

Abstract
Altered RNA metabolism is a key pathophysiological component causing several neurodegenerative diseases. Genetic mutations causing neurodegeneration occur in coding and noncoding regions of seemingly unrelated genes whose products do not always contribute to the gene expression process. Several pathogenic mechanisms may coexist within a single neuronal cell, including RNA/protein toxic gain-of-function and/or protein loss-of-function. Genetic mutations that cause neurodegenerative disorders disrupt healthy gene expression at diverse levels, from chromatin remodelling, transcription, splicing, through to axonal transport and repeat-associated non-ATG (RAN) translation. We address neurodegeneration in repeat expansion disorders [Huntington's disease, spinocerebellar ataxias, C9ORF72-related amyotrophic lateral sclerosis (ALS)] and in diseases caused by deletions or point mutations (spinal muscular atrophy, most subtypes of familial ALS). Some neurodegenerative disorders exhibit broad dysregulation of gene expression with the synthesis of hundreds to thousands of abnormal messenger RNA (mRNA) molecules. However, the number and identity of aberrant mRNAs that are translated into proteins – and how these lead to neurodegeneration – remain unknown. The field of RNA biology research faces the challenge of identifying pathophysiological events of dysregulated gene expression. In conclusion, we discuss current research limitations and future directions to improve our characterization of pathological mechanisms that trigger disease onset and progression.
Keywords: altered gene expression, neurodegeneration, protein loss-of-function, RNA/protein toxic gain-of-function, RNA-mediated diseases
Introduction
RNA-mediated neurodegeneration is implicated in the causes of Huntington's disease (HD), spinocerebellar ataxias (SCAs), spinal muscular atrophy (SMA) and major subtypes of amyotrophic lateral sclerosis (ALS). In contrast, the current neuropathological classification of Parkinson's disease (PD), Alzheimer's disease (AD) and prion disease relates to the abnormal accumulation of misfolded and aggregated proteins in the brain (synucleinopathies, tauopathies and prion protein accumulation, respectively).
HD is a fatal disease initiated by selective death of neurons in the striatum before neurodegeneration spreads to other cerebral regions. Startlingly, some post mortem HD brains may have lost up to 25% of their weight. HD usually develops in patients aged 35–45 years, although onset can occur from childhood to old age. Caucasian populations have a high and likely underestimated prevalence (5–7 cases in 100 000 individuals). The disease progressively affects movement (chorea, particularly impaired swallowing/speech), behaviour and cognitive functions. Neuropsychiatric problems worsen over time and ultimately lead to dementia [1]. HD is caused by an autosomal-dominant glutamine-encoding CAG trinucleotide expansion in the polymorphic exon 1 of the huntingtin gene (HTT). Unaffected individuals normally carry 11–34 CAG repeats while HD patients have >36–250 CAG repeats [2,3].
The SCAs form a large heterogeneous group of autosomal-dominant diseases with typical onset at 30–50 years old. They are caused by repeat expansions in both coding and noncoding regions of multiple genes. Repeat expansions are formed by CAG repeat sequences in most cases; however, some subtypes are characterized by the presence of trinucleotide (CTG), pentanucleotide (ATTCT, TGGAA) or hexanucleotide (GGCCTG) repeats. SCAs are slowly progressive diseases involving neurodegeneration of the cerebellum and spinal cord. They are associated with dysarthria, poor coordination of gait and fine movements, but with retention of cognitive function [4]. They often remain undiagnosed.
SMA is the second most common genetic cause of infant mortality after cystic fibrosis (approximately 1 in 10 000 newborns). Lower motor neurons in the anterior horns of the spinal cord progressively degenerate, leading to muscle atrophy, paralysis and often fatal respiratory failure [5]. Babies affected by aggressive forms of SMA (type 0 and I) never sit and have a very short life expectancy (<2 years). Intermediate SMA type II children sit but do not stand. Types III and IV SMA have impaired gait and mobility; however, their disease does not affect life expectancy. Mental abilities remain unaffected and may be higher than average. This autosomal-recessive neurodegenerative disease is due to homozygous disruption of the survival of motor neuron 1 (SMN1) gene [6] which results in reduced levels of the ubiquitous SMN protein. The greater the reduction in SMN level, the greater severity of disease.
ALS is characterized by relentless degeneration of upper and lower motor neurons, which leads to progressive paralysis and death usually within 3–5 years from symptom onset. ALS is generally an adult disease with age of onset peaking at around 55–60 years of age. It is the most common form of motor neurodegenerative disease and has a prevalence of 6–10 cases per 100 000 individuals, with a lifetime risk of 1 in 400 [7]. Approximately 5% of ALS cases are inherited, usually in an autosomal dominant manner (familial ALS – fALS) while the majority of cases, approximately 95%, occur sporadically (sALS) [8]. ALS is a multifactorial disease in which mutations in multiple genes cause a direct disruption of mRNA metabolism 9–11.
Here, we review the molecular mechanisms by which genetic mutations can alter normal gene expression in neurodegenerative diseases. We also discuss research limitations and future strategies to better understand the functional cellular consequences of widespread RNA dysregulation.
Eukaryotic expression of genes
Eukaryotic gene expression is tightly regulated and integrates activating and repressive mechanisms [12], directionality [13], RNA surveillance [14] and regulated protein degradation [15]. Normal eukaryotic gene expression depends on a very large number of protein-coding mRNAs, protein factors and noncoding RNAs (ribosomal, transfer, small nuclear RNA and regulatory micro-RNAs). RNA molecules associate with RNA-binding proteins to form ribonucleoprotein particles (RNPs). The composition of RNPs dictates the function and fate of the RNA molecules [16]. Most eukaryotic genomes encode hundreds of RNA-binding proteins with diverse biological activities [17]. Mutations that disrupt RNPs are prone to cause disease, whether they affect a particular protein or RNA molecule. In particular, several neurological disorders are associated with transcription and pre-mRNA splicing alterations [18,19], as well as with dysregulation of protein synthesis [20].
Biogenesis/processing of mRNA is orchestrated in separate, but extensively coupled steps within the nucleus [21], including transcription, mRNA processing events (capping, splicing, cleavage/poly-adenylation) and nuclear export [22]. RNA-polymerase II transcribes pre-mRNAs from the genomic DNA [23]. Nascent mRNA transcripts are co-transcriptionally capped at their 5′-end [24]. The spliceosome removes introns and stitches together coding exons in a process called splicing [25]. Alternative splicing allows differential linking of various exons, increasing the repertoire of encoded proteins from a single gene. Finally, processing mRNAs are cleaved and poly-adenylated at the 3′-end [26,27]. Mature mRNAs that have been processed are licensed for nuclear export 28–30. Cytoplasmic mRNAs are circularized before being translated into proteins by the ribosome [31,32]; however, they can also be transported through the axon of neuronal cells [33] to allow localized translation of proteins within the axonal compartment. Figure 1 illustrates the mechanistic steps involved in the neuronal expression of protein-coding genes.
Figure 1.
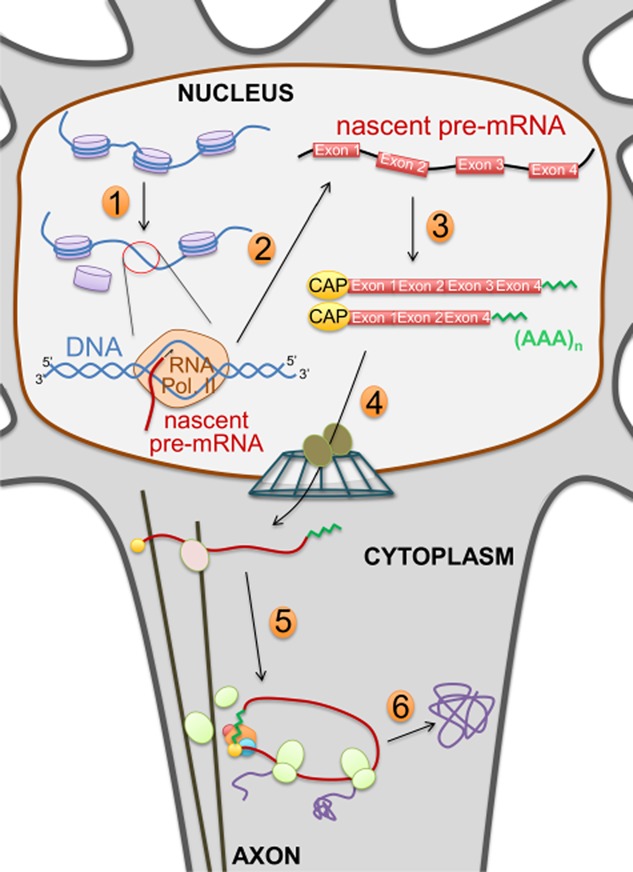
Neuronal expression of protein-coding genes. Diagram highlighting mRNA biogenesis and processing, nuclear export, axonal transport and mRNA translation. (1) Chromatin remodelling; (2) RNA polymerase II (RNA Pol. II) dependent transcription; (3) co-transcriptional processing: 5′-end capping, splicing/alternative spicing, 3′-end cleavage and poly-adenylation; (4) nuclear export of mRNAs; (5) axonal transport of mRNAs; and (6) translation of mRNAs for the biosynthesis of proteins.
HD
The HTT gene encodes a very large protein huntingtin (HTT) of over 3000 amino acids (approximately 350 kDa) which is essential for embryonic development and brain function [34]. The domain structure of HTT does not resemble any known proteins and its precise molecular function still remains unclear.
Poly-glutamine (poly-Q) amino-terminal truncations of HTT, generated through aberrant splicing of HTT in HD [35], inappropriately accumulate within the nucleus through altered interactions with the nuclear pore protein translocated promoter region (Tpr) [36], and form ubiquitinated neuronal intranuclear inclusions in human [37] and mouse [38] HD brains. α-synuclein, a component of Lewy bodies in PD brains, was found in HTT inclusions and independently in cytoplasmic filaments in human and mice HD neurons. Interestingly, the number of HTT inclusions is dependent on α-synuclein expression levels [39], and formation of inclusion bodies was shown to be associated with improved survival rather than death upon live cell imaging [40]. HD pathophysiology is complex, and there are several pathophysiological mechanisms that lead to broadly dysregulated gene expression [41]. Approximately 200 mRNAs are dysregulated in HD brains, and the level of dysregulation correlates with disease severity in the affected brain regions 42–44. Poly-Q expansions trigger both HTT protein loss-of-function and toxic gain-of-function effects [45]. Figure 2 generally illustrates these mechanisms. In addition, the CAG expansion may also contribute to HD pathogenesis via RNA toxic gain-of-function through RNA foci formation and/or partial sequestration of the muscleblind (MBNL1) splicing factor [46] and nucleolin (NCL) [47]. Panels A and B of Figure 3 depict in general these paradigms/mechanisms. NCL sequestration leads to down-regulation of rRNA transcription and nucleolar stress. The CAG-expanded RNA is furthermore thought to cause toxicity by altering miRNA biogenesis [48].
Figure 2.
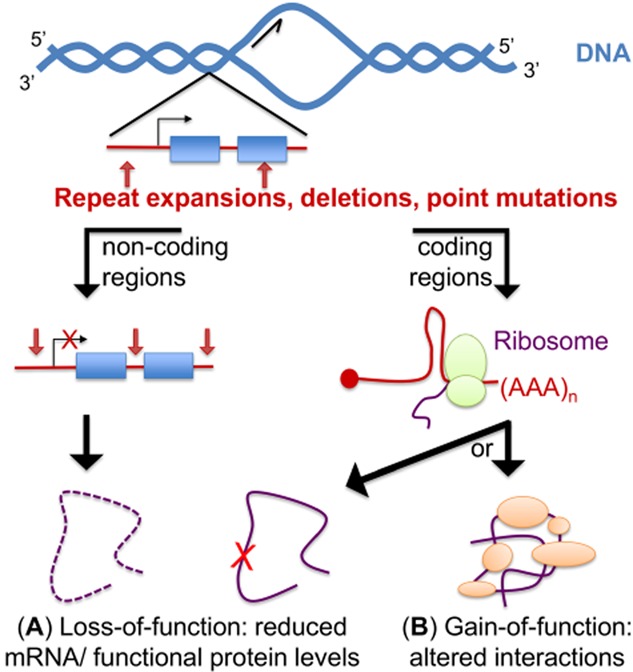
Mechanisms conferring protein loss and toxic gain-of-function effects. The diagram illustrates pathogenic mutations (repeat expansions, deletions, point mutations) that may occur either in noncoding or coding regions of the genome (left and right sides, respectively). (A) Protein loss-of-function. Haploinsufficiency can occur when the level of a particular mRNA is down-regulated due to mutations in noncoding regions of genes such as in promoters/introns, or if the promoter is subjected to histone/DNA modifications (transcriptional repression), but also if mutations in 5′ or 3′ untranslated regions (UTRs) decrease mRNA stability. Protein loss-of-function can also occur when mutations in coding regions alter directly the activity of the mutated protein (misfolding, alteration of the active site). (B) Protein toxic gain-of-functions are caused by mutations in coding regions that either promote abnormal interactions, increase the interaction of the mutated protein with its natural binders and/or promote misfolding/aggregation.
Figure 3.
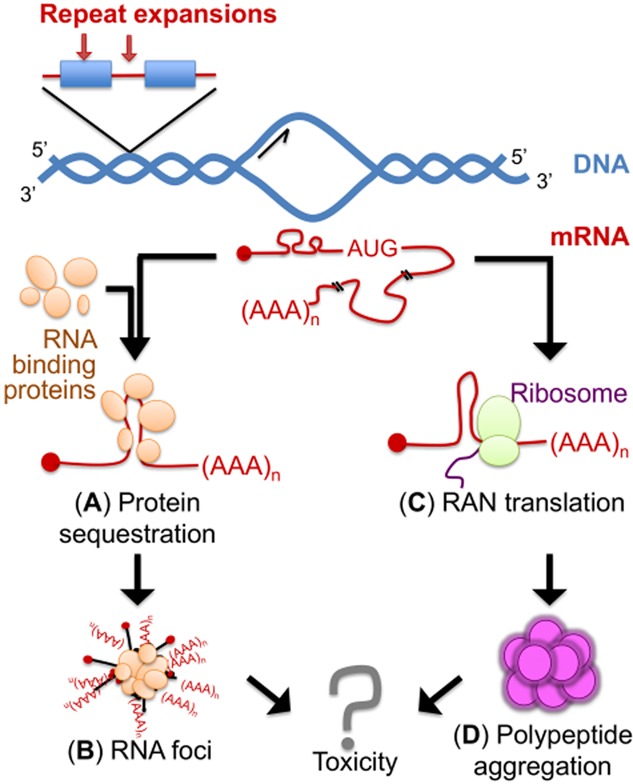
RNA toxic gain-of-function mechanisms. (A) Protein sequestration of RNA-binding proteins that avidly interact with the repeat expanded pre-mRNA/mRNA. (B) Formation of RNA foci. (C) Repeat-associated non-ATG (RNA) translation. (D) RAN translation leads to the formation of repeat-peptide proteins that usually aggregate.
Loss of HTT protein function plays a critical role in HD. HTT is involved in various cellular functions [49], particularly in the nervous system. It protects against excitotoxicity [50] and apoptosis by promoting mitotic spindle formation during neurogenesis [51]. It regulates the axonal transport of vesicles, regulating in turn synaptic transmission [52]. It contributes to miRNA biogenesis [53]. Furthermore, HTT sequesters repressor element 1 silencing transcription (REST) in the cytoplasm of neurons, leading to the transcriptional activation of REST-repressed genes involved in neuronal differentiation and survival [54]. Loss of functional cytoplasmic HTT in HD leads to nuclear accumulation of REST and subsequent down-regulation of REST-regulated neuronal genes [54]. These include brain-derived neurotrophic factor (BDNF) which encodes a cortical pro-survival factor essential for striatal neuron survival [55] and several neuronal miRNAs [56] that regulate the plasticity of the neuronal transcriptome [57,58].
On the other hand, the poly-Q expansions in HTT also trigger several toxic gain-of-functions described below. HTT has the ability to interact with over 100 proteins involved in various cellular functions [49]. Poly-Q expansions either disrupt or cause abnormal HTT: protein interactions, thus affecting many cellular pathways [59,60].
Poly-Q HTT aberrantly interacts and may sequester several transcription factors (TFs) which disrupt transcription and widely affect gene expression. Several TFs have been found in nuclear HTT inclusions, including the general transcription factor TATA box binding protein (TBP) and co-transcriptional activators such as CREB-binding protein (CBP) and specificity protein 1 (SP1) [59,61] which contribute to the establishment of neuronal identity.
Poly-Q HTT directly binds to the promoter of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PPARGC1A), as well as its protein product PGC-1α, a transcriptional master co-regulator that regulates energy homeostasis, mitochondrial biogenesis and antioxidant defences. Reduced PPARGC1A transcription and disrupted PGC-1αfunction are likely to contribute to the mitochondrial dysfunction observed in HD [62], a key pathophysiological component in combination with increased oxidative stress and mitochondrial DNA damage [63]. Interestingly, oxidative stress promotes DNA triplet expansion in HTT [64], highlighting the potential pathophysiological role of DNA damage/repair during neurogenesis. In addition, rescuing PGC-1α function attenuates HD in mice, alleviating both oxidative stress and HTT aggregation proteotoxicity [65].
