

Abstract
Despite the fact, that monoclonal antibodies are the fastest growing group of biopharmaceuticals in development, this is not true for the IgM class, which remains as enigmatic as ever. While more examples of usefulness of IgMs for medical applications are emerging, their recombinant production is still not common. In our study, stable monoclonal IgM producing CHO DG44 and HEK 293 cell lines, expressing two model IgM molecules (IgM-617 and IgM-012) were established. Recombinant cell lines were compared in regard of specific productivity, specific growth rate, maximal achieved antibody titer, gene copy numbers and transcription levels of transgene. IgM-617 cell lines were identified as high while IgM-012 clones were low producers. Although differences in gene copy numbers as well as in transcription levels were observed, they did not seem to be a limitation. Levels of relevant endoplasmic reticulum-stress related proteins were analyzed and no indications of unfolded protein response were detected. This could indicate that the difference in the intrinsic protein stability of our model proteins (as was previously observed on purified samples) might cause lower yields of IgM-012. Transcriptomics and/or proteomics follow up studies might be necessary for identification of potential bottlenecks in IgM producing cell lines.
Keywords: IgM, CHO, HEK293, Recombinant production, ER-stress, IgM assembly
Introduction
Therapeutic proteins are typically produced in mammalian cells, taking advantage of their ability to deliver human-like post translational modifications. Glycoproteins are predominantly produced in Chinese Hamster Ovary (CHO) cells, followed by murine myeloma cells (NS0 and Sp2/0), and Baby Hamster Kidney cells (Kyriakopoulos and Kontoravdi 2012). The largest group of glycoproteins approved by the regulatories (FDA: US Food and Drug Administration; EMA: European Medicines Agency) for pharmaceutical production (Walsh 2010; Reichert 2012) is represented by monoclonal antibodies which require proper expression systems to ensure low immunogenicity, in vivo efficiency and stability of produced antibodies [reviewed in (Jefferis 2009)].
Most of the currently approved therapeutic antibodies are Immunoglobulin G (IgG) class molecules.
Immunoglobulin M (IgM) class, which was up till recently in the marginal area of interest for pharmaceutical production, is one of the most phylogenetically ancient classes and can be found already in the Teleost fish (Castro et al. 2013; Fillatreau et al. 2013). IgMs have important immune and homeostasis related roles, such as (but not only) providing a protection against pathogens, recognition and removal of senescent cells, malignant cells, and other harmful particles.
Natural IgM antibodies have the ability to recognize and bind to tumor-specific post-translationally modified cell surface receptors, and to induce tumor-specific apoptosis (Vollmers and Brandlein 2009), manifesting their potential for anti-cancer therapy.
In 2010, Horn et al. evaluated the effects of a monoclonal IgM antibody employed against Pseudomonas aeruginosa infections (Horn et al. 2010), serving as an example of how IgMs can be used for alternative therapies when facing increasing numbers of antibiotic-resistant microorganisms.
A murine IgM monoclonal antibody binding to alpha beta T cell receptors and its usage in prevention of graft rejection following solid organ transplantation serves as an additional example. The clinical trials in patients after the solid-organ transplant are ongoing and this IgM has been granted the orphan designation by EMA in March 2013 (EMA report 2013).
IgM molecules can be found either as membrane-associated monomers on B-cells or secreted into the human plasma. As is typical for immunoglobulins, each monomer consists of two heavy chains (HC: called μ-chains in the case of IgM) and two light chains (LC). Additionally one joining chain (JC) is present to form a pentameric molecule. On each HC, five glycosylation sites are present (Shimizu et al. 1971) which enable pentameric IgM molecules to become heavily glycosylated. Secreted IgM consists predominantly of pentamers of the size of ~950 kDa. Additionally a small percentage of hexamers (~1,150 kDa) can be found which are formed by six monomers not associated with the JC [reviewed in Klimovich (2011)].
Because of their size, complexity and high level of glycosylation, the recombinant expression of IgMs is still not routinely achieved [reviewed in Mader et al. (2013a)]. However, because of the therapeutic potential of these remarkable molecules, and their potential for broad range of medical applications, elucidation of bottlenecks regarding recombinant production is necessary.
To identify possible bottlenecks in IgM expression, stable IgM producing CHO DG44 and HEK 293 cell lines have been established using two models. The IgM-012, an IgG-switched to IgM, and the IgM-617, a naturally occurring IgM.
Additionally, Calnexin, XBP1, GRP78, and PDI protein levels within established cell lines were also of interest, thanks to their roles in protein folding machinery and unfolded protein response. Calnexin is a type I transmembrane phosphoprotein of 90 kDa, associated with the endoplasmic reticulum (ER) membrane (Wada et al. 1991). It binds incompletely folded glycoproteins, and promotes their early folding as well as their retention within the ER (Ou et al. 1993; Hammond and Helenius 1994; Jackson et al. 1994). XBP1 (X-box binding protein 1) was first identified as a transcription factor binding to the cis-acting X box present in the promoter regions of MHC II genes (Liou et al. 1990). Later it was shown that it plays a vital role in unfolded protein response (Yoshida et al. 2001) and that unfolded protein response proteins may influence the antibody secretion (Gunn et al. 2004). GRP78 (also known as BiP) is the member of HSP70 family, which resides in the lumen of ER and is known to associate with the immunoglobulins heavy chains soon after their translocation into the ER (Melnick et al. 1994). Upon the presence of the light chains, heavy chain is released from GRP78 and forms stable interchain disulfide bridges with the light chain (Feige et al. 2009). In 2005, Borth et al. showed that increased level of GRP78 within antibody producing cells resulted in decrease of antibody production (Borth et al. 2005). PDI (protein disulfide isomerase) is the primary oxidant of cysteine thiols in disulfide bridges containing proteins and one of the most abundant proteins in the ER (Määttänen et al. 2010). Moderately elevated levels of PDI are necessary for the high production rate, but too high an up-regulation of this chaperone results in retention of multiple disulfide bonds containing proteins in the ER and thus decrease of secretion (Borth et al. 2005; Davis et al. 2000). Changes in intracellular levels of above mentioned chaperones may provide useful information about the physiological state of established recombinant cell lines.
In 1987 Munro and Pelham observed that several of the ER present chaperones share a common feature—carboxy-terminal sequence Lys–Asp–Glu–Leu (KDEL; Munro and Pelham 1987), which is now known to be universal retention and retrieval signal for ER resident proteins (Miesenböck and Rothman 1995). According to this information, if increased levels of different chaperones are expected in the ER because of recombinant protein production (or special treatment of cells) it is also expected to detect increased KDEL signal within cells. Increased KDEL signal in cells also suggests for cells to be undergoing ER stress. One of the ways how to cause cells to undergo ER stress is treatment with tunicamycin. Tunicamycin blocks the glycosylation of glycoproteins, which is necessary for their secretion. Glycoproteins are then accumulated in the ER, increasing the ER stress (Hickman et al. 1977). This strategy was used in this work to create a population of stressed cells used as a positive control when assessing the stress level of our recombinant cell lines.
The aim of this study was thereof to compare and evaluate best producing cell lines according to specific productivity (qp), specific growth rate (μ), maximal achieved antibody titer, relative gene copy numbers, transgene transcript levels, intracellular antibody content, and level of stress induced in the ER. Results were compared and conclusions were drawn in regard of existing literature to identify possible bottlenecks in IgM expression in mammalian cells.
