

Abstract
Neutrophils play critical roles in innate immunity and host defense. However, excessive neutrophil accumulation or hyper-responsiveness of neutrophils can be detrimental to the host system. Thus, the response of neutrophils to inflammatory stimuli needs to be tightly controlled. Many cellular processes in neutrophils are mediated by localized formation of an inositol phospholipid, phosphatidylinositol (3,4,5)-trisphosphate (PtdIns(3,4,5)P3), at the plasma membrane. The PtdIns(3,4,5)P3 signaling pathway is negatively regulated by lipid phosphatases and inositol phosphates, which consequently play a critical role in controlling neutrophil function and would be expected to act as ideal therapeutic targets for enhancing or suppressing innate immune responses. Here, we comprehensively review current understanding about the action of lipid phosphatases and inositol phosphates in the control of neutrophil function in infection and inflammation.
Keywords: innate immunity; inositol phosphate; lipid phosphatase; neutrophils; PtdIns(3,4,5)P3
Introduction
Neutrophils migrate from the blood to infected tissues in response to inflammatory stimuli, where they protect their host by engulfing, killing, and digesting invading bacterial and fungal pathogens 1, 2, 3, 4. Neutrophils sense invading pathogens in the extracellular environment via several classes of cell surface receptor: G-protein-coupled receptors (GPCRs) to sense chemoattractants, Fc or complement receptors for phagocytosis, adhesion molecules (such as selectins and integrins), cytokine receptors, and innate immune receptors (such as the Toll-like receptor; TLR). When these receptors are bound by their respective ligands, they activate diverse intracellular signal transduction networks that lead to chemotactic migration, phagocytosis, degranulation, production of reactive oxygen species (ROS), and release of neutrophil extracellular traps 1, 5.
Many cellular processes in neutrophils are mediated by localized formation of an inositol phospholipid, phosphatidylinositol (3,4,5)-trisphosphate (PtdIns(3,4,5)P3), at the plasma membrane (Fig1A). PtdIns(3,4,5)P3 is formed by the phosphoinositide 3-kinase (PI3K) family of enzymes, which transfer the terminal phosphate of adenosine triphosphate (ATP) to PtdIns(4,5)P2 at the D3 position of the inositol ring. PI3Ks are divided class I, II, and III enzymes, but only class I PI3Ks phosphorylate PtdIns(4,5)P2 to form PtdIns(3,4,5)P3 6, 7 (Fig1B). PtdIns(3,4,5)P3 exerts its function by mediating translocation of various proteins via pleckstrin homology (PH) domains, which is a divergent protein module of approximately 120 amino acids found in many proteins involved in signal transduction 8 and that frequently mediates protein–protein or protein–phosphoinositide interactions. A subset of PH domains, including those in Btk, PKB/Akt, PLC-γ, Gab1, PDK1, Grp1, ARAP3, and multiple Rho guanine nucleotide exchange factors (GEFs), drive membrane translocation of their host proteins via specific, high-affinity recognition of PtdIns(3,4,5)P3 6, 9, 10, 11. This membrane translocation is crucial for PtdIns(3,4,5)P3-mediated cellular processes such as cell survival, proliferation, growth, differentiation, polarization, chemotaxis, cytoskeletal rearrangement, and membrane trafficking 12, 13, 14, 15 (Fig1C).
Figure 1.

Inositol phosphates and phosphoinositides
(A) Inositol phosphates contain only inositol and phosphate and are therefore water soluble and cytoplasmic. Ins(1,4,5)P3 can be converted to Ins(1,3,4,5)P4 by InsP3K family members 143, 144, 145. InsP7 is a higher inositol phosphate containing energetic pyrophosphate bonds 128, 139 and is formed by InsP6K family members 130, 136, 177. Phosphoinositides also contain inositol and phosphates, but in addition they contain two hydrophobic fatty acids and are therefore not water soluble and are mainly localized to membrane structures. (B) PI3K isoforms. Class I PI3Ks are heterodimeric proteins with a catalytic subunit and a regulatory subunit. Class IA PI3Ks consist of a catalytic subunit, p110 (α, β, or δ), and an adaptor subunit, p85 (α or β), and they are regulated by receptor tyrosine kinases. The only class IB member is p110γ, which is associated with p101 and p84/p87PIKAP regulatory subunits and is regulated by GPCRs such as chemokine receptors, C5a, and LTB4 6, 9. (C) PtdIns(3,4,5)P3 signaling pathway. PtdIns(3,4,5)P3 exerts its function by mediating protein translocation via binding to their pleckstrin homolog (PH) domains. PH domain is a divergent protein module of approximately 120 amino acids found in many proteins involved in signal transduction 8, 140, 178, 179, 180, 181. There are more than 200 PH domains identified in the human genome. PH domains often mediate protein–protein or protein–phosphoinositide interactions. A subset of PH domains, including those in Btk, protein kinase B (PKB)/Akt, PLC-γ, Gab1, PDK1, and Grp1, drive membrane translocation of their host proteins through specific, high-affinity recognition of PtdIns(3,4,5)P3. Only the PH domain-containing protein molecules on the plasma membrane can be activated, which in turn elicit various signal transduction pathways mediating neutrophil functions.
Negative regulation of PtdIns(3,4,5)P3 signaling in neutrophils
Although neutrophil activation is essential for pathogen killing and clearance, excessive neutrophil accumulation or hyper-responsiveness of neutrophils can be detrimental to the host; the response of neutrophils to inflammatory stimuli therefore needs to be tightly controlled. Over 50 chemokines have been identified in humans, and many of them are constitutively present in blood or tissues. However, neutrophils will only migrate, generate superoxide, or degranulate in response to a strong and stable stimulus, which suggests that inhibitory factors must be present to suppress the positive signals elicited by a weak or unstable chemoattractant signals 16, 17. These intracellular inhibitors establish a threshold for neutrophil responses, and as a result, neutrophils only respond when they receive stimulation that overcomes the negative inhibitory effect. Here, we comprehensively review current understanding about the action of lipid phosphatases and inositol phosphates in the control of neutrophil function in infection and inflammation. The role of PtdIns(3,4,5)P3 in infection and inflammation, including in neutrophil biology, has been thoroughly reviewed elsewhere 18, 19, 20, 21, 22, 23, 24, 25, 26.
Regulation of PtdIns(3,4,5)P3 signaling by lipid phosphatases
PtdIns(3,4,5)P3 signaling is activated in neutrophils by extracellular ligands such as chemokines or growth factors, while PtdIns(3,4,5)P3 synthesis by PI3K is balanced by the action of lipid phosphatases. The tumor suppressor protein phosphatase and tensin homolog (PTEN), a phosphatidylinositol 3′-phosphatase, converts PtdIns(3,4,5)P3 to PtdIns(4,5)P2, antagonizing the effect of PI3K and inhibiting PtdIns(3,4,5)P3 signaling 27, 28. Similarly, SHIP 27, 29 and 5ptase IV 30, 31 convert PtdIns(3,4,5)P3 to PtdIns(3,4)P2, thereby also exerting an inhibitory effect (Fig1C).
PTEN
PTEN is a mammalian phosphatidylinositol 3′-phosphatase known to modulate a range of cellular functions in various cell types, including neutrophils. PtdIns(3,4,5)P3 accumulates at the leading edge of chemotaxing cells 32, 33, 34, 35, 36. Disruption of PTEN in the amoeba Dictyostelium discoideum, a chemotaxis model, results in enhanced and prolonged PtdIns(3,4,5)P3 production, actin polymerization, persistent multiple (or broad) pseudopodia, and impaired directional migration in response to a chemoattractant gradient 33, 37. PTEN's role in hematopoietic cells has been well investigated. Li et al 34 demonstrated that PTEN's intracellular localization and activity can be regulated by chemoattractants via the Rho family of GTPases. In their experiments, PTEN and RhoA co-localized at the back of stimulated neutrophils, and active RhoA increased PTEN phosphatase activity via its downstream effector, Rock, which directly phosphorylated PTEN with Cdc42 acting as an enhancer. PTEN phosphorylation is required for its stability and activity 34, 38. Papakonstanti et al 39 proposed a pathway in which RhoA is activated via p110δ (see Fig1B). Inhibition of RhoA resulted in misorientation of PTEN toward the front of the cell where Cdc42 localizes, and knockdown of PTEN in Jurkat T cells impaired chemotaxis in transwell migration assays. It appears that the RhoA activity at the back of polarizing cells is also regulated by PDZrhoGEF, which contains a Galpha12/13 coupled GRS domain and can be directly activated by G12/13. PDZrhoGEF is localized at the back of chemotaxing cells. Cells depleted of this protein exhibit multiple fronts and long tails 40, 41. It is noteworthy that the COOH terminus of PTEN contains a typical PSD-95/Dlg/ZO-1(PDZ) binding motif. Indeed, PTEN has been reported to associate with several PDZ domain-containing proteins 42, 43, 44, 45, 46, 47. It is intriguing to speculate whether PTEN-PDZ domain interaction may play a role in regulating the biological function of PTEN in neutrophil chemotaxis.
Together, the published results allow us to propose a mechanism for PTEN's function in neutrophils: Localization of PTEN at the uropod locally inhibits PtdIns(3,4,5)P3 production at the back of the cell, thereby localizing PtdIns(3,4,5)P3 production and actin polymerization to the cell's leading edge and mediating directional sensing. Disruption of PTEN would therefore be expected to lead to uncontrolled propagation of PtdIns(3,4,5)P3 at the leading edge and consequent formation of multiple pseudopodia, frequent directional changes, and loss of directionality 48. To investigate the exact role of PTEN in neutrophils, we generated a myeloid-specific PTEN knockout mouse. Stimulation of PTEN−/− mouse neutrophils with chemoattractant resulted in increased PtdIns(3,4,5)P3 synthesis, more exaggerated and sustained F-actin polymerization, elevated sensitivity to chemoattractant, and augmented ROS production 49. Disruption of PTEN also led to enhanced phagocytosis, consistent with the essential role of phospholipids in phagocytosis 50, 51, 52, 53, 54, 55, 56, 57. Single-cell chemotaxis assays showed that PTEN−/− neutrophils had small but significant defects in directionality, consistent with the mechanistic hypothesis that PTEN plays a role in mediating polarized PtdIns(3,4,5)P3 production at the leading edge of chemotaxing cells 34. However, these neutrophils also moved quickly, and, as a result, overall chemotaxis (which is a function of both speed and directionality) was not affected. Similar results have also been observed in PTEN-deficient T cells 58, 59. PTEN's function in directional sensing in Dictyostelium and mammalian cells, including neutrophils, is therefore different. Of note, neutrophils isolated from mice carrying a “knock-in” allele of PI3Kγ showed GPCR-uncoupled PtdIns(3,4,5)P3 accumulation and displayed much greater impairment in directional cell migration in response to chemoattractants. Stimulated mutant macrophages did not polarize PtdIns(3,4,5)P3, and chemoattractant-elicited Rac activation was shortened due to enhanced PI3K-dependent activation of RacGAPs 60.
Consistent with the observed increased responsiveness of PTEN−/− neutrophils in vitro, recruitment of PTEN−/− neutrophils to inflamed mouse peritoneal cavities was significantly enhanced in vivo 49. Using intravital video microscopy, we also observed enhanced neutrophil emigration from cremasteric muscle venules in PTEN knockout mice. PtdIns(3,4,5)P3 is a major downstream target of integrins and chemokine receptors and has been implicated in multiple leukocyte trafficking steps 20, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76. Unexpectedly, leukocyte adhesion was not increased when PtdIns(3,4,5)P3 signaling was enhanced via PTEN depletion, suggesting that PtdIns(3,4,5)P3 signaling is essential for stimuli-elicited cell adhesion but is not a limiting step. Enhanced neutrophil recruitment to the cremaster muscle in response to different stimuli is most likely due to faster neutrophil movement in the vascular bed, across the vascular endothelium, and into the muscle tissue. Neutrophil rolling influx, rolling speed, and the number of firmly adherent neutrophils were not altered in PTEN knockout mice. PTEN therefore acts as a negative regulator of neutrophil trafficking, and enhanced neutrophil recruitment in PTEN knockout mice is mainly caused by augmented transendothelial migration 77.