Poly-Q HTT also binds the acetyltransferase domains of the histone acetyltranferases (HATs) CBP and P/CAF. Post-translational modifications of histones control the accessibility of the chromatin to the transcriptional machineries. Alteration of chromatin remodelling in HD leads to widespread dysregulation of gene transcription in large genomic regions [43]. This occurs through reduction of histone H3/H4 acetylation [66], reduction of histone H3 phosphorylation at the PGC-1α promoter [67] and decreased interaction with the human polycomb-repressive complex 1-like (hPRC1L) E3 ubiquitin ligase complex that promotes mono-ubiquitination of histone H2A [68].
The expanded HTT RNA [48] and protein [53] have furthermore a direct role in the alteration of miRNA biogenesis.
Finally, CBP dysregulation also results in reduced acetylation of the rRNA-specific upstream binding factor 1 (UBF1) which dysregulates rRNA transcription and leads to nucleolar stress [69].
SCAs
There are 36 SCA subtypes identified to date, with a combined prevalence of 5–7 cases per 100 000 people 70–72. Most SCA subtypes are caused by repeat expansion mutations that occur in over 20 known genes. SCAs can be sporadic, or have autosomal recessive or autosomal dominant inheritance dependent on the subtype. The most widely characterized genetic subset are the autosomal dominant cerebellar ataxias (ADCAs), which are subdivided into three broad types based on their clinical presentation [73].
Many of the ADCAs (SCA1-3, SCA6, SCA7, SCA17) are caused by the presence of expanded CAG repeats within exonic regions of genes. These repeats are reminiscent of those found in HD and have led to these SCAs being classified as poly-Q diseases [74]. Misfolded poly-Q-containing proteins or soluble glutamine-rich peptides (created by their cleavage) are thought to cause neuronal toxicity through toxic gain-of-functions, abnormal interactions and/or protein aggregation [75] (Figure 2). The SCA poly-Q proteins are proposed to disrupt the ubiquitin-proteasome system [76,77], alter calcium homeostasis [78] and dysregulate transcription 79–81.
Other SCA subtypes such as SCA10, SCA31 and SCA36 are caused by a variety of different noncoding repeat expansions, which have the potential to cause RNA toxicity important in disease pathogenesis [82] (Figure 3A–C). An ATTCT pentanucleotide repeat expansion in intron 9 of ATXN10 causes SCA10 [83]. Affected individuals have between 800 and 4500 repeats. Intron 9 is spliced out of the ATXN10 pre-mRNA, but the expanded AUUCU RNA is resistant to degradation and aggregates in nuclear and cytoplasmic foci of SCA10 cells and transgenic mouse brain. The expanded AUUCU RNA binds the splicing factor heterogeneous nuclear ribonucleoprotein K (hnRNPK), resulting in hnRNPK sequestration and loss of function. As a result, protein kinase C δ (PKCδ) accumulates in the mitochondria of SCA10 cells, leading to caspase-3 mediated apoptosis [84]. SCA31 is caused by a TGGAA pentameric repeat expansion located within the intron of both brain-expressed associated with NEDD4 (BEAN) and thymidine kinase 2 (TK2), genes that are transcribed in the opposite direction. Affected individuals have over 250 repeats. The expanded UGGAA RNA binds and sequesters the serine arginine-rich splicing factor (SRSF) SRSF1 and SRSF9 in vitro [85]. If these splicing factors are sequestered in vivo, pre-mRNA processing and stability are likely to be disrupted. SCA36 is intriguing because it exhibits similarities with C9ORF72-related ALS. SCA36 patients initially develop cerebellar ataxia and frontal lobe atrophy. However, symptoms typical of ALS follow, with upper and lower motor neuron involvement, including tongue atrophy, skeletal muscular atrophy and fasciculation [86,87]. SCA36 is caused by a GGCCTG hexanucleotide repeat within the first intron of nucleolar protein 56 (NOP56) [88], while C9ORF72-related ALS is caused by an expanded GGGGCC hexanucleotide repeat within intron 1 of C9ORF72 [89,90]. C9ORF72-related ALS and SCA36 are linked not only because of motor neuron dysfunction, but also because both intronic XGGGCC repeat expansions (where X is G or T respectively) interact with SRSF2 and form RNA foci [88,91,92]. The expanded GGCCTG repeat also leads to reduced expression of the neighbouring miR-1292 gene [88].
In some other SCAs, there are contributions from both protein gain-of-function and RNA toxicity. The expanded CAG repeat in SCA3 not only produces poly-Q proteins, but also produces expanded CAG repeat RNA that potentially binds and sequesters RNA-binding proteins. The dysregulated transcription and splicing could contribute to neurodegeneration [93,94]. In addition, SCA8 is caused by contribution from a CTG repeat expansion in ataxin (ATXN) ATXN8OS and a CAG repeat expansion in ATXN8, genes which are transcribed in opposite directions [95]. ATXN8OS is transcribed from the sense strand, and the transcript contains an untranslated CUG repeat, while the CAG repeat expansion within ATXN8 on the antisense strand is translated into an almost pure poly-Q protein. The CUG repeat-containing RNA forms nuclear foci which sequesters the RNA processing factor MBNL1 [96]. MBNL1 sequestration has adverse effects on RNA splicing and other processing events, and is well studied in myotonic dystrophies 1 and 2. Additionally, the CAG repeats are translated into poly-Q proteins, and in a different frame into polyalanine proteins via unconventional repeat-associated non-ATG (RAN) translation in SCA8 mouse model and post mortem human central nervous system (CNS) tissue [97].
SMA
SMA is caused by a drastic reduction of SMN protein levels. The chromosomal 5q13 SMA locus is a duplicated region carrying two inverted copies of almost identical SMN genes that encode the same 294-residue protein. The majority of SMA cases show homozygous deletions, rearrangements or large truncations of the telomeric SMN1 gene copy. However, other cases are caused by short deletions/mutations in the splice sites of SMN1 introns 6 and 7. The SMN1 gene differs from the centromeric SMN2 copy by a few silent base changes [6]. Exon-7 is correctly spliced in only 10–20% of SMN2 transcripts, leaving the vast majority of SMN2 mRNAs as defective transcripts lacking exon-7 [6,98] which are eventually translated into unstable and inactive SMN protein [99,100]. Significantly, SMN2 alterations are not associated with clinical pathology. Healthy motor neurons naturally express lower amounts of fully spliced SMN2 mRNA which may account for the higher vulnerability of motor neurons to SMN1 mRNA loss [101]. Because SMN protein levels are directly linked to disease severity, it is critical to identify the mechanisms that regulate inclusion/exclusion of SMN2 exon-7 for the development of therapeutic approaches. As described below, the intricate regulation of exon-7 splicing involves binding of multiple RNA recognition motif (RRM) containing proteins such as SRSFs and hnRNPs that act directly on binding exon-7 and flanking introns as well as through direct protein : protein interactions.
SRSF1 [98], polypyrimidine tract-binding protein-associated splicing factor (PSF) [102] and hnRNPM [103] interact with exonic splice enhancer (ESE) sequences that promote the inclusion of exon-7 in SMN1, while hnRNPM further stimulates the recruitment of the splicing factor U2AF65 to the flanking intron-7 [103] (Figure 4A). In contrast, several base changes in SMN2 exon-7 and flanking introns inhibit exon-7 inclusion. They alter the +6 ESE sequence leading to reduced interaction with SRSF1 [98,104,105], and form composite exonic splice silencer (ESS) sequences that bind splicing inhibitors hnRNPA1 [106,107] and Sam68 [108], as well as intronic splice silencer (ISS) sequences that interact with a 33 kDa protein p33 [110], hnRNPA1 [112], hnRNPA2 and hnRNPB1 [111]. Inclusion of SMN2 exon-7 is stimulated by ESE-binding of hnRNPQ1 [109], PSF [102], hnRNPM [103] and hTra2-β1 [113], together with hTra2-β1-associated alternative splicing factors Srp30c [114], hnRNPG and RNA binding motif protein chromosome X or Y (RBMX/Y) [115]. Interestingly, a mutation in SMA patients with a mild clinical phenotype provides an ESE for SRSF1 [116] while disrupting an ESS for hnRNPA1 [117] which promote in turn the splicing of exon-7. A schematic of the complex regulation of SMN2 exon-7 splicing is provided in Figure 4B. Notably, SMA patients develop respiratory impairment in the late stages of disease. Hypoxia was reported to induce both hnRNPA1 and Sam68 protein levels, promoting SMN2 exon-7 skipping and further reduction of SMN protein levels. SMA SMNΔ7 mice housed in hyperoxia conditions, in contrast, displayed increased splicing of exon-7 and improved motor function [118].
Figure 4.
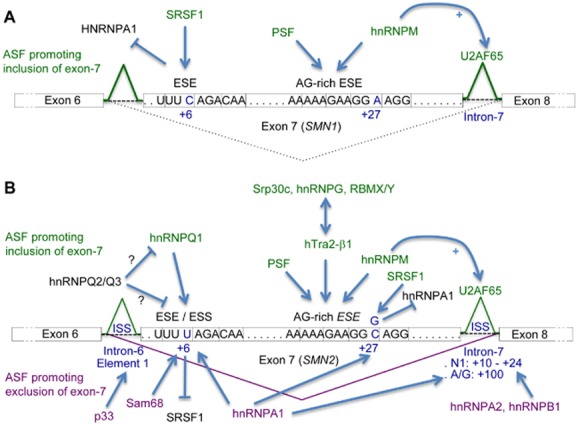
Model for regulation of exon-7 splicing in SMN1 and SMN2. Schematic representation of positive and negative effectors that regulate exon-7 splicing in SMN1 (A) and SMN2 (B) genes. The acronym ASF was used for alternative splicing factors. Exons 7 of SMN1 or SMN2 are represented in boxes that include the DNA sequences of the ESE/ESS motifs. ISS sequences are located in introns flanking exon 7 of SMN2. Arrows represent binding of ASF to the highlighted DNA elements or proteins. Factors that promote or inhibit the inclusion of exon-7 are respectively labelled above or below SMN exons/introns. T lines represent binding inhibition/inhibitory effect of ASF. (A) SRSF1 recognizes a +6 ESE sequence in SMN1 exon-7 promoting inclusion of exon-7. A downstream AG-rich ESE in exon-7 promotes exon-7 inclusion through binding of PSF [102] and hnRNPM [103], which in turn stimulates the recruitment of the splicing factor U2AF65 to the flanking intron-7. (B) The ESE sequence altered by a C/T transition at position +6 in SMN2 exon-7 was initially suggested to reduce exon-7 splicing because of a decreased interaction with SRSF1 [98,104,105]. However, the C/T transition also forms a composite ESS that promotes exon-7 skipping by interaction with the alternative splicing inhibitors hnRNPA1 [106,107] and Sam68 [108]. Furthermore, the activities of both hnRNPQ2 and Q3 antagonize the positive exon-7 splicing role of hnRNPQ1 bound to the +6 ESE [109]. Several base changes in SMN2 introns 6 and 7 also promote SMN2 exon-7 exclusion: (i) an ISS Element 1 in intron-6 (−75 to −89) through binding of p33 [110]; (ii) an ISS-N1 site located in intron-7 (+10 to +24) that provides binding sites for hnRNPA2 and B1 [111]; (iii) an ISS in intron-7 (A/G transition at position +100) that binds hnRNPA1 and inhibits splicing of exon-7 cooperatively with the binding of the same protein to the exon-7 ESS site [112]. In contrast, SMN2 exon-7 inclusion is promoted via two ESE sites: (i) the composite +6 ESE which provides interaction for hnRNPQ1 [109]; and (ii) the AG-rich ESE that provides overlapping binding sites for the splicing factors PSF [102], hnRNPM [103] and hTra2-β1 [113]. The direct interactions of hTra2-β1 with the alternative splicing factors SRp30c [114], hnRNPG and RBMX/Y [115] increase the splicing activity of ESE-bound hTra2-β1, stimulating in turn exon-7 inclusion. Interestingly, a silent C/G transition identified in AG-rich ESE at position +27 (codon Gly287) in some SMA II or III patients which present mild clinical phenotypes, creates an ESE for SRSF1, which in turn promotes exon-7 splicing and the production of full-length SMN2 mRNAs [116]. However, this transition also disrupts a splicing-inhibitory hnRNPA1 binding site indirectly promoting SMN2 exon-7 inclusion [117].
SMN proteins form part of a large oligomeric SMN complex of nine core-proteins composed of SMN (Gemin1), Gemins 2–8 and Unrip in mammals. This complex is found in the nucleus in Gemini of coiled (GEMs) bodies often associated with Cajal bodies where it is thought to play a role in RNA polymerase II dependent transcription and/or pre-mRNA splicing. The SMN complex is required in the cytoplasm for the assembly of uridine-rich small nuclear RNPs (U snRNPs) that compose the spliceosome, providing a binding-platform for the Sm core-domain and the targeted recruitment of snRNA [119,120]. The integrity of the spliceosome is indeed altered in SMA [121], and alternative splicing of several pre-mRNAs is affected in the disease [122]. SMN depletion affects snRNA stoichiometry and promotes widespread pre-mRNA splicing defects [123] altering the alternative splicing of U12-intron-containing genes, including Stasimon (CG8408) [124] and Neurexin2a (Nrxn2a) [125] which are involved in neuromuscular junction transmission and synapse assembly/synaptic transmission, respectively. The SMN complex also plays a key role in axonal mRNP assembly and transport [126] where SMN-containing granules interact with hnRNPR, which in turn binds to the 3′UTR of β-actin mRNA [127], and with Hu-antigen D (HuD) that interacts with the candidate plasticity gene 15 (cpg15) mRNA in neuronal processes [128]. Over 200 neuronal mRNAs are associated with SMN complexes, and approximately one third co-localize in axons and neurites [129]. SMN also binds methylated lysine 79 of histone 3 (H3K79), a post-translational modification marker associated with splicing, suggesting that epigenetic dysregulation may also occur in SMA [130]. In addition, mice deficient for miRNA processing in spinal motor neurons exhibit features of SMA, indicating that miRNA processing plays an essential role in the development and integrity of spinal motor neurons [131].
In SMA, reduction in SMN levels also induces down-regulation of Gemins 2/3, hnRNPQ, hTra2-β1 [132], and promotes SMN2 exon-7 skipping [133], thereby exacerbating the SMA phenotype. In summary, SMA is primarily caused by SMN protein-loss-of function (Figure 2A) that results in broad splicing and axonal mRNP trafficking defects.
ALS
ALS is a multifactorial disease caused by mutations in one of over 20 genes encoding proteins with a variety of functions [11]. Until recently, most understanding of the mechanisms of motor neuron injury emerged from the study of SOD1 mutations in experimental models and human biosamples. In the presence of mutant superoxide dismutase 1 (SOD1) and in sporadic cases of ALS, multiple interacting factors contribute to the neurodegenerative process, including oxidative stress, excitotoxicity, mitochondrial dysfunction, disruption of the cytoskeleton/axonal transport, protein aggregation and altered glial-motor neuron cross-talk [134]. More recently, alteration of mRNA metabolism was identified as a major dysregulated pathway in most common subtypes of ALS. In 2006, the nuclear loss and aggregation of altered forms of the RNA-processing TAR DNA-binding protein 43 (TDP-43) observed in neuronal and glial cells of ALS patients focused attention on altered RNA processing [135,136]. TDP-43 proteinopathy was found to form the hallmark of most familial and sporadic ALS cases. Ubiquitinated, phosphorylated TDP-43 wild type or mutant protein and carboxyl-terminal degradation products constitute major components of intranuclear and cytoplasmic neuronal inclusions that are observed in the majority of ALS variants except for those caused by SOD1 mutations, highlighting alteration of mRNA-processing [137] and mRNA-binding [138] as one of the critical pathophysiological disease mechanisms [10]. Interestingly, oculomotor neurons that are relatively spared during the course of neurodegeneration in ALS show a specific transcriptome profile with decreased susceptibility to excitotoxicity [139], suggesting that selective neuronal death is induced by an inability of affected neurons to cope with increased stress in relation to ALS mutations, environmental factors and/or ageing [140].
The most common genetic causes of familial ALS involve autosomal-dominant mutations in the following four genes: C9ORF72 (uncharacterized) in 40–50% cases [89,90], also most commonly mutated in frontotemporal dementia (FTD), SOD1 [141] in 20% [142], TARDBP (encoding the TDP-43 protein) [143] and FUS (fused in sarcoma) [144,145] in 4% [146] of fALS cases, respectively. These mutations can also be found in a varying proportion of sporadic ALS cases. Widespread dysregulation of gene expression was observed in these ALS subtypes as well as some other less common genetic variants described below.
C9ORF72-related ALS
In contrast to TDP-43 and FUS, the C9ORF72 protein does not display homology to RNA-binding proteins and is not thought to play a direct role in mRNA metabolism. C9ORF72 encodes a protein of uncharacterized function which might belong to the family of DENN-containing proteins, GDP-GTP exchange factors (GEFs) for Rab GTPases domain involved in the regulation of membrane trafficking [147]. ALS mutations are associated with pathological intronic hexanucleotide GGGGCC repeat expansions [89,90] containing from >30 up to several thousand repeats [148,149] in the first intron of the C9ORF72 gene. The C9ORF72 RNA repeat expansions form stable parallel uni and multimeric G-quadruplex structures 150–152 which are well known for avidly interacting with RNA-processing factors. Both sense 89,91,153–155 and anti-sense 152,155–157 intraneuronal RNA foci have been observed in association with the C9ORF72 hexanucleotide expansion and form a pathological hallmark of C9ORF72-related ALS.
