DIF-induced gene expression. These are best understood for a region within the extracellular matrix A (ecmA) promoter, which drives expression in pstO cells. It contains a binding site for the bZIP transcription factor DimB, which was initially identified in a genetic screen for DIF-1–nonresponsive mutants. DimB accumulates in the nuclei of cells induced with DIF-1 and is phosphorylated in DIF-1–treated cells (Yamada et al., 2013). The signaling steps upstream from DimB phosphorylation are entirely unknown.
Studies on ecmA and the closely related ecmB gene showed that DIF-1 activates both promoters, whereas concurrent exposure to extracellular cAMP represses DIF-1–induced ecmB expression but stimulates ecmA expression (Berks and Kay, 1988). Several observations suggest that intracellular Ca2+ has a regulatory role in prestalk differentiation (reviewed in Gross, 2009). There are multiple potential transponders of signaling, but one attractive candidate is the Ca2+/calmodulin–dependent protein phosphatase calcineurin. Reduced calcineurin activity has been linked with incorrect tip and stalk formation and misregulation of stalk cell markers (Horn and Gross, 1996; Boeckeler et al., 2006; Thewes et al., 2013).
In addition to its effects on gene expression, DIF-1 also exerts more rapid effects on other processes. The transcription factor STATc is rapidly phosphorylated after exposure to DIF-1. This occurs by the combined effect of constitutive phosphorylation and the lifting of a balancing repression exerted by the protein tyrosine phosphatase PTP3. This repression of PTP3 activity and the consequent increase in STATc tyrosine phosphorylation correlates with serine phosphorylation of PTP3 (Araki et al., 2008, 2012). These events have been linked to changes in the actin cytoskeleton and cell shape that occur over the same approximate time course as in cells treated with DIF-1 (Araki et al., 2010; Araki and Williams, 2012).
Thus pharmacological and genetic data suggest that there are at least two widely different DIF-1 signaling pathways that direct known phosphorylation events: one signals to STATc, and a second, quite distinct pathway leads to phosphorylation of DimB (Yamada et al., 2013). In addition, there is a pathway that leads to rapid cytoskeletal restructuring and may be part of the STATc pathway (Araki and Williams, 2012). There is other, indirect evidence for earlier functions of DIF-1. At 6 h, the DIF-1 concentration is 2.5 nM (Kay, 1998), which is relatively low, but perhaps it is selectively localized. In addition, the DmtA DIF-1 biosynthesis mutant is delayed in aggregation (Thompson and Kay, 2000; Saito et al., 2008), and DIF-1 inhibits chemotaxis to cAMP (Kuwayama and Kubohara, 2009).
Given the number and variety of early and late responses, multiple signaling intermediates are to be expected, but, aside from the transcription factors, it has not been possible to identify them by orthodox methods (Insall and Kay, 1990). We therefore adopted a new approach, based on the assumption that the rapid responses to DIF-1 involve phosphorylation changes. The optimistic belief that even the receptor(s) may be identifiable using this approach stems from the fact that some receptors, including the G protein–coupled cAMP receptor (cAR1), are rapidly phosphorylated after ligand binding as part of the adaptive response.
Based on recent advances in mass spectrometry–based technologies, the large-scale analysis of phosphoproteome data from complex mixtures has become achievable (Cox and Mann, 2011). Multiplexed stable isotope labeling by amino acids in cell culture (SILAC) techniques have been used to produce time-resolved quantitative data at a site-specific level in response to a variety of specific stimuli (Olsen et al., 2006). To gain insight into early events in the DIF-1 signaling pathways, we applied a triplex SILAC labeling approach to follow phosphorylation and dephosphorylation changes in response to DIF-1 treatment of cells at 5 h of development. Intriguingly, there are many more changes than we expected in a diverse range of signaling pathways that, until now, have not been implicated in DIF-1 signaling. Of importance, we confirm the proteomic data for three key proteins—protein kinase BA (PKBA), protein kinase B–related protein 1 (PKBR1), and ERK2—for which phospho-specific antibodies are available. Antagonistic roles of cAMP and DIF-1 had long been suspected; we present evidence suggesting that DIF-1 has a much more extensive role in the modulation of the cAMP signal than was previously believed.
RESULTS
Experimental design
To identify and quantify phosphorylation changes in the early response to DIF-1, we metabolically labeled Ax2 cells using SILAC (Ong et al., 2002; Sobczyk et al., 2014). Cell populations were labeled to steady state in “light,” “medium,” or “heavy” medium and then starved in shaking culture, mimicking early Dictyostelium development, for 5 h before DIF-1 treatment. Based on knowledge of the phosphorylation changes of STATc and DimB in response to DIF-1 (Fukuzawa, Araki, et al., 2001; Yamada et al., 2013), cells were harvested 1, 5, 8, and 15 min post–DIF-1 treatment to allow a time course of stimulation to be constructed from two overlapping triplex experimental samples, A (0, 1, 8 min) and B (0, 5, 15 min), with each experiment repeated in biological triplicate (Figure 1A).
FIGURE 1:

Overview of phosphoproteomic profiling. (A) Experimental workflow for one replicate experiment. Differently SILAC labeled Dictyostelium Ax2 cells were starved for 5 h before treatment with DIF-1. Cells harvested at 1, 5, 8, and 15 min were pooled with untreated cells as triplex experimental samples. (A and B) Samples were trypsin digested and fractionated by SCX, and phosphopeptides were enriched using TiO2; then each sample was analyzed by LC-MS/MS. Data from triplicate experiments were analyzed using MaxQuant to identify and quantify phosphorylation site changes. (B) Summary of phosphorylation sites identified. (C) Heat-map representation of TPM-based clustering of DIF-1–regulated class I phosphorylation sites. Clusters are separated by horizontal lines and labeled. Numbers in parentheses indicate a four-digit binary label for each cluster based on the change in phosphorylation state: 1, phosphorylation; 0, dephosphorylation. Cluster 1 (1111) has only a single site and is not represented. Averaged temporal profiles (n ≥ 2) for each phosphorylation site in the five most populated clusters are presented.
In a control experiment, DimB and STATc phosphorylation was monitored by immunoblotting. Cells grown in SILAC media gave typical DIF-1 responses compared with cells grown in standard growth media, demonstrating that SILAC labeling had no adverse effect on DIF-1 signaling (unpublished data). Pooled SILAC samples were digested with trypsin and fractionated by strong cation exchange chromatography, and phosphopeptides were enriched using TiO2 beads. Samples were then analyzed in technical duplicate by high-accuracy liquid chromatography tandem mass spectrometry (LC-MS/MS), followed by data analysis using MaxQuant to identify and quantify phosphorylation sites.
Classification of phosphorylation sites
Using a MaxQuant localization probability of ≥0.75 as the cutoff, we identified 3667 phosphorylation sites on 1520 unique phosphoproteins (Supplemental Table S1). In addition to the many phosphorylation sites that have been quantified and show temporal changes in response to DIF-1, there are other phosphorylation sites that are not DIF-1 regulated and are a valuable resource for understanding the role phosphorylation plays in Dictyostelium cellular signaling. The distribution between phosphorylated residues was 73% phosphoserine (pS), 20% phosphothreonine (pT), and 7.1% phosphotyrosine (pY), which is considerably different to that reported for other organisms, including mammalian cells: 86% pS, 12% pT, and 2% pY (Olsen et al. 2006). For unknown reasons, we observe considerably more tyrosine phosphorylation than reported in other organisms, which is unexpected, given the reduced components in the tyrosine phosphorylation “signaling toolkit” of Dictyostelium (Lim and Pawson, 2010). It may, however, reflect the fact that other, much more abundant proteins, such as the actins, are tyrosine phosphorylated in Dictyostelium but not in Metazoa (Howard et al., 1993).
Quantified ratios of phosphorylation fold changes relative to no DIF-1 treatment could be determined for 3005 sites on 1338 proteins. A ratio value of 2, that is, a log2 ratio value of 1, represents a twofold increase in phosphorylation (i.e., phosphorylation), whereas a ratio of 0.5, that is, a log2 ratio value of −1, represents a twofold decrease in phosphorylation (i.e., dephosphorylation). Owing to the stochastic nature of data acquisition, not all sites were observed in all samples or at all time points. To assist with the interpretation of the data, we assigned phosphorylation sites as class I, II, III, or IV, based on their phosphorylation fold change, the number of observations within a biological replicate, and the number of observations across biological replicates. Using increasingly stringent criteria, we defined class IV sites, “DIF-1–regulated phosphorylation sites,” as those with a twofold change in phosphorylation (a ratio of ≥2 or ≤0.5); class III sites as those observed at each time point in a single biological replicate (so a temporal curve can be plotted); class II sites (Supplemental Table S3) as observed in more than one biological replicate at the same time point (average 1.5-fold change); and class I sites (Supplemental Table S2) as observed at each of the four time points in more than one biological replicate (producing an averaged temporal curve). The benchmark class I and II phosphorylation sites are used for the majority of the following discussion. Figure 1B presents a summary of the phosphoproteome data.
DIF-1 causes a major shift toward dephosphorylation
An interesting temporal pattern emerged when class I phosphorylation sites were analyzed by cluster analysis (Saeed et al., 2011) to detect groups of sites that change their phosphorylation with similar temporal profiles. Clusters showing dephosphorylation contained many more sites, indicating that DIF-1 may trigger a net dephosphorylation of cellular proteins (Figure 1C). A similar pattern emerges when class II sites are grouped into either phosphorylation or dephosphorylation events. The balance of phosphorylation versus dephosphorylation events dramatically shifts; at 1 min, just 39% of the class II sites are dephosphorylations, increasing to 94% at 15 min (Figure 2A).
FIGURE 2:

Analysis of class II DIF-1–regulated phosphorylation sites at each time point. (A) Chart summarizing the proportion of class II phosphorylation events vs. dephosphorylation events. Number in bars represents absolute numbers. (B–E) WebLogo sequence logo for motifs identified using Motif-X. Amino acids and color coded according to their hydrophobicity; hydrophobic, black neutral green; hydrophilic, blue. (B) RxxSxxxL motif from phosphorylation sites at 1 min; n = 15, motif score of 29, and 76-fold enrichment. (C) xSDDEx motif from dephosphorylation sites at 8 min; n = 15, motif score of 38, and 120-fold enrichment. (D) xSPRx motif from dephosphorylation sites at 8 min; n = 17, motif score of 26, and 40-fold enrichment. (E) xSPx motif from dephosphorylation sites at 8 min; n = 17, motif score of 16, and 5-fold enrichment.