Recently, Heit et al 78 reported that PTEN is required for prioritizing and integrating responses to multiple chemotactic cues in certain situations. Thus, PTEN's exact function in neutrophils might rely on the number and type of chemoattractants, as well as the relative doses or route used to induce neutrophil inflammatory reactions. In the setting of multiple chemoattractants, neutrophils favor end-target chemoattractants such as fMLP and C5a over intermediary endogenous chemoattractants such as IL-8 and LTB4. Heit et al proposed a hierarchical model in which neutrophil migration toward end-target chemoattractants is mediated by p38 MAPK, whereas migration toward intermediary chemoattractants is mediated by PtdIns(3,4,5)P3. When faced with competing gradients of end-target and intermediary chemoattractants, PtdIns(3,4,5)P3 signaling was significantly reduced by p38 MAPK 79, a cross talk that might be mediated by PTEN. When neutrophils were exposed to an IL8 gradient, PTEN localized to the uropod of chemotaxing cells, thereby contributing to accumulation of PtdIns(3,4,5)P3 at the leading edge. In the presence of opposing fMLP gradients, PTEN was distributed throughout the entire cell circumference, thus inhibiting all PI3K activity and permitting “preferential” migration toward fMLP via phospholipase A2 and p38. As a consequence, PTEN−/− neutrophils cannot prioritize chemoattractants and are “distracted” by chemokines when moving toward bacterial chemoattractants 78. Another recent study showed that the p38 MAPK p38δ and PKD1 regulate PTEN activity in neutrophils, thereby controlling their extravasation and chemotaxis during acute lung inflammation 80.
PTEN is also a key regulator of neutrophil death. Neutrophils normally have a very short life span and readily undergo spontaneous programmed cell death, which plays a crucial role in neutrophil homeostasis and the resolution of inflammation 81. PtdIns(3,4,5)P3 signaling dramatically decreases during neutrophil death, and deactivation of PtdIns(3,4,5)P3 signaling has been identified as a critical event in neutrophil spontaneous death. PTEN-depleted neutrophils live much longer than wild-type neutrophils 82, and SHIP1-null neutrophils, in which the PtdIns(3,4,5)P3 pathway is upregulated, have an extended life span 83. Conversely, activation of SHIP1 by NADPH oxidase-stimulated Lyn leads to enhanced neutrophil apoptosis 84. These results are consistent with reports that neutrophil apoptosis is enhanced in PI3Kγ-deficient mice with reduced Akt activity 85, 86, where Akt is a major survival factor downstream of PtdIns(3,4,5)P3 87. PtdIns(3,4,5)P3 production in apoptotic neutrophils is maintained by chemokines in an autocrine manner, which activate PI3Kγ via GPCRs. PI3Kγ, but not class IA PI3Ks, is negatively regulated by gradually accumulating ROS in apoptotic neutrophils, which suppress PI3Kγ activity by inhibiting an actin-mediated positive feedback loop 88. However, a recent study has shown that granulocyte colony-stimulating factor (G-CSF) activation of Akt is insufficient to prolong neutrophil survival, and neutrophils treated with G-CSF undergo apoptosis even in the presence of high levels of activated Akt. Moreover, inhibition of Akt fails to alter neutrophil survival, suggesting that there are other pathways and factors mediating PtdIns(3,4,5)P3-elicited survival signaling 89.
Of note, some of PTEN's functions may not be mediated by degradation of PtdIns(3,4,5)P3. For instance, Anderson et al recently reported that superoxide production elicited by serum-opsonized bioparticles is mainly mediated by complement and CD18. This process requires class III PI3K and its product, PtdIns(3)P, and is independent of class I PI3K and its product, PtdIns(3,4,5)P3 74, 90. Here, PTEN acts as a lipid phosphatase, removing the D3-positioned phosphate from PtdIns(3,4,5)P3, PtdIns(3,4)P2, and PtdIns(3)P. Therefore, its effect on phagocytosis-associated superoxide production is most likely mediated by its lipid phosphatase activity on PtdIns(3)P.
SHIP
SHIP (or SHIP1) is a 145-kD SH2-containing inositol-5-phosphatase that is widely expressed in hematopoietic cells 27, 29, 91, 92. It was first identified as a tyrosine phosphoprotein associated with Shc in response to numerous cytokines. Hematopoietic cell-specific SHIP1 is responsible for the majority of phosphatidylinositol 5′-phosphatase activity in neutrophils, while the ubiquitously expressed SHIP2 only plays a minor role in hematopoietic cells. SHIP1 blunts PI3K-initiated signaling and is known to negatively regulate various cellular processes, such as phagocytosis, cell migration, degranulation, cell survival, proliferation, differentiation, and sensitivity to chemokines 93, 94, 95, 96. SHIP1-deficient mice exhibit chronic progressive hyperplasia of myeloid cells, perhaps at the expense of B-cell production. SHIP1 knockout myeloid progenitors show enhanced responsiveness to cytokines and are less susceptible to apoptotic stimuli in vitro, and while increased proliferation and decreased apoptosis may contribute to the expanded hematopoietic stem cell (HSC) compartment in SHIP1 knockout mice, these HSCs are also compromised in their ability to home and repopulate 83, 92. SHIP1 expression was initially thought to be restricted to hematopoietic cells, but more recently it has become apparent that most bone marrow (BM) and blood cells express SHIP1, and Hazen et al 97 demonstrated SHIP1 expression and functional activity in non-hematopoietic BM cells. Furthermore, SHIP1 deficiency enables long-term reconstitution of the hematopoietic inductive BM microenvironment 98.
Although excessive PtdIns(3,4,5)P3 production in PTEN−/− neutrophils does not cause significant chemotaxis defects, one report suggests that SHIP1 knockout results in chemotaxis defects 99. Findings from our own lab show that SHIP1 plays a predominant role in cell adhesion, rather than chemoattractant sensing, during cell migration. SHIP1−/− neutrophils were much more adherent than wild-type cells by engaging integrins. In suspension, PtdIns(3,4,5)P3 production in SHIP1−/− neutrophils remained constant when stimulated with chemoattractant, in contrast to adherent SHIP1−/− neutrophils, which showed marked upregulation of PtdIns(3,4,5)P3 when stimulated. As a consequence, actin polymerization was restricted to the leading edge of suspended SHIP1−/− neutrophils, with cell polarity lost upon adhesion 100. During migration, SHIP1 appears to act as a negative regulator of PtdIns(3,4,5)P3 formation at the cell–substratum interface, preventing the formation of top-down PtdIns(3,4,5)P3 polarity and facilitating normal cell attachment and detachment during chemotaxis (Fig2A).
Figure 2.

The role of SHIP1 and PTEN in neutrophil chemotactic migration
(A) SHIP1 and PTEN act via different receptor-regulated processes to control spatial accumulation of PtdIns(3,4,5)P3 and establish a proper anterior–posterior PtdIns(3,4,5)P3 compass. SHIP1 acts as a negative regulator of integrin-mediated cell adhesion in neutrophils. In wild-type neutrophils, integrin-mediated cell adhesion results in PtdIns(3,4,5)P3 production at the sites of cell adhesion. Concurrently, SHIP1 at the cell–substratum interface is engaged, phosphorylated, and activated. This activity is crucial for dephosphorylating the PtdIns(3,4,5)P3 formed during cell adhesion. With the combined actions of both SHIP1 and PTEN, PtdIns(3,4,5)P3 polarity is maintained at the leading edge, neutrophils polarize, and there is effective cell migration. PTEN is localized to the rear end of a migrating cell to facilitate the accumulation of PtdIns(3,4,5)P3 at the anterior end, and SHIP1 is active at the cell–substratum interface to abolish the PtdIns(3,4,5)P3 gradient being formed by integrin activation. With loss of SHIP1, adhesion-mediated PtdIns(3,4,5)P3 formation is uncontrolled, resulting in the formation of “top-down” PtdIns(3,4,5)P3 polarity. Increased PtdIns(3,4,5)P3 levels enhance cell adhesion. This leads to activation of various PtdIns(3,4,5)P3 effector proteins and consequently results in impaired chemotaxis. (B) SHIP1 plays a critical role in neutrophil transendothelial migration by balancing the lateral and dorsal–ventral polarity of transmigrating neutrophils.
In the same study, we also examined neutrophil migration in vivo. Surprisingly, despite the chemotaxis defect, recruitment of SHIP1−/− neutrophils to the inflamed peritoneal cavity was significantly enhanced. We hypothesize that neutrophils passing through the endothelium and entering the tissue control their migration using two independent directional cues: a lateral front-to-back polarity mediated by chemotactic signaling and a dorsal–ventral polarity controlled by adhesion-mediated signaling. SHIP1 activity is regulated by adhesion and plays a critical role in balancing these two types of polarity in neutrophils. Loss of SHIP1 leads to upregulation of dorsal–ventral polarity, preventing neutrophils from effective lateral migration on surfaces. However, this dorsal–ventral polarity promotes top-down migration of neutrophils through soft surfaces such as the gaps between endothelial cells 100 (Fig2B).
Although SHIP1 is thought to be enzymatically active in the cytosol, its activity is ultimately determined by its membrane localization 101. Recruitment of SHIP1 to the plasma membrane is regulated by its association with adapter proteins (e.g. Shc, Grb2, Dok3), scaffolding proteins (e.g. Gab1/2), and by direct association with tyrosine-phosphorylated receptors via its SH2 domain. These interactions require tyrosine phosphorylation of SHIP1 at the NPXY motif 102, 103, 104, 105. We have observed that cell adhesion, but not chemoattractant stimulation, leads to tyrosine phosphorylation of SHIP1. In addition, SHIP1 can interact with FAK and Lyn upon cell adhesion and β3 integrin in both suspension and upon cell adhesion. This indicates that adhesion results in the recruitment of SHIP1 to the membrane where it can act on the PtdIns(3,4,5)P3 produced during cell adhesion 100. It has previously been shown that Lyn, a Src family tyrosine kinase, regulates SHIP1 phosphorylation in integrin αIIbβ3-mediated adhesion and signaling in platelets 106.
Unlike PTEN-deficient neutrophils, in which ROS production is elevated, loss of SHIP1 leads to reduced ROS production in neutrophils in suspension upon stimulation with chemoattractant. In neutrophils, ROS are mainly produced by the phagocyte NADPH oxidase (also known as the NOX2 complex) 107, 108, 109, 110, 111. During cell activation, the cytosolic components of the enzyme, namely p47phox, p67phox, Rac2, and p40phox, are recruited to the membrane to form a complex with its membrane components, p22phox and gp91 complex. Assembly of the NADPH oxidase complex catalyzes the conversion of molecular oxygen to superoxide, which is known to facilitate the destruction of invading pathogens 81, 88, 112, 113, 114. Although PtdIns(3,4,5)P3 and PI3Ks have been identified as key regulators of NADPH oxidase activation, recruitment of cytosolic p47phox and p40phox to the NADPH oxidase complex requires the presence of the class III PI3K product, PtdIns(3)P, and the product of SHIP1, PtdIns(3,4)P2 73, 74, 87, 115, 116, 117, 118, 119, 120, 121, 122, 123. Reduced ROS production in SHIP1−/− neutrophils is primarily due to decreased PtdIns(3,4)P2 levels, which correlates with reduced recruitment of p40phox and p47phox to the NADPH oxidase complex 100. However, when SHIP1−/− neutrophils are primed and permitted to adhere to fibronectin, they produce very high levels of ROS. This is most likely due to the involvement of SHIP1−/− in integrin-mediated PtdIns(3,4,5)P3 signaling, with increased PtdIns(3,4,5)P3 signaling overriding the effect of decreased PtdIns(3,4)P2 levels 100.
The PtdIns(3,4,5)P3 signaling pathway can be negatively regulated by lipid phosphatases SHIP1 and PTEN in neutrophils. The activity and subcellular localization of these lipid phosphatases are tightly controlled. How temporal and spatial regulation contributes to various neutrophil functions remains ill-defined and needs to be further investigated.