Three potential molecular mechanisms of gene expression alteration and neuronal injury have been proposed and may coexist simultaneously: (i) haploinsufficiency due to decreased expression from the altered C9ORF72 allele(s) 89,158–162 (Figure 2A); (ii) RNA mediated gain-of-function toxicity by the GGGGCC-expanded intron that prevents aberrantly bound/sequestered RNA-processing factors from functioning normally in the nucleus 91,92,153–155,163,164; and (iii) protein gain-of-function toxicity by dipeptide repeat proteins (DPRs) aberrantly generated from RAN translation of the expanded intron-containing mRNA [156,165,166] (Figure 3A–C).
Haploinsufficiency was reported by several groups [158,159,161,162] and in a zebrafish model of C9ORF72-related ALS [160]. However, decreased mRNA expression was not observed in induced pluripotent stem cell (iPSC)-derived neurons [154]. Similarly, we [167] and others [161] have also reported that C9ORF72 mRNA steady-state levels are not altered in ALS cases with small GGGGCC repeat lengths. In addition, not a single mutation has been identified in the C9ORF72 coding sequence, suggesting that pathogenicity due to loss-of-function is less likely [168]. On the other hand, a body of evidence is accumulating for RNA gain-of-function toxicity. Antisense oligonucleotides have been shown to rescue the GGGGCC-expanded C9ORF72-mediated RNA toxicity [153,154,156]. We [92] and others 91,153–155,163,164 are proposing RNA mediated gain-of-function toxicity as one contributing mechanism operating to prevent aberrantly bound/sequestered RNA-processing factors from functioning normally in the nucleus, which in turn leads to broad alteration of gene expression in C9ORF72-related ALS. On the other hand, DPRs are abundant in extra-motor areas of the CNS such as the cerebellum and account for the characteristic pathology of p62-positive, TDP-43-negative inclusions seen in the CNS of C9ORF72-related ALS patients. DPRs were recently shown to in vitro alter RNA biogenesis and kill cells [169], as well as causing neurodegeneration in drosophila [170]. However, DPRs seem to be most abundant in areas of the CNS outside the motor system, which is responsible for the key disease-related clinical features, raising also the possibility of a potential beneficial/neuroprotective role of DPR generation.
SOD1-related ALS
SOD1 encodes a ubiquitously expressed free radical scavenging enzyme. ALS SOD1 mutations [141] lead to increased oxidative stress and neurodegeneration associated with mitochondrial dysfunction [140,171]. SOD1 is not thought to play a direct role in RNA metabolism. However, biomarkers of RNA oxidation are detectable in human ALS and as an early indicator of oxidative stress in mutant Sod1 mice. The transcriptomes of dissected spinal cord motor neurons from SOD1-related ALS models [172] were further shown to be significantly altered with up- and down- regulation of over 1000 transcripts whose products are involved in various metabolic pathways, including those controlling neuronal survival and death. In particular, in motor neurons that survived the disease process in human SOD1-related ALS, anti-apoptotic phosphatidylinositol 3-kinase and protein kinase B (AKT3) were up-regulated with a concomitant reduction in the level of phosphatase and tensin homologue (PTEN) which inhibits the pro-survival AKT pathway, suggesting a mechanism for how these intact motor neurons survived the neurodegenerative process. Therapeutic modulation of this pathway is now being developed as a potential neuroprotective approach.
More than 170 missense mutations of SOD1 have been reported in ALS cases (ALSoD Consortium; http://alsod.iop.kcl.ac.uk/). They are distributed over the full length of the 154 amino acids of the human SOD1 protein, suggesting that loss-of-function mutations are unlikely. Furthermore, knock down of Sod1 does not trigger ALS in mice, and several SOD1 mutants, including A4V and G93A, retain virtually normal superoxide dismutase activity. The pathophysiological mechanisms of SOD1 mutations are thought to involve protein toxic gain-of-function (Figure 2B) due to protein misfolding and formation of ubiquitinated intracytoplasmic neuronal and astrocytic inclusions in the CNS of SOD1-related ALS patients and mice [173,174].
TDP-43 and FUS-related ALS
TARDBP and FUS encode ubiquitously expressed DNA/RNA-binding proteins implicated in multiple aspects of gene expression regulation, including transcriptional control, alternative splicing of mRNA [175,176], axonal transport of mRNA [177] and biogenesis of miRNA [178,179]. More recently, TDP-43 was also shown to play additional roles in translation control [180,181]. Several thousand TDP-43 and FUS RNA-binding sites have been characterized on pre-mRNA molecules, including those involved in splicing functions of long pre-mRNAs essential to neuronal development and integrity [176,182,183]. Alternative splicing of pre-mRNAs was found to be broadly altered in TDP-43 and FUS-related ALS, leading to the dysregulation of normal neuronal gene expression with the synthesis of thousands of aberrantly spliced mRNA molecules 182,184–189. In particular, these recent transcriptome studies highlighted an alteration of levels and/or splicing of genes involved in RNA processing, synthesis of neurotrophic factors and synaptic function. Up to one-third of the transcriptome is altered in TDP-43 transgenic mouse models with specific alterations due to the Q331K ALS mutation [186]. Overexpression of FUS in mice also causes ALS, with progressive loss of motor neurons in an age- and dose-dependent response [190]. On the other hand, FUS interacts directly with the SMN complex [191] that associates with GEMs for the biogenesis of snRNPs. GEMs are significantly reduced in SMA, in patient fibroblasts expressing TDP-43 or FUS mutations [191] and in motor neurons of TDP-43 transgenic mice [192]. ALS-associated TDP-43 mutations increase interaction with FUS [193], and spliceosome integrity was found to be affected in TDP-43-related ALS [121]. TDP-43 interacts also with GEMs via SMN [194]. These observations suggest shared defective RNA splicing mechanisms between SMA and TDP-43/FUS related ALS [121,191].
FUS and TDP-43 are hnRNP proteins which shuttle between the nucleus and the cytoplasm. They are composed, respectively, of one and two RRM domains flanked by carboxyl-terminal arginine/glycine (RG)-rich regions, also called the prion-domain because these unstructured regions have a propensity for aggregation. FUS also exhibits an RG-rich region in its amino-terminus. The vast majority of ALS mutations cluster in the RG-rich regions in exon 6 of TDP-43 and in exons 3–6 or 12–15 of FUS. These mutations disrupt protein : protein interactions, including those of TDP-43 with the splicing hnRNPs A1, A2/B1, C1/C2 and A3 [195]. ALS mutations of TDP-43 and FUS also alter the transportin-mediated nuclear localization import, resulting in predominantly cytoplasmic mislocalization and the formation of stress granules [196,197]. It remains unknown whether motor neuron injury is caused by loss of normal nuclear functions of TDP-43/FUS (disrupting transcriptional regulation, pre-mRNA splicing, sorting to distinct cytoplasmic compartments or processing of noncoding RNAs) and/or whether additional toxic gain(s) of function, such as cytoplasmic mislocalization in soluble or aggregated forms, are responsible for disease (Figure 2A,B). However, toxicity does not seem to depend on nuclear localization of mutant TDP-43 or on the formation of intracytoplasmic inclusions, but rather on the increased cytoplasmic mislocalization of soluble ALS-linked TDP-43 mutant proteins [198,199]. Also, the alteration of the axonal mRNA transport is likely to contribute to motor neuron dysfunction in TDP-43 related ALS [200].
Minor subtypes of ALS that exhibit RNA dysregulation
Further evidence of dysfunctional RNA metabolism has been provided by autosomal-dominant mutations in genes encoding senataxin [201] and angiogenin [202,203], two other RNA-binding proteins, involved in rare juvenile familial and adult onset forms of ALS, respectively. The DNA/RNA helicase senataxin (SETX) is predicted to play roles in several steps of gene expression, including functions associated with RNA processing and the maintenance of genome integrity. Angiogenin (ANG), the expression of which is increased during hypoxia to promote angiogenesis, is a tRNA-specific RNase that also regulates the transcription of ribosomal RNA. ALS mutations of ANG are likely to act through a loss of function, as overexpression extends the lifespan of ALS SOD1 mutant mice. The mechanism(s) by which mutations in SETX and ANG cause ALS remains to be determined.
Additional genetic mutations were more recently identified in elongator protein 3 (ELP3) [204], TAF15 [205,206] and Ewing's sarcoma breakpoint region 1 (EWSR1) [207]. Products from these genes are also involved in regulating RNA metabolism. The histone H3/H4 acetyl transferase ELP3 comprises one subunit of the elongator complex that regulates transcription elongation by the RNA Polymerase IIO, post-transcriptional processing of tRNA, as well as acetylation of α-tubulin in microtubules. TAF15 and EWRS1 proteins are functionally and structurally related to FUS. They form the FET family of proteins (FUS-EWS-TAF15) and play a regulatory role in transcription and alternative splicing. Also, poly-Q expansions described in the coding sequence of Ataxin-2 (ATXN2), a gene product involved in the translation control of the circadian rhythm [208,209], were shown to be significant susceptibility factors associated with ALS [210]. ATXN2 localizes with stress granules and TDP-43 in neurons. Products of genes that modulate stress granules are potent modifiers of TDP-43 toxicity in Saccharomyces cerevisiae and Drosophila melanogaster, and TDP-43 interacts with the polyA-binding protein (PABP) [211]. Mutations in the RNA/DNA-binding protein Matrin 3 (MATR3) that also interact with TDP-43 were shown to cause ALS [212]. Furthermore, a mutation causing ALS was found in the prion domain like of hnRNPA1 [213]. The precise mechanisms of neurodegeneration triggered by mutations in genes encoding these various RNA-processing factors remain poorly understood.
RNA-mediated mechanisms of neurodegeneration
Expression level alteration and/or sequestration of proteins involved in the process of gene expression are common mechanisms implicated in neurodegeneration; however, they usually diverge in the targets identified so far across the various RNA-mediated neurodegenerative diseases. Comprehensive transcriptome studies have recently been reviewed in HD [214], SCA [215], SMA [216] and ALS [9,217]. Here, Tables 1 and 2, respectively, focus on the recent transcriptome analysis of pathogenic variants in the nonmotor (HD, SCA) and motor (SMA, ALS) neurodegenerative diseases that are the focus of this review. In common, transcriptome studies of pathogenic mutations found in HTT, ATXN2, ATXN7, SMN, TARDBP/TDP-43, FUS and C9ORF72 all suggest that most changes alter the expression of genes associated with neuronal specificity, plasticity and synaptic function. This provides, in turn, an attractive hypothesis explaining why neurons are preferentially more sensitive than other cell types to the pathogenic mutations.
Table 1.
Recent transcriptome studies of pathogenic mutations in RNA-mediated nonmotor neuron neurodegenerative diseases (HD and SCA)
| Study | Samples | Platform | Measure of differential expression (no. differentially expressed) | Main findings |
|---|---|---|---|---|
| HD: | ||||
| Jacobsen et al. [218] | RNA extracted from mouse embryonic stem cells expressing wild-type huntingtin with 7Q, and knock-in expansions of length 20Q, 50Q, 91Q and 111Q (n = 3–6 replicates per group). | Affymetrix Mouse 430A microarray | Correlation examined between expansion length and gene expression, Pearson coefficient > 0.8, P < 0.001 (25 probes positively correlated, 48 probes negatively correlated) | Genes correlated with expansion size distinct from genes dysregulated by knockdown of huntingtin, but are within similar functional pathways including energy and lipid metabolism. |
| Feyeux et al. [219] | RNA extracted from six embryonic stem cell and corresponding neural stem cell lines expressing huntingtin with 40–51Q expansions and four wild-type lines | Illumina Human WG-6 v3 microarray | Compared with wild type, Limma, P < 0.001 (embryonic stem cell: 163, neural stem cell: 66) | Dysregulated gene expression functionally enriched for energy and lipid metabolism and gene expression; no overlap with data sets analysing gene expression in brain tissue from symptomatic adults |
| Mattis et al. [220] | RNA extracted from neural stem cells and striatal neurons derived via induced pluripotent stem cells from patients with 60–180Q expansions of huntingtin and wild-type controls (n = 2–3 each group) | Human Affymetrix Exon 1.0 ST microarray. | Comparison to wild type, anova, FC > 2 (NSC: 1601; striatal neurons: not stated) | Neural stem cells: Dysregulated genes functionally enriched for signalling, cell cycle, axonal guidance and neural development. Some changes specific to longer or shorter repeat length. Striatal neurons: Dysregulated genes in striatal neurons functionally enriched for proliferation, signalling and cellular assembly |
| Soldati et al. [221] | RNA extracted from striatal neurons obtained and grown from mice expressing two huntingtin alleles with 109Q expansions, nonexpanded 7Q alleles. | Illumina MouseWG-6 v2.0 microarray | Compared with 7Q homozygous cells: Benjamini Hochberg, FDR < 0.02 (1013↑ and 1342↓) | Dysregulated genes functionally enriched for nervous system development and function. Down-regulated genes in these functional categories were enriched for REST binding sites. |
| Lewandowski et al. [222] | RNA extracted from posterior and anterior caudate, and S1 of patient (mean huntingtin expansion 41Q) and control brains (n = 10 each group) | Affymetrix Human U133A 2.0 microarray | Compared with control, repeat measures anova, controlling for region, P < 0.05 (3) | Focused on down-regulation of PPP1R7 which is implicated in neuronal function |
| Lee et al. [223] | RNA extracted from lymphoblastoid cell lines derived from 107 patients with huntingtin expansions of 15–92Q | Affymetrix Human U133A 2.0 microarray | Pearson correlation between probe expression and expansion length used to derive a biomarker of expansion length | Correlated genes involved in ribosomal function, transcription, nucleic acid metabolism, adhesion, energy metabolism, hormone response, synaptic transmission, neurological process |
| SCA7 | ||||
| Chou et al. [224] | RNA extracted from the cerebellum of symptomatic transgenic mice expressing human ataxin 7 with a 52Q expansion, and wild-type mice (n = 4 each group) | Affymetrix Mouse 430A microarray | Compared with wild type, t-test, P < 0.05, FC > 1.4 (10↑ and 34↓) | Down-regulated genes involved in neuronal function, protein processing, heat shock response and glial function. Up-regulated genes include RNA binding proteins. |
| Friedrich et al. [225] | RNA extracted from laser-captured Purkinje neurons from cerebellum of transgenic mice expressing human ataxin 7 with 90Q expansion and wild type mice | Affymetrix Mouse 430A microarray | P7E: Compared with wild type, FDR P < 0.2 (13) | Down-regulated genes involved in vulnerability to excitotoxicity. |
| SCA17 | ||||
| Ren et al. [226] | RNA extracted from heads of transgenic flies expressing human TBP with a 34Q or 80Q expansion, at days 5, 28 and 35. Day 28 is immediately prior to the onset of motor symptoms in the 80Q flies. | Affymetrix Drosophila Genome 2.0 microarray | Compared with 34Q samples at each time point, t-test, P < 0.05; FC > 1.4 (536) | Differentially expressed genes functionally enriched for pathways, including oxidation and mitochondrial function. Pearson correlation analysis implicated transcription factor Su(H) in control of dysregulated gene expression. |
| SCA2 | ||||
| Damrath et al. [227] | RNA extracted from cerebellum, brainstem and liver of transgenic mice expressing ataxin 2 with a 42Q expansion at 6 months (presymptomatic) and 18 months (symptomatic), and wild-type mice (n = 3–4 per group) | Affymetrix Mouse 430A microarray | Compared with wild type, Benjamini Hochberg (6 months: no significant gene expression changes. 18 months: cerebellum 20 genes; brainstem 14 genes, liver 30 genes) |
In cerebellum, identified dysregulation of Adam1a and Fbxw8 which are neighbouring genes of ataxin 2. Fbxw8 implicated in neuronal dendrite formation |
| SCA3 | ||||
| Hsieh et al. [228] | RNA extracted from SK-N-SH cells engineered to stably express ataxin 3 with 26Q or 78Q expansion (n = 2 each group) | ABC Human UniversoChip 8k-1 microarray | Comparison with 26Q expressing cells, genes ranked by fold change | Most differentially expressed gene by fold change was CA11; function of CA11 unknown. |
| SCA28 | ||||
| Mancini et al. [229] | RNA extracted from lymophoblastoid cell lines derived from patients with mutations of AFG3L2 or matched controls (n = 4–6 each group) | Affymetrix Human U133A 2.0 microarray | Compared with controls, rank product, FDR < 0.005 (35↑ and 31↓) | Differentially expressed genes implicated in regulation of cell proliferation, cell death, cell adhesion, oxidative stress and chemical homeostasis |
FC, fold change; FDR, false discovery rate; HD, Huntington's disease; SCA, spinocerebellar ataxia.
Table 2.