Motif extraction and Gene Ontology term analysis of class II DIF-1–regulated phosphorylation
Using Motif-X (Schwartz and Gygi, 2005) to identify protein phosphorylation motifs among the 88 class II sites phosphorylated at 1 min, we identified leucine at P+4 and arginine at P-3 (relative to the phosphorylated residue) as being significantly enriched (Figure 2B). This motif fits the AMP-activated protein kinase (AMPK) substrate recognition site: basic residues (R, K, or H) at P-3 or P-4 and hydrophobic ones (M, V, L, or I) at P+4 and P-5 (Hardie, 2011).
Although dephosphorylation-site motifs rarely determine dephosphorylation by a particular phosphatase (Ubersax and Ferrell, 2007), identification of a phosphorylation motif could reflect inhibition of a kinase rather than activation of a phosphatase. Hence we analyzed class II, DIF-1–induced dephosphorylation sites at 8 min, using Motif-X. We identified eight significantly enriched consensus motifs (Figure 2, C–E; and unpublished data). Four of these motifs—(pS)DDE, (pS)DxD, (pS)DxE, and (pS)ExE—demonstrated that many dephosphorylation sites (sites with net reduction in phosphorylation levels relative to pre–DIF-1 treatment) are in very acidic regions (Figure 2C) that fit the consensus for the acidophilic protein kinase CK2 (casein kinase 2; Meggio and Pinna, 2003). We also identified proline as a prominent residue at P+1 in two motifs—the unique (pS)PR motif and the (pS)P motif (Figure 2, D and E). Most kinases disfavor proline at P+1, except the proline-directed kinases such as extracellular-regulated kinase-2 (ERK2) and cyclin-dependent kinases (Ubersax and Ferrell, 2007).
Using the AmiGO Gene Ontology (GO) term enrichment tool (Boyle et al., 2004), we found significant enrichment of GO terms at 1 and 15 min among class II phosphorylation sites. At 1 min after DIF-1 treatment, 10 GO terms are enriched, including signal transduction, cellular response to stimulus, and cell communication (GO:0050896, GO:0051716, and GO:0007154), and a specific role in GTPase regulator activity (GO:0030695; Supplemental Table S4). Monomeric small GTPases are molecular switches that control a wide variety of cellular processes. They switch between an active, GTP-bound state and an inactive, GDP-bound state and are regulated by activating guanine nucleotide exchange factors (GEFs) and inactivating guanine nucleotide–activating proteins (GAPs). There are many genes encoding small GTPases and their GEFs and GAPs in Dictyostelium (Vlahou and Rivero, 2006; Kortholt and van Haastert, 2008). Members of the Ras and Rho subfamilies are important for transduction of extracellular signals and include RasC and RasG, which are both critical for cAMP-mediated chemotaxis (Kortholt et al., 2013; Table 1). DIF-1–regulated phosphorylation sites at 1 min that are on proteins described by the GO term GTPase regulator activity (GO:0030695) are presented in Table 1. Remarkably, more than half of these sites (14 of 26) are Rho-family GTPase substrates or regulators. These include two RhoGAPs—GacG, a protein kinase B substrate (Tang et al., 2011) and MGEP1, a regulator of the contractile vacuole (Heath and Insall, 2008)—and the RacC substrate WASP, which has a critical role in cell polarization and chemoattractant-induced localization of F-actin (Veltman et al., 2012). Six of the GO term–enriched phosphorylation sites are in Ras-family proteins, including RasG-specific GEFs—GefR and GefD, both involved in cAMP-induced Ras activation at the cell boundary in chemotaxing cells (Kortholt et al., 2013)—and RapGAP3, which regulates Rap1 and has a role in Dictyostelium morphogenesis and cell sorting during apical tip formation (Jeon et al., 2009).
TABLE 1:
Class II phosphorylation sites enriched for GO term GTPase regulator activity (GO:0030695) at 1 min after DIF-1 treatment.
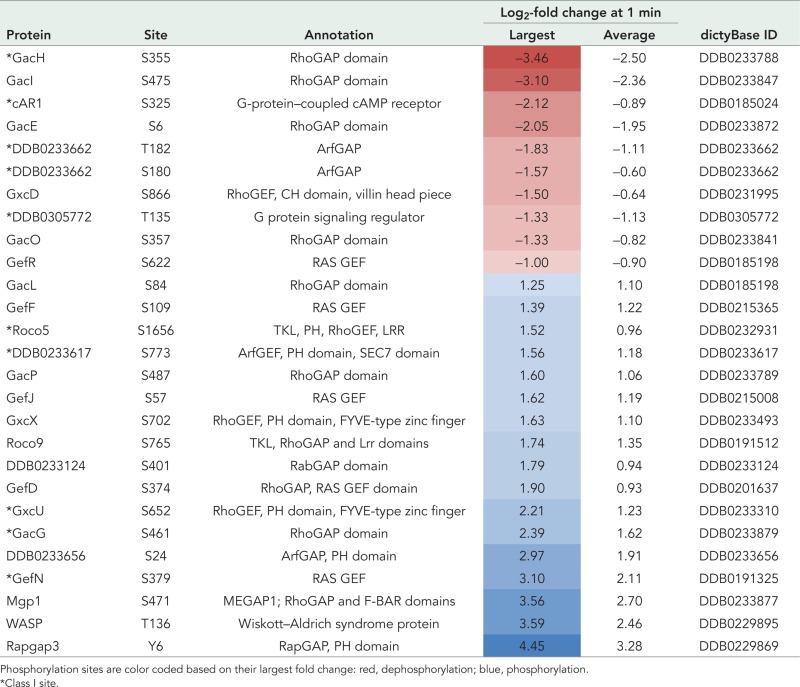 |
Phosphorylation sites are color coded based on their largest fold change: red, dephosphorylation; blue, phosphorylation.
*Class I site.
In summary, we identified a distinct pattern of regulated phosphorylation after DIF-1 treatment—increasing numbers of DIF-1–regulated phosphorylation sites 8 min after treatment, with the majority of these being dephosphorylation events. Evidence from GO term analysis clearly puts DIF-1 upstream of many cellular signaling events and identifies a hitherto unknown role for DIF-1 in regulation of small GTPases.
Calcineurin plays a role in the DIF-1–induced phosphorylation and nuclear localization of DimB
In addition to using GO term analysis to delineate DIF-1 signaling, we investigated selected components that had preexisting links with prestalk differentiation. Several observations suggest that intracellular Ca2+ has a regulatory role in prestalk differentiation (Gross, 2009): 1) Ca2+ levels are highest in prestalk cells (Cubitt et al., 1995); 2) treatment with DIF-1 causes a rise in intracellular Ca2+ (Schaap et al., 1996); 3) BHQ, an agent that liberates Ca2+ from internal stores, causes a slow rise in ecmB expression (Schaap et al., 1996); and 4) treatment of cells with BHQ causes phosphorylation and nuclear localization of STATc (Araki et al., 2010).
We identify class I phosphorylation sites on several proteins that have a direct involvement with Ca2+, and most show dephosphorylation in response to DIF-1 (Supplemental Figure S2). In addition, we observe transient phosphorylation upon four phosphorylation sites in an 18-residue stretch of CanA, the catalytic subunit of the protein phosphatase calcineurin (Figure 3A). Calcineurin is a Ca2+- and calmodulin (CaM)-dependent protein phosphatase that is well conserved from yeast to mammals and is critical to many cellular processes (Rusnak and Mertz, 2000). In its inactive form, calcineurin is a heterodimer of CanA and its regulatory subunit CanB. Increasing Ca2+ allows CaM to bind, displacing the autoinhibitory domain and forming an active calcineurin heterotrimer. In this way, cellular Ca2+ regulates calcineurin phosphatase activity. Ca2+-dependent binding of CaM to Dictyostelium CanA has been demonstrated and putative CaM-binding domains identified, but, unusually, Ca2+/CaM is not essential for Dictyostelium calcineurin activity, which can also be activated by long-chain fatty acids, including arachidonic acid (Kessen et al., 1999; Catalano and O'Day, 2008).
FIGURE 3:

DIF-1–regulated phosphorylation sites on CanA, the catalytic subunit of calcineurin, and its role in the regulation of DimB phosphorylation. (A) Temporal profile for DIF-1–induced phosphorylation changes in class I sites on CanA (DDB0185021). Phosphorylation fold changes are expressed as log2 ratios relative to pretreatment; a log2 ratio of 1 is a twofold increase in phosphorylation, whereas a log2 ratio of −1 represents a twofold decrease in phosphorylation, that is, dephosphorylation. (B) Amino acid sequence 525–559 of CanA. Underlined 529–548 is a putative CaM-binding domain (Catalano and O'Day, 2008). Highlighted and labeled residues are those identified as class I DIF-1 regulated. (C) DimB S590 phosphorylation in response to DIF-1 and gossypol treatment. Ax2 cells starved for 4 h were treated with DIF-1 and/or gossypol and samples collected at the times indicated and then immunoblotted using anti–pS590 DimB and anti–total DimB antibody. (D) Nuclear translocation of DimB in response to DIF-1 and gossypol. Cells developed and treated as in C. Nuclear accumulation of DimB was assayed immunohistochemically at the stated times using anti–total DimB. Ethanol and dimethyl sulfoxide controls (DIF-1 and gossypol, respectively) both showed no nuclear accumulation. Scale bar, 10 μm.
DIF-1–regulated phosphorylation sites in CanA all show a rapid phosphorylation at 1 min, followed by a return to basal phosphorylation levels by 8 min (Figure 3A). Of interest, the clustered sites are within, and adjacent to, a proposed CaM-binding site (Figure 3B; Catalano and O'Day, 2008), suggesting that DIF-1–induced phosphorylation could regulate CaM binding and activity of calcineurin. Dictyostelium calcineurin appears to function throughout growth and development, and studies using calcineurin inhibitors reported growth, cell signaling, and development defects (Horn and Gross, 1996; Catalano and O'Day, 2008). Attempts to knock out calcineurin have been unsuccessful, but there have been successful knockdowns of calcineurin subunits using RNA interference. Strains in which the regulatory and catalytic subunit genes (cnbA and canA) were silenced show aberrant development, including ectopic tip formation, short, incomplete vacuolated stalk cells, and, of importance, reduced expression of the stalk cell marker ecmB and an impaired stress response (Boeckeler et al., 2006; Thewes et al., 2013). Hence we used gossypol, the most specific calcineurin inhibitor available (Baumgrass et al., 2001; Weissenmayer et al., 2005), to investigate the effects of calcineurin on DIF-1–induced changes.
