

Abstract
Human Insulin-like growth factor 1 (hIGF-1) consists of 70 amino acids in a single chain with three intermolecular disulfide bridges possessing valuable therapeutic effects. To date, numerous variants of specifically engineered hIGF-1 have been produced so as to improve hIGF-1 biological activity, stability and stronger binding to IGF-1 receptor. Mecasermin is one of the modified variants with one amino acid substitution near the N-terminal (T4I) approved for the treatment of growth failure diabetes, wound healing, amyotrophic lateral sclerosis and severe primary IGF-1 deficiency. No scientific report for recombinant production of mecasermin in Escherichia coli (E. coli) expression system has been sofar reported. In the present study, we therefore investigated the overexpression of mecasermin in two different E. coli strains in order to obtain higher yield of recombinant protein. To achieve this goal, mecasermin DNA encoding sequence was designed based on polypeptide sequence, optimized according to E. coli codon preference, and cloned in pET15b. Recombinant vector, pET15-mecasermin, transferred into two E. coli strains rigami B (DE3) and BL21 (DE3) and induced for expression in a small scale. Results revealed the E. coli Origami B (DE3) expression system was a preferable host for mecasermin production due to its high expression level being around twice as much as BL21 (DE3). Large scale mecasermin production was performed in batch culture and produced recombinant protein specifically confirmed by western blotting and mass spectroscopy. Since major part of recombinant mecasermin was expressed as inclusion body, isolation and refolding was accomplished through developed purification procedure, and finally recombinant protein was successfully purified by gel filtration chromatography.
Keywords: Mecasermin, Insulin-like growth factor-1, Over-expression, Inclusion body, Purification, Western blotting
INTRODUCTION
Human insulin-like growth factor 1 (hIGF-1) is a 70 amino acid non-glycosylated protein hormone with three intra-molecular disulfide bridges bonds. Because of some biological activities, hIGF-1 is considered for different medical applications (1). Several companies have evaluated native hIGF-1 in clinical trials for a variety of additional indications including type I and II diabetes, amyotrophic lateral sclerosis (ALS aka “Lou Gehrig's Disease”) (2), severe burn injury and myotonic muscular dystrophy (MMD). The clinical trials consequences for IGF-1 therapeutic effect on type I diabetes and type II diabetes showed significant promise in reducing hemoglobin A1C levels, as well as daily insulin consumption (3). The first report relevant to use of recombinant native human IGF-1 therapy in severe insulin resistance was in the early 1990s and largely involved the treatment of patients with type A syndrome (4). In 1991, Schoenle and coworkers were the first to demonstrate that a bolus injection of recombinant native hIGF-I could reduce both insulin and blood glucose levels in patients with severe insulin resistance (4).
Up to now, different variants of engineered recombinant hIGF-1 (rhIGF-1) have been developed to improve hIGF-1 biological activity and drug stability by amino acid substitution in different polypeptide position (5,6,7,8). Mecasermin (Increlex™) is a recombinant variant of hIGF-1 which is approved for the treatment of growth failure diabetes, wound healing and amyotrophic lateral sclerosis and severe primary IGF-1 deficiency (8,9). A company licensed by Genentech (Tercica Inc., Brisbane, CA, USA) obtained orphan drug approval of its rhIGF-I (Increlex™, mecasermin) from the US Food and Drug Administration (FDA) in late 2005 (1,9,10). Increlex as a recombinant form of hIGF-1 has a single chain polypeptide harboring substitution of threonine amino acid at the position 4 with an isoleucine. The sequence modification refines interaction between mecasermin and IGF binding protein-3 (IGFBP3) which leads to a higher stability of IGF-1 in blood. The manipulated sequence of mecasermin divulges higher affinity to IGF-1 receptor, lower tendency to aggregated forms, and consequently reveals more capability as a pharmaceutical product (8,11,12,13,14).
The studies show a wide range of expression systems have been established to produce native hIGF-1 including yeast (15,16), cell-free system (17), transgenic plants, and rabbits (18,19,20); whereas, it is found that the productivity of native hIGF-1 in E. coli is more than other expression systems (21,22,23,24,25).
To our knowledge, there is no scientific report for recombinant production of mecasermin as the FDA approved of rhIGF-1 variant. In this study, we investigated the expression efficiency and inclusion body formation of mecasermin in E. coli expression system. So as to reach maximum expression two different E. coli expression hosts, Origami B (DE3) and BL21 (DE3), were compared. Mecasermin encoding sequence designed according to approved amino acid sequence and commercially constructed in puc57 cloning vector. Coding sequence subsequently subcloned in NcoI frame in pET115 expression vector and then transformed into the expression hosts. The over-expressed recombinant protein in inclusion bodies form was efficiently refolded and subsequently purified through the gel filtration column. The produced recombinant mecasermin was specifically confirmed by western blotting and mass spectroscopy.