Recent transcriptome studies of pathogenic mutations in RNA-mediated neurodegenerative diseases of the motor system (SMA and ALS)
| Study | Samples | Platform | Measure of differential expression (no. differentially expressed) | Main findings |
|---|---|---|---|---|
| SMA | ||||
| Garcia et al. [230] | Smn-null and wild type drosophila lavae (n = 2 each group). RNA extracted when Smn-null lavae first display motor phenotype | Illumina HiSeq 2000 | Gene level: Compared with wild type; Fisher's exact test. Exon level: Compared with wild type, DEXSeq, P < 0.05 (2153 splicing changes); MISO, Bayes factor > 100 (2484 splicing changes) | Comparison of gene level analysis with modENODE data [231] consistent with developmental arrest in the Smn-null animals; splicing changes exceeded normal developmental changes |
| Zhang et al. [232] | Microdissected motor neurons and glial cells from spinal cord of SMN-deficient and wild-type mice at postnatal day 1 before MN pathology develops (n = 2 each group) | Illumina HiSeq 2000 | Compared with wild type. Gene level:, DESeq, P < 0.05, FC > 2 (Motor neurons: 138↑ and 110↓; Glial cells: 125↑ and 87↓). Exon level: MISO, Bayes factor > 10 (Motor neurons: 104; glial cells: 86 splicing changes) | Dysregulation of genes implicated in synaptogenesis. Minimal overlap of transcriptome changes between cell types despite broadly similar genes expressed |
| See et al. [125] | Zebrafish embryos at 48h post-fertilization injected morphilino vs. SMN, Gemin2 or scrambled (n = 6–7 each group) | Custom zebrafish microarray [233] | Compared with scrambled, t-test, P < 0.05, fold change > 2 (1545 transcripts) | Down-regulation of Neurexin2a which is involved in synaptic function. Knockdown of Neurexin2a phenocopied the SMA model in zebrafish |
| Acsadi et al. [234] | NSC34 cells transfected with shRNA vs. SMN or scrambled | StellARray™ microarray NE0100-MM96 | NSC34 cells, vs. scrambled fold change > 2 (5 genes). Note that custom array contains only genes linked to neurodegeneration. | SNCA down-regulation highest fold change. Change confirmed in peripheral and CNS tissue from SMA patients. SNCA is implicated in synaptic function and PD. |
| C9orf72-linked ALS | ||||
| Donnelly et al. [153] | RNA extracted from motor cortex, fibroblasts and iPSNs from C9orf72+ ALS patients and controls (n = 2–4 each group). Followed by transfection of C9orf72+ iPSNs with ASO vs. C9orf72 | Human Affymetrix 1.0 ST Exon microarray. | Compared with control, t-test P < 0.05. (Fibroblasts: 487 genes↑ and 1238 genes↓; iPSNs: 984 genes↑ and 2314 genes↓; motor cortex: 1870 genes↑ and 5114 genes↓) | Significant number of differentially expressed genes in common between C9orf72+ iPSNs and C9orf72+ motor cortex. ASO treatment normalized gene expression in C9orf72+ iPSNs. |
| Sareen et al. [154] | RNA extracted from fibroblasts and iPSNs from C9orf72+ ALS patients and controls (n = 4 each group). Followed by transfection of C9orf72+ iPSNs with ASO vs. C9orf72 or scrambled ASO | Illumina HiSeq. | Compared with control, t-test P < 0.05 | Differentially expressed genes in iPSNs functionally enriched for synaptic transmission. ASO vs. C9orf72 partially corrected gene expression in C9orf72+ iPSNs. |
| Lagier-Tourenne et al. [155] | RNA extracted from fibroblasts of C9orf72+ ALS patients, non-C9orf72 ALS patients and controls (n = 4 each group). Followed by transfection of C9orf72+ samples with ASO vs. C9orf72 | Illumina Hi-Seq | C9orf72+ compared with controls, FDR < 0.05 (122 genes↑, 34 genes↓) | Treatment with ASO vs. C9orf72 failed to reverse expression signature of C9orf72+ fibroblasts. |
| Ismail et al. [235] | RNA extracted from lymphoblastoid cell lines derived from C9orf72+ ALS patients, non-C9orf72 ALS patients and controls (n = 10–16 each group) | Human Affymetrix Genome U133 Plus 2.0 microarray | Compared with controls. PPLR [236], PLikeValue < 0.05 (C9orf72+ vs. control: 319 genes; C9orf72+ vs. non-C9orf72: 294 genes) | Differentially expressed genes functionally enriched for NF-κB activity. Top differentially expressed gene CXCL10 down-regulation in C9orf72+ samples; this gene is controlled by NF-κB and is a positive prognostic marker in sporadic ALS [237]. |
| TDP-43-liked ALS | ||||
| Huang et al. [188] | Astrocytes purified from (GFAP)-tTa/TRE-TDP-43M337v double transgenic rats. RNA extracted after 3, 4 or 6 days of induction of mtTDP-43 expression | Microarray | Compared with baseline FC > 2 at ≥1 time point (449 genes) and progressive change in FC over induction time points | Induction of TDP-43M337V expression altered expression of secreted factors; in particular, expression of neurotrophic factors increased and expression of neurotoxic factors decreased. |
| Arnold et al. [186] | Transgenic mice produced which express TDP-43Q331K, or TDP-43wild-type throughout CNS. RNA was extracted from cortices and spinal cords of 2-month-old mice and compared with nontransgenic mice. | Affymetrix ‘A = chip’ microarray. | Compared with nontransgenic. Absolute separation score < 0.3, q < 0.05 (Cortex: TDP-43wild-type 824 exons alternatively spliced; TDP-43Q331K 1195 exons; Spinal cord: TDP-43wild-type 4462 exons, TDP-43Q331K 4399 exons) | Differentially spliced exons in mutant, but not wild-type transgenic mice enriched for known TDP-43 binding sites. Differentially spliced exons in TDP-43Q331K which overlapped between cortex and spinal cord enriched for synaptic function |
| Highley et al. [189] | RNA extracted from fibroblasts derived from mtTDP-43 ALS patients, mtSOD1 ALS patients, sporadic ALS patients and controls (n = 3–6 each group) | Human Affymetrix Exon 1.0 ST microarray | Compared with controls, ancova P < 0.01 (mtTDP-43: 1070 genes↑ and 433 genes↓, 12 280 exons; mtSOD1: 6335 exons; sporadic ALS: 1140 exons) | Functional enrichment in mtTDP-43 differentially expressed/spliced genes for categories related to RNA processing. Alternative splicing much more abundant in mt-TDP-43 vs. other ALS patients |
ALS, amyotrophic lateral sclerosis; ASO, antisense oligonucleotide; FC, fold change; FDR, false discovery rate; iPSNs, induced pluripotent stem cell-derived motor neurons; SMA, spinal muscular atrophy.
However, it still remains unclear whether widespread gene expression changes are the result of an exclusive alteration of mRNA biogenesis/processing, an indirect effect due to DNA/RNA/protein damage via oxidative stress/mitochondrial dysfunction, or a combination of these exhibiting in turn both loss-of-function effects and toxic gain-of functions via the formation of RNA foci and/or protein aggregates. The apparent lack of convergence in the altered RNA/protein targets does not necessarily imply that there are not common mechanisms involved. The large number of dysregulated gene expression events identified so far and the involvement of redundant pathways, either up- or down-regulated, complicate the interpretation of observations. For example, energy/lipid pathways are similarly dysregulated by either poly-Q HTT expansions or HTT knockdown, but the particular genes whose expression is altered are different [218]. Also, TDP-43 and FUS were shown to bind distinct RNA sites, but their functions overlap in the alternative splicing of pre-mRNAs with long introns, many of which encode genes associated with neuronal integrity [176,182,183].
Expanded pre/mRNA in repeat disorders were reported to form RNA foci and avidly bind/sequester splicing factors such as SRSF1 in SCA31/C9ORF72-related ALS [85,91,92], SRSF2 in SCA36/C9ORF72-related ALS [88,91,92] and MBNL1 in HD/SCA8 [46,96]. Disruption of GEMs is observed in both SMA and TDP-43/FUS-related ALS [191,192], and concordantly, the integrity of spliceosomes was found to be altered in both diseases [121]. Expression level alteration/sequestration of RNA-processing factors such as RRM-containing splicing factors (hnRNPs, SRSFs) further leads to large splicing alterations in SMA 123–125, TDP-43/FUS-related ALS 182,184–189 and C9ORF72-related ALS [153,154].
RAN translation has been observed in SCA8 [97] and C9ORF72-related ALS [156,165,166]. However, how repeat-expanded pre-mRNAs are exported into the cytoplasm remains unknown. Increased binding of mRNA export adaptors (ALYREF, SRSF1, SRSF3, SRSF7) on GGGGCC-expanded C9ORF72 pre-mRNAs [92] may override normal nuclear retention and inappropriately license repeat-expanded pre-mRNAs for nuclear export [28,238], thus allowing RAN translation to occur. On the other hand, repeated sequence expansions such as those observed in HD, SCA and C9ORF72-related ALS are likely to generate transcriptional stress with the formation of R-loops that are more susceptible to DNA damage and may result in increased genome instability (for a recent review, see Skourti-Stathaki & Proudfoot [239]). Interestingly, the ALYREF/THOC4 (THO complex subunit 4) subunit of the TREX complex that links transcription elongation to mRNA nuclear export and genome stability was found to be sequestered by pathogenic C9ORF72 RNA hexanucleotide repeat expansions [92]. Dysregulation of miRNA biogenesis was also reported to play an important role in HD [48,53,56], in SMA [131] and in FUS-related ALS [240].
Whether RNA-mediated neurodegeneration and its various clinical presentations are driven by distinct mechanisms specific to each affected type of neuron or by a common pathophysiological core of dysregulated targets remains to be determined.
Current challenges: identifying altered levels/sequences of RNA molecules and proteins causing neurodegeneration
As described earlier, RNA-mediated neurodegenerative diseases exhibit widespread dysregulation of gene expression with alteration of most RNA classes (mRNA, miRNA, rRNA and tRNA) and disruption of multiple RNA-processing steps. Accordingly, transcriptome studies have revealed dysregulation of hundreds to thousands of RNA molecules in the presence of disease-causing mutations [217].
Transcriptomics as a research methodology has contributed greatly to our understanding of the mechanisms of neuronal cell death [241]. However, there are clear limitations inherent within traditional transcriptomics studies due to both the measurement of steady-state levels and the nature of available samples from which RNA is obtained (i.e. cell/animal models and post mortem tissues enriched for neurons that survived the neurodegenerative process) [242,243]. Identifying events causing neurodegeneration is also challenging because of the widespread and varied components of RNA dysregulation. For example, the large number of reported splicing anomalies constitutes a major hurdle in separating causal molecular events leading to neurodegeneration from those that are downstream consequences of the initial perturbations of gene expression. Another problem lies in the fact that very often, the expression level of a given mRNA transcript is not necessarily representative of the level of its corresponding protein. Counter-intuitively, an increase in the steady-state level of a particular mRNA is often associated with a decrease in the corresponding protein level as the cell tries to counteract the down-regulation of the protein by increasing synthesis/stability of the mRNA [244]. Practically, it is also difficult for researchers to determine which altered RNAs should be considered more or less important in a given biological context. It would be tempting to assume that the RNAs with the greatest relative fold changes would have the highest chance of being pathophysiologically relevant but, as we have seen with the previous example, a significant increase in mRNA level could be misleading, particularly as in most cases, it is the protein product that is more likely to influence cellular health. Additional complexity comes from the fact that intronic repeat sequences such as GGGGCC expansions in C9ORF72 can be translated into toxic proteins [156,165,166,169,170,245] which further extends the repertoire of altered gene expression that can be implicated in neurodegeneration. Furthermore, biomarkers of RNA oxidation are also detectable in human CNS samples from cases with neurodegenerative disorders. Oxidation of RNA molecules triggers a reduction in translation and simultaneously increases translational errors potentially leading to the synthesis of aberrant proteins and mitochondrial dysfunction [140,246]. Lastly, adenosine to inosine editing of miRNA and mRNA molecules by adenosine deaminase act on RNA (ADAR) enzymes is broadly affected during neurodegeneration (ALS, HD, PD, AD), leading in particular to regulatory and translational alterations of edited mRNAs [247]. Like many of the aforementioned changes, these alterations in RNA sequence are difficult to predict in advance and would therefore be unlikely to be accounted for in traditional microarray-based transcriptomics experiments, meaning that ultimately these important changes would be missed.
Concluding remarks and future directions
As yet, the functional consequences of RNA dysregulation for the processes that trigger age-related and selective progression of neuronal cell death remain very poorly understood. The levels, frequency and identity of aberrantly spliced mRNA isoforms that undergo nuclear export and are subsequently translated into aberrant proteins are still largely uncharacterized. These aberrant proteins, many of which are likely to have essential enzymatic or structural activities that impinge upon a multitude of cellular pathways, may have either lost their wild-type biological function or will have acquired new deleterious functions [180]. This complicates the interpretation of dysregulation events as aberrant proteins may in turn trigger cascades of further dysregulation. Given that proteins are ultimately involved in controlling cell fate, identifying altered cytoplasmic levels/sequences of mRNA and the corresponding proteome is becoming crucial for understanding the molecular causes of neurodegeneration. These approaches to OMICS studies therefore represent an essential step in the development of novel neuroprotective therapeutic strategies.
Reliable subcellular fractionation of cytoplasmic RNA, as well as purification of ribosomes for the extraction and next-generation sequencing of actively translating mRNA (i.e. the ‘translatome’) will become essential methodologies that will enable dissection of the molecular events that contribute to neurodegeneration. We anticipate that emerging methodologies investigating gene expression dysregulation at the level of active protein synthesis rather than at RNA/protein steady-state levels will yield biological results of higher relevance to the understanding of disease pathophysiology. Also, the specific sequences of mRNAs undergoing translation would provide rich information on the nature of mutations and/or aberrant splicing variations that are acquired during mRNA processing, and then subsequently translated into aberrant protein expression. Recent advances in molecular biology systems have also allowed the engineering of stable inducible neuronal cell models of neurodegeneration. In these models, cellular insults, such as disease-mutated protein or expanded nucleotide repeat expression, can be turned on as required. Such systems will allow investigators to observe early events in gene expression dysregulation that are more likely to be an upstream cause rather than a consequence of disease. Furthermore, these systems allow for the tracking of the progression of this dysregulation at multiple time points, meaning that it may be possible to define the upstream pathways in the pathophysiology of neuronal injury. Significantly, these types of analysis are prohibitively difficult to perform in animal models and impossible in human post mortem CNS tissue. Single-cell next-generation RNA sequencing [248] on neurons derived from induced pluripotent stem cells (iPScs) produced from patient fibroblasts (which bear the exact genetic makeup that caused disease in an individual) also holds great promise for investigating the functional consequences of RNA dysregulation in primary cells that are directly relevant to the disease being studied.
Widespread dysregulation of RNA metabolism is now clearly recognized as a pathophysiological component triggering neurodegeneration in the neurodegenerative diseases that are the focus of this review. It is very likely that other neurodegenerative diseases involve widespread dysregulation of gene expression. For example, excessive phosphorylation of the ribosomal RPS15 subunit by the PD mutated LRRK2 G2019S kinase was recently shown to trigger a toxic burst in global protein synthesis [249].
Acknowledgments
PJS is supported as an NIHR Senior Investigator. PJS and JK are supported by the MNDA, a European Community's Seventh Framework Programme (FP7/2007–2013) under the Euro- MOTOR project (Grant agreement No: 259867) and by funding from the European Union Joint Programme – Neurodegenerative Disease Research (JPND), Sampling and biomarker OPtimization and Harmonization In ALS and other motor neuron diseases (SOPHIA) and Survival, Trigger and Risk, Epigenetic, eNvironmental and Genetic Targets for motor neuron Health (STRENGTH). These are EU Joint Programme – Neurodegenerative Disease Research (JPND) projects. The projects are supported through the following funding organizations under the aegis of JPND – http://www.jpnd.eu: United Kingdom, Medical Research Council (MRC). JC-K is supported by a Motor Neurone Disease Association (MNDA)/Medical Research Council (MRC) Lady Edith Wolfson Fellowship award [MR/K003771/1].
The authors declare no conflict of interest.