Materials and methods
Generation of antibody expression vectors
IgM-617 is originally expressed by the EBV-transformed B cell line HB617, used for activation of unique population of CD4+ cells capable of tumor recognition and elimination (Jungfer et al. 2003; Jursik et al. 2009). IgM-012 takes its variable region sequence from the 2G12 IgG, famous, broadly neutralizing anti-HIV1 antibody (Kunert et al. 1998). Variable regions of above mentioned antibodies—which are defining the specificity—were cloned into the bicistronic vectors containing either heavy or light chain constant regions of human IgM, as described previously by Wolbank et al. (2003), providing the class-switch in the case of IgM-012.
Variable regions of IgM-617 and IgM-012 antibodies (property of Polymun Scientific GmbH, Klosterneuburg, Austria) were codon-optimized for Cricetulus griseus and synthesized (Life Technologies, Carlsbad, CA, USA), flanked by EcoRI and AgeI restriction sites in case of HC, and by AccIII and BsiWI in case of LC.
For the expression in CHO cells the variable regions of LC and HC were inserted into bicistronic pIRES vectors (Clonetech, Mountain View, CA, USA). Briefly, the variable HCs were inserted into a pIRES vector consisting of a SV40 promoter (Wolbank et al. 2003), a human μ-constant region backbone including leader sequence and the dihydrofolate reductase (DHFR) gene located after the IRES sequence. The LCs were inserted into a pIRES vector consisting of a CMV promoter, a human κ-constant backbone including leader sequence, the human JC located after the IRES sequence and a neomycin resistance cassette under the control of a SV40 promoter. Exchange of variable regions of heavy chain (vH) was done using the restriction enzymes EcoRI and AgeI and exchange of variable regions of light chain (vL) was performed by AccIII and BsiWI restriction enzymes (Fig. 1).
Fig. 1.

Plasmid map of pIRES_HC_DHFR and pIRES_LC_JC. Heavy chain of IgM is expressed under the SV40 promoter and followed by IRES and DHFR sequence. IgM constant region (IgM CH1–CH4) is the same in all HC constructs. Only the variable heavy chain (vH) was exchanged to express the desired antibody molecule. Kappa light chain is expressed under the CMV promoter and followed by IRES and joining chain sequence. Kappa LC constant region is the same in all LC constructs. Only the variable light chain (vL) was exchanged to express the desired antibody molecule. pIRES_LC_JC vector contains also an Neomycine resistance gene
As HEK 293 cells do have their own endogenous DHFR. For expression in these cells, the DHFR coding sequence in the HC containing vector was exchanged for the mutated DHFR sequence (using the restriction enzymes SmaI and XbaI). In mutated DHFR, arginine at position 22 of the enzyme is changed to leucine. Consequently of this, the mutated DHFR demonstrates much lower sensitivity to the presence of MTX compared to wt, thus enabling the gene amplification process also within cells containing endogenous DHFR (Simonsen and Levinson 1983). All of the cloned sequences were codon-optimized for CHO cells only.
Cell lines and transfection methods
CHO-DG44
CHO-DG44 (Life Technologies) DHFR deficient cells were routinely cultivated in suspension in DMEM/Ham’s F12 1:1 (Biochrom AG, Berlin, Germany) with 4 mM l-glutamine (PAA, Pasching, Austria), 0.1 mM hypoxanthine, 0.016 mM thymidine (Life Technologies), 0.1 % Pluronic F-68 (Sigma-Aldrich, St. Louis, MO, USA), and protein-free supplements (Polymun Scientific GmbH). Cells were transfected by DNA/polyethylenimine (PEI) polyplexes (4.8 pg plasmid DNA, 1:1 ratio HC/DHFR vector and LC/JC vector per cell) as described previously (Reisinger et al. 2009; Reinhart et al. 2012). Polyplexes were formed at a DNA:PEI ratio of 1:10 in ProCHO5 chemically defined medium (Lonza, Basel, Switzerland) supplemented with 4 mM l-glutamine (PAA) and 15 mg/l phenol red (Sigma-Aldrich). Selection was started 24 h post transfection by limited dilution subcloning in 96-well plate format using ProCHO5 medium supplemented with 0.5 mg ml−1 G418 (PAA). Gene amplification was initiated at 50 nM methotrexate (MTX) and further increased to 100 nM (MTX; Sigma-Aldrich). CHO clones were screened regularly by ELISA and best ones were again subcloned by the limited dilution method and selected for further analyses. Selected clones were expanded to spinner flasks (50 ml volume) and kept in routine culture in above described ProCHO5 medium with the presence of 100 nM MTX. Cells were seeded at a density of 2 × 105 cells ml−1 and split after reaching 1.6–2.0 × 106 cells ml−1 (after 3–4 days).
HEK-293
HEK-293 cells (ATCC® CRL-1537™) growing adherently were maintained in DMEM/Ham’s F12 1:1 (Biochrom AG) supplemented with 4 mM l-glutamine (PAA, Austria) and 10 % FBS (PAA) and split twice a week. Transfection was done using JetPRIME Transfection Reagent (PolyPlusFrance) according to manufacturer’s instructions. Briefly, 5 × 105 cells were seeded in 60 mm dishes in 5 ml medium 1 day prior to transfection. For transfection, 5 μg of plasmid DNA (1:1 ratio HC/dhfr vector and LC/JC vector) was added to 250 μl NaCl (provided in the kit), and separately 10 μl of JetPRIME reagent was added to 250 μl NaCl. These two preparations were mixed together and added to the seeded cells drop wise. 48 h post transfection selection process was initiated using DMEM/Ham’s medium supplemented with 0.5 mg ml−1 G418 (PAA). Gene amplification process was started with 50 nM MTX (Sigma-Aldrich) and increased stepwise up to 4 μM MTX. Transfectants were screened regularly by ELISA and best clones were subcloned twice by the limited dilution method and selected for further analyses. Selected clones were routinely cultured in T80 flasks in the above described DMEM/Ham’s medium with 4 μM MTX and split every 3–4 days in 1:5 ratio.
Sample preparation
Cell pellets and culture supernatants for determination of specific growth rate and productivity were collected during ten consecutive passages, 3–4 days after splitting. Similarly, samples for gene copy number, transcript, flow cytometric analyses and Western blot analysis were also taken during the exponential phase of the cell culture.
Cell pellets for gene copy number and transcript analysis (qPCR) were centrifuged (188 g 10 min), washed with PBS (PAA), frozen in liquid nitrogen and stored at −80 °C until the analysis. Samples for flow cytometric analyses were centrifuged (188 g 10 min), washed with PBS and fixed in 70 % ethanol and stored at +4 °C until the analysis.
Cell samples for Western blot analysis were centrifuged (188 g 10 min), washed with PBS and used immediately. To obtain cell lysates, cells were lysed in RIPA buffer (Sigma-Aldrich) and BCA Assay was performed to determine the total protein amount per cell using QuantiPro™ BCA Assay Kit (Sigma-Aldrich) according to manufacturer’s instructions. Corresponding cell culture supernatants were also used in Western blot analysis. HEK IgM-012 supernatant was concentrated 20-times using Amicon® Ultra Centrifugal filters (Merck, Darmstadt, Germany).
Determination of specific productivity
The qp of established monoclonal IgM expressing cell lines was calculated as picogram of IgM per cell per day (pg c−1 d−1) as explained in Lattenmayer et al. (2007a):
where ΔP represents the amount of product generated between ti and ti+1:
and CCD (cumulative cell days) represents the accumulation of cells during the process:
xi and xi+1 are cell numbers at time points ti and ti+1 and (ti+1 − ti) is the time interval between passages.