Regulation of PtdIns(3,4,5)P3 signaling by inositol phosphates
PtdIns(3,4,5)P3 signaling was previously thought to be solely dependent on the concentration of PtdIns(3,4,5)P3 in the cell membrane 6. We discovered that two inositol phosphates, InsP7 and Ins(1,3,4,5)P4, compete with PtdIns(3,4,5)P3 for PH domain binding and suppress PH domain translocation, providing a novel mode of regulation for PtdIns(3,4,5)P3 signaling in neutrophils 124, 125 (Fig3).
Figure 3.

Regulation of PtdIns(3,4,5)P3 signaling by inositol phosphates in neutrophils
InsP7 or Ins(1,3,4,5)P4 can compete with PtdIns(3,4,5)P3 for PH domain binding and thus prevent their recruitment onto the plasma membrane and consequently inhibit their activation.
InsP7
InsP7 has been implicated in a variety of cellular functions such as vesicular trafficking, apoptosis, endocytosis, DNA repair/recombination, and maintenance of telomere length 126. InsP7 arises from pyrophosphorylation of InsP6, the most abundant inositol phosphate in mammalian cells 127. The enzymes that catalyze the synthesis of InsP7 comprise a family of InsP6 kinases (InsP6K) including InsP6K1, InsP6K2, and InsP6K3 128, 129 (Fig1). InsP6K1 and InsP6K2 are highly expressed in neutrophils, while InsP6K3 is essentially undetectable. In neutrophils, InsP6K1 and InsP6K2 isoforms have non-redundant roles. InsP6K1 seems to be equally distributed in the nucleus and cytoplasm, while, in contrast, InsP6K2 appears to be almost exclusively nuclear in localization 130. In a recent study, we reported that InsP6K1 could regulate PtdIns(3,4,5)P3 signaling in neutrophils 131; disruption of InsP6K1 enhanced PtdIns(3,4,5)P3 signaling, and, as a consequence, these neutrophils exhibited elevated phagocytic and bactericidal capabilities and amplified NADPH oxidase-mediated superoxide production. These findings established a novel role for InsP7 in the regulation of cellular signal transduction pathways in neutrophils and provided a novel mechanism for modulating PtdIns(3,4,5)P3 signaling in mammalian cells 131.
Consistent with its regulatory role in mediating chemoattractant-elicited signals, InsP7 levels are tightly regulated in neutrophils. Unstimulated neutrophils contain a substantial amount of InsP7, with levels decreasingly markedly and rapidly upon stimulation with chemoattractants 131. Cigarette smoke extract (CSE) and nicotine also reduce InsP7 levels in aging neutrophils, which subsequently leads to suppression of Akt deactivation and delayed neutrophil death. Delayed neutrophil death contributes to the pathogenesis of CS-induced COPD. Disruption of InsP6K1 consistently augments CS-induced neutrophil accumulation and lung damage 132. Recently, Chakraborty et al 133 showed that InsP7 also negatively regulates PtdIns(3,4,5)P3/Akt signaling in glucose homeostasis and protein translation. InsP7 inhibits Akt by acting at the PH domain of Akt to prevent its plasma membrane translocation, phosphorylation, and activation by PDK1 133. Intriguingly, in contrast to our observation that chemoattractant inhibits InsP7 formation, Chakraborty et al showed that growth factors stimulate InsP7 production. Therefore, the inhibition of Akt signaling by InsP7 may be a general signal transduction phenomenon. However, its regulatory mechanism and resulting physiologic consequences might be cell or system specific. The mechanisms by which InsP7 production is suppressed in chemoattractant- and CSE-stimulated neutrophils are still largely unknown and need to be investigated further. However, these mechanisms are likely to involve activation of inositol pyrophosphate phosphatase and/or deactivation of InsP6 kinase.
Although InsP6K1 disruption augments PtdIns(3,4,5)P3 signaling, it fails to augment cell adhesion, sensitivity, or migration speed in neutrophils 131. These results are somewhat different to the migratory phenotypes observed in PTEN-null neutrophils 49. Although overall chemotactic migration is relatively normal, PTEN disruption in neutrophils results in mildly impaired directionality, enhanced sensitivity to chemoattractant stimulation, and slightly increased migration speed 49. These distinct effects are likely to occur as a result of different temporal and spatial regulation of PTEN and InsP6K1 in neutrophils. PTEN activity is increased, and its subcellular localization is altered, after chemoattractant stimulation 34. In contrast, InsP6K1 levels are high in unstimulated neutrophils but are significantly reduced after chemoattractant stimulation. The mechanisms by which PTEN and InsP6K1 regulate PtdIns(3,4,5)P3 signaling are also different; PTEN regulates PtdIns(3,4,5)P3 levels and controls neutrophil function via a number of downstream pathways, while InsP6K1 deletion is not associated with changes in PtdIns(3,4,5)P3 or its downstream signaling cascades, but instead is limited to its action on Akt. In addition, PTEN appears to act as a housekeeping gene, while InsP6K1 exhibits tissue-specific expression, and there is functional redundancy with InsP6K2 and/or InsP6K3 in some cell types 130, 134, 135, 136. These results highlight the complexity of the PtdIns(3,4,5)P3 signaling network and suggest that different temporal and spatial regulation of this signaling molecule can lead to distinct cellular effects.
Ins(1,3,4,5)P4
Ins(1,3,4,5)P4 is the predominant InsP4 isoform in neutrophils 137, 138, intracellular levels of which are regulated by inositol phosphate kinases and phosphatases 128, 139 (Fig4). The major metabolic pathway generating Ins(1,3,4,5)P4 is via Ins(1,4,5)P3, with Ins(1,4,5)P3 formed from the hydrolysis of PtdIns(4,5)P2 by PLC 116, 140, 141, 142. Ins(1,3,4,5)P4 is eventually formed by InsP3Ks, which belong to a highly conserved family of inositol phosphate kinases 128, 136, 139. Members of this family include InsP6Ks, InsP3Ks, and IPMK 143, 144, 145. There are three InsP3K isoforms in mammalian cells, designated A, B, and C 143, 144, 145. The gene encoding InsP3KA is exclusively expressed in specific subpopulations of neurons in the central nervous system and in the testis 143, while isoforms B and C are ubiquitously expressed 143, 144, 145.
Figure 4.
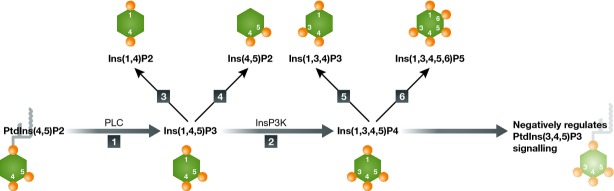
The metabolic pathways controlling intracellular Ins(1,3,4,5)P4 levels
1. Phospholipase C (PLC). 2. Ins(1,4,5)P3 kinase (InsP3K). 3. Ins(1,4,5)P3 5-phosphatase. 4. Ins(1,4,5)P3 1-phosphatase. 5. Inositol polyphosphate 5-phosphatase. 6. Ins(1,3,4,5)P4 6-kinase. All these enzymes are ubiquitously expressed.
The physiologic functions of InsP3K in hematopoietic cells were recently studied using InsP3K knockout mice 146, 147. The InsP3KB isoform appeared to contribute the majority of InsP3K activity in T and B cells 148, 149, 150, 151. Disruption of InsP3KB led to a dramatic decrease in cellular Ins(1,3,4,5)P4 levels, impaired T-cell development, and defective thymocyte selection. Huang et al demonstrated that Ins(1,3,4,5)P4 can bind to the PH domain of the Tec family tyrosine kinase Itk, which plays an important role in TCR signaling. Interestingly, this binding altered the conformation of the Itk PH domain and subsequently promoted, rather than suppressed, PH domain binding to PtdIns(3,4,5)P3 in T cells 152. Disruption of InsP3KB induced B-cell death and impaired B-cell development 153, 154. Miller et al 153 reported that Ins(1,3,4,5)P4 acts by modulating store-operated calcium channels, with elevated calcium influx detected in InsP3KB null B cells. InsP3K has been reported to be a potential modulator of calcium mobilization, since it can decrease the level of Ins(1,4,5)P3, which mediates calcium release from internal stores by converting it to Ins(1,3,4,5)P4. Surprisingly, no substantial defects in Ins(1,4,5)P3 quantity or calcium mobilization were detected in InsP3KB-null T cells 148, 149.
InsP3KB is also the major InsP3K isoform in neutrophils. Disruption of the InsP3KB gene leads to alteration of myelopoiesis, hyperactivation of neutrophils, and dysregulation of innate immunity 125, 155, 156. Ins(1,3,4,5)P4 depletion in InsP3KB-deficient neutrophils enhances membrane translocation of the PtdIns(3,4,5)P3-specific PH domain, thus augmenting PtdIns(3,4,5)P3-mediated downstream signaling. This leads to enhanced sensitivity to chemoattractants, elevated superoxide production, and enhanced neutrophil recruitment to inflamed peritoneal cavities in mice 125. Although loss of InsP3KB leads to elevated neutrophil recruitment and function, bacterial killing was actually reduced in vivo. This was mainly due to B- and T-cell defects, which led to significantly reduced levels of immunoglobulin and impaired opsonization and phago-cytosis 125. Disruption of InsP3KB does not affect overall calcium signaling in the presence of extracellular calcium in neutrophils 125. However, a more detailed investigation revealed significantly decreased calcium release from intracellular stores and enhanced calcium influx through store-operated calcium channels in InsP3KB-null neutrophils stimulated with chemokines. Reduced calcium release from intracellular stores appears to be a result of calcium depletion from the store (Y. Jia and H.R. Luo, unpublished data).
In neutrophils, chemoattractant stimulation triggers a potent and rapid elevation in Ins(1,3,4,5)P4, indicating that Ins(1,3,4,5)P4 levels are regulated by receptor occupancy. InsP3K is not constitutively active in neutrophils, and its activity in unstimulated neutrophils is relatively low. Activity is, however, dramatically increased by chemoattractant stimulation. The low basal InsP3K activity might be necessary for chemoattractant-stimulated neutrophils to generate or maintain an initially high concentration of Ins(1,4,5)P3, which is essential for downstream calcium signaling. Alternatively, the regulation of InsP3K by chemoattractants may simply be another mechanism to finely tune intracellular Ins(1,3,4,5)P4 concentrations.
The molecular mechanism underlying the chemoattractant-induced enhancement of InsP3K activity is still largely unknown. Recent studies have shown that all three InsP3K isoforms contain an F-actin binding domain, and InsP3KB is highly localized to the leading edge of polarized neutrophils 144, 157, 158, 159. Whether the actin–InsP3KB interaction plays any role in modulating InsP3KB activity needs to be determined. In addition, since all three isoforms contain a calmodulin binding motif, the InsP3KB activity in neutrophils may also be regulated by calcium 125.
Targeting PtdIns(3,4,5)P3 signal pathway in infectious and inflammatory diseases
Excessive neutrophil accumulation or hyper-responsiveness of neutrophils can be detrimental to the host system, and the response of neutrophils to inflammatory stimuli needs to be tightly controlled. Neutrophils contribute to the pathogenesis of a number of autoimmune diseases such as rheumatoid arthritis, Crohn's disease, and systemic lupus erythematosus (SLE). Since receptor-mediated signal transduction in neutrophils relies heavily on PI3K isoforms, small molecule inhibitors to class I PI3Ks should counteract the undesirable pro-inflammatory effects of neutrophils in inflammatory conditions. Over the last few years, a number of isoform-specific PI3K inhibitors have been developed, and some of them are already being tested in clinical trials for various inflammatory diseases 68, 75, 160, 161, 162, 163, 164, 165, 166, 167. Since SHIP1 inhibition leads to massive myeloid infiltration of the lungs and progressive inflammation, pharmacological activation of SHIP1 has emerged as a therapeutic strategy for inflammatory pulmonary diseases. A small molecule SHIP1 activator AQX-1125, which binds to the C2 domain of SHIP1 to increase its catalytic activity, is currently in Phase II clinical trials. Since SHIP1 expression is restricted to hematopoietic cells, targeting SHIP1 is expected to limit unwanted off-target side effects. AQX-1125 suppresses Akt phosphorylation, reduces cytokine production, and inhibits neutrophil activation and chemotaxis 168, 169.