References
- 1.Walker FO. Huntington's disease. Lancet. 2007;369:218–228. doi: 10.1016/S0140-6736(07)60111-1. [DOI] [PubMed] [Google Scholar]
- 2.MacDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, Barnes G, Taylor SA, James M, Groot N, MacFarlane H, Jenkins B, Anderson MA, Wexler NS, Gusella JF, Bates GP, Baxendale S, Hummerich H, Kirby S, North M, Youngman S, Mott R, Zehetner G, Sedlacek Z, Poustka A, Frischauf AM, Lehrach H, Buckler AJ, Church D, Doucettestamm L, Odonovan MC, Ribaramirez L, Shah M, Stanton VP, Strobel SA, Draths KM, Wales JL, Dervan P, Housman DE, Altherr M, Shiang R, Thompson L, Fielder T, Wasmuth JJ, Tagle D, Valdes J, Elmer L, Allard M, Castilla L, Swaroop M, Blanchard K, Collins FS, Snell R, Holloway T, Gillespie K, Datson N, Shaw D, Harper PS. A novel gene containing a trinucleotide repeat that is expanded and unstable on huntingtons-disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 3.LaSpada AR, Paulson HL, Fischbeck KH. Trinucleotide repeat expansion in neurological disease. Ann Neurol. 1994;36:814–822. doi: 10.1002/ana.410360604. [DOI] [PubMed] [Google Scholar]
- 4.Whaley NR, Fujioka S, Wszolek ZK. Autosomal dominant cerebellar ataxia type I: a review of the phenotypic and genotypic characteristics. Orphanet J Rare Dis. 2011;6:33. doi: 10.1186/1750-1172-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371:2120–2133. doi: 10.1016/S0140-6736(08)60921-6. [DOI] [PubMed] [Google Scholar]
- 6.Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, Le Paslier D, Frézal J, Cohen D, Weissenbach J, Munnich A, Melki J. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 7.Wood-Allum C, Shaw PJ. Motor neurone disease: a practical update on diagnosis and management. Clin Med. 2010;10:252–258. doi: 10.7861/clinmedicine.10-3-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Logroscino G, Traynor BJ, Hardiman O, Chiò A, Mitchell D, Swingler RJ, Millul A, Benn E, Beghi E EURALS. Incidence of amyotrophic lateral sclerosis in Europe. J Neurol Neurosurg Psychiatry. 2010;81:385–390. doi: 10.1136/jnnp.2009.183525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cooper-Knock J, Kirby J, Ferraiuolo L, Heath PR, Rattray M, Shaw PJ. Gene expression profiling in human neurodegenerative disease. Nat Rev Neurol. 2012;8:518–530. doi: 10.1038/nrneurol.2012.156. [DOI] [PubMed] [Google Scholar]
- 10.Ling S-C, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79:416–438. doi: 10.1016/j.neuron.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Renton AE, Chiò A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014;17:17–23. doi: 10.1038/nn.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malik S, Roeder RG. The metazoan mediator co-activator complex as an integrative hub for transcriptional regulation. Nature. 2010;11:761–772. doi: 10.1038/nrg2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodríguez-Navarro S, Hurt E. Linking gene regulation to mRNA production and export. Curr Opin Cell Biol. 2011;23:302–309. doi: 10.1016/j.ceb.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 14.van Hoof A, Wagner EJ. A brief survey of mRNA surveillance. Trends Biochem Sci. 2011;36:585–592. doi: 10.1016/j.tibs.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwak J, Workman JL, Lee D. The proteasome and its regulatory roles in gene expression. Biochim Biophys Acta. 2011;1809:88–96. doi: 10.1016/j.bbagrm.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 16.Dreyfuss G, Matunis MJ, Piñol-Roma S, Burd CG. hnRNP proteins and the biogenesis of mRNA. Annu Rev Biochem. 1993;62:289–321. doi: 10.1146/annurev.bi.62.070193.001445. [DOI] [PubMed] [Google Scholar]
- 17.Hieronymus H, Silver PA. A systems view of mRNP biology. Genes Dev. 2004;18:2845–2860. doi: 10.1101/gad.1256904. [DOI] [PubMed] [Google Scholar]
- 18.Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell. 2009;136:777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh RK, Cooper TA. Pre-mRNA splicing in disease and therapeutics. Trends Mol Med. 2012;18:472–482. doi: 10.1016/j.molmed.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Le Quesne JPC, Spriggs KA, Bushell M, Willis AE. Dysregulation of protein synthesis and disease. J Pathol. 2010;220:140–151. doi: 10.1002/path.2627. [DOI] [PubMed] [Google Scholar]
- 21.Maniatis T, Reed R. An extensive network of coupling among gene expression machines. Nature. 2002;416:499–506. doi: 10.1038/416499a. [DOI] [PubMed] [Google Scholar]
- 22.Luna R, Gaillard H, González-Aguilera C, Aguilera A. Biogenesis of mRNPs: integrating different processes in the eukaryotic nucleus. Chromosoma. 2008;117:319–331. doi: 10.1007/s00412-008-0158-4. [DOI] [PubMed] [Google Scholar]
- 23.Hsin J-P, Manley JL. The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev. 2012;26:2119–2137. doi: 10.1101/gad.200303.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jurado AR, Tan D, Jiao X, Kiledjian M, Tong L. Structure and function of pre-mRNA 5′-end capping quality control and 3′-end processing. Biochemistry. 2014;53:1882–1898. doi: 10.1021/bi401715v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Will CL. Lührmann R. Spliceosome structure and function. Cold Spring Harb Perspect Biol. 2011;3:a003707. doi: 10.1101/cshperspect.a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tian B, Manley JL. Alternative cleavage and polyadenylation: the long and short of it. Trends Biochem Sci. 2013;38:312–320. doi: 10.1016/j.tibs.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elkon R, Ugalde AP, Agami R. Alternative cleavage and polyadenylation: extent, regulation and function. Nature. 2013;14:496–506. doi: 10.1038/nrg3482. [DOI] [PubMed] [Google Scholar]
- 28.Walsh MJ, Hautbergue GM, Wilson SA. Structure and function of mRNA export adaptors. Biochem Soc Trans. 2010;38:232–236. doi: 10.1042/BST0380232. [DOI] [PubMed] [Google Scholar]
- 29.Müller-McNicoll M, Neugebauer KM. How cells get the message: dynamic assembly and function of mRNA-protein complexes. Nature. 2013;14:275–287. doi: 10.1038/nrg3434. [DOI] [PubMed] [Google Scholar]
- 30.Natalizio BJ, Wente SR. Postage for the messenger: designating routes for nuclear mRNA export. Trends Cell Biol. 2013;23:365–373. doi: 10.1016/j.tcb.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yonath A. Large facilities and the evolving ribosome, the cellular machine for genetic-code translation. J R Soc Interface. 2009;6:S575–585. doi: 10.1098/rsif.2009.0167.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moore PB, Steitz TA. The roles of RNA in the synthesis of protein. Cold Spring Harb Perspect Biol. 2011;3:a003780. doi: 10.1101/cshperspect.a003780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gomes C, Merianda TT, Lee SJ, Yoo S, Twiss JL. Molecular determinants of the axonal mRNA transcriptome. Dev Neurobiol. 2014;74:218–232. doi: 10.1002/dneu.22123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nasir J, Floresco SB, O'Kusky JR, Diewert VM, Richman JM, Zeisler J, Borowski A, Marth JD, Phillips AG, Hayden MR. Targeted disruption of the Huntington's disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81:811–823. doi: 10.1016/0092-8674(95)90542-1. [DOI] [PubMed] [Google Scholar]
- 35.Sathasivam K, Neueder A, Gipson TA, Landles C, Benjamin AC, Bondulich MK, Smith DL, Faull RLM, Roos RAC, Howland D, Detloff PJ, Housman DE, Bates GP. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc Natl Acad Sci USA. 2013;110:2366–2370. doi: 10.1073/pnas.1221891110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cornett J, Cao F, Wang C-E, Ross CA, Bates GP, Li S-H, Li X-J. Polyglutamine expansion of huntingtin impairs its nuclear export. Nat Genet. 2005;37:198–204. doi: 10.1038/ng1503. [DOI] [PubMed] [Google Scholar]
- 37.DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 38.Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 39.Tomás-Zapico C, Díez-Zaera M, Ferrer I, Gómez-Ramos P, Morán MA, Miras-Portugal MT, Díaz-Hernández M, Lucas JJ. α-Synuclein accumulates in huntingtin inclusions but forms independent filaments and its deficiency attenuates early phenotype in a mouse model of Huntington's disease. Hum Mol Genet. 2012;21:495–510. doi: 10.1093/hmg/ddr507. [DOI] [PubMed] [Google Scholar]
- 40.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 41.Moumné L, Betuing S, Caboche J. Multiple aspects of gene dysregulation in Huntington's disease. Front Neurol. 2013;4:127. doi: 10.3389/fneur.2013.00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G, Elliston LA, Hartog C, Goldstein DR, Thu D, Hollingsworth ZR, Collin F, Synek B, Holmans PA, Young AB, Wexler NS, Delorenzi M, Kooperberg C, Augood SJ, Faull RLM, Olson JM, Jones L, Luthi-Carter R. Regional and cellular gene expression changes in human Huntington's disease brain. Hum Mol Genet. 2006;15:965–977. doi: 10.1093/hmg/ddl013. [DOI] [PubMed] [Google Scholar]
- 43.Anderson AN, Roncaroli F, Hodges A, Deprez M, Turkheimer FE. Chromosomal profiles of gene expression in Huntington's disease. Brain. 2008;131:381–388. doi: 10.1093/brain/awm312. [DOI] [PubMed] [Google Scholar]
- 44.Becanovic K, Pouladi MA, Lim RS, Kuhn A, Pavlidis P, Luthi-Carter R, Hayden MR, Leavitt BR. Transcriptional changes in Huntington disease identified using genome-wide expression profiling and cross-platform analysis. Hum Mol Genet. 2010;19:1438–1452. doi: 10.1093/hmg/ddq018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cattaneo E, Zuccato C, Tartari M. Normal huntingtin function: an alternative approach to Huntington's disease. Nat Rev Neurosci. 2005;6:919–930. doi: 10.1038/nrn1806. [DOI] [PubMed] [Google Scholar]
- 46.Krzyzosiak WJ, Sobczak K, Wojciechowska M, Fiszer A, Mykowska A, Kozlowski P. Triplet repeat RNA structure and its role as pathogenic agent and therapeutic target. Nucleic Acids Res. 2012;40:11–26. doi: 10.1093/nar/gkr729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsoi H, Chan HYE. Expression of expanded CAG transcripts triggers nucleolar stress in Huntington's disease. Cerebellum. 2013;12:310–312. doi: 10.1007/s12311-012-0447-6. [DOI] [PubMed] [Google Scholar]
- 48.Bañez-Coronel M, Porta S, Kagerbauer B, Mateu-Huertas E, Pantano L, Ferrer I, Guzmán M, Estivill X, Martí E. A pathogenic mechanism in Huntington's disease involves small CAG-repeated RNAs with neurotoxic activity. PLoS Genet. 2012;8:e1002481. doi: 10.1371/journal.pgen.1002481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goehler H, Lalowski M, Stelzl U, Waelter S, Stroedicke M, Worm U, Droege A, Lindenberg KS, Knoblich M, Haenig C, Herbst M, Suopanki J, Scherzinger E, Abraham C, Bauer B, Hasenbank R, Fritzsche A, Ludewig AH, Büssow K, Buessow K, Coleman SH, Gutekunst C-A, Landwehrmeyer BG, Lehrach H, Wanker EE. A protein interaction network links GIT1, an enhancer of huntingtin aggregation, to Huntington's disease. Mol Cell. 2004;15:853–865. doi: 10.1016/j.molcel.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 50.Leavitt BR, van Raamsdonk JM, Shehadeh J, Fernandes H, Murphy Z, Graham RK, Wellington CL, Raymond LA, Hayden MR. Wild-type huntingtin protects neurons from excitotoxicity. J Neurochem. 2006;96:1121–1129. doi: 10.1111/j.1471-4159.2005.03605.x. [DOI] [PubMed] [Google Scholar]
- 51.Godin JD, Colombo K, Molina-Calavita M, Keryer G, Zala D, Charrin BC, Dietrich P, Volvert M-L, Guillemot F, Dragatsis I, Bellaiche Y, Saudou F, Nguyen L, Humbert S. Huntingtin is required for mitotic spindle orientation and mammalian neurogenesis. Neuron. 2010;67:392–406. doi: 10.1016/j.neuron.2010.06.027. [DOI] [PubMed] [Google Scholar]
- 52.Colin E, Zala D, Liot G, Rangone H, Borrell-Pagès M, Li X-J, Saudou F, Humbert S. Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. EMBO J. 2008;27:2124–2134. doi: 10.1038/emboj.2008.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Savas JN, Makusky A, Ottosen S, Baillat D, Then F, Krainc D, Shiekhattar R, Markey SP, Tanese N. Huntington's disease protein contributes to RNA-mediated gene silencing through association with Argonaute and P bodies. Proc Natl Acad Sci USA. 2008;105:10820–10825. doi: 10.1073/pnas.0800658105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, Cataudella T, Leavitt BR, Hayden MR, Timmusk T, Rigamonti D, Cattaneo E. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet. 2003;35:76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]
- 55.Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- 56.Johnson R, Zuccato C, Belyaev ND, Guest DJ, Cattaneo E, Buckley NJ. A microRNA-based gene dysregulation pathway in Huntington's disease. Neurobiol Dis. 2008;29:438–445. doi: 10.1016/j.nbd.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 57.Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- 58.Kosik KS. The neuronal microRNA system. Nat Rev Neurosci. 2006;7:911–920. doi: 10.1038/nrn2037. [DOI] [PubMed] [Google Scholar]
- 59.Li S-H, Li X-J. Huntingtin-protein interactions and the pathogenesis of Huntington's disease. Trends Genet. 2004;20:146–154. doi: 10.1016/j.tig.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 60.Ratovitski T, Chighladze E, Arbez N, Boronina T, Herbrich S, Cole RN, Ross CA. Huntingtin protein interactions altered by polyglutamine expansion as determined by quantitative proteomic analysis. Cell Cycle. 2012;11:2006–2021. doi: 10.4161/cc.20423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rubinsztein DC, Carmichael J. Huntington's disease: molecular basis of neurodegeneration. Expert Rev Mol Med. 2003;5:1–21. doi: 10.1017/S1462399403006549. [DOI] [PubMed] [Google Scholar]
- 62.Johri A, Chandra A, Beal MF. PGC-1α, mitochondrial dysfunction, and Huntington's disease. Free Radic Biol Med. 2013;62:37–46. doi: 10.1016/j.freeradbiomed.2013.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ayala-Peña S. Role of oxidative DNA damage in mitochondrial dysfunction and Huntington's disease pathogenesis. Free Radic Biol Med. 2013;62:102–110. doi: 10.1016/j.freeradbiomed.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jonson I, Ougland R, Klungland A, Larsen E. Oxidative stress causes DNA triplet expansion in Huntington's disease mouse embryonic stem cells. Stem Cell Res. 2013;11:1264–1271. doi: 10.1016/j.scr.2013.08.010. [DOI] [PubMed] [Google Scholar]
- 65.Tsunemi T, Ashe TD, Morrison BE, Soriano KR, Au J, Roque RAV, Lazarowski ER, Damian VA, Masliah E, La Spada AR. PGC-1α rescues Huntington's disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci Transl Med. 2012;4:142ra97. doi: 10.1126/scitranslmed.3003799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- 67.Martin E, Betuing S, Pages C, Cambon K, Auregan G, Deglon N, Roze E, Caboche J. Mitogen- and stress-activated protein kinase 1-induced neuroprotection in Huntington's disease: role on chromatin remodeling at the PGC-1-alpha promoter. Hum Mol Genet. 2011;20:2422–2434. doi: 10.1093/hmg/ddr148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim M-O, Chawla P, Overland RP, Xia E, Sadri-Vakili G, Cha J-HJ. Altered histone monoubiquitylation mediated by mutant huntingtin induces transcriptional dysregulation. J Neurosci. 2008;28:3947–3957. doi: 10.1523/JNEUROSCI.5667-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee J, Hwang YJ, Ryu H, Kowall NW, Ryu H. Nucleolar dysfunction in Huntington's disease. Biochim Biophys Acta. 2014;1842:785–790. doi: 10.1016/j.bbadis.2013.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.van de Warrenburg BPC, Sinke RJ, Verschuuren-Bemelmans CC, Scheffer H, Brunt ER, Ippel PF, Maat-Kievit JA, Dooijes D, Notermans NC, Lindhout D, Knoers NVAM, Kremer HPH. Spinocerebellar ataxias in the Netherlands: prevalence and age at onset variance analysis. Neurology. 2002;12:702–708. doi: 10.1212/wnl.58.5.702. [DOI] [PubMed] [Google Scholar]
- 71.Brusco A, Gellera C, Cagnoli C, Saluto A, Castucci A, Michielotto C, Fetoni V, Mariotti C, Migone N, Di Donato S, Taroni F. Molecular genetics of hereditary spinocerebellar ataxia: mutation analysis of spinocerebellar ataxia genes and CAG/CTG repeat expansion detection in 225 Italian families. Arch Neurol. 2004;61:727–733. doi: 10.1001/archneur.61.5.727. [DOI] [PubMed] [Google Scholar]
- 72.Craig K, Keers SM, Archibald K, Curtis A, Chinnery PF. Molecular epidemiology of spinocerebellar ataxia type 6. Ann Neurol. 2004;55:752–755. doi: 10.1002/ana.20110. [DOI] [PubMed] [Google Scholar]
- 73.Harding AE. Clinical features and classification of inherited ataxias. Adv Neurol. 1993;61:1–14. [PubMed] [Google Scholar]