Cell number was determined by Z2™Coulter Counter® (Beckman Coulter, Brea, CA, USA) and concentration of IgM-012 and IgM-617 antibodies was analyzed by ELISA as previously described (Vorauer-Uhl et al. 2010). Briefly, immunosorbent plates (Thermo Scientific, Waltham, MA, USA) were coated with 1 μg ml−1 anti-human μ-chain antibody and anti-human kappa-chain directed antibody conjugated to HRP (both Sigma-Aldrich) was used as secondary antibody. Purified IgM from human serum (Sigma-Aldrich) was used as reference with a starting concentration of 200 ng ml−1. Reactions were visualized with o-phenylenediamine and H2O2 (Merck). Plates were measured with a microplate reader (Tecan, Männedorf, Switzerland) at 492 nm with a reference wavelength of 620 nm.
Western blot
To investigate oligomeric status of produced IgMs, an optimized electrophoretic method reported by Vorauer-Uhl et al. (2010) was used. Briefly, cell culture supernatant and cell lysates were loaded onto non-reducing NuPAGE gradient 3–12 % Bis–Tris gels (Life Technologies), run at 200 V for 60 min in Tris–Acetate SDS buffer (Life Technologies), and stained by Silver staining (Switzer et al. 1979). Protein marker used was NativeMark™ Unstained Protein Standard (Life Technologies). Semi-wet Western blots were prepared using the XCell II™ Blot Module (Life Technologies) with blotting time 60 min at 140 mA. IgMs were detected with anti-human μ-chain (1:5,000) and anti-human kappa-chain (1:5,000) antibodies which were HRP- or AP-conjugated (all of them from Sigma-Aldrich). JC was detected with primary rabbit anti-human J-chain (1:1,000) antibody (Antibodies-online, Aachen, Germany) and secondary anti-rabbit HRP-conjugated antibody (1:5,000; Sigma-Aldrich). HRP conjugated antibodies were detected by Super Signal® West Pico Chemiluminescent Substrate (Thermo Scientific) on the FUSION FX7™ chemiluminescence reader (Peqlab, Erlangen, Germany). AP conjugated antibodies were stained with NBT/BCIP (Promega, US-WI).
The presence of GRP78, calnexin, and beta-actin within the cell lysates was determined by 10 % PAGE and Western blot analyses. Antibodies used for detection of protein in Western blot were rabbit anti-calnexin (1:2,000) and rabbit anti-GRP78 (1:1,000) primary antibodies (all Abcam, Cambridge, UK) followed by the incubation of the membranes with polyclonal anti-rabbit HRP conjugated antibody (Sigma-Aldrich). Mouse anti-beta-actin antibody (1:2,000) was used to detect beta-actin levels within the cells.
Gene copy number and transgene transcript analyses
Preparation of genomic DNA (gDNA), complementary DNA (cDNA) and qPCR were performed according to Mader et al. (2013b). 10 ng of either gDNA or cDNA was used per reaction. Sequences of primers and probes (synthesized by Sigma Aldrich) are summarized in Table 1. Cricetulus griseus ACTB gene (GenBank U20114.1) and Homo sapiens ACTB gene (Chromosome 7 GRCh37.p13 primary assembly; NCBI refseq. NC_000007.13) share 82 % identities, therefore only one set of beta-actin primers was used for both cell lines. Primers and probes targeting protein disulfide isomerase (PDI: GenBank AF364317.1), xbox binding protein (XBP1: NCBI refseq: NM001244047.1; this primer probe set was designed to detect the spliced variant only) and 78 kDa glucose-regulated protein (GRP78: GenBank M171169.1; also known as binding immunoglobulin protein, BiP; summarized in Table 1) were used in qPCR analysis on a MiniOpticon™ System (BioRad, Hercules, CA, USA), using TaqMan probe principle. Calculation of gene copy numbers and transgene transcript levels was done relative to beta-actin housekeeping gene (Pfaffl 2001; Sommeregger et al. 2013).
Table 1.
Primers and probes for qPCR analysis
| β-actin s | TGAGCGCAAGTACTCTGTG |
| β-actin as | TTGCTGATCCACATCTCCTG |
| β-actin probe | [6FAM]CCATCCTGGCCTCACTGTCCACCT[TAM] |
| μ-chain s | GAGGAGGAGTGGAACACAGG |
| μ-chain as | ACGTTGTACAGGGTAGGTTTGC |
| μ-chain probe | [6FAM]CACGAGGCCCTGCCTAA[TAM] |
| kappa-chain s | TGTGCCTGCTGAACAACTTC |
| kappa-chain as | AGGCGTACACCTTGTGCTTC |
| kappa-chain probe | [6FAM]AGCAGCACCCTGACCCTGTCCAA[TAM] |
| J-chain s | CTCTGAACAACCGGGAGAAC |
| J-chain as | GTTCCGGTCGTAGGTGTAGC |
| J-chain probe | [6FAM]CACCTGTCCGACCTGTGCAAGAA[TAM] |
| PDI s | AGCACAACCAACTGCCTTTG |
| PDI as | GCTGCTTTCTTGAAGTTGCC |
| PDI probe | [6FAM]TCACTGAACAGACAGCCCCGA[TAM] |
| XBP1 s | TCCAAGGGAAATGGAGTAAGGC |
| XBP1 as | ATGTTCTGGGGAGGTGACAAC |
| XBP1 probe | [6FAM]CGGGTCTGCTGAGTCCGCAG[TAM] |
| GRP78 s | TGCAGAGAAGTTTGCTGAGG |
| GRP78 as | AGTTTACCGCCCAGCTTTTC |
| GRP78 probe | [6FAM]AGCGCATTGATACCAGGAACGAGT[TAM] |
Flow cytometry
Fixed cells were stained with anti-human μ-chain-FITC, anti-human kappa chain-FITC (both Sigma-Aldrich) or anti-GRP78, and anti-Calnexin (both Abcam) as primary and anti-rabbit-FITC (Sigma-Aldrich) as secondary antibodies. CHO DG44 cells were used as a negative control. As positive control, host cells were treated with 2 μg ml−1 tunicamycin (Sigma-Aldrich) for 12.5 h. Tunicamycin treated cells were fixed in 70 % ethanol incubated with anti-KDEL antibody (ELS AG, Lausen, Switzerland) and stained with anti-mouse-FITC (Sigma-Aldrich). Analysis was done using Gallios™ flow cytometer (Beckman Coulter). There were 10,000 events measured per sample.
Results
Evaluation of cell culture performance
Monoclonal CHO DG44 and HEK 293 cell lines expressing IgM-012 and IgM-617 were established by co-transfection of pIRES plasmids harboring either the “HC IRES DHFR” cassette (in case of CHO) or the “HC IRES DHFRmut” cassette (in case of HEK293) and the pIRES plasmid containing the “LC IRES JC” cassette. After screening and subcloning rounds the best producer of each cell line was chosen for further characterization.
Cell culture specific parameters are summarized in Table 2. Specific growth rate was equal for both recombinant CHO clones during routine culturing in spinner flasks. Maximal cell densities reached in spinner culture conditions were not differing significantly, however qp of CHO IgM-617 (25 pg c−1 d−1) cell line was almost seven times higher than the qp reached by CHO IgM-012 (3.59 pg c−1 d−1). There was also approximately seven-fold difference in volumetric titer values of “high producer” (CHO IgM-617) and “low producer” (CHO IgM-012).
Table 2.