There are also cases in which enhanced neutrophil recruitment and function would be expected to be clinically beneficial. For example, neutropenia and related infections are the most important dose-limiting toxicities of cytotoxic anti-cancer chemo- and radio-therapies, which impact on both the quality and quantity of life. One obvious strategy for treating neutropenia-related infections is to administer broad-spectrum antibiotics to neutropenic patients. However, not all patients respond to antibiotic treatments, and this method carries the risk of inducing antibiotic resistance. An alternative approach is G-CSF (filgrastim) treatment 170, which is now used clinically to restore neutrophil counts in neutropenia-related pneumonia patients via stimulation of the BM to produce more neutrophils. However, this therapy does not always work if the BM has not recovered from chemotherapy, and treatment is associated with side effects such as bone pain, headache, fatigue, and nausea. Long-term use of G-CSF might also increase the risk of leukemia 170, 171, 172, 173. Therefore, directly elevating innate immunity by promoting neutrophil recruitment and function is a reasonable alternative strategy under these circumstances. We have recently shown that neutrophil function can be enhanced in neutropenic conditions by activating intracellular PtdIns(3,4,5)P3 signaling 174; significantly, more neutrophils were recruited to inflamed lungs during neutropenia-associated pneumonia in myeloid-specific PTEN knockout mice. Using an adoptive transfer technique, we demonstrated that enhanced neutrophil recruitment was caused directly by PTEN depletion in neutrophils. In addition, disruption of PTEN increased recruitment of macrophages and elevated levels of pro-inflammatory cytokines/chemokines in the inflamed lungs, which might also have contributed to enhanced neutrophil recruitment. Finally, we provided direct evidence that the enhanced neutrophil function caused by elevated PtdIns(3,4,5)P3 signaling can alleviate pneumonia-associated lung damage and decrease pneumonia-elicited mortality. Furthermore, we showed that activation of PtdIns(3,4,5)P3 signaling by PTEN disruption or using the SF1670 PTEN inhibitor increased the efficacy of granulocyte transfusion in neutropenia-related pneumonia 175. It is noteworthy that although we consistently observed enhanced neutrophil migration in PTEN-deficient mice suffering from E. coli pneumonia, Schabbauer et al recently reported that the absence of myeloid cell-associated PTEN dampens pulmonary inflammation, reduces neutrophil influx, and augments the phagocytic properties of macrophages, which ultimately resulted in decreased tissue injury and improved survival during murine pneumococcal pneumonia. PTEN may differentially regulate the attraction of neutrophils depending on the inducing agent 176.
Elevating PtdIns(3,4,5)P3 can augment neutrophil recruitment and function in host defense responses. However, hyperactivation of neutrophils can lead to unwanted tissue damage and inflammation. In addition, some activators of PtdIns(3,4,5)P3 signaling may not be appropriate therapeutic targets for clinical intervention due to their carcinogenetic or other harmful effects. This balance between benefit and harm remains a challenge in targeting PtdIns(3,4,5)P3 signaling in inflammatory conditions. The PtdIns(3,4,5)P3 signaling pathway is negatively regulated by lipid phosphatases and inositol phosphates, which consequently play a critical role in controlling neutrophil function and thus would be expected to act as ideal therapeutic targets for enhancing or suppressing innate immune responses.
Sidebar A: In need of answers.
How are the activity and subcellular localization of PTEN and SHIP1 regulated in neutrophils? How do temporal and spatial regulation contribute to various neutrophil functions?
What molecular mechanism underpins chemoattractant-induced enhancement of InsP3KB activity in neutrophils? Do actin–InsP3KB interactions play any role in modulating InsP3KB activity?
The mechanisms by which InsP7 production is suppressed in chemoattractant- and CSE-stimulated neutrophils.
When targeting PtdIns(3,4,5)P3 signaling in the clinical setting, how can a balance between the beneficial and detrimental effects be achieved?
Conflict of interest
The authors declare that they have no conflict of interest.
Glossary
- 5ptase IV
phosphoinositide-specific inositol polyphosphate 5-phosphatase IV
- COPD
chronic obstructive pulmonary disease
- CSE
cigarette smoke extract
- G-CSF
granulocyte colony-stimulating factor
- GEF
guanine nucleotide exchange factors
- GPCR
G-protein coupled receptor
- InsP3K
inositol trisphosphate kinase
- InsP6K
inositol hexakisphosphate kinase
- InsP7
diphosphoinositol pentakisphosphate
- IPMK
inositol phosphate multi-kinase
- PH-domains
pleckstrin homolog domain
- PI3K
phosphatidylinositol 3′-kinases
- PLC
phosphatidylinositol-specific phospholipase C
- PtdIns(3,4,5)P3
phosphatidylinositol 3, 4, 5 trisphosphate
- PtdIns(4,5)P2
phosphatidylinositol 4,5-bisphosphate
- PTEN
phosphatase and tensin homologue deleted on chromosome ten
- Rock
RhoA-associated kinase
- ROS
reactive oxygen species
- SHIP1
SH2-containing inositol-5′-phosphatase-1
- TLR
Toll-like receptor
References
- Nauseef WM, Borregaard N. Neutrophils at work. Nat Immunol. 2014;15:602–611. doi: 10.1038/ni.2921. [DOI] [PubMed] [Google Scholar]
- Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. 2006;6:173–182. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- Phillipson M, Kubes P. The neutrophil in vascular inflammation. Nat Med. 2011;17:1381–1390. doi: 10.1038/nm.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159–175. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- Dale DC, Boxer L, Liles WC. The phagocytes: neutrophils and monocytes. Blood. 2008;112:935–945. doi: 10.1182/blood-2007-12-077917. [DOI] [PubMed] [Google Scholar]
- Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol. 2012;13:195–203. doi: 10.1038/nrm3290. [DOI] [PubMed] [Google Scholar]
- Lemmon MA. Phosphoinositide recognition domains. Traffic. 2003;4:201–213. doi: 10.1034/j.1600-0854.2004.00071.x. [DOI] [PubMed] [Google Scholar]
- Cantrell DA. Phosphoinositide 3-kinase signalling pathways. J Cell Sci. 2001;114:1439–1445. doi: 10.1242/jcs.114.8.1439. [DOI] [PubMed] [Google Scholar]
- Krugmann S, Anderson KE, Ridley SH, Risso N, McGregor A, Coadwell J, Davidson K, Eguinoa A, Ellson CD, Lipp P, et al. Identification of ARAP3, a novel PI3K effector regulating both Arf and Rho GTPases, by selective capture on phosphoinositide affinity matrices. Mol Cell. 2002;9:95–108. doi: 10.1016/s1097-2765(02)00434-3. [DOI] [PubMed] [Google Scholar]
- Enomoto A, Murakami H, Asai N, Morone N, Watanabe T, Kawai K, Murakumo Y, Usukura J, Kaibuchi K, Takahashi M. Akt/PKB regulates actin organization and cell motility via Girdin/APE. Dev Cell. 2005;9:389–402. doi: 10.1016/j.devcel.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Rickert P, Weiner OD, Wang F, Bourne HR, Servant G. Leukocytes navigate by compass: roles of PI3Kgamma and its lipid products. Trends Cell Biol. 2000;10:466–473. doi: 10.1016/s0962-8924(00)01841-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmings BA. PtdIns(3,4,5)P3 gets its message across. Science. 1997;277:534. doi: 10.1126/science.277.5325.534. [DOI] [PubMed] [Google Scholar]
- Corvera S, Czech MP. Direct targets of phosphoinositide 3-kinase products in membrane traffic and signal transduction. Trends Cell Biol. 1998;8:442–446. doi: 10.1016/s0962-8924(98)01366-x. [DOI] [PubMed] [Google Scholar]
- Downward J. Mechanisms and consequences of activation of protein kinase B/Akt. Curr Opin Cell Biol. 1998;10:262–267. doi: 10.1016/s0955-0674(98)80149-x. [DOI] [PubMed] [Google Scholar]
- Foxman EF, Campbell JJ, Butcher EC. Multistep navigation and the combinatorial control of leukocyte chemotaxis. J Cell Biol. 1997;139:1349–1360. doi: 10.1083/jcb.139.5.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zigmond SH. Chemotactic response of neutrophils. Am J Respir Cell Mol Biol. 1989;1:451–453. doi: 10.1165/ajrcmb/1.6.451. [DOI] [PubMed] [Google Scholar]
- Sapey E, Greenwood H, Walton G, Mann E, Love A, Aaronson N, Insall RH, Stockley RA, Lord JM. Phosphoinositide 3-kinase inhibition restores neutrophil accuracy in the elderly: toward targeted treatments for immunosenescence. Blood. 2014;123:239–248. doi: 10.1182/blood-2013-08-519520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson GJ, Milne L, Kulkarni S, Sasaki T, Walker S, Andrews S, Crabbe T, Finan P, Jones G, Jackson S, et al. PI(3)Kgamma has an important context-dependent role in neutrophil chemokinesis. Nat Cell Biol. 2007;9:86–91. doi: 10.1038/ncb1517. [DOI] [PubMed] [Google Scholar]
- Heit B, Liu L, Colarusso P, Puri KD, Kubes P. PI3K accelerates, but is not required for, neutrophil chemotaxis to fMLP. J Cell Sci. 2008;121:205–214. doi: 10.1242/jcs.020412. [DOI] [PubMed] [Google Scholar]
- Andrews S, Stephens LR, Hawkins PT. PI3K class IB pathway in neutrophils. Sci STKE. 2007;2007:cm3. doi: 10.1126/stke.4072007cm3. [DOI] [PubMed] [Google Scholar]
- Hawkins PT, Stephens LR, Suire S, Wilson M. PI3K signaling in neutrophils. Curr Top Microbiol Immunol. 2010;346:183–202. doi: 10.1007/82_2010_40. [DOI] [PubMed] [Google Scholar]
- Hannigan MO, Huang CK, Wu DQ. Roles of PI3K in neutrophil function. Curr Top Microbiol Immunol. 2004;282:165–175. doi: 10.1007/978-3-642-18805-3_6. [DOI] [PubMed] [Google Scholar]
- Cadwallader KA, Condliffe AM, McGregor A, Walker TR, White JF, Stephens LR, Chilvers ER. Regulation of phosphatidylinositol 3-kinase activity and phosphatidylinositol 3,4,5-trisphosphate accumulation by neutrophil priming agents. J Immunol. 2002;169:3336–3344. doi: 10.4049/jimmunol.169.6.3336. [DOI] [PubMed] [Google Scholar]
- Condliffe AM, Hawkins PT, Stephens LR, Haslett C, Chilvers ER. Priming of human neutrophil superoxide generation by tumour necrosis factor-alpha is signalled by enhanced phosphatidylinositol 3,4,5-trisphosphate but not inositol 1,4,5-trisphosphate accumulation. FEBS Lett. 1998;439:147–151. doi: 10.1016/s0014-5793(98)01358-1. [DOI] [PubMed] [Google Scholar]