We treated cells with gossypol and monitored the phosphorylation state of the transcription factors DimB and STATc, both phosphorylated in response to DIF-1, by immunoblotting (Fukuzawa et al., 2001; Yamada et al., 2013). We were surprised to find that not only did gossypol cause DimB phosphorylation, but also the effects of DIF-1 and gossypol were additive (Figure 3C). Gossypol treatment also caused weaker and sporadic phosphorylation of STATc (unpublished data). Phosphorylation of DimB and STATc leads to their nuclear localization, and treatment with gossypol caused nuclear localization of DimB (Figure 3D) and also STATc, although after 15 min, and the response was lower than with DIF-1 alone (unpublished data). Gossypol has been shown to inhibit calcineurin at multiple sites, and this could explain the additive behavior of DIF-1 and gossypol (Carruthers et al., 2007).
DIF-1 regulates many cAMP-responsive proteins, including the cAMP receptor cAR1
Surprisingly, many of the DIF-1–regulated phosphorylation sites lie in cAMP signaling pathways (Figure 4). Extracellular cAMP signals are perceived and transferred into the cell by cAR1. Binding of extracellular cAMP to cAR1 stimulates many signaling events and phosphorylation at multiple residues on its cytoplasmic tail (Hereld et al., 1994). We identify 10 DIF-1–regulated phosphorylation sites on cAR1; eight are the same residues phosphorylated in response to cAMP binding (Figure 5A). Four of the five class I sites (S299, S303, S304, S324, and S325) show a major and consistent dephosphorylation (Figure 5B), contrasting with the phosphorylation that occurs in response to cAMP.
FIGURE 4:

Schematic summary of signaling pathways downstream of cAMP and cAR1 during early development, which control chemotaxis and cAMP relay. This depiction of signaling events is not an attempt to summarize all cAMP signaling but is a representation to demonstrate enrichment of DIF-1–regulated sites in cAMP signaling pathways. Proteins components are color coded based on the presence of a DIF-1–regulated phosphorylation site; see key. For simplicity, pathways involved in the regulation of PIP3 are not fully shown.
FIGURE 5:
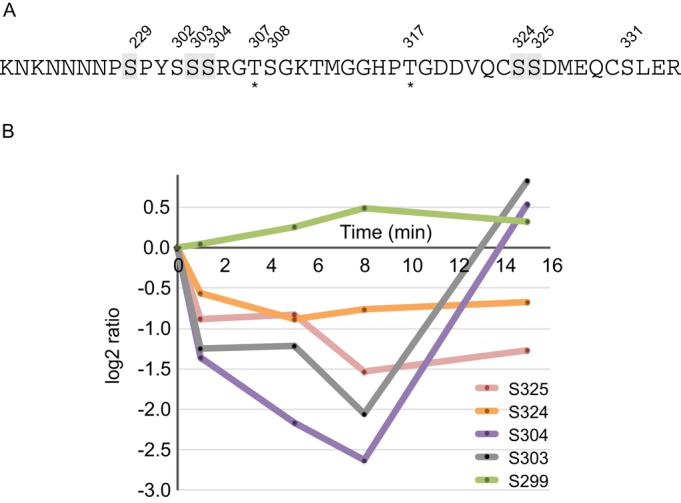
DIF-1–regulated phosphorylation sites on cAR1. (A) Phosphorylation sites identified on residues 291–334 of cAR1 (clusters 1 and 2 from Hereld et al., 1994). Numbered residues indicate class I–IV sites; those highlighted in gray are class I sites. Asterisks indicate phosphorylation sites that have not been previously identified. (B) Temporal profile for DIF-1– induced phosphorylation changes in class I sites on cAR1 (DDB0185024). Phosphorylation fold changes are expressed as log2 ratios relative to pretreatment; a log2 ratio of 1 is a twofold increase in phosphorylation, whereas a log2 ratio of –1 represents a twofold decrease in phosphorylation.
DIF-1 leads to a rapid dephosphorylation of PKB and its substrates
PKB plays a key role in multiple cellular processes and in Dictyostelium is central to cAMP responsiveness during chemotaxis (King and Insall, 2009). Dictyostelium has two PKB homologues. PKBA has a mammalian PKB-like domain structure, with kinase and regulatory domains plus a PH domain allowing phosphatidylinositol (3,4,5)-triphosphate (PIP3)–dependent membrane recruitment. The second PKB homologue in Dictyostelium has an atypical structure, missing a PH domain. PKBR1 is instead myristoylated, keeping it associated with the membrane independently of PIP3 (Cai and Devreotes, 2011). Activation of PKB is by phosphorylation in its activation loop (AL), promoting phosphorylation by the upstream target of rapamycin complex 2 (TORC2) kinase complex at a hydrophobic motif (HM). Both HM and AL in PKBA (T435 and T278, respectively) and PKBR1 (T470 and T309) are phosphorylated in response to cAMP.
We identified two closely spaced phosphorylation sites on PKBR1 (T442 and S449), both of which are dephosphorylated in response to DIF-1 (Figure 6A). Averaged temporal data for the class I site, S449, shows consistent dephosphorylation after DIF-1 treatment (Figure 6B). Nine PKBA/PKBR1 substrates have been identified (Kamimura et al., 2008; Liao et al., 2010; Tang et al., 2011), and, significantly, we identified class I phosphorylation sites in no fewer than four of these proteins (Figure 6, A and C): GacG, a RhoGAP; GefN, a RasGEF; PakA, an STE20-family, P21-activated protein kinase; and most, strikingly, SHAPS, which undergoes a 19-fold dephosphorylation (log2 ratio, −4.3) after just 1 min of DIF-1 treatment (Figure 6B).
FIGURE 6:

DIF-1 induced dephosphorylation of PKB homologues and their substrates. (A) Summary of DIF-1–regulated phosphorylation sites in PKBA, PKBR1, and their substrates. (B) Temporal profile for DIF-1–induced phosphorylation changes in class I sites on PKBA, PKBR1, and their substrate SHAPS (DDB0306661). Solid lines represent averaged data for class I sites and dashed lines are from class III sites from a single experiment. (C) Heat-map representation of the temporal phosphorylation changes of class I DIF-1–regulated phosphorylation sites in PKB substrates. Averaged values, n ≥ 2. (D) PKB AL phosphorylation in response to DIF-1. Ax2 cells starved for 5 h were treated ± 100 nM DIF-1. Results are representative of at least three independent experiments. (E) Antagonism between cAMP and DIF-1. Cells were starved as in D and then treated with 1 μM cAMP ± 100 nM DIF-1. Samples collected at the times indicated and then immunoblotted as in D. (F) The response of PKB substrate phosphorylation to DIF-1. Samples from D were immunoblotted with anti-phosphospecific PKB substrate antibody. Blots were stripped and reprobed with anti-actin antibody as a loading control.
Having observed dephosphorylation of PKBR1 and several PKB substrates in response to DIF-1, we reasoned that PKBR1 S449, although not the regulatory site, might be associated with phosphorylation changes in the AL or HM. We used antibodies that specifically recognize the phosphorylated AL in PKBR1 and PKBA, which are both phosphorylated in response to cAMP (Liao et al., 2010), to monitor phosphorylation changes. DIF-1 caused a rapid dephosphorylation of the AL of PKBR1 and PKBA (T309 and T278, respectively; Figure 6D). Antagonism of the cAMP-induced phosphorylation state of AL in PKBR1 and PKBA by DIF-1 was also directly analyzed using immunoblotting. Although DIF-1 had no effect on the initial cAMP-induced phosphorylation, DIF-1 caused accelerated dephosphorylation of the AL after the first burst of phosphorylation (Figure 6E). This is one of four experiments, all of which showed accelerated dephosphorylation by DIF-1 after the initial, rapid cAMP response. Presumably the lack of an effect on the initial extremely fast cAMP response reflects the need for DIF-1 to access its intracellular targets, but we did not investigate this further.
The phosphorylation state of the HM in Dictyostelium PKBs has also been monitored using antibodies, but in our hands, these failed to produce acceptable results. We were therefore unable to determine whether dephosphorylation of the AL was accompanied by dephosphorylation of the HM. The HM residue of PKBA (T435) was identified, however, as a class III phosphorylation site and showed major dephosphorylation (19-fold dephosphorylation after 1 min; Figure 6B).
We confirmed that there is an effect of DIF-1 on PKB substrates by Western blotting using a phospho-PKB substrate antibody that recognizes the sequence RXRXXpS/T and has been used to identify potential PKB substrates in Dictyostelium (Kamimura et al., 2008; Liao et al., 2010; Tang et al., 2011). We detect bands in cell extracts, and several of these show dephosphorylation after DIF-1 treatment. These phosphorylation changes parallel the changes we see in PKBA/PKBR1 phosphorylation. We see three potential PKB substrate bands on immunoblots—p75/82, p65, and p62 (Figure 6F). PHAPS was originally identified as p78, a PKB substrate phosphorylated at T102 (Liao et al., 2010). Given that we identify it from the proteomics, it is presumably the same protein as our p75/82 band. We identified net dephosphorylation of PHAPS (DDB0307127) in response to DIF-1 at class III site T102 (8.0-fold maximum dephosphorylation at 5 min; Supplemental Table S1). Similarly, GacQ, a RhoGAP, was identified as a PKB substrate, as p65/67 (Kamimura et al., 2008), the p62 or p65 immunoblot band that we identify as being dephosphorylated in response to DIF-1 (Table 2 and Figure 6F), could be the same phosphoprotein.