MATERIALS AND METHODS
Materials
Antibodies were from Santa Cruz Biotechnology Inc, USA. All restriction enzymes and T4-DNA ligase were purchased from Thermo Scientific, USA. Bacterial strains (E. coli DH5α, Origami B and BL21) and pET15 expression vector were provided by pasture institute, Iran. All chemicals were procured from Sigma (Germany). Gene synthesis and DNA sequencing were performed by GeneScript Inc (USA) and Macrogen Inc. (Korea) respectively. Protein marker was purchased from Santa Cruz Biotechnology Inc, USA. DNA standard size marker was from Thermo Scientific.
Mecasermin DNA optimization
Mecasermin DNA encoding sequence was designed based on FDA approved polypeptide sequence (Drug Bank accession no. DB01277). Codon optimization was performed according to E. coli codon preference. The designed DNA sequence was commercially synthesized in puc57 cloning vector between NcoI and BamHI restriction sites by GeneScript Inc. (USA).
pET15-mecasermin construction
Recombinant puc57 vector was digested by NcoI and BamHI restriction enzymes (Thermo Scientific, USA) according to manufacturer. Released fragment was excised from agarose gel and purified using DNA extraction kit (Thermo Scientific, USA). Purified DNA fragment was ligated to pET15b expression vector linearized with NcoI and BamHI restriction enzymes to give pET15-mecasermin according to Maniaties and coworkers (26).
The construct was transformed into E. coli DH5α and sequenced in both strand. The confirmed plasmid was extracted from DH5α according to alkaline lysis method (26) and transformed into Origami B (DE3) and BL21 (DE3) expression hosts.
Protein expression
Transformants were grown overnight in 5 mL Luria-Bertani medium in 200 mL flask to an OD600 of around 0.7. Both E. coli BL21 and Origami B cultures were supplemented with 100 μM ampicillin. Additionally, 12.5 μM tetracycline was applied for Origami B (DE3). The bacterial cultures were induced by 100 μM isopropyl-beta-D-thiogalactopyranoside (IPTG) and allowed to grow for further 4 h at 37 °C. Cells were harvested by centrifugation at 7000 g for 5 min in 4 °C and kept at -20 °C until use.
Batch cultivation
To prepare the inoculum, a bacterial culture containing the pET15-mecasermin expression vector was inoculated into 200 mL of Lauria-Bertani (LB) culture medium containing 100 μM ampicillin and grown overnight at 37 °C. The inoculum was transferred into a 2.3-L BioFlo 110 fermenter (New Brunswick, USA) containing 1300 ml of sterilized Terrific Broth (TB) medium and 100 μM of ampicillin. The inoculated fermenter was maintained at pH 7.0 and 37 °C with agitation at 500 rpm and 1.1 min air. Dissolved oxygen was controlled at 30-40% of air saturation. To control foaming during the process silicon oil was applied. The culture was induced at OD600 of 1 with the addition of 100 μM IPTG. The fermentation was stopped 4 h post-induction and cells were harvested from the culture with centrifugation at 7000 g for 5 min.
Protein extraction and refolding
To isolate the cytoplasmic insoluble protein the fermented broth was centrifuged at 8000 g for 30 min at 4 °C and the pellet was washed twice with 50 mM phosphate buffer pH 7.4. The wet cells (4 g) were resuspended in 20 ml of lysis buffer (50 mM Tris–HCl containing 1 mM EDTA, 1 mM PMSF). The cells were broken using homogenizer (NIRO-SOAVI) at 1200 bar four times at 4 °C. The disrupted cells were centrifuged at 6000 g for 30 min at 4 °C. The supernatant was discarded and the pellet was washed twice with washing buffer (1 M urea in 50 mM Tris–HCl pH 8.0 containing 5 mM EDTA and 1 mM PMSF) for 40 min at room temperature and centrifuged for 15 min at 11000 g at 25 °C.
To solubilized washed inclusion bodies, solubilization urea buffer (30 mM Tris/HCl, 1 mM EDTA, 100 mM GSH, 8 M Urea, pH:12) was used. Initially, 50 mg inclusion bodies were dissolved in 3 ml buffer and incubated for 12 h at room temperature and then centrifuged for 20 min at room temperature.