Cell culture performance and intracellular HC/LC analysis
| μ (d−1) | qp (pg c−1 d−1) | vol. Titer (mg l−1) | MFI | ||
|---|---|---|---|---|---|
| HC | LC | ||||
| CHO/IgM-617 | 0.50 (±0.12) | 25.00 (±5.15) | 75.25 (±25.64) | 27.14 (±6.96) | 16.45 (±4.02) |
| CHO/IgM-012 | 0.51 (±0.10) | 3.59 (±0.81) | 9.94 (±3.51) | 28.31 (±3.34) | 16.10 (±2.62) |
| HEK/IgM-617 | 0.34 (±0.09) | 4.60 (±1.71) | 23.44 (±11.28) | 23.75 (±1.35) | 52.97 (±7.19) |
| HEK/IgM-012 | 0.33 (±0.08) | 0.21 (±0.08) | 0.92 (±0.31) | 10.86 (±0.17) | 34.61 (±2.00) |
Standard deviation in brackets represent seven consecutive passages (for μ, qp and vol titer) and three biological samples for flow cytometry analysis (MFI)
Recombinant HEK cell lines had more than 20-fold difference in qp as well as in volumetric titer values between “high producer” HEK IgM-617 (4.60 pg c−1 d−1) and “low producer” HEK IgM-012 (0.21 pg c−1 d−1), whereas there was no difference in specific growth rate (Table 2).
Genomic and transcript level analyses of recombinant cell lines
To identify the cause of this difference in productivity and for more detailed characterization of established cell lines, qPCR analysis was performed, in which gene copy number as well as transcriptional level of HC, LC, and JC were addressed.
CHO IgM-617 cell line had on a chromosomal level 37-times more HC, 31-times more LC and 21-times more JC than CHO IgM-012 cell line. HEK IgM-617 cell line had 4-times more HC and the same amounts of LC and JC as HEK IgM-012 cell line (data not shown).
This huge difference in gene copy numbers between high producers and low producers in case of CHO recombinant clones is not reflected at the transcription level. CHO IgM-617 cells had only 3.1 fold more transcript of HC, while for LC and JC there was no statistical significant difference (p = <0.001) between CHO IgM-012 and CHO IgM-617. HEK IgM-617 recombinant cells had slightly lower amounts of HC but a 1.9-fold higher LC and a 1.4-fold higher JC transcript level, compared to HEK IgM-012 cell line (Fig. 2).
Fig. 2.
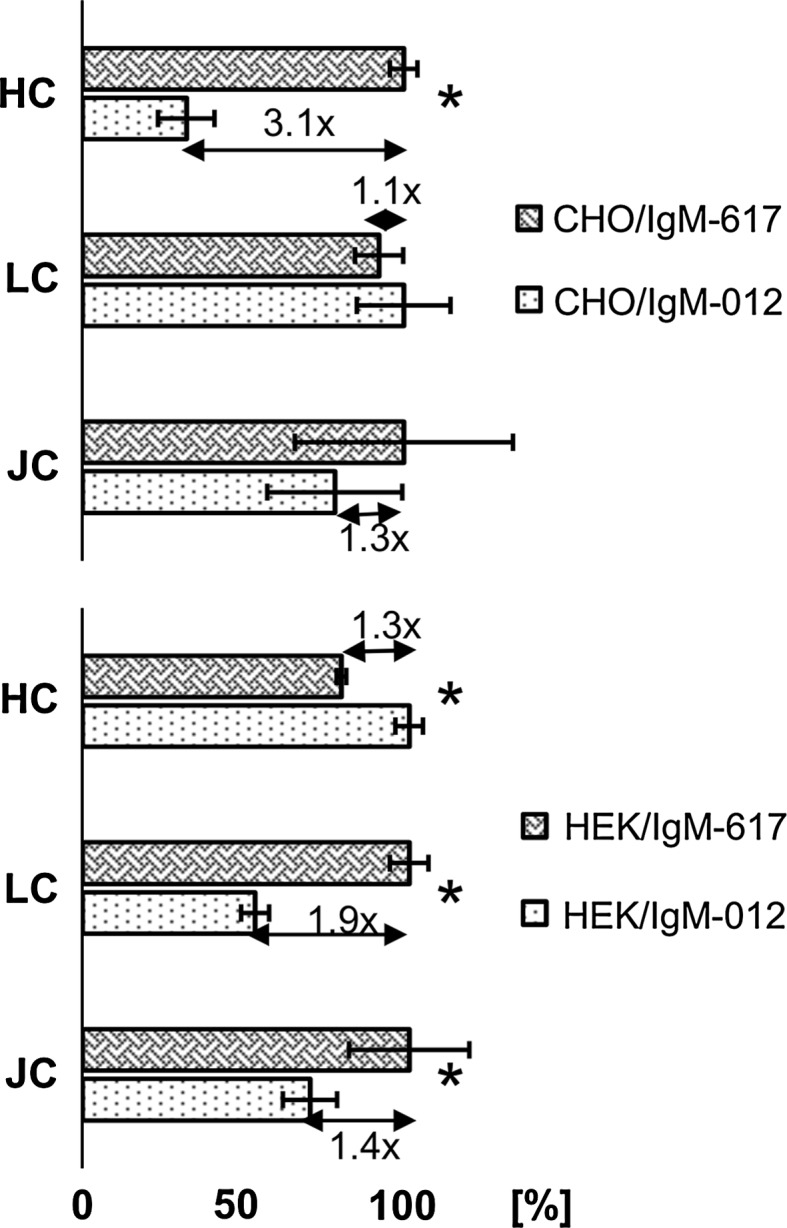
Transcriptional level of HC, LC and JC of recombinant CHO as well as HEK cells was addressed by the qPCR. Values were calculated relative to beta-actin housekeeping gene and normalized. Formula used for calculation: ratio = [ECq(ref)(ref)]:[ECq(target)(target)]. In CHO recombinant cells, the levels of LC and JC transcripts were not statistically significantly different. CHO IgM-617 cell line had 3.1-times more HC transcript than CHO IgM-012 cell line. Situation was different in recombinant HEK cells: HEK IgM-617 cell line had 1.3-times more HC, 1.9-times more LC and 1.4-times more JC transcript than HEK IgM-012 cell line. Statistically significant differences (t test) were tested at a confidence interval of p = <0.001 and are marked with an asterisk (*). Error bars indicate coefficient of variation (%CV)
Evaluation of intracellular antibody content
The intracellular content of HC and LC was analyzed using flow cytometry and Western blot. In flow cytometric analyses no differences could be observed between IgM-617 and IgM-012 producing CHO cells in terms of mean fluorescence intensity (MFI; Table 2). In case of HEK cells the fluorescence peak for HC in IgM-012 cells was broad and probably contains different subpopulations (data not shown). Despite of that, intracellular content of HC and LC was lower in HEK IgM-012 than in HEK IgM-617 (Table 2).
Additionally to the differences in qp, a difference in product quality between the IgM-012 and IgM-617 was observed. The predominant oligomeric IgM form in IgM-617 supernatant was the pentamer while it seemed that in IgM-012 lower oligomeric structures (trimers) were more pronounced (Fig. 3 lane 1, 3, 6 and 8). To explore the intracellular IgM content of all the recombinant clones and to be able to directly compare it with the oligomeric status of IgMs present in cell culture supernatants, samples were taken from the culture in the exponential phase and lysed in RIPA buffer. Equal amounts (7 μg) of total protein out of each prepared cell lysate were analyzed. Supernatants were used in their original state except for HEK IgM-012 supernatant, which was concentrated 20-times prior to use.
Fig. 3.