- Germena G, Hirsch E. PI3Ks and small GTPases in neutrophil migration: two sides of the same coin. Mol Immunol. 2013;55:83–86. doi: 10.1016/j.molimm.2012.10.004. [DOI] [PubMed] [Google Scholar]
- Di Cristofano A, Pandolfi PP. The multiple roles of PTEN in tumor suppression. Cell. 2000;100:387–390. doi: 10.1016/s0092-8674(00)80674-1. [DOI] [PubMed] [Google Scholar]
- Maehama T, Dixon JE. PTEN: a tumour suppressor that functions as a phospholipid phosphatase. Trends Cell Biol. 1999;9:125–128. doi: 10.1016/s0962-8924(99)01519-6. [DOI] [PubMed] [Google Scholar]
- Rauh MJ, Kalesnikoff J, Hughes M, Sly L, Lam V, Krystal G. Role of Src homology 2-containing-inositol 5′-phosphatase (SHIP) in mast cells and macrophages. Biochem Soc Trans. 2003;31:286–291. doi: 10.1042/bst0310286. [DOI] [PubMed] [Google Scholar]
- Kisseleva MV, Cao L, Majerus PW. Phosphoinositide-specific inositol polyphosphate 5-phosphatase IV inhibits Akt/protein kinase B phosphorylation and leads to apoptotic cell death. J Biol Chem. 2002;277:6266–6272. doi: 10.1074/jbc.M105969200. [DOI] [PubMed] [Google Scholar]
- Astle MV, Horan KA, Ooms LM, Mitchell CA. The inositol polyphosphate 5-phosphatases: traffic controllers, waistline watchers and tumour suppressors? Biochem Soc Symp. 2007;74:161–181. doi: 10.1042/BSS0740161. [DOI] [PubMed] [Google Scholar]
- Iijima M, Devreotes P. Tumor suppressor PTEN mediates sensing of chemoattractant gradients. Cell. 2002;109:599–610. doi: 10.1016/s0092-8674(02)00745-6. [DOI] [PubMed] [Google Scholar]
- Funamoto S, Meili R, Lee S, Parry L, Firtel RA. Spatial and temporal regulation of 3-phosphoinositides by PI 3-kinase and PTEN mediates chemotaxis. Cell. 2002;109:611–623. doi: 10.1016/s0092-8674(02)00755-9. [DOI] [PubMed] [Google Scholar]
- Li Z, Dong X, Wang Z, Liu W, Deng N, Ding Y, Tang L, Hla T, Zeng R, Li L, et al. Regulation of PTEN by Rho small GTPases. Nat Cell Biol. 2005;7:399–404. doi: 10.1038/ncb1236. . [DOI] [PubMed] [Google Scholar]
- Wang F, Herzmark P, Weiner OD, Srinivasan S, Servant G, Bourne HR. Lipid products of PI(3)Ks maintain persistent cell polarity and directed motility in neutrophils. Nat Cell Biol. 2002;4:513–518. doi: 10.1038/ncb810. [DOI] [PubMed] [Google Scholar]
- Suire S, Condliffe AM, Ferguson GJ, Ellson CD, Guillou H, Davidson K, Welch H, Coadwell J, Turner M, Chilvers ER, et al. Gbetagammas and the Ras binding domain of p110gamma are both important regulators of PI(3)Kgamma signalling in neutrophils. Nat Cell Biol. 2006;8:1303–1309. doi: 10.1038/ncb1494. [DOI] [PubMed] [Google Scholar]
- Iijima M, Huang YE, Devreotes P. Temporal and spatial regulation of chemotaxis. Dev Cell. 2002;3:469–478. doi: 10.1016/s1534-5807(02)00292-7. [DOI] [PubMed] [Google Scholar]
- Vemula S, Shi J, Hanneman P, Wei L, Kapur R. ROCK1 functions as a suppressor of inflammatory cell migration by regulating PTEN phosphorylation and stability. Blood. 2010;115:1785–1796. doi: 10.1182/blood-2009-08-237222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papakonstanti EA, Ridley AJ, Vanhaesebroeck B. The p110delta isoform of PI 3-kinase negatively controls RhoA and PTEN. EMBO J. 2007;26:3050–3061. doi: 10.1038/sj.emboj.7601763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong K, Van Keymeulen A, Bourne HR. PDZRhoGEF and myosin II localize RhoA activity to the back of polarizing neutrophil-like cells. J Cell Biol. 2007;179:1141–1148. doi: 10.1083/jcb.200706167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Wang F, Van Keymeulen A, Herzmark P, Straight A, Kelly K, Takuwa Y, Sugimoto N, Mitchison T, Bourne HR. Divergent signals and cytoskeletal assemblies regulate self-organizing polarity in neutrophils. Cell. 2003;114:201–214. doi: 10.1016/s0092-8674(03)00555-5. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Morales FC, Kreimann EL, Georgescu MM. PTEN tumor suppressor associates with NHERF proteins to attenuate PDGF receptor signaling. EMBO J. 2006;25:910–920. doi: 10.1038/sj.emboj.7600979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Dowbenko D, Spencer S, Laura R, Lee J, Gu Q, Lasky LA. Interaction of the tumor suppressor PTEN/MMAC with a PDZ domain of MAGI3, a novel membrane-associated guanylate kinase. J Biol Chem. 2000;275:21477–21485. doi: 10.1074/jbc.M909741199. [DOI] [PubMed] [Google Scholar]
- Wu X, Hepner K, Castelino-Prabhu S, Do D, Kaye MB, Yuan XJ, Wood J, Ross C, Sawyers CL, Whang YE. Evidence for regulation of the PTEN tumor suppressor by a membrane-localized multi-PDZ domain containing scaffold protein MAGI-2. Proc Natl Acad Sci USA. 2000;97:4233–4238. doi: 10.1073/pnas.97.8.4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adey NB, Huang L, Ormonde PA, Baumgard ML, Pero R, Byreddy DV, Tavtigian SV, Bartel PL. Threonine phosphorylation of the MMAC1/PTEN PDZ binding domain both inhibits and stimulates PDZ binding. Cancer Res. 2000;60:35–37. [PubMed] [Google Scholar]
- Valiente M, Andres-Pons A, Gomar B, Torres J, Gil A, Tapparel C, Antonarakis SE, Pulido R. Binding of PTEN to specific PDZ domains contributes to PTEN protein stability and phosphorylation by microtubule-associated serine/threonine kinases. J Biol Chem. 2005;280:28936–28943. doi: 10.1074/jbc.M504761200. [DOI] [PubMed] [Google Scholar]
- Wu H, Feng W, Chen J, Chan LN, Huang S, Zhang M. PDZ domains of Par-3 as potential phosphoinositide signaling integrators. Mol Cell. 2007;28:886–898. doi: 10.1016/j.molcel.2007.10.028. [DOI] [PubMed] [Google Scholar]
- Billadeau DD. PTEN gives neutrophils direction. Nat Immunol. 2008;9:716–718. doi: 10.1038/ni0708-716. [DOI] [PubMed] [Google Scholar]
- Subramanian KK, Jia Y, Zhu D, Simms BT, Jo H, Hattori H, You J, Mizgerd JP, Luo HR. Tumor suppressor PTEN is a physiologic suppressor of chemoattractant-mediated neutrophil functions. Blood. 2007;109:4028–4037. doi: 10.1182/blood-2006-10-055319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- Yeung T, Ozdamar B, Paroutis P, Grinstein S. Lipid metabolism and dynamics during phagocytosis. Curr Opin Cell Biol. 2006;18:429–437. doi: 10.1016/j.ceb.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Vieira OV, Botelho RJ, Rameh L, Brachmann SM, Matsuo T, Davidson HW, Schreiber A, Backer JM, Cantley LC, Grinstein S. Distinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation. J Cell Biol. 2001;155:19–25. doi: 10.1083/jcb.200107069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox D, Berg JS, Cammer M, Chinegwundoh JO, Dale BM, Cheney RE, Greenberg S. Myosin X is a downstream effector of PI(3)K during phagocytosis. Nat Cell Biol. 2002;4:469–477. doi: 10.1038/ncb805. [DOI] [PubMed] [Google Scholar]
- Ai J, Maturu A, Johnson W, Wang Y, Marsh CB, Tridandapani S. The inositol phosphatase SHIP-2 down-regulates FcgammaR-mediated phagocytosis in murine macrophages independently of SHIP-1. Blood. 2006;107:813–820. doi: 10.1182/blood-2005-05-1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horan KA, Watanabe K, Kong AM, Bailey CG, Rasko JE, Sasaki T, Mitchell CA. Regulation of FcgammaR-stimulated phagocytosis by the 72-kDa inositol polyphosphate 5-phosphatase: SHIP1, but not the 72-kDa 5-phosphatase, regulates complement receptor 3 mediated phagocytosis by differential recruitment of these 5-phosphatases to the phagocytic cup. Blood. 2007;110:4480–4491. doi: 10.1182/blood-2007-02-073874. [DOI] [PubMed] [Google Scholar]
- Botelho RJ, Teruel M, Dierckman R, Anderson R, Wells A, York JD, Meyer T, Grinstein S. Localized biphasic changes in phosphatidylinositol-4,5-bisphosphate at sites of phagocytosis. J Cell Biol. 2000;151:1353–1368. doi: 10.1083/jcb.151.7.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairn GD, Ogata K, Botelho RJ, Stahl PD, Anderson RA, De Camilli P, Meyer T, Wodak S, Grinstein S. An electrostatic switch displaces phosphatidylinositol phosphate kinases from the membrane during phagocytosis. J Cell Biol. 2009;187:701–714. doi: 10.1083/jcb.200909025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Wange RL, Zhang N, Oppenheim JJ, Howard OM. Negative regulation of CXCR4-mediated chemotaxis by the lipid phosphatase activity of tumor suppressor PTEN. Blood. 2005;106:2619–2626. doi: 10.1182/blood-2004-08-3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacalle RA, Gomez-Mouton C, Barber DF, Jimenez-Baranda S, Mira E, Martinez AC, Carrera AC, Manes S. PTEN regulates motility but not directionality during leukocyte chemotaxis. J Cell Sci. 2004;117:6207–6215. doi: 10.1242/jcs.01545. [DOI] [PubMed] [Google Scholar]
- Costa C, Barberis L, Ambrogio C, Manazza AD, Patrucco E, Azzolino O, Neilsen PO, Ciraolo E, Altruda F, Prestwich GD, et al. Negative feedback regulation of Rac in leukocytes from mice expressing a constitutively active phosphatidylinositol 3-kinase gamma. Proc Natl Acad Sci USA. 2007;104:14354–14359. doi: 10.1073/pnas.0703175104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totani L, Piccoli A, Manarini S, Federico L, Pecce R, Martelli N, Cerletti C, Piccardoni P, Lowell CA, Smyth SS, et al. Src-family kinases mediate an outside-in signal necessary for beta2 integrins to achieve full activation and sustain firm adhesion of polymorphonuclear leucocytes tethered on E-selectin. Biochem J. 2006;396:89–98. doi: 10.1042/BJ20051924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mocsai A, Abram CL, Jakus Z, Hu Y, Lanier LL, Lowell CA. Integrin signaling in neutrophils and macrophages uses adaptors containing immunoreceptor tyrosine-based activation motifs. Nat Immunol. 2006;7:1326–1333. doi: 10.1038/ni1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowell CA, Fumagalli L, Berton G. Deficiency of Src family kinases p59/61hck and p58c-fgr results in defective adhesion-dependent neutrophil functions. J Cell Biol. 1996;133:895–910. doi: 10.1083/jcb.133.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mocsai A, Ligeti E, Lowell CA, Berton G. Adhesion-dependent degranulation of neutrophils requires the Src family kinases Fgr and Hck. J Immunol. 1999;162:1120–1126. [PubMed] [Google Scholar]