To refold solubilized protein, 6 ml of refolding buffer (30 mM Tris / HCl (pH 5), 2 mM GSSG-2 mM GSH, 3 M urea) was gradually added to the solution and protein concentration was reached 5 mg/ml. After refolding, the pH of the resulting suspension was adjusted to 5 by 2 mM citric acid and incubated for two h. The sample was centrifuged at 9000 g for 20 min at 4 °C and filtered (0.22 μm) before chromatography to remove cellular debris and aggregated proteins.
Purification
Extracted refolded protein was purified by gel filtration chromatography and loaded onto Hi-Load 16/600 Superdex 75 prep grade column. The column was washed with 150 ml buffer (50 mM Sodium acetate, 100 mM NaCl, pH 5.4) and flow rate of 1 ml/min at room temperature. The Bradford assay was used for protein quantification at 595 nm.
The analysis of protein fractions were performed on 16% Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Samples were resuspended in 1 × protein loading buffer (60 mM Tris-HCl, 2% SDS, Glycerol 10 %, 0.01 % bromophenol blue, 5 mM DTT) and gels were stained with coomassie Brilliant Blue R-250 (26).
Western blotting
Western blot analysis was performed using IGF-1 polyclonal antibody (Cat. Nr: Sc-9013, Santa Cruz Biotechnology Inc.) as primary antibody, Goat anti-rabbit antibody (IgG-HRP: Cat. Nr: sc-2030, Santa Cruz Biotechnology Inc.) as secondary antibody and DAB, 50X: (Cat. Nr: sc-24982, Santa Cruz Biotechnology Inc.). SDS-PAGE migrated protein bands was transferred into poly-vinylidene fluoride (PVDF) membrane (Roche Diagnostic, USA) in order to identify the accurate production of mecasermin in E. coli. The first incubation of membrane was accomplished with bovine serum albumin (BSA) 3% (w/v) in tris-buffered saline and Tween 20 (TBS-T) solutions for one h to block the membrane. The second incubation was held by anti-IGF-1 polyclonal antibody (Santa Cruz Biotechnology, USA) added at a dilution of 1:500 in TBS-T solution. Anti-IgG (Santa Cruz Biotechnology) at a dilution of 1:1000 in TBS-T solution was used in third incubation. Subsequently incubation was performed with diamino-benzidine solution (DAB; Santa Cruz Biotechnology) for 5 min (0.5 mg/ml DAB, 0.1% H2O2).
Mass spectrometry
Mass spectrometry was carried out by electrospray soft ionization in the positive mode with a linear ion trap mass spectrometer (Shimadzu- 2010 EV, Japan). Measurements were done by selective reaction monitoring of the transitions from [M+6]6+ 1278.
RESULTS
Gene design and construction of mecasermin
Mecasermin DNA encoding sequence was designed based on FDA approved polypeptide sequence (Drug Bank accession no. DB01277). Codon optimization was performed according to E. coli codon preference and open reading frame (ORF) was inserted between NcoI and BamHI in pET15b restriction site. We used ATG sequence in NcoI site as start codon and designed TAA sequence before BamHI as stop codon.
At first pET15-mecasermin sequence was verified by double digestion of purified plasmid using NcoI and BamHI restriction enzymes. Digested fragment was analyzed by agarose gel electrophoresis (Fig. 1) and results confirmed the length of cloned fragment (219 bp). Finally, the sequence of the insert was determined on both strands.
Fig. 1.
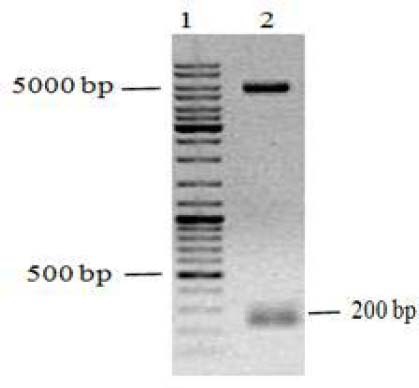
Agarose gel analysis for verification of cloned fragment (mecasermin DNA encoding sequence) in pET15b vector by restriction enzyme digestion. Lane 1; DNA size marker, lane 2; enzymatic digestion of pET15-mecasermin by NcoI and BamHI.
Small scale expression
The expression of mecasermin polypeptide was investigated in two strains of E. coli including Bl21 (DE3) and Origami B (DE3). The recombinant polypeptide was effectively expressed in both strains of the E. coli transformants after induction with 100 μM IPTG.
The theoretical molecular mass of mecasermin without initial methionine was 7.6 kDa. SDS-PAGE analysis for equivalent amount of two culture broth showed a prominent polypeptide band in agreement with the expected molecular mass (Fig. 2, lane 2 and 3). However, on the basis of SDS-PAGE results, the intensity of band corresponding to recombinant mecasermin in Origami B (DE3) was significantly more than the produced mecasermin in Bl21 (DE3) which indicated higher level expression (Fig. 2).