- 74.Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Annu Rev Neurosci. 2000;23:217–247. doi: 10.1146/annurev.neuro.23.1.217. [DOI] [PubMed] [Google Scholar]
- 75.Dueñas AM, Goold R, Giunti P. Molecular pathogenesis of spinocerebellar ataxias. Brain. 2006;129:1357–1370. doi: 10.1093/brain/awl081. [DOI] [PubMed] [Google Scholar]
- 76.Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10:S10–17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- 77.Park Y, Hong S, Kim S-J, Kang S. Proteasome function is inhibited by polyglutamine-expanded ataxin-1, the SCA1 gene product. Mol Cells. 2005;19:23–30. [PubMed] [Google Scholar]
- 78.Matsuyama Z, Yanagisawa NK, Aoki Y, Black JL, Lennon VA, Mori Y, Imoto K, Inuzuka T. Polyglutamine repeats of spinocerebellar ataxia 6 impair the cell-death-preventing effect of CaV2.1 Ca2+ channel–loss-of-function cellular model of SCA6. Neurobiol Dis. 2004;17:198–204. doi: 10.1016/j.nbd.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 79.Shimohata T, Nakajima T, Yamada M, Uchida C, Onodera O, Naruse S, Kimura T, Koide R, Nozaki K, Sano Y, Ishiguro H, Sakoe K, Ooshima T, Sato A, Ikeuchi T, Oyake M, Sato T, Aoyagi Y, Hozumi I, Nagatsu T, Takiyama Y, Nishizawa M, Goto J, Kanazawa I, Davidson I, Tanese N, Takahashi H, Tsuji S. Expanded polyglutamine stretches interact with TAFII130, interfering with CREB-dependent transcription. Nat Genet. 2000;26:29–36. doi: 10.1038/79139. [DOI] [PubMed] [Google Scholar]
- 80.Li F, Macfarlan T, Pittman RN, Chakravarti D. Ataxin-3 is a histone-binding protein with two independent transcriptional corepressor activities. J Biol Chem. 2002;277:45004–45012. doi: 10.1074/jbc.M205259200. [DOI] [PubMed] [Google Scholar]
- 81.Tsai C-C, Kao H-Y, Mitzutani A, Banayo E, Rajan H, McKeown M, Evans RM. Ataxin 1, a SCA1 neurodegenerative disorder protein, is functionally linked to the silencing mediator of retinoid and thyroid hormone receptors. Proc Natl Acad Sci USA. 2004;101:4047–4052. doi: 10.1073/pnas.0400615101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wojciechowska M, Krzyzosiak WJ. Cellular toxicity of expanded RNA repeats: focus on RNA foci. Hum Mol Genet. 2011;20:3811–3821. doi: 10.1093/hmg/ddr299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lin X, Ashizawa T. Recent progress in spinocerebellar ataxia type-10 (SCA10) Cerebellum. 2005;4:37–42. doi: 10.1080/14734220510007897. [DOI] [PubMed] [Google Scholar]
- 84.White MC, Gao R, Xu W, Mandal SM, Lim JG, Hazra TK, Wakamiya M, Edwards SF, Raskin S, Teive HAG, Zoghbi HY, Sarkar PS, Ashizawa T. Inactivation of hnRNP K by expanded intronic AUUCU repeat induces apoptosis via translocation of PKCdelta to mitochondria in spinocerebellar ataxia 10. PLoS Genet. 2010;6:e1000984. doi: 10.1371/journal.pgen.1000984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sato N, Amino T, Kobayashi K, Asakawa S, Ishiguro T, Tsunemi T, Takahashi M, Matsuura T, Flanigan KM, Iwasaki S, Ishino F, Saito Y, Murayama S, Yoshida M, Hashizume Y, Takahashi Y, Tsuji S, Shimizu N, Toda T, Ishikawa K, Mizusawa H. Spinocerebellar ataxia type 31 is associated with ‘inserted’ penta-nucleotide repeats containing (TGGAA)n. Am J Hum Genet. 2009;85:544–557. doi: 10.1016/j.ajhg.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ohta Y, Hayashi T, Nagai M, Okamoto M, Nagotani S, Nagano I, Ohmori N, Takehisa Y, Murakami T, Shoji M, Kamiya T, Abe K. Two cases of spinocerebellar ataxia accompanied by involvement of the skeletal motor neuron system and bulbar palsy. Intern Med. 2007;46:751–755. doi: 10.2169/internalmedicine.46.6261. [DOI] [PubMed] [Google Scholar]
- 87.Abe K, Ikeda Y, Kurata T, Ohta Y, Manabe Y, Okamoto M, Takamatsu K, Ohta T, Takao Y, Shiro Y, Shoji M, Kamiya T, Kobayashi H, Koizumi A. Cognitive and affective impairments of a novel SCA/MND crossroad mutation Asidan. Eur J Neurol. 2012;19:1070–1078. doi: 10.1111/j.1468-1331.2012.03669.x. [DOI] [PubMed] [Google Scholar]
- 88.Kobayashi H, Abe K, Matsuura T, Ikeda Y, Hitomi T, Akechi Y, Habu T, Liu W, Okuda H, Koizumi A. Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am J Hum Genet. 2011;89:121–130. doi: 10.1016/j.ajhg.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung G-YR, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita V-M, Kaivorinne A-L, Hölttä-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chiò A, Restagno G, Borghero G, Sabatelli M ITALSGEN Consortium. Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lee Y-B, Chen H-J, Peres JN, Gomez-Deza J, Attig J, Štalekar M, Troakes C, Nishimura AL, Scotter EL, Vance C, Adachi Y, Sardone V, Miller JW, Smith BN, Gallo J-M, Ule J, Hirth F, Rogelj B, Houart C, Shaw CE. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013;5:1178–1186. doi: 10.1016/j.celrep.2013.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cooper-Knock J, Walsh MJ, Higginbottom A, Highley JR, Dickman MJ, Edbauer D, Ince PG, Wharton SB, Wilson SA, Kirby J, Hautbergue GM, Shaw PJ. Sequestration of multiple RNA recognition motif-containing proteins by C9ORF72 repeat expansions. Brain. 2014;137:2040–2051. doi: 10.1093/brain/awu120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nalavade R, Griesche N, Ryan DP, Hildebrand S, Krauss S. Mechanisms of RNA-induced toxicity in CAG repeat disorders. Cell Death Dis. 2013;4:e752. doi: 10.1038/cddis.2013.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Evers MM, Toonen LJA, van Roon-Mom WMC. Ataxin-3 protein and RNA toxicity in spinocerebellar ataxia type 3: current insights and emerging therapeutic strategies. Mol Neurobiol. 2014;49:1513–1531. doi: 10.1007/s12035-013-8596-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Moseley ML, Zu T, Ikeda Y, Gao W, Mosemiller AK, Daughters RS, Chen G, Weatherspoon MR, Clark HB, Ebner TJ, Day JW, Ranum LPW. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat Genet. 2006;38:758–769. doi: 10.1038/ng1827. [DOI] [PubMed] [Google Scholar]
- 96.Daughters RS, Tuttle DL, Gao W, Ikeda Y, Moseley ML, Ebner TJ, Swanson MS, Ranum LPW. RNA gain-of-function in spinocerebellar ataxia type 8. PLoS Genet. 2009;5:e1000600. doi: 10.1371/journal.pgen.1000600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MAC, Nan Z, Forster C, Low WC, Schoser B, Somia NV, Clark HB, Schmechel S, Bitterman PB, Gourdon G, Swanson MS, Moseley M, Ranum LPW. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci USA. 2011;108:260–265. doi: 10.1073/pnas.1013343108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cartegni L, Krainer AR. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet. 2002;30:377–384. doi: 10.1038/ng854. [DOI] [PubMed] [Google Scholar]
- 99.Burnett BG, Muñoz E, Tandon A, Kwon DY, Sumner CJ, Fischbeck KH. Regulation of SMN protein stability. Mol Cell Biol. 2009;29:1107–1115. doi: 10.1128/MCB.01262-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cho S, Dreyfuss G. A degron created by SMN2 exon 7 skipping is a principal contributor to spinal muscular atrophy severity. Genes Dev. 2010;24:438–442. doi: 10.1101/gad.1884910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ruggiu M, McGovern VL, Lotti F, Saieva L, Li DK, Kariya S, Monani UR, Burghes AHM, Pellizzoni L. A role for SMN exon 7 splicing in the selective vulnerability of motor neurons in spinal muscular atrophy. Mol Cell Biol. 2012;32:126–138. doi: 10.1128/MCB.06077-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cho S, Moon H, Loh TJ, Oh HK, Williams DR, Liao DJ, Zhou J, Green MR, Zheng X, Shen H. PSF contacts exon 7 of SMN2 pre-mRNA to promote exon 7 inclusion. Biochim Biophys Acta. 2014;1839:517–525. doi: 10.1016/j.bbagrm.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cho S, Moon H, Loh TJ, Oh HK, Cho S, Choy HE, Song WK, Chun J-S, Zheng X, Shen H. hnRNP M facilitates exon 7 inclusion of SMN2 pre-mRNA in spinal muscular atrophy by targeting an enhancer on exon 7. Biochim Biophys Acta. 2014;1839:306–315. doi: 10.1016/j.bbagrm.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 104.Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA. 1999;96:6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Monani UR, Lorson CL, Parsons DW, Prior TW, Androphy EJ, Burghes AH, McPherson JD. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet. 1999;8:1177–1183. doi: 10.1093/hmg/8.7.1177. [DOI] [PubMed] [Google Scholar]
- 106.Kashima T, Manley JL. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat Genet. 2003;34:460–463. doi: 10.1038/ng1207. [DOI] [PubMed] [Google Scholar]
- 107.Kashima T, Rao N, David CJ, Manley JL. hnRNP A1 functions with specificity in repression of SMN2 exon 7 splicing. Hum Mol Genet. 2007;16:3149–3159. doi: 10.1093/hmg/ddm276. [DOI] [PubMed] [Google Scholar]
- 108.Pedrotti S, Bielli P, Paronetto MP, Ciccosanti F, Fimia GM, Stamm S, Manley JL, Sette C. The splicing regulator Sam68 binds to a novel exonic splicing silencer and functions in SMN2 alternative splicing in spinal muscular atrophy. EMBO J. 2010;29:1235–1247. doi: 10.1038/emboj.2010.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chen H-H, Chang J-G, Lu R-M, Peng T-Y, Tarn W-Y. The RNA binding protein hnRNP Q modulates the utilization of exon 7 in the survival motor neuron 2 (SMN2) gene. Mol Cell Biol. 2008;28:6929–6938. doi: 10.1128/MCB.01332-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Miyajima H, Miyaso H, Okumura M, Kurisu J, Imaizumi K. Identification of a cis-acting element for the regulation of SMN exon 7 splicing. J Biol Chem. 2002;277:23271–23277. doi: 10.1074/jbc.M200851200. [DOI] [PubMed] [Google Scholar]
- 111.Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet. 2008;82:834–848. doi: 10.1016/j.ajhg.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kashima T, Rao N, Manley JL. An intronic element contributes to splicing repression in spinal muscular atrophy. Proc Natl Acad Sci USA. 2007;104:3426–3431. doi: 10.1073/pnas.0700343104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hofmann Y, Lorson CL, Stamm S, Androphy EJ, Wirth B. Htra2-beta 1 stimulates an exonic splicing enhancer and can restore full-length SMN expression to survival motor neuron 2 (SMN2) Proc Natl Acad Sci USA. 2000;9:9618–9623. doi: 10.1073/pnas.160181697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Young PJ, DiDonato CJ, Hu D, Kothary R, Androphy EJ, Lorson CL. SRp30c-dependent stimulation of survival motor neuron (SMN) exon 7 inclusion is facilitated by a direct interaction with hTra2 beta 1. Hum Mol Genet. 2002;11:577–587. doi: 10.1093/hmg/11.5.577. [DOI] [PubMed] [Google Scholar]
- 115.Hofmann Y, Wirth B. hnRNP-G promotes exon 7 inclusion of survival motor neuron (SMN) via direct interaction with Htra2-beta1. Hum Mol Genet. 2002;11:2037–2049. doi: 10.1093/hmg/11.17.2037. [DOI] [PubMed] [Google Scholar]
- 116.Prior TW, Krainer AR, Hua Y, Swoboda KJ, Snyder PC, Bridgeman SJ, Burghes AHM, Kissel JT. A positive modifier of spinal muscular atrophy in the SMN2 gene. Am J Hum Genet. 2009;85:408–413. doi: 10.1016/j.ajhg.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Vezain M, Saugier-Veber P, Goina E, Touraine R, Manel V, Toutain A, Fehrenbach S, Frébourg T, Pagani F, Tosi M, Martins A. A rare SMN2 variant in a previously unrecognized composite splicing regulatory element induces exon 7 inclusion and reduces the clinical severity of spinal muscular atrophy. Hum Mutat. 2010;31:E1110–1125. doi: 10.1002/humu.21173. [DOI] [PubMed] [Google Scholar]
- 118.Bebee TW, Dominguez CE, Samadzadeh-Tarighat S, Akehurst KL, Chandler DS. Hypoxia is a modifier of SMN2 splicing and disease severity in a severe SMA mouse model. Hum Mol Genet. 2012;21:4301–4313. doi: 10.1093/hmg/dds263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kolb SJ, Battle DJ, Dreyfuss G. Molecular functions of the SMN complex. J Child Neurol. 2007;22:990–994. doi: 10.1177/0883073807305666. [DOI] [PubMed] [Google Scholar]
- 120.Fischer U, Englbrecht C, Chari A. Biogenesis of spliceosomal small nuclear ribonucleoproteins. Wiley Interdiscip Rev RNA. 2011;2:718–731. doi: 10.1002/wrna.87. [DOI] [PubMed] [Google Scholar]
- 121.Tsuiji H, Iguchi Y, Furuya A, Kataoka A, Hatsuta H, Atsuta N, Tanaka F, Hashizume Y, Akatsu H, Murayama S, Sobue G, Yamanaka K. Spliceosome integrity is defective in the motor neuron diseases ALS and SMA. EMBO Mol Med. 2013;5:221–234. doi: 10.1002/emmm.201202303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Liu H, Shafey D, Moores JN, Kothary R. Neurodevelopmental consequences of Smn depletion in a mouse model of spinal muscular atrophy. J Neurosci Res. 2010;88:111–122. doi: 10.1002/jnr.22189. [DOI] [PubMed] [Google Scholar]
- 123.Zhang Z, Lotti F, Dittmar K, Younis I, Wan L, Kasim M, Dreyfuss G. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell. 2008;133:585–600. doi: 10.1016/j.cell.2008.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lotti F, Imlach WL, Saieva L, Beck ES, Hao LT, Li DK, Jiao W, Mentis GZ, Beattie CE, McCabe BD, Pellizzoni L. An SMN-dependent U12 splicing event essential for motor circuit function. Cell. 2012;151:440–454. doi: 10.1016/j.cell.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.See K, Yadav P, Giegerich M, Cheong PS, Graf M, Vyas H, Lee SGP, Mathavan S, Fischer U, Sendtner M, Winkler C. SMN deficiency alters Nrxn2 expression and splicing in zebrafish and mouse models of spinal muscular atrophy. Hum Mol Genet. 2014;23:1754–1770. doi: 10.1093/hmg/ddt567. [DOI] [PubMed] [Google Scholar]
- 126.Fallini C, Bassell GJ, Rossoll W. Spinal muscular atrophy: the role of SMN in axonal mRNA regulation. Brain Res. 2012;1462:81–92. doi: 10.1016/j.brainres.2012.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rossoll W, Jablonka S, Andreassi C, Kröning A-K, Karle K, Monani UR, Sendtner M. Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J Cell Biol. 2003;163:801–812. doi: 10.1083/jcb.200304128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Akten B, Kye MJ, Hao LT, Wertz MH, Singh S, Nie D, Huang J, Merianda TT, Twiss JL, Beattie CE, Steen JAJ, Sahin M. Interaction of survival of motor neuron (SMN) and HuD proteins with mRNA cpg15 rescues motor neuron axonal deficits. Proc Natl Acad Sci USA. 2011;108:10337–10342. doi: 10.1073/pnas.1104928108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Rage F, Boulisfane N, Rihan K, Neel H, Gostan T, Bertrand E, Bordonné R, Soret J. Genome-wide identification of mRNAs associated with the protein SMN whose depletion decreases their axonal localization. RNA. 2013;19:1755–1766. doi: 10.1261/rna.040204.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sabra M, Texier P, Maalouf El J, Lomonte P. The Tudor protein survival motor neuron (SMN) is a chromatin-binding protein that interacts with methylated lysine 79 of histone H3. J Cell Sci. 2013;126:3664–3677. doi: 10.1242/jcs.126003. [DOI] [PubMed] [Google Scholar]
- 131.Haramati S, Chapnik E, Sztainberg Y, Eilam R, Zwang R, Gershoni N, McGlinn E, Heiser PW, Wills A-M, Wirguin I, Rubin LL, Misawa H, Tabin CJ, Brown R, Chen A, Hornstein E. miRNA malfunction causes spinal motor neuron disease. Proc Natl Acad Sci USA. 2010;107:13111–13116. doi: 10.1073/pnas.1006151107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Helmken C, Hofmann Y, Schoenen F, Oprea G, Raschke H, Rudnik-Schöneborn S, Zerres K, Wirth B. Evidence for a modifying pathway in SMA discordant families: reduced SMN level decreases the amount of its interacting partners and Htra2-beta1. Hum Genet. 2003;114:11–21. doi: 10.1007/s00439-003-1025-2. [DOI] [PubMed] [Google Scholar]
- 133.Jodelka FM, Ebert AD, Duelli DM, Hastings ML. A feedback loop regulates splicing of the spinal muscular atrophy-modifying gene, SMN2. Hum Mol Genet. 2010;19:4906–4917. doi: 10.1093/hmg/ddq425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ferraiuolo L, Kirby J, Grierson AJ, Sendtner M, Shaw PJ. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat Rev Neurol. 2011;7:616–630. doi: 10.1038/nrneurol.2011.152. [DOI] [PubMed] [Google Scholar]
- 135.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 136.Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 137.Ince PG, Highley JR, Kirby J, Wharton SB, Takahashi H, Strong MJ, Shaw PJ. Molecular pathology and genetic advances in amyotrophic lateral sclerosis: an emerging molecular pathway and the significance of glial pathology. Acta Neuropathol. 2011;122:657–671. doi: 10.1007/s00401-011-0913-0. [DOI] [PubMed] [Google Scholar]
- 138.Ihara R, Matsukawa K, Nagata Y, Kunugi H, Tsuji S, Chihara T, Kuranaga E, Miura M, Wakabayashi T, Hashimoto T, Iwatsubo T. RNA binding mediates neurotoxicity in the transgenic Drosophila model of TDP-43 proteinopathy. Hum Mol Genet. 2013;22:4474–4484. doi: 10.1093/hmg/ddt296. [DOI] [PubMed] [Google Scholar]