TABLE 2:
cAMP-regulated phosphorylation sites also shown to be DIF-1 regulated.
| Protein | Site | Annotation | Largest fold change (log2)a | Site class | Reference | dictyBase ID |
|---|---|---|---|---|---|---|
| cAR1 | S304 | G-protein–coupled cAMP receptor | –3.39 | I | Hereld et al. (1994) | DDB0185024 |
| cAR1 | S303 | G-protein–coupled cAMP receptor | −3.39 | I | Hereld et al. (1994) | DDB0185024 |
| cAR1 | S325 | G-protein–coupled cAMP receptor | −2.92 | I | Hereld et al. (1994) | DDB0185024 |
| cAR1 | S324 | G-protein–coupled cAMP receptor | −1.33 | I | Hereld et al. (1994) | DDB0185024 |
| cAR1 | S299 | G-protein–coupled cAMP receptor | 1.26 | I | Hereld et al. (1994) | DDB0185024 |
| STATa | Y702 | Signal transducer and transcriptional activator | −1.97 | I | Kawata et al. (1997) | DDB0215388 |
| MhcA | T1833 | Myosin II heavy chain | −2.37 | I | Vaillancourt et al. (1988) | DDB0191444 |
| MlcR | S13 | Regulatory myosin light chain | −2.25 | I | Griffith et al. (1987) | DDB0185146 |
| MyoIB | S332 | Myosin IB | −1.59 | I | Tsiavaliaris et al. (2008) | DDB0191351 |
| cAR1 | S302 | G-protein–coupled cAMP receptor | −2.69 | II | Hereld et al. (1994) | DDB0185024 |
| cAR1 | S331 | G-protein–coupled cAMP receptor | −1.72 | II | Hereld et al. (1994) | DDB0185024 |
| cAR1 | S308 | G-protein–coupled cAMP receptor | 1.23 | II | Hereld et al. (1994) | DDB0185024 |
| PHAPS | T102 | PKB substrate | −3.00 | III | Liao et al. (2010) | DDB0307127 |
| ERK2 | Y178 | Extracellular response kinase | −3.18 | III | Kosaka and Pears (1997) | DDB0191457 |
| ERK2 | T176 | Extracellular response kinase | −2.62 | III | Kosaka and Pears (1997) | DDB0191457 |
| PKBA | T435 | Protein kinase B | –4.26 | III | Liao et al. (2010) | DDB0191195 |
| MhcA | T1823 | Myosin II heavy chain | −2.22 | IV | Egelhoff et al. (1993) | DDB0191444 |
| MhcA | T2029 | Myosin II heavy chain | −1.87 | IV | Vaillancourt et al. (1998) | DDB0191444 |
aNegative values indicate dephosphorylation.
We also identified other cAMP-stimulated signaling proteins upstream of PKB with DIF-1–regulated sites These include members of the upstream TORC2 kinase complex, which phosphorylate PKB, and members of the Sca1 complex, which regulate RasC and lie upstream of PKB (Supplemental Figure S3). In summary, we identified multiple DIF-1–induced dephosphorylation events in the Dictyostelium PKB pathway and confirmed DIF-1–induced phosphorylation changes in PKBA, PKBR1, and potential PKB substrates by immunoblotting.
DIF-1 leads to a rapid dephosphorylation of ERK2 and its substrate EppA
Mitogen-activated protein kinase (MAPK) cascades are conserved signaling pathways that transfer extracellular signals to a range of regulatory pathways. ERK2, one of the two Dictyostelium MAPKs, has a role in cAMP relay and chemotaxis and lies downstream of the cAR1 receptor. ERKs are regulated by phosphorylation of a highly conserved TEY sequence in the activation loop of the kinase. Residues T176 and Y178 are phosphorylated in response to cAMP (Maeda et al., 1996; Kosaka and Pears, 1997). We identified T176 and Y178 in ERK2 as class III phosphorylation sites, and both are rapidly dephosphorylated in response to DIF-1 (Y178, maximally ninefold dephosphorylated at 1 min; Figure 7A). We confirmed these ERK2 phosphorylation changes using a phospho-specific antibody designed to recognize T202 and Y204 of human ERK2. We used caffeine treatment to inhibit endogenous cAMP production (Brenner and Thoms, 1984), which temporarily blocks the fluctuating phosphorylation of ERK2. We stimulated cells with cAMP to cause rapid phosphorylation of ERK2 and then monitored the phosphorylation state of T176/Y178 in response to DIF-1. The late decrease in phosphorylation in control cells seems very likely to be due to slow hydrolysis of cAMP. However, within just 30 s of DIF-1 addition, ERK2 T176/Y178 was completely dephosphorylated before levels returned to baseline within 2–3 min (Figure 7, A and B). These data are consistent with DIF-1 causing a transient accelerated dephosphorylation of ERK2.
FIGURE 7:

DIF-1–regulated phosphorylation sites in ERK2 and its substrate EppA. (A) Temporal profile for DIF-1–induced phosphorylation changes in class I sites on ERK2 (DDB0191457) and EppA (DDB0233660). Solid lines represent averaged data for class I sites and dashed lines from class III sites from a single experiment. (B) ERK2 T176/Y178 phosphorylation in response to DIF-1. Ax2 cells starved for 5 h in KK2 were treated with 5 mM caffeine (10 min) and then washed out before cells (1 × 107 cells/ml) were treated with 1 μM cAMP for 2 min before addition of 100 nM DIF-1. Samples were collected at the times indicated and then immunoblotted using anti–phospho p44/42 MAPK antibody. Blots were stripped and reprobed with anti-actin antibody as a loading control. Results are representative of at least three independent experiments.
EppA is an ERK2 substrate and is phosphorylated in response to cAMP; like ERK2, it has a role in cAMP relay and chemotaxis (Chen and Segall, 2006). We found a more than fivefold dephosphorylation of EppA (S325) after 5 min of DIF-1 treatment (Figure 7A). S325 was not identified as a cAMP-induced phosphorylation site, but it is one of the four predicted ERK2 phosphorylated sites in EppA and is a critical residue; overexpression of EppA with an S325A mutation in wild-type cells causes a growth defect (Chen and Segall, 2006).
In summary, we identified S325 on the ERK2 substrate EppA and cAMP-regulated residues in ERK2 as being dephosphorylated in response to DIF-1 and confirmed these effects on ERK2 using phospho-specific antibodies. There is a mysterious link between ERK2 and DIF-1; erkB-null cells fail to undergo multicellular morphogenesis but can be rescued by the addition of DIF-1 (Kuwayama et al., 2000; Tsujioka et al., 2004). How this result might relate to ours is unclear.
Many other cAMP-responsive proteins contain DIF-1–regulated phosphorylation sites
In addition to cAMP-regulated phosphorylation sites in cAR1, PKB, and ERK2, we identified sites on other proteins known to be stimulated in response to cAMP and that change their phosphorylation state in response to DIF-1. For example, proteins implicated as regulators of heterotrimeric G proteins, such as the phosducin-like protein Phlp1 and regulator of G protein signaling (RGS) domain–containing proteins (Supplemental Figure S3), which transduce the cAMP signaling immediately downstream of cAR1, have DIF-1–regulated phosphorylation sites
ElmoE is an essential component of the chemotaxis machinery and forms a direct link between cAMP-activated cAR1 and the actin cytoskeleton (Figure 4). It includes seven DIF-1–regulated class I sites. ElmoE is believed likely to exert its regulation by association with heterotrimeric G proteins and DOCK-like GEF proteins, regulating actin polymerization at the leading edge of migrating cells (Yan et al., 2012). All sites on ElmoE show consistent dephosphorylation in response to DIF-1 (Figure 8A).
FIGURE 8:

DIF-1–regulated phosphorylation sites in other cAMP-stimulated proteins. (A) Temporal profile for DIF-1–induced phosphorylation changes in class I sites on ElmoE (DDB0233920). (B) Heat-map representation of the temporal phosphorylation changes of class I DIF-1–regulated phosphorylation sites in myosin II components and regulators and other myosin heavy chains and their regulators. Averaged values, n ≥ 2. Asterisk indicates site previously identified as a cAMP-regulated phosphorylation site; dagger indicates TEDS site of phosphorylation on myosin I. (C) Temporal profile for class I DIF-1–induced phosphorylation changes to cAMP-regulated phosphorylation sites in myosin II components.
Actin and myosin both have critical roles during cell motility. Actin polymerization drives pseudopod formation at the leading edge of chemotaxing cells, whereas actinomyosin filaments generate contractile forces at the sides and rear (Cai and Devreotes, 2011). In addition to cAMP signaling proteins upstream of actin components, we identify many DIF-1–regulated phosphorylation sites in components of the myosin machinery.
Myosins are a ubiquitous family of eukaryotic proteins whose structure and function are highly conserved, all having the basic properties of actin binding, ATP hydrolysis, and force transduction and being involved in a wide range of motility processes. Dictyostelium myosin II is rapidly redistributed in response to cAMP from the cytoplasm to the cortex during chemotaxis. This is accompanied by phosphorylation changes in both its heavy and regulatory light chains (MhcA and MlcR, respectively). We observe multiple DIF-1–regulated phosphorylation sites on myosin II heavy and light chains, heavy chain kinases (MhkB, C, and D), and their regulator, PakA (Figure 8B). These include MlcR (S13) and MhcA (T1833), which are both phosphorylated in response to cAMP, we show dephosphorylation in response to DIF-1. The temporal phosphorylation changes in MlcR (S13) and MhcA (T1833) are remarkably consistent (Figure 8C) and suggest that DIF-1 has a prominent role in myosin II regulation. Intriguingly, the time courses of phosphorylation changes for all class I sites on myosin heavy chains cluster and are rapidly dephosphorylated between 5 and 8 min after DIF-1 treatment (Supplemental Figure S4A).
In addition to phosphorylation sites on myosin II, we observe DIF-1–regulated phosphorylation sites in other cAMP-regulated myosins. Myosin I heavy chains are single- headed, non–filament-forming, actin-based motor proteins that function in membrane cytoskeletal interactions involved in exocytosis, endocytosis, and cell migration (de la Roche and Cote, 2001; McConnell and Tyska, 2010). Conserved phosphorylation sites (TEDS sites) often regulate myosin I motor activity. The TEDS site on Dictyostelium MyoIB (S332) is phosphorylated in response to cAMP (Tsiavaliaris et al., 2008), and we identify it as a class I site, which is dephosphorylated in response to DIF-1 (Figure 8B). DIF-1–regulated TEDS sites on other myosin I heavy chains were also identified, as well as sites dephosphorylated in PAKs, which often regulate myosin I via their TEDS sites (Supplemental Figure S4B). We also identify DIF-1–regulated phosphorylation sites in two orphan myosins, including a class I site, S200 in MyoG (Figure 8B and Supplemental Figure S4B). MyoG is required for cell polarization and chemotaxis to cAMP (Breshears et al., 2010).