Western blot analysis of the intracellular and extracellular IgM content of recombinant CHO and HEK cells (7 μg of total cell lysate per lane). Intracellular IgM content of CHO IgM-617 (lanes 2 and 7, upper blot) and CHO IgM-012 (lanes 4 and 9, upper blot) as well as HEK IgM-617 (lanes 2 and 7, bottom blot) cells mostly consists of lower IgM oligomers. No IgM signal could be detected in HEK IgM-012 cell lysates (lanes 4 and 9, bottom blot). In extracellular fractions of CHO IgM-617 (lanes 1 and 6, upper blot), CHO IgM-012 (lanes 3 and 8, upper blot), HEK IgM-617 (lanes 1 and 6, bottom blot), and HEK IgM-012 (lanes 3 and 8, bottom blot), IgM pentamers can be observed. HEK IgM-012 supernatant was concentrated 20-times prior to use. IgM from human serum was used as the positive control (lanes 5 and 10)
The intracellular IgM content of CHO (Fig. 3 cell lysates lane 2, 4, 7, and 9, upper blots) and HEK293 (Fig. 3 cell lysates lane 2, 4, 7, and 9, bottom blots) cell lines was mainly composed of monomeric, dimeric, and trimeric IgM molecules. Additionally to above mentioned IgM fractions, a mixture of higher molecular weight forms was present. In case of CHO IgM-617 cells it seemed that these higher oligomeric structures were more pronounced (Fig. 3 lane 2 and 7, upper blots) than in the CHO IgM-012 cell line (Fig. 3 lanes 4 and 9, upper blots). This might be the consequence of higher productivity (and therefore higher “protein flow” in cells) of IgM-617 cell line, assuming the higher molecular weight fraction consisted of various folding and glycosylation IgM intermediates. Comparison of intra- (Fig. 3 lane 2, 4, 7, and 9) and extracellular (Fig. 3 lane 1, 3, 6, 8) IgM content showed that the pentameric IgM fraction is most exclusively present in the extracellular fraction, indicating that the majority of the pentameric IgMs were secreted. Additionally to the clearly visible pentameric form in the cells supernatants, trimeric, tetrameric IgMs and free antibody chains (LC, LC dimers, HC dimers) could be found. The trimeric IgM molecules in the extracellular fraction are of slightly higher molecular weight than the trimers from the intracellular fraction. This might be caused by incomplete glycosylation status of the intracellular trimers. Higher abundance of pentameric IgM was evident in IgM-617 expressing cell line (Fig. 3 lanes 1 and 6) compared to IgM-012 producers (Fig. 3 lanes 3 and 8). Except for the stronger band intensity of IgM-617 cell lines, which is expected for the high producing cell line, the overall intra- and extracellular fractionation pattern was similar for IgM-012 (low producer) as well as for IgM-617 (high producer) cell lines. As positive control IgM from the human serum was used (Fig. 3 lanes 5 and 10).
The evaluation of intra- and extracellular IgM content in HEK recombinant cells was more complex. The intracellular content of IgMs within these cells was so low, it almost gave no visible signal, when keeping the same amount of total cell protein loaded as was used for CHO clones (~7 μg). Additionally, the presence of fetal serum in the culture medium was distorting the shape of the bands on the gel and might be the source of unspecific signals in the areas of the blot corresponding to lower molecular weights (Fig. 3 lanes 3 and 8, bottom blot). This is especially evident in supernatant samples of HEK IgM-012, which was 20-times concentrated prior to use.
Despite of this assay-dependent inhomogeneity, similarly to recombinant CHO cells analyses, weak signals corresponding to monomers and trimers were present in the intracellular fraction of HEK IgM-617 (Fig. 3 lane 2 and 7, bottom blot). There was no IgM signal detected in HEK IgM-012 intracellular fraction (Fig. 3 lane 4 and 9, bottom blot). Pentamers were exclusively present only in extracellular fractions (Fig. 3 lane 1, 3, 6, and 9, bottom blot) and were more abundant in case of HEK IgM-617 than in HEK IgM-012. Additionally to the pentameric fraction, tetrameric and trimeric molecules could be found in HEK IgM-617 extracellular fraction (Fig. 3 lane 1 and 6, bottom blot). They are not clearly distinguishable in HEK IgM-012 (Fig. 3 lane 3 and 8, bottom blot), probably because of the presence of the serum. In both HEK IgM producing cell lines, free LCs were present in the extracellular fraction.
In all four recombinant cell lines (CHO and HEK), JC could only be detected extracellularly, but not inside the cells (data not shown).
Analysis of ER-stress
As a next step, we addressed presence of ER stress within our CHO IgM producing, recombinant cells. Relative amounts of PDI, XBP1, and GRP78 transcript levels were inspected by qPCR and none of the above mentioned genes in recombinant CHO cells was exceeding the amounts present in the CHO host cell line (Fig. 4). To evaluate if differences could be observed on the protein level flow cytometric and Western blot analysis detecting GRP78, calnexin and total chaperones (identified by the ER-retention signal KDEL) were performed (Fig. 5).
Fig. 4.

Transcriptional level of GRP78, XBP1 and PDI of recombinant CHO cells addressed by qPCR. Values were calculated relative to beta-actin housekeeping gene and normalized. Formula used for calculation: ratio = [ECq(ref)(ref)]:[ECq(target)(target)]. None of the ER-stress related proteins exceeded the values of the host CHO DG44 cells, indicating there is no ER-stress present in the recombinant cell lines
Fig. 5.

Presence of ER stress within CHO IgM producing cells: No significant difference between the host and recombinant cell lines in a GRP78 and b calnexin protein levels were observed (host cell line in black, IgM-012 in green, IgM-617 in orange colors). Measured 10,000 events per sample; y-axis % gates, 1 = 100 %. c Western blot analysis confirmed observed results—intensity of calnexin and GRP78 bands was similar for host as well as recombinant CHO cell lines. Beta-actin was used as internal control. d To explore overall chaperone level, ER-retention signal (KDEL) inside the cells were analyzed by flow cytometry. Host cell line was treated with 2 μg ml−1 tunicamycin for 12.5 h, to induce ER-stress (red color). Recombinant IgM-expressing CHO clones did not show increased chaperone levels (CHO host untreated in black, CHO, IgM-012 in green, and IgM-617 in orange color). (Color figure online)
Analysis of GRP78 revealed no significant difference between recombinant and host CHO cell lines in flow cytometry (Fig. 5a). The host cell line showed slightly increased signal compared to IgM producers indicating a slightly higher amount of GRP78 than in the recombinant cell lines. Similar results were obtained when testing for calnexin protein levels. However, in general the difference between the recombinant and host cells is not significant (Fig. 5b). Western blot analysis confirmed results from flow cytometry. The intensity of bands was similar in all recombinant and host CHO cells (Fig. 5c). To additionally explore the overall chaperone level in the ER, proteins harboring the ER retention signal “KDEL” were analyzed by flow cytometry. Positive control for ER-stress was induced by treatment of the host cell line with tunicamycin. Thereby 30 % of the CHO host cells population had increased chaperone levels, indicating ER stress. The MFI of the CHO host cells treated with tunicamycin (MFICHO+TM: 25.76) was increased twofold when compared to untreated CHO host cells (MFICHO−TM: 13.26). Recombinant CHO clones expressing either IgM-617 (MFI: 15.04) or IgM-012 (MFI: 13.69) did not show increased chaperone levels, indicating no ER stress present within these cell lines (Fig. 5d).