Fig. 2.
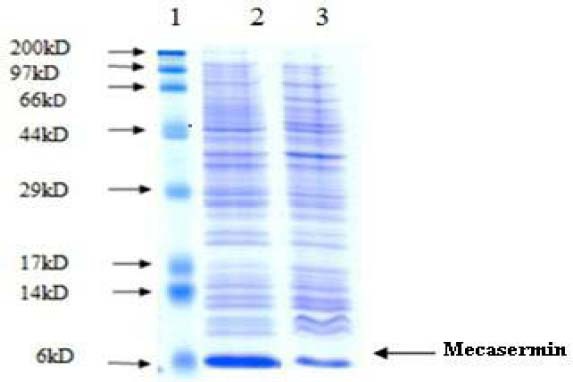
Sodium dodecyl sulfate polyacrylamide gel electrophoresis analysis for overexpression of mecasermin in E. coli Origami B and BL21. Lane1; Molecular weight marker, lane 2; total protein after addition of isopropyl-beta-D-thiogalactopyranoside in Origami B (DE3) transformant, lane 3; total protein after addition of IPTG in BL21 (DE3) transformant.
However, on the basis of SDS-PAGE results, the intensity of band corresponding to recombinant mecasermin in Origami B (DE3) was significantly more than the produced mecasermin in Bl21 (DE3) which indicated higher level expression (Fig. 2). Also, western blotting analysis confirmed specifically produced mecasermin in E. coli (Fig. 3). Therefore, in this study Origami B (DE3) strain was recognized as efficient host to produce mecasermin and used for next steps.
Fig. 3.
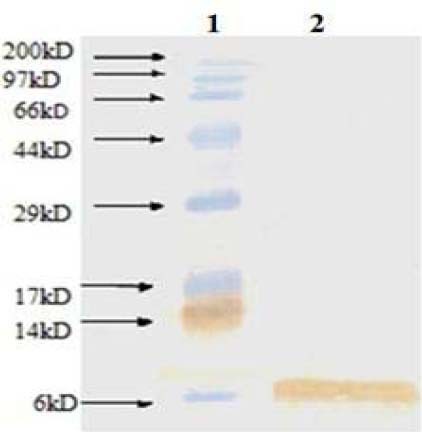
Western blotting of recombinant mecasermin. Lane 1; molecular weight marker, lane 2; purified mecasermin.
Batch fermentation
To achieve high amount of recombinant protein, batch fermentation was done by final recombinant Origami B strain harboring pET15-mecasermin. Specific growth rate and dry cell weight of desired strain were calculated during fermentation process. In this process, the final dry cell density reached 1.4 g/L in 5 h fermentation (Fig. 4). Also, SDS- PAGE analysis well confirmed the high level expression of mecasermin in batch cultivation (Fig. 5).
Fig. 4.
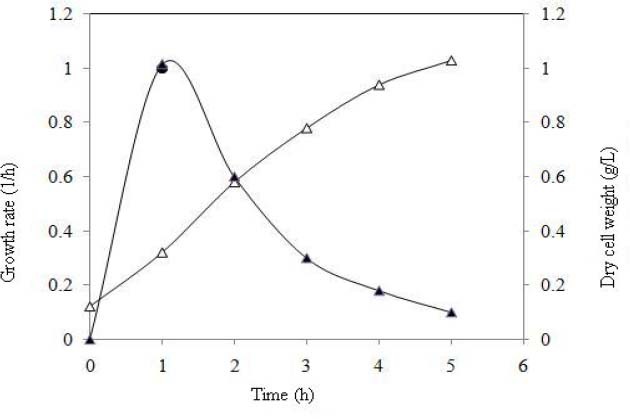
Specific growth rate and dry cell weight curves of Origami B harboring pET15-mecasermin in batch culture. Δ) Dry cell weight, (▲) growth rate.
Fig. 5.
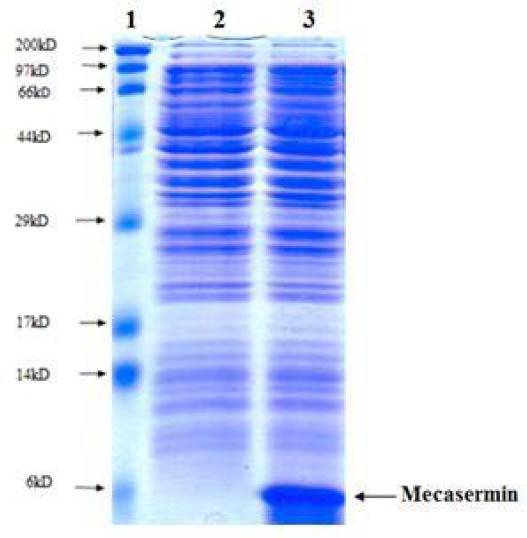
Sodium dodecyl sulfate polyacrylamide gel electrophoresis analysis for over-expression of mecasermin in batch culture. Lane 1; molecular weight marker, lane 2; total protein from Origami B harboring pET15-mecasermin before addition of isopropyl-beta-D-thiogalactopyranoside, lane 3; total protein from Origami B harboring pET15-mecasermin 4 h after addition of IPTG.