Other proteins with multiple DIF-1–regulated phosphorylation sites
We identified DIF-1–regulated phosphorylation sites on many other non–cAMP-regulated proteins with unknown roles in signaling. Of the 270 proteins that contain class I sites (Supplemental Table S2), 29 are protein kinases, four are phosphatases, and 23 are small GTPases or their regulating GEF/GAPs. Considering just class I sites, we see many proteins with multiple sites, including seven sites in ElmoE, five sites in cAR1 and MhcA, and four sites in CanA (Figures 8A, 5, 8B, and 3A, respectively), but the protein with the most DIF-1–regulated sites is the nontransporter, uncharacterized ABC protein AbcF4 (Anjard et al., 2002). The temporal dynamics of all nine class I sites on AbcF4 strongly cluster (Supplemental Figure S5A). We knocked out the abcf4 gene in Ax2, but the strain shows no gross developmental defects or abnormal DIF-1–induced changes (unpublished data); however, due to the large number of ABC transporters in Dictyostelium (Anjard et al., 1998), this could reflect a functional redundancy.
Other interesting proteins that contain multiple class I sites include the polyketide synthase PKS16; the uncharacterized protein DDB0232240, which contains PH, CH, actin-binding, and Ras association domains; and the protein kinase NdrB, which shows three sites consistently phosphorylated rapidly after DIF-1 (Supplemental Figure S5, B–D).
DISCUSSION
A link between dephosphorylation and kinase consensus motifs
There is a distinct shift in the balance of phosphorylation and dephosphorylation in the time after DIF-1 treatment. Clustering analysis of class I sites shows a prominent dephosphorylation event between 5 and 8 min after DIF-1 treatment, with many apparently unrelated phosphorylation sites showing net dephosphorylation. This same dephosphorylation pattern is observed within several proteins with multiple phosphorylation sites, such as ElmoE, myosins and their regulators, and Abcf4. We suggest that the pattern of increasing dephosphorylation and the clustered dephosphorylation event at 5–8 min is consistent with DIF-1 inhibiting acidophilic protein kinases such as CK2 and/or the proline-directed ERK2 or CDK kinases, thus shifting the balance toward dephosphorylation of many proteins. In support of this, we identify ERK2 and the ERK2 substrate EppA as being dephosphorylated in response to DIF-1.
Calcineurin has a role in DIF-1 signaling to DimB
We identified several DIF-1–regulated sites in Ca2+/CaM–regulated proteins, including the protein phosphatase calcineurin. We showed that DIF-1 causes phosphorylation of multiple sites close to a predicted CaM-binding site on CanA. DIF-1 triggers the phosphorylation and nuclear localization of DimB, and the calcineurin inhibitor gossypol mimics the effect of DIF-1 on DimB. This leads us to propose a model in which DIF-1 treatment leads to phosphorylation of CanA at a Ca2+/CaM–binding site. Calcineurin is activated by Ca2+/CaM binding to CanA at the site, and we propose that phosphorylation adjacent to the site could disrupt Ca2+/CaM binding and activation of calcineurin. This would cause inactivation of calcineurin's phosphatase activity, leading to DimB phosphorylation and its movement to the nucleus. Regulation of calcineurin by phosphorylation adjacent to CaM-binding sites has been reported in other species (Kume et al., 2011; Juvvadi et al., 2013).
All four phosphorylation sites on CanA fit the AMPK consensus motif, suggesting that AMPK could be responsible for the early DIF-1–induced phosphorylation changes of CanA. AMPK is an essential sensor and regulator of cellular energy status, and work on the Dictyostelium homologue demonstrates that AMPK signaling has an important role in mitochondrial disease (Francione et al., 2011). We also identified AMPK consensus sites in many other proteins 1 min after DIF-1 treatment, which could be consistent with AMPK acting as a transponder of the early DIF-1 response.
DIF-1 has an extensive role in cellular signaling
Previous evidence showed that in tight aggregate cells, DIF-1 signals to key prestalk-specific transcription factors (Fukuzawa et al., 2003; Thompson et al., 2004; Huang et al., 2006; Zhukovskaya et al., 2006; Keller and Thompson, 2008), but the data presented here, at least for earlier cells, revealed that DIF-1 has a much more pervasive role in cellular signaling. For example, 10% of the Dictyostelium kinome is DIF-1 regulated. In addition to potential control of cellular signaling via protein kinase regulation, we identified a previously unknown role for DIF-1 in the regulation of small GTPases. Our data provide compelling evidence of DIF-1's role in the regulation of many GTPases via changes in their phosphorylation state and that of their GEFs and GAPs. Although there is no previous evidence for phosphorylation regulating GEF or GAP function in Dictyostelium, regulation by phosphorylation of mammalian small GTPases is well established; for example, the RhoGEF Vav1 has been linked to regulation in several human diseases and cancers (reviewed by Bustelo, 2000).
DIF-1/cAMP antagonism
cAMP has critical roles during early development, and we showed that many of the cognate cAMP-regulated proteins contain DIF-1–regulated phosphorylation sites. These include cAR1, ERK2, PKB, PKB substrates, heterotrimeric G protein regulators, ElmoE, and myosins and their regulators. Specific cAMP-induced phosphorylation sites have been identified, and many of these same sites are DIF-1 regulated, but unlike cAMP, which predominantly causes phosphorylation, we find dephosphorylation in response to DIF-1 (Table 2). Significantly, given that SILAC is a relatively new technology, we were able to use phospho-specific antibody against PKBA and PKBR1 to confirm the effect of DIF.
An antagonistic role of cAMP and DIF-1 has long been suspected; DIF-1 is a prestalk inducer, whereas cAMP is a stalk repressor (Berks and Kay, 1990). DIF-1 has also been shown to disrupt cAMP signaling during early development (Wurster and Kay, 1990). Another indicator that DIF-1 and cAMP may be antagonistic comes from recent work showing phosphorylation of a GATA transcription factor, GtaC, that, in response to cAMP, shuttles out of the nucleus and functions as a developmental timer (Cai et al., 2014). GtaC has been shown to shuttle into the nucleus in response to DIF-1 (Keller and Thompson, 2008), and we identified S349 as a class III phosphorylation site on GtaC (Supplemental Table S1), which is dephosphorylated in response to DIF-1 (6.2-fold maximum dephosphorylation at 8 min) and shows strong similarity to the dynamics of DIF-1–induced GtaC nuclear translocation (Keller and Thompson, 2008).
One mechanistic explanation for the broad array of cAMP targets containing DIF-1–regulated phosphorylation sites is that DIF-1 is moderating cAMP signaling via effects on cAR1 itself. Indeed, there is evidence that DIF-1 directly affects cAR1; DIF-1 causes inhibition of cAMP binding to its receptor by decreasing the affinity of cAR1 for cAMP (Wang et al., 1986). Binding of ligand to G-protein–coupled receptors typically causes conformational change, activation of associated heterotrimeric G proteins, and then initiation of downstream signaling. The accompanying phosphorylation of the receptor triggers an adaptive response via receptor sequestration and loss of ligand binding. Binding of extracellular cAMP to cAR1 activates many signaling events and phosphorylation at multiple residues on its cytoplasmic tail (Hereld et al., 1994), but phosphorylation of cAR1 was believed to be nonessential (Kim et al., 1997). The fact that the kinetics of cAMP-induced phosphorylation changes in cAR1 are different from TORC2 phosphorylation kinetics and those of other downstream targets (Brzostowski et al., 2013) also argues against cAR1 phosphorylation having a universal role in signaling pathways downstream of cAR1. However, there is a functional consequence to the tail phosphorylation for selected cAR1-regulated events, such as its membrane localization, which may have a role in progress through development (Serge et al., 2011), the coregulation of chemotaxis, actin reorganization, and signal relay (Brzostowski et al., 2013). Alternatively, since the majority of cAMP signaling occurs via heterotrimeric G proteins, DIF-1 could be having its effect on cAMP signaling via G protein regulation, perhaps via the DIF-1–regulated phosphorylation sites we identified on two RGS domain–containing proteins and/or the phosducin-like protein Phlp1.
DIF-1's role in shape change and cytoskeletal dynamics
Many of the proteins we identify that contain DIF-1–regulated dephosphorylation sites have roles in the cAMP-regulated cytoskeletal restructuring that drives chemotaxis. Consistent with this, previous work showed that DIF-1 has a negative effect on chemotaxis, probably acting through its effects on intracellular GMP (Kuwayama and Kubohara, 2009). There was also a report linking DIF-1 signaling and the cell shape changes that accompany endogenous cAMP pulsing (Wurster and Kay, 1990). More recently, we showed that DIF-1 induces a highly transient polymerization of F-actin at the cortex and accompanying cell rounding. We also used drugs that affect actin polymerization to show that, in some way, this leads to STATc activation (Araki and Williams, 2012). Many of the DIF-1–regulated proteins identified here seem likely to be involved in cAMP-stimulated/DIF-1–down-regulated actinomyosin changes, including the PKB substrate SHAPS, which is rapidly dephosphorylated in response to DIF-1. SHAPS contains a BAR domain, which in other contexts is involved in membrane shaping or sensing (Qualmann et al., 2011). MEGAP1 is a RhoGAP that is also rapidly dephosphorylated in response to DIF-1, has an F-BAR domain, and belongs to family of proteins linking membrane curvature and cytoskeletal reorganization (Heath and Insall, 2008). In addition, we identified large numbers of DIF-1–regulated phosphorylation sites in Rho GTPases, and these are considered master regulators of the actin cytoskeleton (Etienne-Manneville and Hall, 2002). We are therefore led to consider a link between cell shape change, actin recycling, and activation of STATc.
MATERIALS AND METHODS
Cell culture and SILAC labeling
D. discoideum Ax2 cells (Gerisch isolate) were grown axenically as described previously (Sugden et al., 2010). Ax2 cells before SILAC labeling were grown in SIH medium (without arginine and lysine; Formedium #SIH1001 [Huntstaton, UK]) supplemented with Arg and Lys at 3 and 4.5 mM, respectively, for at least three generations before switching cells into three batches in SIH medium supplemented with labeled Arg and Lys at 3 and 4.5 mM, respectively, for at least eight generations. The labeled amino acids used were as follows: light, R0K0 natural abundance l-Arg and l-Lys); medium, R6K4 (U-13C6 l-Arg,4,4,5,5-2H4 l-Lys); and heavy, R10K8 (U-13C6, 15N4 l-Arg, U-13C6, 15N4 l-Lys). All labeled amino acids were from Cambridge Isotope Laboratories (Cambridge, UK). Light-, medium-, and heavy-labeled cells were harvested while growing at (1–4) × 106 cells/ml, washed in KK2 (20 mM K2HPO4/KH2PO4, pH 6.2), resuspended, and shaken (200 rpm) at 1 × 107 cells/ml for 5 h in preparation for DIF-1 stimulation. Dictyostelium cells were successfully SILAC labeled, and efficient incorporation of label into the proteome and no significant arginine-to-proline transition were demonstrated under identical growth conditions (Sobczyk et al., 2014).
