

Abstract
Pneumocyte injury is a characteristic of pulmonary interstitial pneumonias (IPs). Histological markers of pneumocyte injury and inflammation include pneumocyte necrosis, erosion, hyaline membrane and fibrin exudation with subsequent intraluminal granulation tissue formation. We found that intracytoplasmic inclusions in pneumocytes are ubiquitin-positive (Ub+) and that the number of Ub+ pneumocytes shows positive correlation with the extent of diffuse alveolar damage (DAD). To determine the role of Ub+ pneumocytes and inclusions in IPs, we studied their relationship with pathological and clinical features of DAD, usual interstitial pneumonia (UIP) and organizing pneumonia (OP), including airspace enlargement with fibrosis (AEF). We analysed Ub+ pneumocytes, inclusions, erosions and intraluminal granulation tissue in relation to pneumocyte injury. The numbers of immunohistochemically identified Ub+ inclusions in each IP were higher than the number of inclusions detected by light microscopy. The inclusions detected by Ub+ immunostaining were identical to the inclusions observed by light microscopy. UIP and DAD had many Ub+ inclusions, while OP and AEF had fewer Ub+ inclusions. These results suggest that the extent of Ub+ inclusions reflects the severity of pneumocyte injury among IPs. Thus, Ub+ inclusions are a histological marker of pneumocyte injury that may be helpful in determining the severity and prognosis of IPs.
Keywords: eosinophilic inclusion body (Mallory body), interstitial fibrosis, interstitial lung disease, pneumocyte injury, ubiquitin
Introduction
Pneumocyte injury may induce pneumocyte necrosis, exfoliation and erosion, followed by hyaline membrane formation in a diffuse alveolar damage (DAD) pattern, intra-alveolar fibrin exudation, and subsequent intraluminal granulation tissue formation. Pneumocyte injury is induced by chemicals or drugs,1 irradiation,2 oxidative stress3 and cobalt,4 among others.5 Pneumocyte injury causes various types of interstitial pneumonias (IPs) and other interstitial lung diseases (ILDs), including airspace enlargement with fibrosis (AEF).
The major histological classifications of idiopathic interstitial pneumonias (IIPs)6–9 are usual interstitial pneumonia (UIP), non-specific interstitial pneumonia (NSIP), organizing pneumonia (OP), DAD, respiratory bronchiolitis (RB) and desquamative interstitial pneumonia (DIP).10 These IP patterns can also be caused by drugs, collagen vascular diseases and hypersensitivity pneumonia.
Biomarkers have been reported to be a sensitive marker for interstitial lung diseases.11–13 Epithelial necrosis, erosion, fibrin exudation and other characteristics in the alveolar spaces are histological markers of pneumocyte injury. Intracytoplasmic eosinophilic inclusion bodies (inclusions) or Mallory bodies have been found in the pneumocytes of patients with asbestosis,14 DAD and other IPs.15–17 These inclusions were originally termed hyaline globules or spherical intracytoplasmic eosinophilic droplets. The inclusions contain aggregations of intermediate filament proteins (including cytokeratin), with a filamentous or granular amorphous appearance.18
The presence of ubiquitin-positive (Ub+) pneumocytes may represent another histological parameter of pneumocyte injury in IPs with various clinical backgrounds. We reviewed the roles of inclusions, Ub+ pneumocytes, pneumocyte injury and fibrosis in IPs and the relationships among these factors.
Interstitial pneumonias and the ubiquitin-proteasome system
The pathology of IPs is mediated by an inflammatory process, including cellular injury with mediators in the pulmonary interstitium as well as vessels outside the alveolar spaces. The inflammatory process leads to the development of interstitial fibrosis of variable extent and localization in the lungs. The mixture of inflammation and fibrosis with distorted lung structure complicates the entity of IPs through the long course of the inflammatory process, with inflammatory cell infiltration and release of chemical mediators. Cytokines and other substances are released by inflammatory cells, stimulating the production of collagen and subsequently leading to the development of fibrosis.19 There are many factors in the inflammatory process that cause pneumocyte injury.
The ubiquitin–proteasome system (UPS) was first described by Ciechanover et al.,20,21 and has emerged as a key pathway that defines cellular protein turnover and, consequently, the levels and activity of numerous intracellular proteins. Ub is a small, ubiquitously expressed protein that has been highly conserved throughout evolution. The conjugation of Ub to protein substrates involves a series of hierarchically organized steps. The proteasome is located in both the cytoplasm and the nucleus. Protein degradation by the UPS modulates transforming growth factor beta 1 (TGF-β1) signalling at multiple steps.22 Thus, proteasomal inhibition could modulate TGF-β1 signalling at several potential targets and might abrogate the development of tissue fibrosis. The UPS plays a key role in proteolysis through the ubiquitination of target proteins,23,24 and the Ub gene is expressed in all eukaryotic cells.25 In addition to its role in removing damaged proteins, the UPS has been shown to play a critical role in the regulation of proteins that affect inflammatory processes, cell growth and differentiation. UPS inhibition caused by pneumocyte injury results in the accumulation of unresolved ubiquitinated proteins in the pneumocyte, which are detected as inclusions by light microscopy. UPS inhibition by pneumocyte injury may accelerate the progression of idiopathic pulmonary fibrosis (IPF). Further research is needed to elucidate the detailed relationship between UPS inhibition and IPF.
Characteristics of inclusions
Ub+ inclusions with a skein-like appearance were identified initially in the nerve cells and other cells of patients with diseases of the central nervous system (CNS).26–28 Ub was found in the inclusions,29,30 which were induced with UPS inhibition.31 Inclusion-like structures have also been found in some tumour cells18,32 and in the macrophages of smokers.33 Pneumocyte injury inhibits the UPS pathway and induces an accumulation of Ub proteins.4 Nevertheless, Ub+ inclusions have been found in only a few patients with specific tumour types18 or alcoholic liver damage,34,35 in addition to patients with diseases of the CNS. The formation of ubiquitinated intermediate filament/alphaB crystallin inclusions is most probably a common cellular response to chronic lethal injury, and is passively cytoprotective.36,37
Typically shaped inclusions with a distinct border were observed in the cytoplasm of pneumocytes adjacent to areas of immature or mature organization (Figure1). They stained with haematoxylin and eosin (H&E), and were also weakly stained by periodic acid-Schiff, both with and without diastase. The histological features of these inclusions by H&E staining were similar to the features of inclusions in the patients with IPs. The inclusions varied in shape from eosinophilic filamentous to globular with a distinct border in the cytoplasm (Figure2). They could be differentiated from viral-associated inclusion bodies (cytomegalovirus, herpes simplex virus38 and varicella zoster virus39) by in their size, shape and location.
Figure 1.
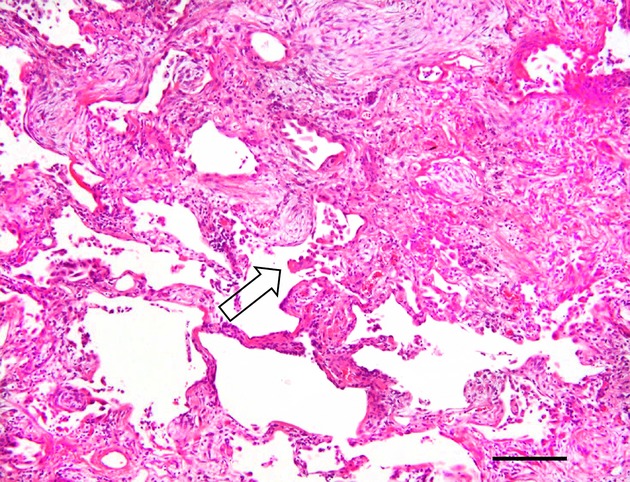
Localization of inclusions. Photomicrograph showing the histology of interstitial fibrosis from a patient with non-specific interstitial pneumonia (H&E staining), with an eosinophilic inclusion at the interstitial fibrosis front (arrow); details of the inclusion are unclear in this field due to the low magnification. Bar, 200 μm.
Figure 2.