- 139.Brockington A, Ning K, Heath PR, Wood E, Kirby J, Fusi N, Lawrence N, Wharton SB, Ince PG, Shaw PJ. Unravelling the enigma of selective vulnerability in neurodegeneration: motor neurons resistant to degeneration in ALS show distinct gene expression characteristics and decreased susceptibility to excitotoxicity. Acta Neuropathol. 2013;125:95–109. doi: 10.1007/s00401-012-1058-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 141.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 142.Gaudette M, Hirano M, Siddique T. Current status of SOD1 mutations in familial amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:83–89. doi: 10.1080/14660820050515377. [DOI] [PubMed] [Google Scholar]
- 143.Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kwiatkowski TJ, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 145.Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mackenzie IRA, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010;9:995–1007. doi: 10.1016/S1474-4422(10)70195-2. [DOI] [PubMed] [Google Scholar]
- 147.Zhang D, Iyer LM, He F, Aravind L. Discovery of novel DENN proteins: implications for the evolution of eukaryotic intracellular membrane structures and human disease. Front Genet. 2012;3:283. doi: 10.3389/fgene.2012.00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Stewart H, Rutherford NJ, Briemberg H, Krieger C, Cashman N, Fabros M, Baker M, Fok A, DeJesus-Hernandez M, Eisen A, Rademakers R, Mackenzie IRA. Clinical and pathological features of amyotrophic lateral sclerosis caused by mutation in the C9ORF72 gene on chromosome 9p. Acta Neuropathol. 2012;123:409–417. doi: 10.1007/s00401-011-0937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cooper-Knock J, Hewitt C, Highley JR, Brockington A, Milano A, Man S, Martindale J, Hartley J, Walsh T, Gelsthorpe C, Baxter L, Forster G, Fox M, Bury J, Mok K, McDermott CJ, Traynor BJ, Kirby J, Wharton SB, Ince PG, Hardy J, Shaw PJ. Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain. 2012;135:751–764. doi: 10.1093/brain/awr365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Fratta P, Mizielinska S, Nicoll AJ, Zloh M, Fisher EMC, Parkinson G, Isaacs AM. C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci Rep. 2012;2:1016. doi: 10.1038/srep01016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Reddy K, Zamiri B, Stanley SYR, Macgregor RB, Pearson CE. The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length-dependent uni- and multimolecular RNA G-quadruplex structures. J Biol Chem. 2013;288:9860–9866. doi: 10.1074/jbc.C113.452532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Haeusler AR, Donnelly CJ, Periz G, Simko EAJ, Shaw PG, Kim M-S, Maragakis NJ, Troncoso JC, Pandey A, Sattler R, Rothstein JD, Wang J. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014;507:195–200. doi: 10.1038/nature13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Donnelly CJ, Zhang P-W, Pham JT, Heusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM, Maragakis N, Tienari PJ, Petrucelli L, Traynor BJ, Wang J, Rigo F, Bennett CF, Blackshaw S, Sattler R, Rothstein JD. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80:415–428. doi: 10.1016/j.neuron.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sareen D, O'Rourke JG, Meera P, Muhammad AKMG, Grant S, Simpkinson M, Bell S, Carmona S, Ornelas L, Sahabian A, Gendron T, Petrucelli L, Baughn M, Ravits J, Harms MB, Rigo F, Bennett CF, Otis TS, Svendsen CN, Baloh RH. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med. 2013;5:208ra149. doi: 10.1126/scitranslmed.3007529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li H-R, Jiang J, Watt AT, Chun S, Katz M, Qiu J, Sun Y, Ling S-C, Zhu Q, Polymenidou M, Drenner K, Artates JW, McAlonis-Downes M, Markmiller S, Hutt KR, Pizzo DP, Cady J, Harms MB, Baloh RH, Vandenberg SR, Yeo GW, Fu X-D, Bennett CF, Cleveland DW, Ravits J. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci USA. 2013;110:E4530–4539. doi: 10.1073/pnas.1318835110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zu T, Liu Y, Bañez-Coronel M, Reid T, Pletnikova O, Lewis J, Miller TM, Harms MB, Falchook AE, Subramony SH, Ostrow LW, Rothstein JD, Troncoso JC, Ranum LPW. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci USA. 2013;110:E4968–4977. doi: 10.1073/pnas.1315438110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Mizielinska S, Lashley T, Norona FE, Clayton EL, Ridler CE, Fratta P, Isaacs AM. C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol. 2013;126:845–857. doi: 10.1007/s00401-013-1200-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G, Janssens J, Bettens K, Van Cauwenberghe C, Pereson S, Engelborghs S, Sieben A, De Jonghe P, Vandenberghe R, Santens P, De Bleecker J, Maes G, Bäumer V, Dillen L, Joris G, Cuijt I, Corsmit E, Elinck E, Van Dongen J, Vermeulen S, Van den Broeck M, Vaerenberg C, Mattheijssens M, Peeters K, Robberecht W, Cras P, Martin J-J, De Deyn PP, Cruts MR, Van Broeckhoven C. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 2012;1:54–65. doi: 10.1016/S1474-4422(11)70261-7. [DOI] [PubMed] [Google Scholar]
- 159.Belzil VV, Bauer PO, Prudencio M, Gendron TF, Stetler CT, Yan IK, Pregent L, Daughrity L, Baker MC, Rademakers R, Boylan K, Patel TC, Dickson DW, Petrucelli L. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol. 2013;126:895–905. doi: 10.1007/s00401-013-1199-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ciura S, Lattante S, Le Ber I, Latouche M, Tostivint H, Brice A, Kabashi E. Loss of function of C9orf72 causes motor deficits in a zebrafish model of Amyotrophic Lateral Sclerosis. Ann Neurol. 2013;74:180–187. doi: 10.1002/ana.23946. [DOI] [PubMed] [Google Scholar]
- 161.Xi Z, Zinman L, Moreno D, Schymick J, Liang Y, Sato C, Zheng Y, Ghani M, Dib S, Keith J, Robertson J, Rogaeva E. Hypermethylation of the CpG island near the G4C2 repeat in ALS with a C9orf72 expansion. Am J Hum Genet. 2013;92:981–989. doi: 10.1016/j.ajhg.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Waite AJ, Bäumer D, East S, Neal J, Morris HR, Ansorge O, Blake DJ. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol Aging. 2014;35:1779. doi: 10.1016/j.neurobiolaging.2014.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Mori K, Lammich S, Mackenzie IRA, Forné I, Zilow S, Kretzschmar H, Edbauer D, Janssens J, Kleinberger G, Cruts MR, Herms J, Neumann M, Van Broeckhoven C, Arzberger T, Haass C. hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol. 2013;125:413–423. doi: 10.1007/s00401-013-1088-7. [DOI] [PubMed] [Google Scholar]
- 164.Xu Z, Poidevin M, Li X, Li Y, Shu L, Nelson DL, Li H, Hales CM, Gearing M, Wingo TS. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci USA. 2013;110:7778–7783. doi: 10.1073/pnas.1219643110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ash PEA, Bieniek KF, Gendron TF, Caulfield T, Lin W-L, DeJesus-Hernandez M, van Blitterswijk MM, Jansen-West K, Paul JW, Rademakers R, Boylan KB, Dickson DW, Petrucelli L. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77:639–646. doi: 10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Mori K, Weng S-M, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts MR, Van Broeckhoven C, Haass C, Edbauer D. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339:1335–1338. doi: 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- 167.Cooper-Knock J, Higginbottom A, Connor-Robson N, Bayatti N, Bury JJ, Kirby J, Ninkina N, Buchman VL, Shaw PJ. C9ORF72 transcription in a frontotemporal dementia case with two expanded alleles. Neurology. 2013;81:1719–1721. doi: 10.1212/01.wnl.0000435295.41974.2e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Harms MB, Cady J, Zaidman C, Cooper P, Bali T, Allred P, Cruchaga C, Baughn M, Libby RT, Pestronk A, Goate A, Ravits J, Baloh RH. Lack of C9ORF72 coding mutations supports a gain of function for repeat expansions in amyotrophic lateral sclerosis. Neurobiol Aging. 2013;34:2234. doi: 10.1016/j.neurobiolaging.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kwon I, Xiang S, Kato M, Wu L, Theodoropoulos P, Wang T, Kim J, Yun J, Xie Y, McKnight SL. Poly-dipeptides encoded by the C9ORF72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science. 2014;345:1139–1145. doi: 10.1126/science.1254917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Mizielinska S, Grönke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, Moens T, Norona FE, Woollacott IOC, Pietrzyk J, Cleverley K, Nicoll AJ, Pickering-Brown S, Dols J, Cabecinha M, Hendrich O, Fratta P, Fisher EMC, Partridge L, Isaacs AM. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science. 2014;345:1192–1194. doi: 10.1126/science.1256800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Barber SC, Mead RJ, Shaw PJ. Oxidative stress in ALS: a mechanism of neurodegeneration and a therapeutic target. Biochim Biophys Acta. 2006;1762:1051–1067. doi: 10.1016/j.bbadis.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 172.Kirby J, Ning K, Ferraiuolo L, Heath PR, Ismail A, Kuo S-W, Valori CF, Cox L, Sharrack B, Wharton SB, Ince PG, Shaw PJ, Azzouz M. Phosphatase and tensin homologue/protein kinase B pathway linked to motor neuron survival in human superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain. 2011;134:506–517. doi: 10.1093/brain/awq345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 174.Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281:1851–1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- 175.Honda D, Ishigaki S, Iguchi Y, Fujioka Y, Udagawa T, Masuda A, Ohno K, Katsuno M, Sobue G. The ALS/FTLD-related RNA-binding proteins TDP-43 and FUS have common downstream RNA targets in cortical neurons. FEBS Open Bio. 2013;4:1–10. doi: 10.1016/j.fob.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Nakaya T, Alexiou P, Maragkakis M, Chang A, Mourelatos Z. FUS regulates genes coding for RNA-binding proteins in neurons by binding to their highly conserved introns. RNA. 2013;19:498–509. doi: 10.1261/rna.037804.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Yoshimura A, Fujii R, Watanabe Y, Okabe S, Fukui K, Takumi T. Myosin-Va facilitates the accumulation of mRNA/protein complex in dendritic spines. Curr Biol. 2006;16:2345–2351. doi: 10.1016/j.cub.2006.10.024. [DOI] [PubMed] [Google Scholar]
- 178.Kawahara Y, Mieda-Sato A. TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc Natl Acad Sci USA. 2012;109:3347–3352. doi: 10.1073/pnas.1112427109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Morlando M, Dini Modigliani S, Torrelli G, Rosa A, Di Carlo V, Caffarelli E, Bozzoni I. FUS stimulates microRNA biogenesis by facilitating co-transcriptional Drosha recruitment. EMBO J. 2012;31:4502–4510. doi: 10.1038/emboj.2012.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Fiesel FC, Weber SS, Supper J, Zell A, Kahle PJ. TDP-43 regulates global translational yield by splicing of exon junction complex component SKAR. Nucleic Acids Res. 2012;40:2668–2682. doi: 10.1093/nar/gkr1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Higashi S, Kabuta T, Nagai Y, Tsuchiya Y, Akiyama H, Wada K. TDP-43 associates with stalled ribosomes and contributes to cell survival during cellular stress. J Neurochem. 2013;126:288–300. doi: 10.1111/jnc.12194. [DOI] [PubMed] [Google Scholar]
- 182.Lagier-Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, Huelga SC, Clutario KM, Ling S-C, Liang TY, Mazur C, Wancewicz E, Kim AS, Watt A, Freier S, Hicks GG, Donohue JP, Shiue L, Bennett CF, Ravits J, Cleveland DW, Yeo GW. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci. 2012;15:1488–1497. doi: 10.1038/nn.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Rogelj B, Easton LE, Bogu GK, Stanton LW, Rot G, Curk T, Zupan B, Sugimoto Y, Modic M, Haberman N, Tollervey J, Fujii R, Takumi T, Shaw CE, Ule J. Widespread binding of FUS along nascent RNA regulates alternative splicing in the brain. Sci Rep. 2012;2:603. doi: 10.1038/srep00603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, König J, Hortobágyi T, Nishimura AL, Župunski V. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci. 2011;14:452–458. doi: 10.1038/nn.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Xiao S, Sanelli T, Dib S, Sheps D, Findlater J, Bilbao J, Keith J, Zinman L, Rogaeva E, Robertson J. RNA targets of TDP-43 identified by UV-CLIP are deregulated in ALS. Mol Cell Neurosci. 2011;47:167–180. doi: 10.1016/j.mcn.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 186.Arnold ES, Ling S-C, Huelga SC, Lagier-Tourenne C, Polymenidou M, Ditsworth D, Kordasiewicz HB, McAlonis-Downes M, Platoshyn O, Parone PA, Da Cruz S, Clutario KM, Swing D, Tessarollo L, Marsala M, Shaw CE, Yeo GW, Cleveland DW. ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc Natl Acad Sci USA. 2013;110:E736–745. doi: 10.1073/pnas.1222809110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Fujioka Y, Ishigaki S, Masuda A, Iguchi Y, Udagawa T, Watanabe H, Katsuno M, Ohno K, Sobue G. FUS-regulated region- and cell-type-specific transcriptome is associated with cell selectivity in ALS/FTLD. Sci Rep. 2013;3:2388. doi: 10.1038/srep02388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Huang C, Huang B, Bi F, Yan LH, Tong J, Huang J, Xia X-G, Zhou H. Profiling the genes affected by pathogenic TDP-43 in astrocytes. J Neurochem. 2014;129:932–939. doi: 10.1111/jnc.12660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Highley JR, Kirby J, Jansweijer JA, Webb PS, Hewamadduma CA, Heath PR, Higginbottom A, Raman R, Ferraiuolo L, Cooper-Knock J, McDermott CJ, Wharton SB, Shaw PJ, Ince PG. Loss of nuclear TDP-43 in ALS causes altered expression of splicing machinery and widespread dysregulation of RNA splicing in motor neurons. Neuropathol Appl Neurobiol. 2014;40:670–685. doi: 10.1111/nan.12148. doi: 10.1111/nan.12148. [DOI] [PubMed] [Google Scholar]
- 190.Mitchell JC, McGoldrick P, Vance C, Hortobágyi T, Sreedharan J, Rogelj B, Tudor EL, Smith BN, Klasen C, Miller CCJ, Cooper JD, Greensmith L, Shaw CE. Overexpression of human wild-type FUS causes progressive motor neuron degeneration in an age- and dose-dependent fashion. Acta Neuropathol. 2013;125:273–288. doi: 10.1007/s00401-012-1043-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Yamazaki T, Chen S, Yu Y, Yan B, Haertlein TC, Carrasco MA, Tapia JC, Zhai B, Das R, Lalancette-Hebert M, Sharma A, Chandran S, Sullivan G, Nishimura AL, Shaw CE, Gygi SP, Shneider NA, Maniatis T, Reed R. FUS-SMN protein interactions link the motor neuron diseases ALS and SMA. Cell Rep. 2012;2:799–806. doi: 10.1016/j.celrep.2012.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Shan X, Chiang P-M, Price DL, Wong PC. Altered distributions of Gemini of coiled bodies and mitochondria in motor neurons of TDP-43 transgenic mice. Proc Natl Acad Sci USA. 2010;107:16325–16330. doi: 10.1073/pnas.1003459107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ling SC, Albuquerque CP, Han JS, Lagier-Tourenne C, Tokunaga S, Zhou H, Cleveland DW. ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proc Natl Acad Sci USA. 2010;107:13318–13323. doi: 10.1073/pnas.1008227107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Wang I-F, Reddy NM, Shen C-KJ. Higher order arrangement of the eukaryotic nuclear bodies. Proc Natl Acad Sci USA. 2002;99:13583–13588. doi: 10.1073/pnas.212483099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Buratti E, Brindisi A, Giombi M, Tisminetzky S, Ayala YM, Baralle FE. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J Biol Chem. 2005;280:37572–37584. doi: 10.1074/jbc.M505557200. [DOI] [PubMed] [Google Scholar]
- 196.Lagier-Tourenne C, Cleveland DW. Rethinking ALS: the FUS about TDP-43. Cell. 2009;136:1001–1004. doi: 10.1016/j.cell.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Lattante S, Rouleau GA, Kabashi E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum Mutat. 2013;34:812–826. doi: 10.1002/humu.22319. [DOI] [PubMed] [Google Scholar]
- 198.Barmada SJ, Skibinski G, Korb E, Rao EJ, Wu JY, Finkbeiner S. Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J Neurosci. 2010;30:639–649. doi: 10.1523/JNEUROSCI.4988-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Kabashi E, Lin L, Tradewell ML, Dion PA, Bercier V, Bourgouin P, Rochefort D, Bel Hadj S, Durham HD, Vande Velde C, Rouleau GA, Drapeau P. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum Mol Genet. 2010;19:671–683. doi: 10.1093/hmg/ddp534. [DOI] [PubMed] [Google Scholar]