Isolation, refolding and purification of mecasermin
Major recombinant mecasermin was found in insoluble fraction of E. coli and isolation was performed by releasing and washing mecasermin inclusion bodies. Since during extraction process impurities act as an obstacle in protein refolding, they could considerably impress the process yield. As a result, using efficient washing buffers help to reduce extra impurities and avoid losing target protein (20). Here we used washing buffer containing 1 M urea for initial recovery of mecasermin inclusion bodies according to the previous studies (20). The SDS-PAGE gel electrophoresis displayed a high purity of mecasermin in washed inclusion bodies estimated over 90% (Fig. 6). The results well confirmed the efficiency of washing step in developed purification process whichfollowed by next steps in inclusion body extraction.
Fig. 6.
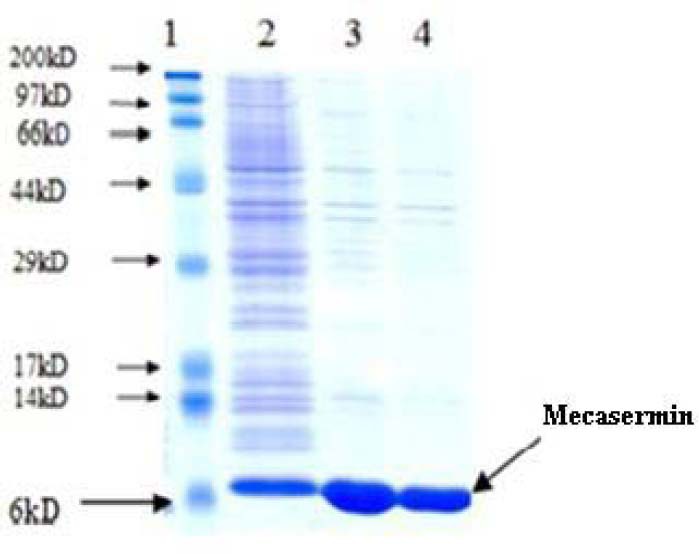
Sodium dodecyl sulfate polyacrylamide gel electrophoresis analysis for mecasermin inclusion bodies. Lane 1; molecular weight marker, lane 2; total protein 4 h after induction, lane 3; total protein after addition of washing buffer, lane 4; total protein after addition of 1 mM urea.
Protein aggregation is another imperative problem in solubilizing and refolding inclusion bodies. In this study mild solubilization was recruited to overcome this trouble (27). After refolding inclusion bodies, final purification step was followed. With respect to mecasermin pharmaceutical application, we didn’t use fusion tag strategy for target protein purification. Hence overexpressed mecasermin was successfully purified trough gel filtration column in one step and purified polypeptide analyzed on SDS-PAGE (Fig. 7). The recombinant protein was gathered from all fractions, but greater amount of the purified recombinant protein was observed in the first fraction as shown in Fig. 7, lane 1
Fig. 7.
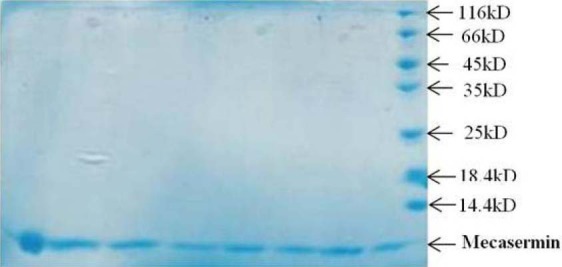
Sodium dodecyl sulfate polyacrylamide gel electrophoresis analysis for different fraction of purified mecasermin by gel filtration column. Lanes 1-8: different fractions, lane 9; molecular weight marker.
The concentration of purified recombinant mecasermin was determined by A280 and Beer-lambert law with molar extinction coefficient 4560 M−1cm−1 and Bradford assay. The absorption spectrum was recorded using 1.68 mg/ml protein. Finally, the purified mecasermin mass was confirmed by mass spectroscopy technique. Obtained data were in agreement with the theoretical mass of protein without initial methionine 7665 Da.