Cell stimulation, lysis, and digestion
Light labeled cells were harvested before stimulation. Medium- and heavy-labeled cells were treated with 100 nM DIF-1 (Enzo Life Sciences) with shaking at 200 rpm. Each experiment contained two triplex samples, A and B, each sample a mixture of three differently labeled cells (light, medium, and heavy) stimulated with DIF-1 for different times. Sample A, light 0 min, medium 1 min, and heavy 8 min. Sample B, light 0 min, medium 5 min, and heavy 15 min. The experiment was repeated in biological triplicate. For each sample, 1.5 × 108 cells were pooled, 5 × 107 from each label/DIF-1 treatment condition, and precipitated by the addition of and equal volume of 20% trichloroacetic acid for 20 min on ice with occasional mixing by inversion. The precipitated pellet was collected by centrifugation (10,000 × g for 15 min at 4°C), washed twice in ice-cold acetone (10,000 × g for 5 min at 4°C), and then air-dried. The dried pellet was resuspended in 2 ml of 8 M urea in 100 mM Tris-HCl (pH 8 at 22°C) with occasional sonication, and then the resuspended cellular proteins were reduced by the addition of 5 mM dithiothreitol for 25 min at 56°C, followed by alkylation with 14 mM iodoacetamide for 30 min at 22°C and protected from light and then quenched by the further addition of 5 mM dithiothreitol (22°C for 15 min, protected from light). The total protein at this stage, typically 10 mg, was diluted in 100 mM Tris-Cl (pH 8 at 22°C) to obtain a concentration of 1.6 M urea, and 80 μg of sequencing- grade trypsin (Promega, Essex, UK) was added and the solution incubated at 37°C for 18 h.
Phosphopeptide enrichment
Tryptic peptides were acidified with trifluoroacetic acid to 0.4% (vol/vol) and then desalted using 500 mg of tC18 Sep-Pak cartridges (500 mg; Waters, Dartford, UK) and lyophilized. Strong cation exchange (SCX) was performed on an Akta Purifier chromatography system using a 1-ml Resource S column (1 ml; GE Healthcare, Amersham, UK) with a flow rate of 1 ml/min and detection at 280 nm. Peptides were resuspended in buffer A (7 mM KH2PO4, pH 2.7, 30% [vol/vol] acetonitrile) and separated by a salt gradient consisting of 4 min of buffer A and a 4-min gradient to buffer A plus 350 mM KCl, followed by 3 min at buffer A plus 350 mM KCl. Fractions were collected throughout the run and combined into six or seven fractions. Combined fractions were lyophilized, desalted using 5-mg tC18 Sep-Pak cartridges) enriched for phosphopeptides using TiO2 according to Wilson-Grady et al. (2008), and lyophilized again before analysis.
LC-MS/MS analysis
LC-MS/MS was performed by the FingerPrints Proteomics facility at the University of Dundee. TiO2-enriched SCX fractions were analyzed by LC-MS/MS with multistage acquisition in technical duplicate on an Orbitrap Velos (Thermo Scientific) connected with an Ultimate3000 (Dionex) HPLC. The samples were first separated with a nano C18 column (Acclaim PepMap100, 15 cm × 75 μm, 3 μm). A linear gradient (buffer B: 2% acetonitrile, 0.1% formic acid; buffer C: 90% acetonitrile, 0.08% formic acid) from 5% buffer C to 40% buffer C over 65 min was used to separate the peptides, with a flow rate of 0.3 μl/min. The MS data were acquired in data-dependent mode: a full MS scan (mass range, 335–1800; resolution 60,000), followed by top-10 MS/MS scan in which the most intense 10 ions were selected for MS/MS by linear ion trap. The detection of a neutral loss of −97.9, −48.9, −32.6, and 24.5 triggers an additional MS3 scan (Ulintz et al., 2009). The precursor ion isolation window was 2 Da, and the dynamic exclusion time was 45 s.
Data processing
The combined data from a total of 76 LC-MS/MS runs were analyzed using MaxQuant (version 1.3.0.5), which incorporates the Andromeda search engine (Cox and Mann, 2008). Proteins were identified by searching the protein sequence database from D. discoideum (version 02/04/2013 from dictyBase, http://dictyBase.org/; Eichinger et al., 2005) supplemented with frequently observed contaminates and reverse sequences. Search parameters specified an MS tolerance of 20 ppm, an MS/MS tolerance of 0.5 Da, and full trypsin specificity, allowing for up to two missed cleavages. Carbamidomethylation of cysteine was set as a fixed modification, with oxidation of methionine, N-terminal protein acetylation, and phosphorylation of serine, threonine, and tyrosine allowed as variable modifications. Peptides were required to be at least seven amino acids in length and a MaxQuant score of ≥5, with false discovery rates or mapped to a given protein group and required a minimum of two SILAC pairs. To account for any errors in the counting or handling of cells mixed, the distribution of SILAC ratios was normalized within MaxQuant at the peptide level so the median of log2 ratios is zero (Cox and Mann, 2008). For examples of phosphorylation site location from tandem MS data, see Supplemental Figure S1.
Further analysis
Of the 3667 identified phosphorylation sites, 1234 were annotated with GO information. Using these as background data, we looked for GO term enrichment using the AmiGO GO Term Enrichment tool (Boyle et al., 2004). Cluster analysis of class I phosphorylation sites with similar temporal profiles were performed using a temporal pattern mining (TPM) algorithm (Saeed et al., 2011). Motif-X (Schwartz and Gygi, 2005) was used to identify protein phosphorylation motifs, using the Dictyostelium primary protein sequence (dictyBase release 17/09/12) as background. The mass spectrometry proteomics data were deposited at the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the data set identifier PXD001555.
Reagents and immunochemical analysis
All standard lab chemicals were from Sigma-Aldrich (Munich, Germany) unless stated otherwise. Gossypol was from Santa Cruz Biotechnology. Immunostaining of DimB was performed as described previously (Yamada et al., 2011). Western analysis was performed as described by Sugden et al. (2010) using antibodies anti–pS590 DimB and anti–total DimB (Yamada et al., 2013). From Cell Signaling Technology (Cambridge, UK) were anti–phospho PKC (pan) antibody (190D10), to detect the phosphorylation of the activation loops of PKBA and PKBR1; anti–phosphospecific PKB substrate antibody (110B7E), which recognizes RXRXXS/TXX; and anti–phospho p44/42 MAPK (Erk1/2) antibody (D13.14.4E), to detect the threonine and tyrosine phosphorylation of the ERK2 activation loop. Anti-actin antibody (sc-1615; Santa Cruz Biotechnology) was used as loading control.
Supplementary Material
Acknowledgments
We thank Rob Kay and John Nichols for invaluable help with the SILAC method and David Martin for early discussions. We acknowledge the PRIDE team for the deposition of our data to the ProteomeXchange Consortium. This work was supported by a Wellcome Trust program grant to J.G.W.
Abbreviations used:
- AL
activation loop
- AMPK
AMP-activated protein kinase
- DIF-1
differentiation-inducing factor-1
- GAP
guanine-activating protein
- GEF
guanine exchange factor
- GO
Gene Ontology
- HM
hydrophobic motif
- LC-MS/MS
liquid chromatography tandem mass spectrometry
- PKB
protein kinase B
- PKBR1
protein kinase B–related protein 1
- RGS
regulator of G protein signaling
- SILAC
stable isotope labeling by amino acids in cell culture
- TORC2
target of rapamycin complex 2
- TPM
temporal pattern mining.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-08-1319) on December 17, 2014.