- Carbo C, Duerschmied D, Goerge T, Hattori H, Sakai J, Cifuni SM, White GC, 2nd, Chrzanowska-Wodnicka M, Luo HR, Wagner DD. Integrin-independent role of CalDAG-GEFI in neutrophil chemotaxis. J Leukoc Biol. 2010;88:313–319. doi: 10.1189/jlb.0110049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash-Koney M, Deevi RK, McFarlane C, Dib K. Exchange protein directly activated by cAMP 1 (Epac1) is expressed in human neutrophils and mediates cAMP-dependent activation of the monomeric GTPase Rap1. J Leukoc Biol. 2011;90:741–749. doi: 10.1189/jlb.0211108. [DOI] [PubMed] [Google Scholar]
- Lefort CT, Rossaint J, Moser M, Petrich BG, Zarbock A, Monkley SJ, Critchley DR, Ginsberg MH, Fassler R, Ley K. Distinct roles for talin-1 and kindlin-3 in LFA-1 extension and affinity regulation. Blood. 2012;119:4275–4282. doi: 10.1182/blood-2011-08-373118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camps M, Rückle T, Ji H, Ardissone V, Rintelen F, Shaw J, Ferrandi C, Chabert C, Gillieron C, Françon B, et al. Blockade of PI3Kgamma suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat Med. 2005;11:936–943. doi: 10.1038/nm1284. [DOI] [PubMed] [Google Scholar]
- Smith DF, Deem TL, Bruce AC, Reutershan J, Wu D, Ley K. Leukocyte phosphoinositide-3 kinase {gamma} is required for chemokine-induced, sustained adhesion under flow in vivo. J Leukoc Biol. 2006;80:1491–1499. doi: 10.1189/jlb.0306227. [DOI] [PubMed] [Google Scholar]
- Liu L, Puri KD, Penninger JM, Kubes P. Leukocyte PI3Kgamma and PI3Kdelta have temporally distinct roles for leukocyte recruitment in vivo. Blood. 2007;110:1191–1198. doi: 10.1182/blood-2006-11-060103. [DOI] [PubMed] [Google Scholar]
- Puri KD, Doggett TA, Douangpanya J, Hou Y, Tino WT, Wilson T, Graf T, Clayton E, Turner M, Hayflick JS, et al. Mechanisms and implications of phosphoinositide 3-kinase delta in promoting neutrophil trafficking into inflamed tissue. Blood. 2004;103:3448–3456. doi: 10.1182/blood-2003-05-1667. [DOI] [PubMed] [Google Scholar]
- Puri KD, Doggett TA, Huang CY, Douangpanya J, Hayflick JS, Turner M, Penninger J, Diacovo TG. The role of endothelial PI3Kgamma activity in neutrophil trafficking. Blood. 2005;106:150–157. doi: 10.1182/blood-2005-01-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Irie-Sasaki J, Jones RG, Oliveira-dos-Santos AJ, Stanford WL, Bolon B, Wakeham A, Itie A, Bouchard D, Kozieradzki I, et al. Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science. 2000;287:1040–1046. doi: 10.1126/science.287.5455.1040. [DOI] [PubMed] [Google Scholar]
- Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, Sozzani S, Mantovani A, Altruda F, Wymann MP. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science. 2000;287:1049–1053. doi: 10.1126/science.287.5455.1049. [DOI] [PubMed] [Google Scholar]
- Barber DF, Bartolomé A, Hernandez C, Flores JM, Redondo C, Fernandez-Arias C, Camps M, Rückle T, Schwarz MK, Rodríguez S, et al. PI3Kgamma inhibition blocks glomerulonephritis and extends lifespan in a mouse model of systemic lupus. Nat Med. 2005;11:933–935. doi: 10.1038/nm1291. [DOI] [PubMed] [Google Scholar]
- Sadhu C, Masinovsky B, Dick K, Sowell CG, Staunton DE. Essential role of phosphoinositide 3-kinase delta in neutrophil directional movement. J Immunol. 2003;170:2647–2654. doi: 10.4049/jimmunol.170.5.2647. [DOI] [PubMed] [Google Scholar]
- Sarraj B, Massberg S, Li Y, Kasorn A, Subramanian K, Loison F, Silberstein LE, von Andrian U, Luo HR. Myeloid-specific deletion of tumor suppressor PTEN augments neutrophil transendothelial migration during inflammation. J Immunol. 2009;182:7190–7200. doi: 10.4049/jimmunol.0802562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heit B, Robbins SM, Downey CM, Guan Z, Colarusso P, Miller BJ, Jirik FR, Kubes P. PTEN functions to “prioritize” chemotactic cues and prevent “distraction” in migrating neutrophils. Nat Immunol. 2008b;9:743–752. doi: 10.1038/ni.1623. [DOI] [PubMed] [Google Scholar]
- Heit B, Tavener S, Raharjo E, Kubes P. An intracellular signaling hierarchy determines direction of migration in opposing chemotactic gradients. J Cell Biol. 2002;159:91–102. doi: 10.1083/jcb.200202114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner A, Block H, Reichel CA, Varjosalo M, Gehart H, Sumara G, Gstaiger M, Krombach F, Zarbock A, Ricci R. Regulation of PTEN activity by p38delta-PKD1 signaling in neutrophils confers inflammatory responses in the lung. J Exp Med. 2012;209:2229–2246. doi: 10.1084/jem.20120677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo HR, Loison F. Constitutive neutrophil apoptosis: mechanisms and regulation. Am J Hematol. 2008;83:288–295. doi: 10.1002/ajh.21078. [DOI] [PubMed] [Google Scholar]
- Zhu D, Hattori H, Jo H, Jia Y, Subramanian KK, Loison F, You J, Le Y, Honczarenko M, Silberstein L, et al. Deactivation of phosphatidylinositol 3,4,5-trisphosphate/Akt signaling mediates neutrophil spontaneous death. Proc Natl Acad Sci USA. 2006;103:14836–14841. doi: 10.1073/pnas.0605722103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Sasaki T, Kozieradzki I, Wakeham A, Itie A, Dumont DJ, Penninger JM. SHIP is a negative regulator of growth factor receptor-mediated PKB/Akt activation and myeloid cell survival. Genes Dev. 1999;13:786–791. doi: 10.1101/gad.13.7.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardai S, Whitlock BB, Helgason C, Ambruso D, Fadok V, Bratton D, Henson PM. Activation of SHIP by NADPH oxidase-stimulated Lyn leads to enhanced apoptosis in neutrophils. J Biol Chem. 2002;277:5236–5246. doi: 10.1074/jbc.M110005200. [DOI] [PubMed] [Google Scholar]
- Yang KY, Arcaroli J, Kupfner J, Pitts TM, Park JS, Strasshiem D, Perng RP, Abraham E. Involvement of phosphatidylinositol 3-kinase gamma in neutrophil apoptosis. Cell Signal. 2003;15:225–233. doi: 10.1016/s0898-6568(02)00063-3. [DOI] [PubMed] [Google Scholar]
- Webb PR, Wang KQ, Scheel-Toellner D, Pongracz J, Salmon M, Lord JM. Regulation of neutrophil apoptosis: a role for protein kinase C and phosphatidylinositol-3-kinase. Apoptosis. 2000;5:451–458. doi: 10.1023/a:1009601220552. [DOI] [PubMed] [Google Scholar]
- Chen J, Tang H, Hay N, Xu J, Ye RD. Akt isoforms differentially regulate neutrophil functions. Blood. 2010;115:4237–4246. doi: 10.1182/blood-2009-11-255323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Loison F, Luo HR. Neutrophil spontaneous death is mediated by down-regulation of autocrine signaling through GPCR, PI3Kgamma, ROS, and actin. Proc Natl Acad Sci USA. 2010;107:2950–2955. doi: 10.1073/pnas.0912717107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza LR, Silva E, Calloway E, Cabrera C, McLemore ML. G-CSF activation of AKT is not sufficient to prolong neutrophil survival. J Leukoc Biol. 2013;93:883–893. doi: 10.1189/jlb.1211591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KE, Boyle KB, Davidson K, Chessa TA, Kulkarni S, Jarvis GE, Sindrilaru A, Scharffetter-Kochanek K, Rausch O, Stephens LR, et al. CD18-dependent activation of the neutrophil NADPH oxidase during phagocytosis of Escherichia coli or Staphylococcus aureus is regulated by class III but not class I or II PI3Ks. Blood. 2008;112:5202–5211. doi: 10.1182/blood-2008-04-149450. [DOI] [PubMed] [Google Scholar]
- Damen JE, Liu L, Rosten P, Humphries RK, Jefferson AB, Majerus PW, Krystal G. The 145-kDa protein induced to associate with Shc by multiple cytokines is an inositol tetraphosphate and phosphatidylinositol 3,4,5-triphosphate 5-phosphatase. Proc Natl Acad Sci USA. 1996;93:1689–1693. doi: 10.1073/pnas.93.4.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helgason CD, Damen JE, Rosten P, Grewal R, Sorensen P, Chappel SM, Borowski A, Jirik F, Krystal G, Humphries RK. Targeted disruption of SHIP leads to hemopoietic perturbations, lung pathology, and a shortened life span. Genes Dev. 1998;12:1610–1620. doi: 10.1101/gad.12.11.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber M, Helgason CD, Damen JE, Liu L, Humphries RK, Krystal G. The src homology 2-containing inositol phosphatase (SHIP) is the gatekeeper of mast cell degranulation. Proc Natl Acad Sci USA. 1998;95:11330–11335. doi: 10.1073/pnas.95.19.11330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sly LM, Ho V, Antignano F, Ruschmann J, Hamilton M, Lam V, Rauh MJ, Krystal G. The role of SHIP in macrophages. Front Biosci. 2007;12:2836–2848. doi: 10.2741/2276. [DOI] [PubMed] [Google Scholar]
- Leung WH, Tarasenko T, Bolland S. Differential roles for the inositol phosphatase SHIP in the regulation of macrophages and lymphocytes. Immunol Res. 2009;43:243–251. doi: 10.1007/s12026-008-8078-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry RV, Harris SJ, Ward SG. Fine tuning T lymphocytes: a role for the lipid phosphatase SHIP-1. Biochim Biophys Acta. 2010;1804:592–597. doi: 10.1016/j.bbapap.2009.09.019. [DOI] [PubMed] [Google Scholar]
- Hazen AL, Smith MJ, Desponts C, Winter O, Moser K, Kerr WG. SHIP is required for a functional hematopoietic stem cell niche. Blood. 2009;113:2924–2933. doi: 10.1182/blood-2008-02-138008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang OD, Lu J, Nombela-Arrieta C, Zhong J, Zhao L, Pivarnik G, Mondal S, Chai L, Silberstein LE, Luo HR. Deficiency of lipid phosphatase SHIP enables long-term reconstitution of hematopoietic inductive bone marrow microenvironment. Dev Cell. 2013;25:333–349. doi: 10.1016/j.devcel.2013.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishio M, Watanabe K, Sasaki J, Taya C, Takasuga S, Iizuka R, Balla T, Yamazaki M, Watanabe H, Itoh R, et al. Control of cell polarity and motility by the PtdIns(3,4,5)P3 phosphatase SHIP1. Nat Cell Biol. 2007;9:36–44. doi: 10.1038/ncb1515. [DOI] [PubMed] [Google Scholar]
- Mondal S, Subramanian KK, Sakai J, Bajrami B, Luo HR. Phosphoinositide lipid phosphatase SHIP1 and PTEN coordinate to regulate cell migration and adhesion. Mol Biol Cell. 2012;23:1219–1230. doi: 10.1091/mbc.E11-10-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phee H, Jacob A, Coggeshall KM. Enzymatic activity of the Src homology 2 domain-containing inositol phosphatase is regulated by a plasma membrane location. J Biol Chem. 2000;275:19090–19097. doi: 10.1074/jbc.M001093200. [DOI] [PubMed] [Google Scholar]
- Tu Z, Ninos JM, Ma Z, Wang JW, Lemos MP, Desponts C, Ghansah T, Howson JM, Kerr WG. Embryonic and hematopoietic stem cells express a novel SH2-containing inositol 5′-phosphatase isoform that partners with the Grb2 adapter protein. Blood. 2001;98:2028–2038. doi: 10.1182/blood.v98.7.2028. [DOI] [PubMed] [Google Scholar]