DISCUSSION
The reported studies about wide variety of specifically engineered IGFs and insulin analogues have been provided useful information about specific residues playing a vital role in ligand-receptor interactions (6,8). Among different hIGF analogues, some of them are the basis of designing therapeutic agents in the treatment of IGF-related diseases. Mecasermin, a commercial name for the hIGF-1 (T4I) mutant form, have FDA approval which is noticeably applied as therapeutic agent (3). In the present research, this mutant form of hIGF-1 was considered for production due to more efficient interactions provided by specific residues. Up to now, a wide range of expression systems have been reported to produce native hIGF-1 such as yeast (15,16), cell-free system (17), transgenic plants, and rabbits (18,19,20) whereas, it is found that E. coli host is known as more powerful expression system for recombinant production of native hIGF-1 protein (21,22,23,24,28).
Generally, E. coli expression systems are the most widely used host to produce industrial and pharmaceutical heterologous proteins owing to rapid growth, high cell density, high productivity, and low costs (22,29). The E. coli cytoplasm is considered to be the first choice in heterologous protein production because of high efficiency (23,30), while inclusion body formation frequently occurs as a result of protein over-expression in the cytoplasm which greatly encumbered the subsequent purification of bioactive proteins. The foremost privilege of inclusion bodies is facilitating purification procedure (28,31). Human IGF-1 as native analogue of mecasermin has been expressed as inclusion body in E. coli cytoplasm (28) but achieving bioactive refolded hIGF-1 from inclusion bodies sounds difficult (22,24,32). To overcome this problem some strategies such as linking the hIGF-1 polypeptide to a soluble fusion protein (21), periplasmic expression (22,30), co-expression of chaperons (33), co-expression of down regulated genes (34), or genes involve in oxido-reductases pathways (22) have been used.
However, these strategies may have some drawbacks; for example the addition of fusion partner facilitate refolding process but decreases overall yield of recombinant protein (31), or co-expression of some chaperons significantly consume cellular energy and decrease target protein translation (33).
In some reports optimization of some factors such as bacterial strain and temperature in expression process has been another applied strategy to achieve more bioactive protein (24,31).
Similar to native hIGF-1, mecasermin possesses three disulfide bonds in its structure. In this report we investigated mecasermin production in two different E. coli strain Origami and BL21 under control of T7 promoter. To over-produce mecasermin, we used pET15b plasmid containing the powerful T7 promoter being more efficient for over- expression of recombinant proteins (29). In spite of our prediction that Origami B oxidative environment leads to decrease inclusion bodies formation, our results surprisingly indicated that overall mecasermin production in Origami B (DE3) strain was nearly two fold higher than BL21 (Fig. 2). It meant not only inclusion bodies were not reduced but also overall expression significantly increased.
Moreover, Origami B (DE3) strain was capable of supportimg proper refolding of recombinant protein. E. coli Origami strain is one of the commercially available hosts that highly recommended for heterologous expression of cysteine rich proteins (29). Unlike the other E. coli strain (BL21), Origami cytoplasm provides oxidative environment for proteins involving disulfide bonds, because Trx/Grx mutations are associated with inactivation of both glutaredoxin and thioredoxin pathways (31,35). So expression of specific recombinant proteins under oxidative conditions in Origami strains cytoplasm is a strategy to achieve more active protein (24,32,36).
These results well confirmed with previous works on mecasermin analogue (hIGF-1) that enhanced disulfide isomerization which is responsible for the substantial increase in hIGF-I yield (22). In this report, transient overexpression of either DsbA or DsbC can double the yield of hIGF-1 in E. coli (22).
Since the major part of overexpressed recombinant protein located at insoluble phase, the effective method with high efficiency (as described in pervious section) was recruited in extracting, solubilizing and refolding the produced inclusion bodies (27); therefore, the maximum active form of recombinant protein was gathered to purification step (Fig. 6).
The produced protein was considered to be pharmaceutical agent; preferably no fusion tag strategy was applied to target protein purification. So the recombinant protein was successfully purified by gel filtration method (29) as shown in Fig. 7 and final protein concentration was calculated 1.68 mg/ml. Also, identification of the produced recombinant protein was specifically confirmed by western blotting and mass spectrometry techniques.
CONCLUSION
Here, we introduce the first scientific report in production of recombinant mecasermin in E.coli expression system. This study investigates expression level between two E.coli host strains with application of efficient method in refolding and purifying over-expressed recombinant protein to achieve high yield of mecasermin. Future studies could be focused on optimization of overproduction mecasermin at industrial scale regarding its high potential as a pharmaceutical agent.
ACKNOWLEDGMENTS
The present work was supported by the grant (No.190008) from Iranian Council of Stem Cell Technology and Isfahan University of medical sciences.