Discussion
In this study we describe the generation and characterization of stable recombinant CHO DG44 and HEK 293 cell lines producing model IgM antibodies IgM-012 and IgM-617, from which one (IgM-012) is class switched from IgG, and the other one (IgM-617) was generated from an IgM producing lymphocyte. In our work, we included two different cell lines: while focusing more on the data set obtained from examination of CHO recombinant clones, established HEK clones provided us with the proof, that our observations are not cell line-specific. In general, HEK cells demonstrated several times lower recombinant protein yields and specific productivity than CHO cells. This might be caused by alternative codon usage compared to CHO cells, not sufficient gene amplification, by cultivation media used, or just by the intrinsic production abilities of the cells. However, while being aware of this, the study is focusing more on the differences between IgM-012 and IgM-617 producing cells within the same cell line than on the interspecies comparison. Therefore also the gene amplification process was stopped at 100 nM MTX (CHO cells) and was not pursued further. In both, CHO as well as HEK cells, IgM-012 producing cell line showed several times lower specific productivity and antibody titer levels than IgM-617 producing cell line (sevenfold difference in CHO cells, 22-fold difference in HEK cells). Although CHO and HEK IgM-617 cell lines manifested same or higher gene copy numbers for HC, LC, and JC coding genes than IgM-012 cell lines (with dramatically higher values for CHO IgM-617 cells), this fact is not reflected at the transgene transcript and specific productivity level, similarly as shown previously (Lattenmayer et al. 2007b; Reisinger et al. 2008). These studies as well as our results indicated that many of coding gene copies are probably incorporated into places, which are not as favorable for transcription initiation or are not active at all. It can be therefore concluded, that observed specific productivities of our recombinant cell lines are not dependent on gene copy numbers.
When having a closer look at relative transgene transcript levels in recombinant cells obtained by qPCR, it could be seen that CHO IgM-617 cells have approximately 3-times more HC mRNA, than CHO IgM-012 cells. LC and JC mRNA levels are similar. On the other hand, HEK IgM-617 cells have slightly lower amounts of HC but slightly higher JC and almost twofold excess of LC mRNA when compared to the HEK IgM-012 cell line (Fig. 2). These results reveal that neither HC nor LC mRNA excess is necessary for superior expression titers and in our experiments also intracellular levels of HC and LC are comparable in CHO IgM-012 and CHO IgM-617 cell lines according to flow cytometric analysis as shown in Table 2 despite secretion rates differing significantly.
Additionally to the differences in qp also a difference in product quality of secreted IgMs between the IgM-012 and IgM-617 could be observed. The cell culture supernatant of IgM-617 is more abundant in pentameric IgMs while lower oligomeric IgM structures were present in the supernatant of IgM-012 (Fig. 3). This phenomenon was previously reported by Vorauer-Uhl et al. (2010). Following the hypothesis, that maybe the IgM-012 protein has some folding problems, which would explain the lower specific productivity, as well as the difference in abundance of pentamers in supernatants in IgM-012 producing cell lines, signs for any kind of ER stress (elevated levels of GRP78, PDI, XBP-1 and calnexin proteins and their transcript levels) were analyzed.
The GRP78, also known as BiP, is the only member of the Hsp70 protein family present in the ER (Munro and Pelham 1986). It is known to interact with the immunoglobulins HCs and prevent their aggregation before LCs are attached (Vanhove et al. 2001). It was observed previously, that with increasing specific mAb productivity in recombinant cells, number of molecular chaperones known to directly interact with nascent immunoglobulins exhibit a significant increase in abundance as well. Among these are GRP78 and PDI (Alete et al. 2005). GRP78 is also often coordinately up regulated together with GRP94 upon ER-stress as part of homeostatic response (Eletto et al. 2012).
PDI, the protein disulfide isomerase, catalyzes the formation of disulfide bonds between cysteine residues in immunoglobulins (and other proteins; Roth and Pierce 1987) and therefore plays important role as a chaperone in the ER. It was shown before, that when recombinant, antibody-producing CHO cells are transfected with the exogenous PDI, specific productivity increases significantly (Borth et al. 2005). Because IgM molecules are rich in disulfide bonds (Hendershot and Sitia 2005), it’s reasonable to assume, that the PDI level would be influenced by the over-expression of IgM molecules too.
XBP-1 is a vital part of the unfolded protein response in the ER and helps to ensure that appropriate adjustments of ER capacity are taking place (McGehee et al. 2009). It was argued before, that the XBP-1 driven differentiation of B cells to plasma cells (naturally expressing IgMs) is necessary for the cells to be able to cope with the excess of unfolded IgMs (Calfon et al. 2002). Later it was shown, that XBP-1 deficient plasma cells do not have problem with correctly folding neither secretory nor membrane-bound IgMs, however the absence of this protein is influencing the N-glycosylation.
Calnexin (as well as calreticulin) has an important function of binding unfolded and misfolded glycosylated and non-glycosylated proteins, keeping them in the ER and preventing their aggregation (Saito et al. 1999).
We assume if the folding is indeed the problem, we would be able to detect at least one of these chaperones in higher amounts. However, according to qPCR analysis, none of the tested genes in recombinant CHO cells exceeded the level of transcripts present in host cell line (Fig. 4). Same result was obtained, when flow cytometric analysis (detecting GRP78 and calnexin) and Western blotting was employed (Fig. 5).
The distribution of protein bands on the Western blot comparing supernatants and cell lysates of recombinant cells (Fig. 3) seem to differ slightly between high and low producer. It seems that IgM-012 expresses more trimeric IgM molecules than IgM-617 cell line. LCs are not the limiting factor, as they can be found as free LCs in both supernatants. There is no evidence for any protein folding problem in low producing cell line IgM-012 and the same is true also for high producing cell line, despite the presumptive higher IgM flow in the IgM-617 producing cells, as described in the results. It is possible, if the selection and amplification pressure was pushed further, and productivities of the recombinant cell lines would increase as well, that clear correlation between the antibody production and the relative mRNA amount of calnexin (or calreticulin), GRP78 and XBP-1 would be detectable as shown by Kober et al. (2012). Our “high producer” is probably still not producing enough to overwhelm the capacity of the secretory machinery inside the cells.
Finally we analyzed all of the ER resident proteins (this means also all of the chaperones), with an anti-KDEL specific antibody, as KDEL is the ER retention signal. Again, we could not observe any difference between the recombinant CHO and host cell line.
So far, we were not able to detect and identify the bottleneck causing several times different specific production rate between the low producer and the high producer. Nevertheless, Bentley and co-workers showed that different antibodies are expressed with characteristic efficiencies, that can vary up to 200-fold between antibodies (Bentley et al. 1998). This might be well possible also the case of our model IgM antibodies. Since both model IgMs contain the same IgM backbone structure (forecasting same N-glycosylation pattern), the production profile is then defined by the variable region sequence. As Ewert and colleagues showed, stability and yield of antibodies is limited by the least stable domain and by the strength of its interactions with surrounding domains (Ewert et al. 2003).
Another possibility is that lower secretion in case of IgM-012 producing cell lines is accounted for by slower assembly kinetics of subunits. But what is causing this? Is it the primary sequence of the antibody itself? If yes, which cellular factors are influenced? Or are more general factors responsible for changes in cellular physiology? It was shown before that silencing of pERp1 protein in plasma cells is lowering the secretion rate of IgM antibodies. This decrease in IgM secretion was accounted for by slower assembly kinetics of the subunits. However, it was also shown, pERp1 protein expression is tissue specific with highest level in lymphocyte specific tissues (van Anken et al. 2009).
Understanding physiological processes causing bottlenecks in IgM-producing cells and overcoming challenges in recombinant production of this class of immunoglobulins might provide entirely new set of therapeutic antibodies addressing different aspects of the immune system than what we already know from the work with IgGs for the medical use.
Acknowledgments
Funded by the PhD program “BioToP - Biomolecular Technology of Proteins” (Austrian Science Funds, FWF Project W1224). We thank Emilio Casanova (Ludwig Boltzmann Institute for Cancer Research, Vienna) for critical reading.