- Zhang S, Mantel C, Broxmeyer HE. Flt3 signaling involves tyrosyl-phosphorylation of SHP-2 and SHIP and their association with Grb2 and Shc in Baf3/Flt3 cells. J Leukoc Biol. 1999;65:372–380. doi: 10.1002/jlb.65.3.372. [DOI] [PubMed] [Google Scholar]
- Lamkin TD, Walk SF, Liu L, Damen JE, Krystal G, Ravichandran KS. Shc interaction with Src homology 2 domain containing inositol phosphatase (SHIP) in vivo requires the Shc-phosphotyrosine binding domain and two specific phosphotyrosines on SHIP. J Biol Chem. 1997;272:10396–10401. doi: 10.1074/jbc.272.16.10396. [DOI] [PubMed] [Google Scholar]
- Lemay S, Davidson D, Latour S, Veillette A. Dok-3, a novel adapter molecule involved in the negative regulation of immuno-receptor signaling. Mol Cell Biol. 2000;20:2743–2754. doi: 10.1128/mcb.20.8.2743-2754.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell MJ, Yuan Y, Anderson KE, Hibbs ML, Salem HH, Jackson SP. SHIP1 and Lyn kinase negatively regulate integrin alpha IIb beta 3 signaling in platelets. J Biol Chem. 2004;279:32196–32204. doi: 10.1074/jbc.M400746200. [DOI] [PubMed] [Google Scholar]
- Henderson LM, Chappel JB. NADPH oxidase of neutrophils. Biochim Biophys Acta. 1996;1273:87–107. doi: 10.1016/0005-2728(95)00140-9. [DOI] [PubMed] [Google Scholar]
- Heyworth PG, Cross AR, Curnutte JT. Chronic granulomatous disease. Curr Opin Immunol. 2003;15:578–584. doi: 10.1016/s0952-7915(03)00109-2. [DOI] [PubMed] [Google Scholar]
- Babior BM, Lambeth JD, Nauseef W. The neutrophil NADPH oxidase. Arch Biochem Biophys. 2002;397:342–344. doi: 10.1006/abbi.2001.2642. [DOI] [PubMed] [Google Scholar]
- Segal BH, Leto TL, Gallin JI, Malech HL, Holland SM. Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine. 2000;79:170–200. doi: 10.1097/00005792-200005000-00004. [DOI] [PubMed] [Google Scholar]
- Dinauer MC. Chronic granulomatous disease and other disorders of phagocyte function. Hematology Am Soc Hematol Educ Program. 2005;2005:89–95. doi: 10.1182/asheducation-2005.1.89. [DOI] [PubMed] [Google Scholar]
- Subramanian KK, Luo HR. Non-classical roles of NADPH-oxidase dependent reactive oxygen species in phagocytes. In: Hagg R, Kohlund S, editors. Granulocytes: Classification, toxic materials produced and pathology. Hauppauge NY: Nova Science Publishers, Inc; 2009. pp. 137–148. [Google Scholar]
- Dahlgren C, Karlsson A. Respiratory burst in human neutrophils. J Immunol Methods. 1999;232:3–14. doi: 10.1016/s0022-1759(99)00146-5. [DOI] [PubMed] [Google Scholar]
- Hattori H, Subramanian KK, Sakai J, Jia Y, Li Y, Porter TF, Loison F, Sarraj B, Kasorn A, Jo H, et al. Small-molecule screen identifies reactive oxygen species as key regulators of neutrophil chemotaxis. Proc Natl Acad Sci USA. 2010;107:3546–3551. doi: 10.1073/pnas.0914351107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AW, Kim C, Zhen L, Lowe JB, Kapur R, Petryniak B, Spaetti A, Pollock JD, Borneo JB, Bradford GB, et al. Deficiency of the hematopoietic cell-specific Rho family GTPase Rac2 is characterized by abnormalities in neutrophil function and host defense. Immunity. 1999;10:183–196. doi: 10.1016/s1074-7613(00)80019-9. [DOI] [PubMed] [Google Scholar]
- Li Z, Jiang H, Xie W, Zhang Z, Smrcka AV, Wu D. Roles of PLC-beta2 and -beta3 and PI3Kgamma in chemoattractant-mediated signal transduction. Science. 2000;287:1046–1049. doi: 10.1126/science.287.5455.1046. [DOI] [PubMed] [Google Scholar]
- Ding J, Vlahos CJ, Liu R, Brown RF, Badwey JA. Antagonists of phosphatidylinositol 3-kinase block activation of several novel protein kinases in neutrophils. J Biol Chem. 1995;270:11684–11691. doi: 10.1074/jbc.270.19.11684. [DOI] [PubMed] [Google Scholar]
- Vlahos CJ, Matter WF, Brown RF, Traynor-Kaplan AE, Heyworth PG, Prossnitz ER, Ye RD, Marder P, Schelm JA, Rothfuss KJ, et al. Investigation of neutrophil signal transduction using a specific inhibitor of phosphatidylinositol 3-kinase. J Immunol. 1995;154:2413–2422. [PubMed] [Google Scholar]
- Sue AQAK, Fialkow L, Vlahos CJ, Schelm JA, Grinstein S, Butler J, Downey GP. Inhibition of neutrophil oxidative burst and granule secretion by wortmannin: potential role of MAP kinase and renaturable kinases. J Cell Physiol. 1997;172:94–108. doi: 10.1002/(SICI)1097-4652(199707)172:1<94::AID-JCP11>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Condliffe AM, Davidson K, Anderson KE, Ellson CD, Crabbe T, Okkenhaug K, Vanhaesebroeck B, Turner M, Webb L, Wymann MP, et al. Sequential activation of class IB and class IA PI3K is important for the primed respiratory burst of human but not murine neutrophils. Blood. 2005;106:1432–1440. doi: 10.1182/blood-2005-03-0944. [DOI] [PubMed] [Google Scholar]
- DeLeo FR, Renee J, McCormick S, Nakamura M, Apicella M, Weiss JP, Nauseef WM. Neutrophils exposed to bacterial lipopolysaccharide upregulate NADPH oxidase assembly. J Clin Invest. 1998;101:455–463. doi: 10.1172/JCI949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch HC, Condliffe AM, Milne LJ, Ferguson GJ, Hill K, Webb LM, Okkenhaug K, Coadwell WJ, Andrews SR, Thelen M, et al. P-Rex1 regulates neutrophil function. Curr Biol. 2005;15:1867–1873. doi: 10.1016/j.cub.2005.09.050. [DOI] [PubMed] [Google Scholar]
- Lemmon MA. Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol. 2008;9:99–111. doi: 10.1038/nrm2328. [DOI] [PubMed] [Google Scholar]
- Luo HR, Huang YE, Chen JC, Saiardi A, Iijima M, Ye K, Huang Y, Nagata E, Devreotes P, Snyder SH. Inositol pyrophosphates mediate chemotaxis in Dictyostelium via pleckstrin homology domain-PtdIns(3,4,5)P3 interactions. Cell. 2003;114:559–572. doi: 10.1016/s0092-8674(03)00640-8. [DOI] [PubMed] [Google Scholar]
- Jia Y, Subramanian KK, Erneux C, Pouillon V, Hattori H, Jo H, You J, Zhu D, Schurmans S, Luo HR. Inositol 1,3,4,5-tetrakisphosphate negatively regulates phosphatidylinositol-3,4,5- trisphosphate signaling in neutrophils. Immunity. 2007;27:453–467. doi: 10.1016/j.immuni.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiardi A. Cell signalling by inositol pyrophosphates. Subcell Biochem. 2012;59:413–443. doi: 10.1007/978-94-007-3015-1_14. [DOI] [PubMed] [Google Scholar]
- Shears SB, Weaver JD, Wang H. Structural insight into inositol pyrophosphate turnover. Adv Biol Regul. 2013;53:19–27. doi: 10.1016/j.jbior.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shears SB. How versatile are inositol phosphate kinases? Biochem J. 2004;377:265–280. doi: 10.1042/BJ20031428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton A, Hu X, Saiardi A. Are inositol pyrophosphates signalling molecules? J Cell Physiol. 2009;220:8–15. doi: 10.1002/jcp.21763. [DOI] [PubMed] [Google Scholar]
- Saiardi A, Nagata E, Luo HR, Snowman AM, Snyder SH. Identification and characterization of a novel inositol hexakisphosphate kinase. J Biol Chem. 2001;276:39179–39185. doi: 10.1074/jbc.M106842200. [DOI] [PubMed] [Google Scholar]
- Prasad A, Jia Y, Chakraborty A, Li Y, Jain SK, Zhong J, Roy SG, Loison F, Mondal S, Sakai J, et al. Inositol hexakisphosphate kinase 1 regulates neutrophil function in innate immunity by inhibiting phosphatidylinositol-(3,4,5)-trisphosphate signaling. Nat Immunol. 2011;12:752–760. doi: 10.1038/ni.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Li H, Bajrami B, Kwak H, Cao S, Liu P, Zhou J, Zhou Y, Zhu H, Ye K, et al. Cigarette smoke (CS) and nicotine delay neutrophil spontaneous death via suppressing production of diphosphoinositol pentakisphosphate. Proc Natl Acad Sci USA. 2013;110:7726–7731. doi: 10.1073/pnas.1302906110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty A, Koldobskiy MA, Bello NT, Maxwell M, Potter JJ, Juluri KR, Maag D, Kim S, Huang AS, Dailey MJ, et al. Inositol pyrophosphates inhibit Akt signaling, thereby regulating insulin sensitivity and weight gain. Cell. 2010;143:897–910. doi: 10.1016/j.cell.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CF, Voglmaier SM, Bembenek ME, Saiardi A, Snyder SH. Identification and purification of diphosphoinositol pentakisphosphate kinase, which synthesizes the inositol pyrophosphate bis(diphospho)inositol tetrakisphosphate. Biochemistry. 1998;37:14998–15004. doi: 10.1021/bi981920l. [DOI] [PubMed] [Google Scholar]
- Saiardi A, Erdjument-Bromage H, Snowman AM, Tempst P, Snyder SH. Synthesis of diphosphoinositol pentakisphosphate by a newly identified family of higher inositol polyphosphate kinases. Curr Biol. 1999;9:1323–1326. doi: 10.1016/s0960-9822(00)80055-x. [DOI] [PubMed] [Google Scholar]
- Saiardi A, Caffrey JJ, Snyder SH, Shears SB. The inositol hexakisphosphate kinase family. Catalytic flexibility and function in yeast vacuole biogenesis. J Biol Chem. 2000;275:24686–24692. doi: 10.1074/jbc.M002750200. [DOI] [PubMed] [Google Scholar]
- Stuart JA, Anderson KL, French PJ, Kirk CJ, Michell RH. The intracellular distribution of inositol polyphosphates in HL60 promyeloid cells. Biochem J. 1994;303:517–525. doi: 10.1042/bj3030517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford PG, Rubin RP. Quantitative changes in inositol 1,4,5-trisphosphate in chemoattractant-stimulated neutrophils. J Biol Chem. 1986;261:15644–15647. [PubMed] [Google Scholar]
- Irvine RF, Schell MJ. Back in the water: the return of the inositol phosphates. Nat Rev Mol Cell Biol. 2001;2:327–338. doi: 10.1038/35073015. [DOI] [PubMed] [Google Scholar]
- Rhee SG. Regulation of phosphoinositide-specific phospholipase C. Annu Rev Biochem. 2001;70:281–312. doi: 10.1146/annurev.biochem.70.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Huang CK, Jiang H. Roles of phospholipid signaling in chemoattractant-induced responses. J Cell Sci. 2000b;113(17):2935–2940. doi: 10.1242/jcs.113.17.2935. (Pt. [DOI] [PubMed] [Google Scholar]
- Cicchetti G, Allen PG, Glogauer M. Chemotactic signaling pathways in neutrophils: from receptor to actin assembly. Crit Rev Oral Biol Med. 2002;13:220–228. doi: 10.1177/154411130201300302. [DOI] [PubMed] [Google Scholar]
- Choi KY, Kim HK, Lee SY, Moon KH, Sim SS, Kim JW, Chung HK, Rhee SG. Molecular cloning and expression of a complementary DNA for inositol 1,4,5-trisphosphate 3-kinase. Science. 1990;248:64–66. doi: 10.1126/science.2157285. [DOI] [PubMed] [Google Scholar]