REFERENCES
- 1.Rosenbloom AL. Mecasermin (recombinant human insulin-like growth factor I) Adv Ther. 2009;26:40–54. doi: 10.1007/s12325-008-0136-5. [DOI] [PubMed] [Google Scholar]
- 2.Vaught JL, Contreras PC, Glicksman MA, Neff NT. Potential utility of rhIGF-1 in neuromuscular and/or degenerative disease. Ciba Found Symp. 1996;196:18–27. doi: 10.1002/9780470514863.ch3. [DOI] [PubMed] [Google Scholar]
- 3.McDonald A, Williams RM, Regan FM, Semple RK, Dunger DB. IGF-I treatment of insulin resistance. Eur J Endocrinol. European Society of Endocrinology. 2007;157:S51–6. doi: 10.1530/EJE-07-0271. [DOI] [PubMed] [Google Scholar]
- 4.Schoenle EJ, Zenobi PD, Torresani T, Werder EA, Zachmann M, Froesch ER. Recombinant human insulin-like growth factor I (rhIGF I) reduces hyperglycaemia in patients with extreme insulin resistance. Diabetologia. 1991;34:675–9. doi: 10.1007/BF00400998. [DOI] [PubMed] [Google Scholar]
- 5.Glass DJ. inventors, Modified IGF1 polypeptides with increased stability and potency. US Patent 7,355,018. 2008 [Google Scholar]
- 6.Glass D, Fornaro M. inventors, Stabilized insulin-like growth factor polypeptides. US Patent. 20,130,059,779. 2013 [Google Scholar]
- 7.Dubaquie Y, Fielder P. inventors, IGF-I protein variants for treating IGFBP-1-related disorders. US Patent 8097587. 2012 [Google Scholar]
- 8.Rajapaksha H, Alvino C, McCarthy P, Forbes BE. The insulin-like growth factor mutation database (IGFmdb) Growth Horm IGF Res. 2012;22:158–66. doi: 10.1016/j.ghir.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 9.Rosenbloom AL. The role of recombinant insulin-like growth factor I in the treatment of the short child. Curr Opin Pediatr. 2007;19:458–64. doi: 10.1097/MOP.0b013e3282094126. [DOI] [PubMed] [Google Scholar]
- 10.Keating GM. Mecasermin. BioDrugs. 2008;22:177–88. doi: 10.2165/00063030-200822030-00004. [DOI] [PubMed] [Google Scholar]
- 11.Zhu J, Kahn CR. Analysis of a peptide hormone-receptor interaction in the yeast two-hybrid system. Proc Natl Acad Sci USA. 1997;94:13063–13068. doi: 10.1073/pnas.94.24.13063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Denley A, Cosgrove LJ, Booker GW, Wallace JC, Forbes BE. Molecular interactions of the IGF system. Cytokine Growth Factor Rev. 2005;16:421–39. doi: 10.1016/j.cytogfr.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 13.Clark RG, Yeung DA, Oeswein JQ. Formulated IGF-I composition. US Patent, 5,681,814. 1997 [Google Scholar]
- 14.Spencer EM, Talkington-verser C. Human somatomedin carrier protein subunits and process for producing them; recombinant DNA molecules, hosts, processes and human somatomedin carrier protein-like polypeptides. US Patent., 5,200,509. 1991 [Google Scholar]
- 15.Gill R, Verma C, Wallach B, Ursø B, Pitts J, Wollmer A, et al. Modelling of the disulphide- swapped isomer of human insulin-like growth factor-1: implications for receptor binding. Protein Eng. 1999;12:297–303. doi: 10.1093/protein/12.4.297. [DOI] [PubMed] [Google Scholar]
- 16.Vai M, Brambilla L, Orlandi I, Rota N, Ranzi BM, Alberghina L, et al. Improved secretion of native human insulin-like growth factor 1 from gas1 mutant Saccharomyces cerevisiae cells. Appl Environ Microbiol. 2000;66:1–4. doi: 10.1128/aem.66.12.5477-5479.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Swartz J. Developing cell-free biology for industrial applications. J Ind Microbiol Biotechnol. 2006;33:476–85. doi: 10.1007/s10295-006-0127-y. [DOI] [PubMed] [Google Scholar]
- 18.Panahi M, Alli Z, Cheng X, Belbaraka L, Belgoudi J, Sardana R, et al. Recombinant protein expression plasmids optimized for industrial E. coli fermentation and plant systems produce biologically active human insulin-like growth factor-1 in transgenic rice and tobacco plants. Transgenic Res. 2004;13:245–59. doi: 10.1023/b:trag.0000034619.21613.d0. [DOI] [PubMed] [Google Scholar]