References
- Alete DE, Racher AJ, Birch JR, Stansfield SH, James DC, Smales CM. Proteomic analysis of enriched microsomal fractions from GS-NS0 murine myeloma cells with varying secreted recombinant monoclonal antibody productivities. Proteomics. 2005;5:4689–4704. doi: 10.1002/pmic.200500019. [DOI] [PubMed] [Google Scholar]
- Bentley KJ, Gewert R, Harris WJ. Differential efficiency of expression of humanized antibodies in transient transfected mammalian cells. Hybridoma. 1998;17:559–567. doi: 10.1089/hyb.1998.17.559. [DOI] [PubMed] [Google Scholar]
- Borth N, Mattanovich D, Kunert R, Katinger H. Effect of increased expression of protein disulfide isomerase and heavy chain binding protein on antibody secretion in a recombinant CHO cell line. Biotechnol Prog. 2005;21:106–111. doi: 10.1021/bp0498241. [DOI] [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- Castro R, Jouneau L, Pham HP, Bouchez O, Giudicelli V, Lefranc MP, Quillet E, Benmansour A, Cazals F, Six A, Fillatreau S, Sunyer O, Boudinot P. Teleost fish mount complex clonal IgM and IgT responses in spleen upon systemic viral infection. PLoS Pathog. 2013;9:e1003098. doi: 10.1371/journal.ppat.1003098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis R, Schooley K, Rasmussen B, Thomas J, Reddy P. Effect of PDI overexpression on recombinant protein secretion in CHO cells. Biotechnol Prog. 2000;16:736–743. doi: 10.1021/bp000107q. [DOI] [PubMed] [Google Scholar]
- Eletto D, Maganty A, Eletto D, Dersh D, Makarewich C, Biswas C, Paton JC, Paton AW, Doroudgar S, Glembotski CC, Argon Y. Limitation of individual folding resources in the ER leads to outcomes distinct from the unfolded protein response. J Cell Sci. 2012;125:4865–4875. doi: 10.1242/jcs.108928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewert S, Hubert T, Honegger A, Plückthun A. Biophysical properties of human antibody variable domains. J Mol Biol. 2003;325:531–553. doi: 10.1016/S0022-2836(02)01237-8. [DOI] [PubMed] [Google Scholar]
- Feige MJ, Groscurth S, Marcinowski M, Shimizu Y, Kessler H, Hendershot LM, Buchner J. An unfolded CH1 domain controls the assembly and secretion of IgG antibodies. Mol Cell. 2009;34:569–579. doi: 10.1016/j.molcel.2009.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillatreau S, Six A, Magadan S, Castro R, Sunyer JO, Boudinot P. The astonishing diversity of Ig classes and B cell repertoires in teleost fish. Front Immunol. 2013;4:28. doi: 10.3389/fimmu.2013.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn KE, Gifford NM, Mori K, Brewer JW. A role for the unfolded protein response in optimizing antibody secretion. Mol Immunol. 2004;41:919–927. doi: 10.1016/j.molimm.2004.04.023. [DOI] [PubMed] [Google Scholar]
- Hammond C, Helenius A. Quality control in the secretory pathway: retention of a misfolded viral membrane glycoprotein involves cycling between the ER, intermediate compartment and Golgi apparatus. J Cell Biol. 1994;126:41–52. doi: 10.1083/jcb.126.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendershot LM, Sitia R (2005) Molecular biology of B cells. In Honjo TAF NM (ed) Molecular biology of B cells. Elsevier, Amsterdam, pp 261–273
- Hickman S, Kulczycki A, Lynch RG, Kornfeld S. Studies of the mechanism of IgA and IgE secretion by plasma cells. J Biol Chem. 1977;252:4402–4408. [PubMed] [Google Scholar]
- Horn MP, Zuercher AW, Imboden MA, Rudolf MP, Lazar H, Wu H, Hoiby N, Fas SC, Lang AB. Preclinical in vitro and in vivo characterization of the fully human monoclonal IgM antibody KBPA101 specific for Pseudomonas aeruginosa serotype IATS-O11. Antimicrob Agents Chemother. 2010;54:2338–2344. doi: 10.1128/AAC.01142-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MR, Cohen-Doyle MF, Peterson PA, Williams DB. Regulation of MHC class I transport by the molecular chaperon, calnexin (p88, IP90) Science. 1994;263:384–387. doi: 10.1126/science.8278813. [DOI] [PubMed] [Google Scholar]
- Jefferis R. Glycosylation as a strategy to improve antibody-based therapeutics. Nat Rev Drug Discov. 2009;8:226–234. doi: 10.1038/nrd2804. [DOI] [PubMed] [Google Scholar]
- Jungfer H, Barchet H, Albert W, Widle U (2003) Tumoricidal T lymphocytes. US patent no. 6576466B2, filed Dec 04 1995 and published Jun 10 2003
- Jursik C, Prchal M, Grillari-Voglauer R, Drbal K, Fuertbauer E, Jungfer H, Albert WH, Steinhuber E, Hemetsberger T, Grillari J, Stockinger H, Katinger H. Large-scale production and characterization of novel CD4+ cytotoxic T cells with broad tumor specificity for immunotherapy. Mol Cancer Res. 2009;7:339–353. doi: 10.1158/1541-7786.MCR-07-2208. [DOI] [PubMed] [Google Scholar]
- Klimovich VB. IgM and its receptors: structural and functional aspects. Biochem Biokhimiia. 2011;76:534–549. doi: 10.1134/S0006297911050038. [DOI] [PubMed] [Google Scholar]
- Kober L, Zehe C, Bode J. Development of a novel ER stress based selection system for the isolation of highly productive clones. Biotechnol Bioeng. 2012;109:2599–2611. doi: 10.1002/bit.24527. [DOI] [PubMed] [Google Scholar]
- Kunert R, Rüker F, Katinger H. Molecular characterization of five neutralizing anti-HIV type I antibodies: identification of nonconventional D segments in the human monoclonal antibodies 2G12 and 2F5. AIDS Res Hum Retrovir. 1998;14:1115–1128. doi: 10.1089/aid.1998.14.1115. [DOI] [PubMed] [Google Scholar]
- Kyriakopoulos S, Kontoravdi C. Analysis of the landscape of biologically-derived pharmaceuticals in Europe: dominant production systems, molecule types on the rise and approval trends. Eur J Pharm Sci. 2012;48:428–441. doi: 10.1016/j.ejps.2012.11.016. [DOI] [PubMed] [Google Scholar]
- Lattenmayer C, Loeschel M, Schriebl K, Steinfellner W, Sterovsky T, Trummer E, Vorauer-Uhl K, Muller D, Katinger H, Kunert R. Protein-free transfection of CHO host cells with an IgG-fusion protein: selection and characterization of stable high producers and comparison to conventionally transfected clones. Biotechnol Bioeng. 2007;96:1118–1126. doi: 10.1002/bit.21183. [DOI] [PubMed] [Google Scholar]
- Lattenmayer C, Trummer E, Schriebl K, Vorauer-Uhl K, Mueller D, Katinger H, Kunert R. Characterisation of recombinant CHO cell lines by investigation of protein productivities and genetic parameters. J Biotechnol. 2007;128:716–725. doi: 10.1016/j.jbiotec.2006.12.016. [DOI] [PubMed] [Google Scholar]
- Liou HC, Boothby MR, Finn PW, Davidon R, Nabavi N, Zeleznik-Le NJ, Ting JP, Glimcher LH. A new member of the leucine zipper class of proteins that binds to the HLA DR_promoter. Science. 1990;247:1581–1584. doi: 10.1126/science.2321018. [DOI] [PubMed] [Google Scholar]