- Nalaskowski MM, Bertsch U, Fanick W, Stockebrand MC, Schmale H, Mayr GW. Rat inositol 1,4,5-trisphosphate 3-kinase C is enzymatically specialized for basal cellular inositol trisphosphate phosphorylation and shuttles actively between nucleus and cytoplasm. J Biol Chem. 2003;278:19765–19776. doi: 10.1074/jbc.M211059200. [DOI] [PubMed] [Google Scholar]
- Dewaste V, Roymans D, Moreau C, Erneux C. Cloning and expression of a full-length cDNA encoding human inositol 1,4,5-trisphosphate 3-kinase B. Biochem Biophys Res Commun. 2002;291:400–405. doi: 10.1006/bbrc.2002.6456. [DOI] [PubMed] [Google Scholar]
- Sauer K, Cooke MP. Regulation of immune cell development through soluble inositol-1,3,4,5-tetrakisphosphate. Nat Rev Immunol. 2010;10:257–271. doi: 10.1038/nri2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer K, Park E, Siegemund S, French AR, Wahle JA, Sternberg L, Rigaud S, Jonsson AH, Yokoyama WM, Huang YH. Inositol tetrakisphosphate limits NK cell effector functions by controlling PI3K signaling. Blood. 2013;121:286–297. doi: 10.1182/blood-2012-05-429241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouillon V, Hascakova-Bartova R, Pajak B, Adam E, Bex F, Dewaste V, Van Lint C, Leo O, Erneux C, Schurmans S. Inositol 1,3,4,5-tetrakisphosphate is essential for T lymphocyte development. Nat Immunol. 2003;4:1136–1143. doi: 10.1038/ni980. [DOI] [PubMed] [Google Scholar]
- Wen BG, Pletcher MT, Warashina M, Choe SH, Ziaee N, Wiltshire T, Sauer K, Cooke MP. Inositol (1,4,5) trisphosphate 3 kinase B controls positive selection of T cells and modulates Erk activity. Proc Natl Acad Sci USA. 2004;101:5604–5609. doi: 10.1073/pnas.0306907101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marechal Y, Queant S, Polizzi S, Pouillon V, Schurmans S. Inositol 1,4,5-trisphosphate 3-kinase B controls survival and prevents anergy in B cells. Immunobiology. 2011;216:103–109. doi: 10.1016/j.imbio.2010.03.012. [DOI] [PubMed] [Google Scholar]
- Pouillon V, Marechal Y, Frippiat C, Erneux C, Schurmans S. Inositol 1,4,5-trisphosphate 3-kinase B (Itpkb) controls survival, proliferation and cytokine production in mouse peripheral T cells. Adv Biol Regul. 2013;53:39–50. doi: 10.1016/j.jbior.2012.08.001. [DOI] [PubMed] [Google Scholar]
- Huang YH, Grasis JA, Miller AT, Xu R, Soonthornvacharin S, Andreotti AH, Tsoukas CD, Cooke MP, Sauer K. Positive regulation of Itk PH domain function by soluble IP4. Science. 2007;316:886–889. doi: 10.1126/science.1138684. [DOI] [PubMed] [Google Scholar]
- Miller AT, Sandberg M, Huang YH, Young M, Sutton S, Sauer K, Cooke MP. Production of Ins(1,3,4,5)P4 mediated by the kinase Itpkb inhibits store-operated calcium channels and regulates B cell selection and activation. Nat Immunol. 2007;8:514–521. doi: 10.1038/ni1458. [DOI] [PubMed] [Google Scholar]
- Marechal Y, Pesesse X, Jia Y, Pouillon V, Pérez-Morga D, Daniel J, Izui S, Cullen PJ, Leo O, Luo HR, et al. Inositol 1,3,4,5-tetrakisphosphate controls proapoptotic Bim gene expression and survival in B cells. Proc Natl Acad Sci USA. 2007;104:13978–13983. doi: 10.1073/pnas.0704312104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y, Loison F, Hattori H, Li Y, Erneux C, Park SY, Gao C, Chai L, Silberstein LE, Schurmans S, et al. Inositol trisphosphate 3-kinase B (InsP3KB) as a physiological modulator of myelopoiesis. Proc Natl Acad Sci USA. 2008;105:4739–4744. doi: 10.1073/pnas.0800218105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y, Schurmans S, Luo HR. Regulation of innate immunity by inositol 1,3,4,5-tetrakisphosphate. Cell Cycle. 2008;7:2803–2808. doi: 10.4161/cc.7.18.6688. [DOI] [PubMed] [Google Scholar]
- Schell MJ, Erneux C, Irvine RF. Inositol 1,4,5-trisphosphate 3-kinase A associates with F-actin and dendritic spines via its N terminus. J Biol Chem. 2001;276:37537–37546. doi: 10.1074/jbc.M104101200. [DOI] [PubMed] [Google Scholar]
- Schell MJ, Irvine RF. Calcium-triggered exit of F-actin and IP(3) 3-kinase A from dendritic spines is rapid and reversible. Eur J Neurosci. 2006;24:2491–2503. doi: 10.1111/j.1460-9568.2006.05125.x. [DOI] [PubMed] [Google Scholar]
- Dewaste V, Moreau C, De Smedt F, Bex F, De Smedt H, Wuytack F, Missiaen L, Erneux C. The three isoenzymes of human inositol-1,4,5-trisphosphate 3-kinase show specific intracellular localization but comparable Ca2+ responses on transfection in COS-7 cells. Biochem J. 2003;374:41–49. doi: 10.1042/BJ20021963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billottet C, Grandage VL, Gale RE, Quattropani A, Rommel C, Vanhaesebroeck B, Khwaja A. A selective inhibitor of the p110delta isoform of PI 3-kinase inhibits AML cell proliferation and survival and increases the cytotoxic effects of VP16. Oncogene. 2006;25:6648–6659. doi: 10.1038/sj.onc.1209670. [DOI] [PubMed] [Google Scholar]
- Rommel C, Camps M, Ji H. PI3K delta and PI3K gamma: partners in crime in inflammation in rheumatoid arthritis and beyond? Nat Rev Immunol. 2007;7:191–201. doi: 10.1038/nri2036. [DOI] [PubMed] [Google Scholar]
- Ito K, Caramori G, Adcock IM. Therapeutic potential of phosphatidylinositol 3-kinase inhibitors in inflammatory respiratory disease. J Pharmacol Exp Ther. 2007;321:1–8. doi: 10.1124/jpet.106.111674. [DOI] [PubMed] [Google Scholar]
- Park SJ, Min KH, Lee YC. Phosphoinositide 3-kinase delta inhibitor as a novel therapeutic agent in asthma. Respirology. 2008;13:764–771. doi: 10.1111/j.1440-1843.2008.01369.x. [DOI] [PubMed] [Google Scholar]
- Venable JD, Ameriks MK, Blevitt JM, Thurmond RL, Fung-Leung WP. Phosphoinositide 3-kinase gamma (PI3Kgamma) inhibitors for the treatment of inflammation and autoimmune disease. Recent Pat Inflamm Allergy Drug Discov. 2010;4:1–15. doi: 10.2174/187221310789895603. [DOI] [PubMed] [Google Scholar]
- Blajecka K, Borgstrom A, Arcaro A. Phosphatidylinositol 3-kinase isoforms as novel drug targets. Curr Drug Targets. 2011;12:1056–1081. doi: 10.2174/138945011795677773. [DOI] [PubMed] [Google Scholar]
- Blunt MD, Ward SG. Targeting PI3K isoforms and SHIP in the immune system: new therapeutics for inflammation and leukemia. Curr Opin Pharmacol. 2012;12:444–451. doi: 10.1016/j.coph.2012.02.015. [DOI] [PubMed] [Google Scholar]
- Foster JG, Blunt MD, Carter E, Ward SG. Inhibition of PI3K signaling spurs new therapeutic opportunities in inflammatory/autoimmune diseases and hematological malignancies. Pharmacol Rev. 2012;64:1027–1054. doi: 10.1124/pr.110.004051. [DOI] [PubMed] [Google Scholar]
- Stenton GR, Mackenzie LF, Tam P, Cross JL, Harwig C, Raymond J, Toews J, Wu J, Ogden N, MacRury T, et al. Characterization of AQX-1125, a small-molecule SHIP1 activator: part 1. Effects on inflammatory cell activation and chemotaxis in vitro and pharmacokinetic characterization in vivo. Br J Pharmacol. 2013;168:1506–1518. doi: 10.1111/bph.12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenton GR, Mackenzie LF, Tam P, Cross JL, Harwig C, Raymond J, Toews J, Chernoff D, MacRury T, Szabo C. Characterization of AQX-1125, a small-molecule SHIP1 activator: part 2. Efficacy studies in allergic and pulmonary inflammation models in vivo. Br J Pharmacol. 2013;168:1519–1529. doi: 10.1111/bph.12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderlini P, Przepiorka D, Champlin R, Korbling M. Biologic and clinical effects of granulocyte colony-stimulating factor in normal individuals. Blood. 1996;88:2819–2825. [PubMed] [Google Scholar]
- Joos L, Tamm M. Breakdown of pulmonary host defense in the immunocompromised host: cancer chemotherapy. Proc Am Thorac Soc. 2005;2:445–448. doi: 10.1513/pats.200508-097JS. [DOI] [PubMed] [Google Scholar]
- Viscoli C, Varnier O, Machetti M. Infections in patients with febrile neutropenia: epidemiology, microbiology, and risk stratification. Clin Infect Dis. 2005;40(Suppl. 4):S240–S245. doi: 10.1086/427329. [DOI] [PubMed] [Google Scholar]
- Leung AN, Gosselin MV, Napper CH, Braun SG, Hu WW, Wong RM, Gasman J. Pulmonary infections after bone marrow transplantation: clinical and radiographic findings. Radiology. 1999;210:699–710. doi: 10.1148/radiology.210.3.r99mr39699. [DOI] [PubMed] [Google Scholar]
- Li Y, Jia Y, Pichavant M, Loison F, Sarraj B, Kasorn A, You J, Robson BE, Umetsu DT, Mizgerd JP, et al. Targeted deletion of tumor suppressor PTEN augments neutrophil function and enhances host defense in neutropenia-associated pneumonia. Blood. 2009;113:4930–4941. doi: 10.1182/blood-2008-06-161414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Prasad A, Jia Y, Roy SG, Loison F, Mondal S, Kocjan P, Silberstein LE, Ding S, Luo HR. Pretreatment with phosphatase and tensin homolog deleted on chromosome 10 (PTEN) inhibitor SF1670 augments the efficacy of granulocyte transfusion in a clinically relevant mouse model. Blood. 2011;117:6702–6713. doi: 10.1182/blood-2010-09-309864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schabbauer G, Matt U, Günzl P, Warszawska J, Furtner T, Hainzl E, Elbau I, Mesteri I, Doninger B, Binder BR, et al. Myeloid PTEN promotes inflammation but impairs bactericidal activities during murine pneumococcal pneumonia. J Immunol. 2010;185:468–476. doi: 10.4049/jimmunol.0902221. [DOI] [PubMed] [Google Scholar]
- Schell MJ, Letcher AJ, Brearley CA, Biber J, Murer H, Irvine RF. PiUS (Pi uptake stimulator) is an inositol hexakisphosphate kinase. FEBS Lett. 1999;461:169–172. doi: 10.1016/s0014-5793(99)01462-3. [DOI] [PubMed] [Google Scholar]
- Lemmon MA, Ferguson KM. Signal-dependent membrane targeting by pleckstrin homology (PH) domains. Biochem J. 2000;350(1):1–18. (Pt. [PMC free article] [PubMed] [Google Scholar]
- Maffucci T, Falasca M. Specificity in pleckstrin homology (PH) domain membrane targeting: a role for a phosphoinositide-protein co-operative mechanism. FEBS Lett. 2001;506:173–179. doi: 10.1016/s0014-5793(01)02909-x. [DOI] [PubMed] [Google Scholar]
- Rebecchi MJ, Scarlata S. Pleckstrin homology domains: a common fold with diverse functions. Annu Rev Biophys Biomol Struct. 1998;27:503–528. doi: 10.1146/annurev.biophys.27.1.503. [DOI] [PubMed] [Google Scholar]
- Kavran JM, Klein DE, Lee A, Falasca M, Isakoff SJ, Skolnik EY, Lemmon MA. Specificity and promiscuity in phosphoinositide binding by pleckstrin homology domains. J Biol Chem. 1998;273:30497–30508. doi: 10.1074/jbc.273.46.30497. . [DOI] [PubMed] [Google Scholar]