- 19.Brem G, Hartl P, Besenfelder U, Wolf E, Zinovieva N, Pfaller R. Expression of synthetic cDNA sequences encoding human insulin-like growth factor-1 (IGF-1) in the mammary gland of transgenic rabbits. Gene. 1994;149:351–5. doi: 10.1016/0378-1119(94)90175-9. [DOI] [PubMed] [Google Scholar]
- 20.Zinovieva N, Lassnig C, Schams D, Besenfelder U, Wolf E, Müller S, et al. Stable production of human insulin-like growth factor 1 (IGF-1) in the milk of hemi- and homozygous transgenic rabbits over several generations. Transgenic Res. 1998;7:437–47. doi: 10.1023/a:1008831028620. [DOI] [PubMed] [Google Scholar]
- 21.Kim SO, Lee YI. High-level expression and simple purification of recombinant human insulin-like growth factor I. J Biotechnol. 1996;48:97–105. doi: 10.1016/0168-1656(96)01402-2. [DOI] [PubMed] [Google Scholar]
- 22.Joly JC, Leung WS, Swartz JR. Overexpression of Escherichia coli oxidoreductases increases recombinant insulin-like growth factor-I accumulation. Proc Natl Acad Sci USA. 1998;95:2773–7. doi: 10.1073/pnas.95.6.2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chung BH, Choi YJ, Yoon SH, Lee SY, Lee YI. Process development for production of recombinant human insulin-like growth factor-I in Escherichia coli. J Ind Microbiol Biotechnol. 2000;24:94–9. [Google Scholar]
- 24.Zhang D, Wei P, Fan L, Lian J, Huang L, Cai J, et al. High-level soluble expression of hIGF-1 fusion protein in recombinant Escherichia coli. Process Biochem. 2010;45:1401–5. [Google Scholar]
- 25.Samuelsson E, Moks T, Uhlen M, Nilsson B. Enhanced in vitro refolding of insulin-like growth factor I using a solubilizing fusion partner. Biochemistry. 1994;33:4207–4211. doi: 10.1021/bi00180a013. [DOI] [PubMed] [Google Scholar]
- 26.Sambrook J, EF F, Maniatie T. Vol. 1. New York: Cold spring harbor laboratory press; 2001. Molecular cloning: A Laboratory Mannul; pp. 31–162. [Google Scholar]
- 27.Glass D. Modified IGF1 polypeptides with increased stability and potency. US Patent, 7,355,018. 2008 [Google Scholar]
- 28.Samuelsson E, Moks T, Nilsson B, Uhlen M. Refolding of Insulin-like Growth Factor. 1994;4:207–11. doi: 10.1021/bi00180a013. [DOI] [PubMed] [Google Scholar]
- 29.Sørensen HP, Mortensen KK. Advanced genetic strategies for recombinant protein expression in Escherichia coli. J Biotechnol. 2005;115:113–128. doi: 10.1016/j.jbiotec.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 30.Georgiou G, Segatori L. Preparative expression of secreted proteins in bacteria: status report and future prospects. Curr Opin Biotechnol. 2005;16:538–545. doi: 10.1016/j.copbio.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 31.De Marco A. Strategies for successful recombinant expression of disulfide bond-dependent proteins in Escherichia coli. Microb Cell Fact. 2009;8:26. doi: 10.1186/1475-2859-8-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singh SM, Panda AK. Solubilization and refolding of bacterial inclusion body proteins. J Biosci Bioeng. 2005;99:303–310. doi: 10.1263/jbb.99.303. [DOI] [PubMed] [Google Scholar]
- 33.Gething M, Sambrook J. Protein folding in the cell. Nature. 1992;355:33–45. doi: 10.1038/355033a0. [DOI] [PubMed] [Google Scholar]
- 34.Choi JH, Lee SJ, Lee SJ, Lee SY. Enhanced production of insulin-like growth factor i fusion protein in Escherichia coli by coexpression of the down-regulated genes identified by transcriptome profiling. Appl Environ Microbiol. 2003;69:4737–4742. doi: 10.1128/AEM.69.8.4737-4742.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hatahet F, Nguyen VD, Salo KEH, Ruddock LW. Disruption of reducing pathways is not essential for efficient disulfide bond formation in the cytoplasm of E. coli. Microb Cell. 2010;9:67. doi: 10.1186/1475-2859-9-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jungbauer A, Kaar W. Current status of technical protein refolding. J Biotechnol. 2007;128:587–596. doi: 10.1016/j.jbiotec.2006.12.004. [DOI] [PubMed] [Google Scholar]