- Määttänen P, Gehring K, Bergeron JJM, Thomas DY. Protein quality control in the ER: the recognition of misfolded proteins. Semin Cell Dev Biol. 2010;21:500–511. doi: 10.1016/j.semcdb.2010.03.006. [DOI] [PubMed] [Google Scholar]
- Mader A, Chromikova V, Kunert R. Recombinant IgM expression in mammalian cells: a target protein challenging biotechnological production. Adv Biosci Biotechnol. 2013;4:38–43. doi: 10.4236/abb.2013.44A006. [DOI] [Google Scholar]
- Mader A, Prewein B, Zboray K, Casanova E, Kunert R (2013b) Exploration of BAC versus plasmid expression vectors in recombinant CHO cells. Appl Microbiol Biotechnol 97(9):4049-4054 [DOI] [PubMed]
- McGehee AM, Dougan SK, Klemm EJ, Shui G, Park B, Kim YM, Watson N, Wenk MR, Ploegh HL, Hu CC. XBP-1-deficient plasmablasts show normal protein folding but altered glycosylation and lipid synthesis. J Immunol. 2009;183:3690–3699. doi: 10.4049/jimmunol.0900953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnick J, Dul JL, Argon Y. Sequential interaction of chaperones BiP and GRP94 with immunoglobulin chains in the endoplasmic reticulum. Nature. 1994;370:373–375. doi: 10.1038/370373a0. [DOI] [PubMed] [Google Scholar]
- Miesenböck G, Rothman JE. The capacity to retrieve escaped ER proteins extends to the trans-most cisterna of the Golgi stack. J Cell Biol. 1995;129:309–319. doi: 10.1083/jcb.129.2.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro SP, Pelham HRB. An Hsp70-like protein in the ER: identity with the 78 kd glucose-regulated protein and immunoglobulin heavy chain binding protein. Cell. 1986;46:291–300. doi: 10.1016/0092-8674(86)90746-4. [DOI] [PubMed] [Google Scholar]
- Munro SP, Pelham HRB. A C-terminal signal prevents secretion of luminal ER proteins. Cell. 1987;48:899–907. doi: 10.1016/0092-8674(87)90086-9. [DOI] [PubMed] [Google Scholar]
- Ou WJ, Cameron PH, Thomas DY, Bergeron JJM. Association of folding intermediates of glycoproteins with calnexin during protein maturation. Nature. 1993;364:771–776. doi: 10.1038/364771a0. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichert JM (2012) Which are the antibodies to watch in 2012? mAbs 4:1–3 [DOI] [PMC free article] [PubMed]
- Reinhart D, Weik R, Kunert R. Recombinant IgA production: single step affinity purification using camelid ligands and product characterization. J Immunol Methods. 2012;378:95–101. doi: 10.1016/j.jim.2012.02.010. [DOI] [PubMed] [Google Scholar]
- Reisinger H, Steinfellner W, Stern B, Katinger H, Kunert R. The absence of effect of gene copy number and mRNA level on the amount of mAb secretion from mammalian cells. Appl Microbiol Biotechnol. 2008;81:701–710. doi: 10.1007/s00253-008-1701-1. [DOI] [PubMed] [Google Scholar]
- Reisinger H, Steinfellner W, Katinger H, Kunert R. Serum-free transfection of CHO cells with chemically defined transfection systems and investigation of their potential for transient and stable transfection. Cytotechnology. 2009;60:115–123. doi: 10.1007/s10616-009-9224-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EMA report (2013) Public summary of opinion on orphan designation. EMA/COMP/105743/2013 http://www.ema.europa.eu/docs/en_GB/document_library/Orphan_designation/2013/04/WC500141551.pdf. European Medicines Agency
- Roth RA, Pierce SB. In vivo cross-linking of protein disulfide isomerase to immunoglobulins. Biochemistry. 1987;26:4179–4182. doi: 10.1021/bi00388a001. [DOI] [PubMed] [Google Scholar]
- Saito Y, Ihara Y, Leach MR, Cohen-Doyle MF, Williams DB. Calreticulin functions in vitro as a molecular chaperone for both glycosylated and non-glycosylated proteins. EMBO J. 1999;18:6718–6729. doi: 10.1093/emboj/18.23.6718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu A, Putnam FW, Paul C, Clamp JR, Johnson I. Structure and role of the five glycopeptides of human IgM immunoglobulins. Nature. 1971;231:73–76. doi: 10.1038/newbio231073a0. [DOI] [PubMed] [Google Scholar]
- Simonsen CC, Levinson AD. Isolation and expression of an altered mouse dihydrofolate reductase cDNA. Proc Natl Acad Sci USA. 1983;80:2495–2499. doi: 10.1073/pnas.80.9.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommeregger W, Prewein B, Reinhart D, Mader A, Kunert R (2013) Transgene copy number comparison in recombinant mammalian cell lines: critical reflection of quantitative real-time PCR evaluation. Cytotechnology 65(5):811–818 [DOI] [PMC free article] [PubMed]
- Switzer RC, III, Merril CR, Shifrin S. A highly sensitive silver stain for detecting proteins and peptides in polyacrylamide gels. Anal Biochem. 1979;98:231–237. doi: 10.1016/0003-2697(79)90732-2. [DOI] [PubMed] [Google Scholar]
- van Anken E, Pena F, Hafkemeijer N, Christis C, Romijn EP, Grauschopf U, Oorschot VM, Pertel T, Engels S, Ora A, Lastun V, Glockshuber R, Klumperman J, Heck AJ, Luban J, Braakman I. Efficient IgM assembly and secretion require the plasma cell induced endoplasmic reticulum protein pERp1. Proc Natl Acad Sci USA. 2009;106:17019–17024. doi: 10.1073/pnas.0903036106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhove M, Usherwood YK, Hendershot LM. Unassembled Ig heavy chains do not cycle from BiP in vivo but require light chains to trigger their release. Immunity. 2001;15:105–114. doi: 10.1016/S1074-7613(01)00163-7. [DOI] [PubMed] [Google Scholar]
- Vollmers HP, Brandlein S. Natural antibodies and cancer. New Biotechnol. 2009;25:294–298. doi: 10.1016/j.nbt.2009.03.016. [DOI] [PubMed] [Google Scholar]
- Vorauer-Uhl K, Wallner J, Lhota G, Katinger H, Kunert R. IgM characterization directly performed in crude culture supernatants by a new simple electrophoretic method. J Immunol Methods. 2010;359:21–27. doi: 10.1016/j.jim.2010.05.003. [DOI] [PubMed] [Google Scholar]
- Wada I, Rindress D, Cameron PH, Ou WJ, Doherty JJ, Louvard D, Bell AW, Dignard D, Thomas DY, Bergeron JJM. SSRα and associated calnexin are major calcium binding proteins of the endoplasmic reticulum membrane. J Biol Chem. 1991;266:19599–19610. [PubMed] [Google Scholar]
- Walsh G. Biopharmaceutical benchmarks 2010. Nat Biotechnol. 2010;28:917–924. doi: 10.1038/nbt0910-917. [DOI] [PubMed] [Google Scholar]
- Wolbank S, Kunert R, Stiegler G, Katinger H. Characterization of human class-switched polymeric (immunoglobulin M [IgM] and IgA) anti-human immunodeficiency virus type 1 antibodies 2F5 and 2G12. J Virol. 2003;77:4095–4103. doi: 10.1128/JVI.77.7.4095-4103.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/S0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]