- 200.Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SSW, Kiskinis E, Winborn B, Freibaum BD, Kanagaraj A, Clare AJ, Badders NM, Bilican B, Chaum E, Chandran S, Shaw CE, Eggan KC, Maniatis T, Taylor JP. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron. 2014;81:536–543. doi: 10.1016/j.neuron.2013.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Chen Y-Z, Bennett CL, Huynh HM, Blair IP, Puls I, Irobi J, Dierick I, Abel A, Kennerson ML, Rabin BA, Nicholson GA, Auer-Grumbach M, Wagner K, De Jonghe P, Griffin JW, Fischbeck KH, Timmerman V, Cornblath DR, Chance PF. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4) Am J Hum Genet. 2004;74:1128–1135. doi: 10.1086/421054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Greenway MJ, Alexander MD, Ennis S, Traynor BJ, Corr B, Frost E, Green A, Hardiman O. A novel candidate region for ALS on chromosome 14q11.2. Neurology. 2004;63:1936–1938. doi: 10.1212/01.wnl.0000144344.39103.f6. [DOI] [PubMed] [Google Scholar]
- 203.Greenway MJ, Andersen PM, Russ C, Ennis S, Cashman S, Donaghy C, Patterson V, Swingler R, Kieran D, Prehn J, Morrison KE, Green A, Acharya KR, Brown RH, Hardiman O. ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat Genet. 2006;38:411–413. doi: 10.1038/ng1742. [DOI] [PubMed] [Google Scholar]
- 204.Simpson CL, Lemmens R, Miskiewicz K, Broom WJ, Hansen VK, van Vught PWJ, Landers JE, Sapp P, Van Den Bosch L, Knight J, Neale BM, Turner MR, Veldink JH, Ophoff RA, Tripathi VB, Beleza A, Shah MN, Proitsi P, Van Hoecke A, Carmeliet P, Horvitz HR, Leigh PN, Shaw CE, van den Berg LH, Sham PC, Powell JF, Verstreken P, Brown RH, Robberecht W, Al-Chalabi A. Variants of the elongator protein 3 (ELP3) gene are associated with motor neuron degeneration. Hum Mol Genet. 2009;18:472–481. doi: 10.1093/hmg/ddn375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Couthouis J, Hart MP, Shorter J, DeJesus-Hernandez M, Erion R, Oristano R, Liu AX, Ramos D, Jethava N, Hosangadi D, Epstein J, Chiang A, Diaz Z, Nakaya T, Ibrahim F, Kim H-J, Solski JA, Williams KL, Mojsilovic-Petrovic J, Ingre C, Boylan K, Graff-Radford NR, Dickson DW, Clay-Falcone D, Elman L, McCluskey L, Greene R, Kalb RG, Lee VMY, Trojanowski JQ, Ludolph A, Robberecht W, Andersen PM, Nicholson GA, Blair IP, King OD, Bonini NM, Van Deerlin V, Rademakers R, Mourelatos Z, Gitler AD. A yeast functional screen predicts new candidate ALS disease genes. Proc Natl Acad Sci USA. 2011;108:20881–20890. doi: 10.1073/pnas.1109434108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Ticozzi N, Vance C, Leclerc AL, Keagle P, Glass JD, McKenna-Yasek D, Sapp PC, Silani V, Bosco DA, Shaw CE, Brown RH, Landers JE. Mutational analysis reveals the FUS homolog TAF15 as a candidate gene for familial amyotrophic lateral sclerosis. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:285–290. doi: 10.1002/ajmg.b.31158. [DOI] [PubMed] [Google Scholar]
- 207.Couthouis J, Hart MP, Erion R, King OD, Diaz Z, Nakaya T, Ibrahim F, Kim H-J, Mojsilovic-Petrovic J, Panossian S, Kim CE, Frackelton EC, Solski JA, Williams KL, Clay-Falcone D, Elman L, McCluskey L, Greene R, Hakonarson H, Kalb RG, Lee VMY, Trojanowski JQ, Nicholson GA, Blair IP, Bonini NM, Van Deerlin VM, Mourelatos Z, Shorter J, Gitler AD. Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum Mol Genet. 2012;21:2899–2911. doi: 10.1093/hmg/dds116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Lim C, Allada R. ATAXIN-2 activates PERIOD translation to sustain circadian rhythms in Drosophila. Science. 2013;340:875–879. doi: 10.1126/science.1234785. [DOI] [PubMed] [Google Scholar]
- 209.Zhang Y, Ling J, Yuan C, Dubruille R, Emery P. A role for Drosophila ATX2 in activation of PER translation and circadian behavior. Science. 2013;340:879–882. doi: 10.1126/science.1234746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Elden AC, Kim H-J, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM, Padmanabhan A, Clay-Falcone D, McCluskey L, Elman L, Juhr D, Gruber PJ, Rüb U, Auburger G, Trojanowski JQ, Lee VMY, Van Deerlin VM, Bonini NM, Gitler AD. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466:1069–1075. doi: 10.1038/nature09320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Kim H-J, Raphael AR, LaDow ES, McGurk L, Weber RA, Trojanowski JQ, Lee VMY, Finkbeiner S, Gitler AD, Bonini NM. Therapeutic modulation of eIF2α phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat Genet. 2014;46:152–160. doi: 10.1038/ng.2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Johnson JO, Pioro EP, Boehringer A, Chia R, Feit H, Renton AE, Pliner HA, Abramzon Y, Marangi G, Winborn BJ, Gibbs JR, Nalls MA, Morgan S, Shoai M, Hardy J, Pittman A, Orrell RW, Malaspina A, Sidle KC, Fratta P, Harms MB, Baloh RH, Pestronk A, Weihl CC, Rogaeva E, Zinman L, Drory VE, Borghero G, Mora G, Calvo A, Rothstein JD ITALSGEN. Drepper C, Sendtner M, Singleton AB, Taylor JP, Cookson MR, Restagno G, Sabatelli M, Bowser R, Chiò A, Traynor BJ. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci. 2014;17:664–666. doi: 10.1038/nn.3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Kim HJ, Kim NC, Wang Y-D, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, Kanagaraj AP, Carter R, Boylan KB, Wojtas AM, Rademakers R, Pinkus JL, Greenberg SA, Trojanowski JQ, Traynor BJ, Smith BN, Topp S, Gkazi A-S, Miller J, Shaw CE, Kottlors M, Kirschner J, Pestronk A, Li YR, Ford AF, Gitler AD, Benatar M, King OD, Kimonis VE, Ross ED, Weihl CC, Shorter J, Taylor JP. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467–473. doi: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Valor LM. Transcription, epigenetics and ameliorative strategies in Huntington's disease: a genome-wide perspective. Mol Neurobiol. 2014 doi: 10.1007/s12035-014-8715-8. doi: 10.1007/s12035-014-8715-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Bettencourt C, Ryten M, Forabosco P, Schorge S, Hersheson J, Hardy J, Houlden H United Kingdom Brain Expression Consortium. Insights from cerebellar transcriptomic analysis into the pathogenesis of ataxia. JAMA Neurol. 2014;71:831–839. doi: 10.1001/jamaneurol.2014.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Wirth B, Garbes L, Riessland M. How genetic modifiers influence the phenotype of spinal muscular atrophy and suggest future therapeutic approaches. Curr Opin Genet Dev. 2013;23:330–338. doi: 10.1016/j.gde.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 217.Tanaka F, Ikenaka K, Yamamoto M, Sobue G. Neuropathology and omics in motor neuron diseases. Neuropathology. 2012;32:458–462. doi: 10.1111/j.1440-1789.2011.01281.x. [DOI] [PubMed] [Google Scholar]
- 218.Jacobsen JC, Gregory GC, Woda JM, Thompson MN, Coser KR, Murthy V, Kohane IS, Gusella JF, Seong IS, MacDonald ME, Shioda T, Lee J-M. HD CAG-correlated gene expression changes support a simple dominant gain of function. Hum Mol Genet. 2011;20:2846–2860. doi: 10.1093/hmg/ddr195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Feyeux M, Bourgois-Rocha F, Redfern A, Giles P, Lefort N, Aubert S, Bonnefond C, Bugi A, Ruiz M, Deglon N, Jones L, Peschanski M, Allen ND, Perrier AL. Early transcriptional changes linked to naturally occurring Huntington's disease mutations in neural derivatives of human embryonic stem cells. Hum Mol Genet. 2012;21:3883–3895. doi: 10.1093/hmg/dds216. [DOI] [PubMed] [Google Scholar]
- 220.HD iPSC Consortium. Induced pluripotent stem cells from patients with Huntington's disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell. 2012;11:264–278. doi: 10.1016/j.stem.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Soldati C, Bithell A, Johnston C, Wong K-Y, Stanton LW, Buckley NJ. Dysregulation of REST-regulated coding and non-coding RNAs in a cellular model of Huntington's disease. J Neurochem. 2013;124:418–430. doi: 10.1111/jnc.12090. [DOI] [PubMed] [Google Scholar]
- 222.Lewandowski NM, Bordelon Y, Brickman AM, Angulo S, Khan U, Muraskin J, Griffith EY, Wasserman P, Menalled L, Vonsattel JP, Marder K, Small SA, Moreno H. Regional vulnerability in Huntington's disease: fMRI-guided molecular analysis in patients and a mouse model of disease. Neurobiol Dis. 2013;52:84–93. doi: 10.1016/j.nbd.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Lee J-M, Galkina EI, Levantovsky RM, Fossale E, Anne Anderson M, Gillis T, Srinidhi Mysore J, Coser KR, Shioda T, Zhang B, Furia MD, Derry J, Kohane IS, Seong IS, Wheeler VC, Gusella JF, MacDonald ME. Dominant effects of the Huntington's disease HTT CAG repeat length are captured in gene-expression data sets by a continuous analysis mathematical modeling strategy. Hum Mol Genet. 2013;22:3227–3238. doi: 10.1093/hmg/ddt176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Chou A-H, Chen C-Y, Chen S-Y, Chen W-J, Chen Y-L, Weng Y-S, Wang H-L. Polyglutamine-expanded ataxin-7 causes cerebellar dysfunction by inducing transcriptional dysregulation. Neurochem Int. 2010;56:329–339. doi: 10.1016/j.neuint.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 225.Friedrich B, Euler P, Ziegler R, Kuhn A, Landwehrmeyer BG, Luthi-Carter R, Weiller C, Hellwig S, Zucker B. Comparative analyses of Purkinje cell gene expression profiles reveal shared molecular abnormalities in models of different polyglutamine diseases. Brain Res. 2012;1481:37–48. doi: 10.1016/j.brainres.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 226.Ren J, Jegga AG, Zhang M, Deng J, Liu J, Gordon CB, Aronow BJ, Lu LJ, Zhang B, Ma J. A Drosophila model of the neurodegenerative disease SCA17 reveals a role of RBP-J/Su(H) in modulating the pathological outcome. Hum Mol Genet. 2011;20:3424–3436. doi: 10.1093/hmg/ddr251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Damrath E, Heck MV, Gispert S, Azizov M, Nowock J, Seifried C, Rüb U, Walter M, Auburger G. ATXN2-CAG42 sequesters PABPC1 into insolubility and induces FBXW8 in cerebellum of old ataxic knock-in mice. PLoS Genet. 2012;8:e1002920. doi: 10.1371/journal.pgen.1002920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Hsieh M, Chang W-H, Hsu C-F, Nishimori I, Kuo C-L, Minakuchi T. Altered expression of carbonic anhydrase-related protein XI in neuronal cells expressing mutant ataxin-3. Cerebellum. 2013;12:338–349. doi: 10.1007/s12311-012-0430-2. [DOI] [PubMed] [Google Scholar]
- 229.Mancini C, Roncaglia P, Brussino A, Stevanin G, Buono Lo N, Krmac H, Maltecca F, Gazzano E, Bartoletti Stella A, Calvaruso MA, Iommarini L, Cagnoli C, Forlani S, Le Ber I, Durr A, Brice A, Ghigo D, Casari G, Porcelli AM, Funaro A, Gasparre G, Gustincich S, Brusco A. Genome-wide expression profiling and functional characterization of SCA28 lymphoblastoid cell lines reveal impairment in cell growth and activation of apoptotic pathways. BMC Med Genomics. 2013;6:22. doi: 10.1186/1755-8794-6-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Garcia EL, Lu Z, Meers MP, Praveen K, Matera AG. Developmental arrest of Drosophila survival motor neuron (Smn) mutants accounts for differences in expression of minor intron-containing genes. RNA. 2013;19:1510–1516. doi: 10.1261/rna.038919.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, Artieri CG, van Baren MJ, Boley N, Booth BW, Brown JB, Cherbas L, Davis CA, Dobin A, Li R, Lin W, Malone JH, Mattiuzzo NR, Miller D, Sturgill D, Tuch BB, Zaleski C, Zhang D, Blanchette M, Dudoit S, Eads B, Green RE, Hammonds A, Jiang L, Kapranov P, Langton L, Perrimon N, Sandler JE, Wan KH, Willingham A, Zhang Y, Zou Y, Andrews J, Bickel PJ, Brenner SE, Brent MR, Cherbas P, Gingeras TR, Hoskins RA, Kaufman TC, Oliver B, Celniker SE. The developmental transcriptome of Drosophila melanogaster. Nature. 2011;471:473–479. doi: 10.1038/nature09715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Zhang Z, Pinto AM, Wan L, Wang W, Berg MG, Oliva I, Singh LN, Dengler C, Wei Z, Dreyfuss G. Dysregulation of synaptogenesis genes antecedes motor neuron pathology in spinal muscular atrophy. Proc Natl Acad Sci USA. 2013;110:19348–19353. doi: 10.1073/pnas.1319280110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Mathavan S, Lee SGP, Mak A, Miller LD, Murthy KRK, Govindarajan KR, Tong Y, Wu YL, Lam SH, Yang H, Ruan Y, Korzh V, Gong Z, Liu ET, Lufkin T. Transcriptome analysis of zebrafish embryogenesis using microarrays. PLoS Genet. 2005;1:260–276. doi: 10.1371/journal.pgen.0010029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Acsadi G, Li X, Murphy KJ, Swoboda KJ, Parker GC. Alpha-synuclein loss in spinal muscular atrophy. J Mol Neurosci. 2011;43:275–283. doi: 10.1007/s12031-010-9422-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Ismail A, Cooper-Knock J, Highley JR, Milano A, Kirby J, Goodall E, Lowe J, Scott I, Constantinescu CS, Walters SJ, Price S, McDermott CJ, Sawcer S, Compston DAS, Sharrack B, Shaw PJ. Concurrence of multiple sclerosis and amyotrophic lateral sclerosis in patients with hexanucleotide repeat expansions of C9ORF72. J Neurol Neurosurg Psychiatry. 2013;84:79–87. doi: 10.1136/jnnp-2012-303326. [DOI] [PubMed] [Google Scholar]
- 236.Liu X, Milo M, Lawrence ND, Rattray M. Probe-level measurement error improves accuracy in detecting differential gene expression. Bioinformatics. 2006;22:2107–2113. doi: 10.1093/bioinformatics/btl361. [DOI] [PubMed] [Google Scholar]
- 237.Tateishi T, Yamasaki R, Tanaka M, Matsushita T, Kikuchi H, Isobe N, Ohyagi Y, Kira J-I. CSF chemokine alterations related to the clinical course of amyotrophic lateral sclerosis. J Neuroimmunol. 2010;222:76–81. doi: 10.1016/j.jneuroim.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 238.Hautbergue GM, Hung M-L, Golovanov AP, Lian L-Y, Wilson SA. Mutually exclusive interactions drive handover of mRNA from export adaptors to TAP. Proc Natl Acad Sci USA. 2008;105:5154–5159. doi: 10.1073/pnas.0709167105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Skourti-Stathaki K, Proudfoot NJ. A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 2014;28:1384–1396. doi: 10.1101/gad.242990.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Dini Modigliani S, Morlando M, Errichelli L, Sabatelli M, Bozzoni I, An ALS. associated mutation in the FUS 3′-UTR disrupts a microRNA-FUS regulatory circuitry. Nat Commun. 2014;5:4335. doi: 10.1038/ncomms5335. [DOI] [PubMed] [Google Scholar]
- 241.Heath PR, Kirby J, Shaw PJ. Investigating cell death mechanisms in amyotrophic lateral sclerosis using transcriptomics. Front Cell Neurosci. 2013;7:259. doi: 10.3389/fncel.2013.00259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Henriques A, De Aguilar J-LG. Can transcriptomics cut the gordian knot of amyotrophic lateral sclerosis? Curr Genomics. 2011;12:506. doi: 10.2174/138920211797904043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Mitchell AC, Mirnics K. Gene expression profiling of the brain: pondering facts and fiction. Neurobiol Dis. 2012;45:3–7. doi: 10.1016/j.nbd.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Domínguez-Sánchez MS, Sáez C, Japón MA, Aguilera A, Luna R. Differential expression of THOC1 and ALY mRNP biogenesis/export factors in human cancers. BMC Cancer. 2011;11:77. doi: 10.1186/1471-2407-11-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Mori K, Arzberger T, Grässer FA, Gijselinck I, May S, Rentzsch K, Weng S-M, Schludi MH, van der Zee J, Cruts MR, Van Broeckhoven C, Kremmer E, Kretzschmar HA, Haass C, Edbauer D. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 2013;126:881–893. doi: 10.1007/s00401-013-1189-3. [DOI] [PubMed] [Google Scholar]
- 246.Tanaka M, Chock PB, Stadtman ER. Oxidized messenger RNA induces translation errors. Proc Natl Acad Sci USA. 2007;104:66–71. doi: 10.1073/pnas.0609737104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Singh M. Dysregulated A to I RNA editing and non-coding RNAs in neurodegeneration. Front Genet. 2012;3:326. doi: 10.3389/fgene.2012.00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Shapiro E, Biezuner T, Linnarsson S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nature. 2013;14:618–630. doi: 10.1038/nrg3542. [DOI] [PubMed] [Google Scholar]
- 249.Martin I, Kim JW, Lee BD, Kang HC, Xu J-C, Jia H, Stankowski J, Kim M-S, Zhong J, Kumar M, Andrabi SA, Xiong Y, Dickson DW, Wszolek ZK, Pandey A, Dawson TM, Dawson VL. Ribosomal protein s15 phosphorylation mediates LRRK2 neurodegeneration in Parkinson's disease. Cell. 2014;157:472–485. doi: 10.1016/j.cell.2014.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]