REFERENCES
- Anjard C, Loomis WF. Evolutionary analyses of ABC transporters of Dictyostelium discoideum. Eukaryot Cell. 2002;1:643–652. doi: 10.1128/EC.1.4.643-652.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anjard C, Zeng C, Loomis WF, Nellen W. Signal transduction pathways leading to spore differentiation in Dictyostelium discoideum. Dev Biol. 1998;193:146–155. doi: 10.1006/dbio.1997.8804. [DOI] [PubMed] [Google Scholar]
- Araki T, Kawata T, Williams JG. Identification of the kinase that activates a nonmetazoan STAT gives insights into the evolution of phosphotyrosine-SH2 domain signaling. Proc Natl Acad Sci USA. 2012;109:E1931–E1937. doi: 10.1073/pnas.1202715109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki T, Langenick J, Gamper M, Firtel RA, Williams JG. Evidence that DIF-1 and hyper-osmotic stress activate a Dictyostelium STAT by inhibiting a specific protein tyrosine phosphatase. Development. 2008;135:1347–1353. doi: 10.1242/dev.009936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki T, van Egmond WN, van Haastert PJ, Williams JG. Dual regulation of a Dictyostelium STAT by cGMP and Ca2+ signalling. J Cell Sci. 2010;123:837–841. doi: 10.1242/jcs.064436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki T, Williams JG. Perturbations of the actin cytoskeleton activate a Dictyostelium STAT signalling pathway. Eur J Cell Biol. 2012;91:420–425. doi: 10.1016/j.ejcb.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgrass R, Weiwad M, Erdmann F, Liu JO, Wunderlich D, Grabley S, Fischer G. Reversible inhibition of calcineurin by the polyphenolic aldehyde gossypol. J Biol Chem. 2001;276:47914–47921. doi: 10.1074/jbc.M103273200. [DOI] [PubMed] [Google Scholar]
- Berks M, Kay RR. Cyclic AMP is an inhibitor of stalk cell differentiation in Dictyostelium discoideum. Dev Biol. 1988;126:108–114. doi: 10.1016/0012-1606(88)90244-8. [DOI] [PubMed] [Google Scholar]
- Berks M, Kay RR. Combinatorial control of cell differentiation by cAMP and DIF-1 during development of Dictyostelium discoideum. Development. 1990;110:977–984. doi: 10.1242/dev.110.3.977. [DOI] [PubMed] [Google Scholar]
- Boeckeler K, Tischendorf G, Mutzel R, Weissenmayer B. Aberrant stalk development and breakdown of tip dominance in Dictyostelium cell lines with RNAi-silenced expression of calcineurin B. BMC Dev Biol. 2006;6:12. doi: 10.1186/1471-213X-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle EI, Weng S, Gollub J, Jin H, Botstein D, Cherry JM, Sherlock G. GO::TermFinder–open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics. 2004;20:3710–3715. doi: 10.1093/bioinformatics/bth456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner M, Thoms SD. Caffeine blocks activation of cyclic AMP synthesis in Dictyostelium discoideum. Dev Biol. 1984;101:136–146. doi: 10.1016/0012-1606(84)90124-6. [DOI] [PubMed] [Google Scholar]
- Breshears LM, Wessels D, Soll DR, Titus MA. An unconventional myosin required for cell polarization and chemotaxis. Proc Natl Acad Sci USA. 2010;107:6918–6923. doi: 10.1073/pnas.0909796107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzostowski JA, Sawai S, Rozov O, Liao XH, Imoto D, Parent CA, Kimmel AR. Phosphorylation of chemoattractant receptors regulates chemotaxis, actin reorganization and signal relay. J Cell Sci. 2013;126:4614–4626. doi: 10.1242/jcs.122952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustelo XR. Regulatory and signaling properties of the Vav family. Mol Cell Biol. 2000;20:1461–1477. doi: 10.1128/mcb.20.5.1461-1477.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Devreotes PN. Moving in the right direction: how eukaryotic cells migrate along chemical gradients. Semin Cell Dev Biol. 2011;22:834–841. doi: 10.1016/j.semcdb.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Katoh-Kurasawa M, Muramoto T, Santhanam B, Long Y, Li L, Ueda M, Iglesias PA, Shaulsky G, Devreotes PN. Nucleocytoplasmic shuttling of a GATA transcription factor functions as a development timer. Science. 2014;343:1329–1339. doi: 10.1126/science.1249531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carruthers NJ, Dowd MK, Stemmer PM. Gossypol inhibits calcineurin phosphatase activity at multiple sites. Eur J Pharmacol. 2007;555:106–114. doi: 10.1016/j.ejphar.2006.10.046. [DOI] [PubMed] [Google Scholar]
- Catalano A, O'Day DH. Calmodulin-binding proteins in the model organism Dictyostelium: a complete & critical review. Cell Signal. 2008;20:277–291. doi: 10.1016/j.cellsig.2007.08.017. [DOI] [PubMed] [Google Scholar]
- Chen ZH, Schaap P. The prokaryote messenger c-di-GMP triggers stalk cell differentiation in Dictyostelium. Nature. 2012;488:680–683. doi: 10.1038/nature11313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Segall JE. EppA; a putative substrate of DdERK2; regulates cyclic AMP relay and chemotaxis in Dictyostelium discoideum. Eukaryot Cell. 2006;5:1136–1146. doi: 10.1128/EC.00383-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Cox J, Mann M. Quantitative, high-resolution proteomics for data-driven systems biology. Annu Rev Biochem. 2011;80:273–299. doi: 10.1146/annurev-biochem-061308-093216. [DOI] [PubMed] [Google Scholar]
- Cubitt AB, Firtel RA, Fischer G, Jaffe LF, Miller AL. Patterns of free calcium in multicellular stages of Dictyostelium expressing jellyfish apoaequorin. Development. 1995;121:2291–2301. doi: 10.1242/dev.121.8.2291. [DOI] [PubMed] [Google Scholar]
- de la Roche MA, Cote GP. Regulation of Dictyostelium myosin I and II. Biochim Biophys Acta. 2001;1525:245–261. doi: 10.1016/s0304-4165(01)00110-6. [DOI] [PubMed] [Google Scholar]
- Egelhoff TT, Lee RJ, Spudich JA. Dictyostelium myosin heavy chain phosphorylation sites regulate myosin filament assembly and localization in vivo. Cell. 1993;75:363–371. doi: 10.1016/0092-8674(93)80077-r. [DOI] [PubMed] [Google Scholar]
- Eichinger L, Pachebat JA, Glockner G, Rajandream MA, Sucgang R, Berriman M, Song J, Olsen R, Szafranski K, Xu Q, et al. The genome of the social amoeba Dictyostelium discoideum. Nature. 2005;435:43–57. doi: 10.1038/nature03481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- Francione LM, Annesley SJ, Carilla-Latorre S, Escalante R, Fisher PR. The Dictyostelium model for mitochondrial disease. Semin Cell Dev Biol. 2011;22:120–130. doi: 10.1016/j.semcdb.2010.11.004. [DOI] [PubMed] [Google Scholar]
- Fukuzawa M, Abe T, Williams JG. The Dictyostelium prestalk cell inducer DIF regulates nuclear accumulation of a STAT protein by controlling its rate of export from the nucleus. Development. 2003;130:797–804. doi: 10.1242/dev.00303. [DOI] [PubMed] [Google Scholar]
- Fukuzawa M, Araki T, Adrian I, Williams JG. Tyrosine phosphorylation-independent nuclear translocation of a Dictyostelium STAT in response to DIF signaling. Mol Cell. 2001;7:779–788. doi: 10.1016/s1097-2765(01)00222-2. [DOI] [PubMed] [Google Scholar]
- Griffith LM, Downs SM, Spudich JA. Myosin light chain kinase and myosin light chain phosphatase from Dictyostelium: effects of reversible phosphorylation on myosin structure and function. J Cell Biol. 1987;104:1309–1323. doi: 10.1083/jcb.104.5.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross JD. Acidic Ca2+ stores, excitability and cell patterning in Dictyostelium discoideum. Eukaryot Cell. 2009;8:696–702. doi: 10.1128/EC.00360-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 2011;25:1895–1908. doi: 10.1101/gad.17420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath RJ, Insall RH. Dictyostelium MEGAPs: F-BAR domain proteins that regulate motility and membrane tubulation in contractile vacuoles. J Cell Sci. 2008;121:1054–1064. doi: 10.1242/jcs.021113. [DOI] [PubMed] [Google Scholar]
- Hereld D, Vaughan R, Kim JY, Borleis J, Devreotes P. Localization of ligand-induced phosphorylation sites to serine clusters in the C-terminal domain of the Dictyostelium cAMP receptor, cAR1. J Biol Chem. 1994;269:7036–7044. [PubMed] [Google Scholar]
- Horn F, Gross J. A role for calcineurin in Dictyostelium discoideum development. Differentiation. 1996;60:269–275. doi: 10.1046/j.1432-0436.1996.6050269.x. [DOI] [PubMed] [Google Scholar]
- Howard PK, Sefton BM, Firtel RA. Tyrosine phosphorylation of actin in Dictyostelium associated with cell-shape changes. Science. 1993;259:241–244. doi: 10.1126/science.7678470. [DOI] [PubMed] [Google Scholar]
- Huang EY, Blagg SL, Keller T, Katoh M, Shaulsky G, Thompson CRL. bZlP transcription factor interactions regulate DIF responses in Dictyostelium. Development. 2006;133:449–458. doi: 10.1242/dev.02240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insall R, Kay RR. A specific DIF binding protein in Dictyostelium. EMBO J. 1990;9:3323–3328. doi: 10.1002/j.1460-2075.1990.tb07532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon TJ, Lee S, Weeks G, Firtel RA. Regulation of Dictyostelium morphogenesis by RapGAP3. Dev Biol. 2009;328:210–220. doi: 10.1016/j.ydbio.2009.01.016. [DOI] [PubMed] [Google Scholar]
- Juvvadi PR, Gehrke C, Fortwendel JR, Lamoth F, Soderblom EJ, Cook EC, Hast MA, Asfaw YG, Moseley MA, Creamer TP, Steinbach WJ. Phosphorylation of Calcineurin at a novel serine-proline rich region orchestrates hyphal growth and virulence in Aspergillus fumigatus. PLoS Pathog. 2013;9:e1003564. doi: 10.1371/journal.ppat.1003564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamimura Y, Xiong Y, Iglesias PA, Hoeller O, Bolourani P, Devreotes PN. PIP3-independent activation of TorC2 and PKB at the cell's leading edge mediates chemotaxis. Curr Biol. 2008;18:1034–1043. doi: 10.1016/j.cub.2008.06.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawata T, Shevchenko A, Fukuzawa M, Jermyn KA, Totty NF, Zhukovskaya NV, Sterling AE, Mann M, Williams JG. SH2 signaling in a lower eukaryote: a STAT protein that regulates stalk cell differentiation in dictyostelium. Cell. 1997;89:909–916. doi: 10.1016/s0092-8674(00)80276-7. [DOI] [PubMed] [Google Scholar]
- Kay RR. The biosynthesis of differentiation-inducing factor, a chlorinated signal molecule regulating Dictyostelium development. J Biol Chem. 1998;273:2669–2675. doi: 10.1074/jbc.273.5.2669. [DOI] [PubMed] [Google Scholar]
- Keller T, Thompson CR. Cell type specificity of a diffusible inducer is determined by a GATA family transcription factor. Development. 2008;135:1635–1645. doi: 10.1242/dev.020883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessen U, Schaloske R, Aichem A, Mutzel R. Ca2+/calmodulin-independent activation of calcineurin from Dictyostelium by unsaturated long chain fatty acids. J Biol Chem. 1999;274:37821–37826. doi: 10.1074/jbc.274.53.37821. [DOI] [PubMed] [Google Scholar]
- Kim JY, Soede RDM, Schaap P, Valkema R, Borleis JA, van Haastert PJM, Devreotes PN, Hereld D. Phosphorylation of chemoattractant receptors is not essential for chemotaxis or termination of G-protein-mediated responses. J Biol Chem. 1997;272:27313–27318. doi: 10.1074/jbc.272.43.27313. [DOI] [PubMed] [Google Scholar]
- King JS, Insall RH. Chemotaxis: finding the way forward with Dictyostelium. Trends Cell Biol. 2009;19:523–530. doi: 10.1016/j.tcb.2009.07.004. [DOI] [PubMed] [Google Scholar]
- Kortholt A, Keizer-Gunnink I, Kataria R, Van Haastert PJ. Ras activation and symmetry breaking during Dictyostelium chemotaxis. J Cell Sci. 2013;126:4502–4513. doi: 10.1242/jcs.132340. [DOI] [PubMed] [Google Scholar]
- Kortholt A, van Haastert PJ. Highlighting the role of Ras and Rap during Dictyostelium chemotaxis. Cell Signal. 2008;20:1415–1422. doi: 10.1016/j.cellsig.2008.02.006. [DOI] [PubMed] [Google Scholar]
- Kosaka C, Pears CJ. Chemoattractants induce tyrosine phosphorylation of ERK2 in Dictyostelium discoideum by diverse signalling pathways. Biochem J. 1997;324:347–352. doi: 10.1042/bj3240347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kume K, Koyano T, Kanai M, Toda T, Hirata D. Calcineurin ensures a link between the DNA replication checkpoint and microtubule-dependent polarized growth. Nat Cell Biol. 2011;13:234–242. doi: 10.1038/ncb2166. [DOI] [PubMed] [Google Scholar]
- Kuwayama H, Kubohara Y. Differentiation-inducing factor-1 and -2 function also as modulators for Dictyostelium chemotaxis. PLoS One. 2009;4:e6658. doi: 10.1371/journal.pone.0006658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwayama H, Oyama M, Kubohara Y, Maeda M. A novel role of differentiation-inducing factor-1 in Dictyostelium development, assessed by the restoration of a developmental defect in a mutant lacking mitogen-activated protein kinase ERK2. Dev Growth Differ. 2000;42:531–538. doi: 10.1046/j.1440-169x.2000.00537.x. [DOI] [PubMed] [Google Scholar]
- Liao XH, Buggey J, Kimmel AR. Chemotactic activation of Dictyostelium AGC-family kinases AKT and PKBR1 requires separate but coordinated functions of PDK1 and TORC2. J Cell Sci. 2010;123:983–992. doi: 10.1242/jcs.064022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim WA, Pawson T. Phosphotyrosine signaling: evolving a new cellular communication system. Cell. 2010;142:661–667. doi: 10.1016/j.cell.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda M, Aubry L, Insall R, Gaskins C, Devreotes PN, Firtel RA. Seven helix chemoattractant receptors transiently stimulate mitogen-activated protein kinase in Dictyostelium—role of heterotrimeric G proteins. J Biol Chem. 1996;271:3351–3354. doi: 10.1074/jbc.271.7.3351. [DOI] [PubMed] [Google Scholar]
- McConnell RE, Tyska MJ. Leveraging the membrane-cytoskeleton interface with myosin-1. Trends Cell Biol. 2010;20:418–426. doi: 10.1016/j.tcb.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meggio F, Pinna LA. One-thousand-and-one substrates of protein kinase CK2. FASEB J. 2003;17:349–368. doi: 10.1096/fj.02-0473rev. [DOI] [PubMed] [Google Scholar]
- Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127:635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- Qualmann B, Koch D, Kessels MM. Let's go bananas: revisiting the endocytic BAR code. EMBO J. 2011;30:3501–3515. doi: 10.1038/emboj.2011.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusnak F, Mertz P. Calcineurin: form and function. Physiol Rev. 2000;80:1483–1521. doi: 10.1152/physrev.2000.80.4.1483. [DOI] [PubMed] [Google Scholar]
- Saeed F, Pisitkun T, Knepper M, Hoffert JD. Mining temporal patterns from iTRAQ mass spectrometry (LC-MS/MS) data. In: Al-Mubaid H, editor. In: 3rd International Conference on Bioinformatics and Computational Biology. Winona, MN: International Society for Computers and Their Applications; 2011. pp. 152–159. [Google Scholar]
- Saito T, Kato A, Kay RR. DIF-1 induces the basal disc of the Dictyostelium fruiting body. Dev Biol. 2008;317:444–453. doi: 10.1016/j.ydbio.2008.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaap P, Nebl T, Fisher PR. A slow sustained increase in cytosolic Ca2 +levels mediates stalk gene induction by differentiation inducing factor in Dictyostelium. EMBO J. 1996;15:5177–5183. [PMC free article] [PubMed] [Google Scholar]
- Schwartz D, Gygi SP. An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat Biotechnol. 2005;23:1391–1398. doi: 10.1038/nbt1146. [DOI] [PubMed] [Google Scholar]
- Serge A, de Keijzer S, Van Hemert F, Hickman MR, Hereld D, Spaink HP, Schmidt T, Snaar-Jagalska BE. Quantification of GPCR internalization by single-molecule microscopy in living cells. Integr Biol (Camb) 2011;3:675–683. doi: 10.1039/c0ib00121j. [DOI] [PubMed] [Google Scholar]
- Sobczyk GJ, Wang J, Weijer CJ. SILAC-based proteomic quantification of chemoattractant-induced cytoskeleton dynamics on a second to minute timescale. Nat Commun. 2014;5:3319. doi: 10.1038/ncomms4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugden C, Ross S, Bloomfield G, Ivens A, Skelton J, Mueller-Taubenberger A, Williams JG. Two novel Src homology 2 domain proteins interact to regulate dictyostelium gene expression during growth and early development. J Biol Chem. 2010;285:22927–22935. doi: 10.1074/jbc.M110.139733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M, Iijima M, Kamimura Y, Chen L, Long Y, Devreotes P. Disruption of PKB signaling restores polarity to cells lacking tumor suppressor PTEN. Mol Biol Cell. 2011;22:437–447. doi: 10.1091/mbc.E10-06-0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thewes S, Schubert SK, Park K, Mutzel R. Stress and development in Dictyostelium discoideum: the involvement of the catalytic calcineurin A subunit. J Basic Microbiol. 2013;54:607–613. doi: 10.1002/jobm.201200574. [DOI] [PubMed] [Google Scholar]
- Thompson CRL, Fu Q, Buhay C, Kay RR, Shaulsky G. A bZIP/bRLZ transcription factor required for DIF signaling in Dictyostelium. Development. 2004;131:513–523. doi: 10.1242/dev.00939. [DOI] [PubMed] [Google Scholar]
- Thompson CRL., Kay RR. The role of DIF-1 signaling in Dictyostelium development. Mol Cell. 2000;6:1509–1514. doi: 10.1016/s1097-2765(00)00147-7. [DOI] [PubMed] [Google Scholar]
- Tsiavaliaris G, Fujita-Becker S, Durrwang U, Diensthuber RP, Geeves MA, Manstein DJ. Mechanism, regulation, and functional properties of Dictyostelium myosin-1B. J Biol Chem. 2008;283:4520–4527. doi: 10.1074/jbc.M708113200. [DOI] [PubMed] [Google Scholar]
- Tsujioka M, Yamamoto T, Thompson CR, Kay RR, Maeda M. Novel development rescuing factors (DRFs) secreted by the developing Dictyostelium cells, that are involved in the restoration of a mutant lacking MAP-kinase ERK2. Zool Sci. 2004;21:829–834. doi: 10.2108/zsj.21.829. [DOI] [PubMed] [Google Scholar]
- Ubersax JA, Ferrell JE., Jr Mechanisms of specificity in protein phosphorylation. Nat Rev Mol Cell Biol. 2007;8:530–541. doi: 10.1038/nrm2203. [DOI] [PubMed] [Google Scholar]
- Ulintz PJ, Yocum AK, Bodenmiller B, Aebersold R, Andrews PC, Nesvizhskii AI. Comparison of MS(2)-only, MSA, and MS(2)/MS(3) methodologies for phosphopeptide identification. J Proteome Res. 2009;8:887–899. doi: 10.1021/pr800535h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaillancourt JP, Lyons C, Côté GP. Identification of two phosphorylated threonines in the tail region of Dictyostelium myosin II. J Biol Chem. 1988;263:10082–10087. [PubMed] [Google Scholar]
- Veltman DM, King JS, Machesky LM, Insall RH. SCAR knockouts in Dictyostelium: WASP assumes SCAR's position and upstream regulators in pseudopods. J Cell Biol. 2012;198:501–508. doi: 10.1083/jcb.201205058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlahou G, Rivero F. Rho GTPase signaling in Dictyostelium discoideum: insights from the genome. Eur J Cell Biol. 2006;85:947–959. doi: 10.1016/j.ejcb.2006.04.011. [DOI] [PubMed] [Google Scholar]
- Wang M, van Haastert PJM, Schaap P. Multiple effects of differentiation-inducing factor on prespore differentiation and cyclic-AMP signal transduction in Dictyostelium. Differentiation. 1986;33:24–28. [Google Scholar]
- Weissenmayer B, Boeckeler K, Lahrz A, Mutzel R. The calcineurin inhibitor gossypol impairs growth, cell signalling and development in Dictyostelium discoideum. FEMS Microbiol Lett. 2005;242:19–25. doi: 10.1016/j.femsle.2004.10.035. [DOI] [PubMed] [Google Scholar]
- Wilson-Grady JT, Villen J, Gygi SP. Phosphoproteome analysis of fission yeast. J Proteome Res. 2008;7:1088–1097. doi: 10.1021/pr7006335. [DOI] [PubMed] [Google Scholar]
- Wurster B, Kay RR. New roles for DIF? Effects on early development in Dictyostelium. Dev Biol. 1990;140:189–195. doi: 10.1016/0012-1606(90)90066-r. [DOI] [PubMed] [Google Scholar]
- Yamada Y, Kubohara Y, Kikuchi H, Oshima Y, Wang HY, Ross S, Williams JG. The Dictyostelium prestalk inducer DIF-1 directs phosphorylation of a bZIP transcription factor. Int J Dev Biol. 2013;57:375–381. doi: 10.1387/ijdb.130046jw. [DOI] [PubMed] [Google Scholar]
- Yamada Y, Nunez-Corcuera B, Williams JG. DIF-1 regulates Dictyostelium basal disc differentiation by inducing the nuclear accumulation of a bZIP transcription factor. Dev Biol. 2011;354:77–86. doi: 10.1016/j.ydbio.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Mihaylov V, Xu X, Brzostowski JA, Li H, Liu L, Veenstra TD, Parent CA, Jin T. A Gbetagamma effector, ElmoE, transduces GPCR signaling to the actin network during chemotaxis. Dev Cell. 2012;22:92–103. doi: 10.1016/j.devcel.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhukovskaya NV, Fukuzawa M, Yamada Y, Araki T, Williams JG. The Dictyostelium bZIP transcription factor DimB regulates prestalk-specific gene expression. Development. 2006;133:439–448. doi: 10.1242/dev.02190. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.