Histology of inclusions. H&E staining (left) shows eosinophilic inclusions (arrows) in pneumocytes. The image on the right shows another pneumocyte containing a Ub-positive inclusion (arrow), with intense immunoreactivity and an irregularly-shaped border (Ub immunostaining). Bar, 50 μm.
Inclusions in the pneumocytes are immunohistochemically positive for Ub.40 Furthermore, a granular pattern of Ub, not representative of inclusions (Figure3), was also found in some monocyte-derived macrophages and pneumocytes (Table1).41 Ub+ cells have also been reported in patients with severe fibrosis in ILD.42 The mechanisms of the UPS have been clarified, and Ub+ inclusions can be a histological hallmark of pneumocyte injury.36 We found a relationship between Ub+ pneumocytes and the degree or severity of hyaline membrane formation, based on analyses of the accumulation of Ub+-reactive products in pneumocytes and inclusions from individuals with DAD.43 Next, various IPs were studied to clarify the relationship between Ub+ inclusions and pneumocyte injury among cases of IP, AEF and centrilobular emphysema (CLE).9,44 Our data indicated that the frequency of inclusions was strongly suggestive of the severity of lung injury and/or response to lung injury in ILDs (Table2).
Figure 3.

Pneumocytes with granular Ub+ staining. The photomicrograph on the left with H&E staining shows fragmented inclusions in the pneumocyte (arrows). That on the right shows a weakly Ub+ pneumocyte with a granular pattern of cytoplasmic staining, of non-inclusion type (Ub immunostaining). Bar, 30 μm.
Table 1.
Inclusions and ubiquitin (Ub) positive pneumocytes
| Light microscope | Ub immunostaining |
|---|---|
| 1. Pneumocyte with inclusion | Ub+ pneumocyte (Ub+ inclusion) |
| (a) Typical inclusion form (skein-like appearance) | Ub+ |
| (b) Indistinct form (fragmented inclusion) | Ub+ (fragmented or granular staining pattern) |
| 2. Pneumocyte without inclusion (non-inclusion type) | Ub+ pneumocyte (fine granular staining) |
| Ub– pneumocyte |
Table 2.
Summary of Ub+ inclusions and pneumocyte injury among interstitial pneumonias and other lung diseases
| DAD | UIP | NSIP | OP | AEF | CLE | |
|---|---|---|---|---|---|---|
| Light microscope | ||||||
| Inclusions (no. of cases) | 22/54 | 26/35 | 5/28 | 2/34 | 1/9 | 0/5 |
| Mean numbers of inclusions/slide | 27 | 11.8 | 2.3 | 1.5 | 0.1 | 0 |
| Immunostaining | ||||||
| Ub+ inclusions (no. of cases) | 26 | 28 | 11 | 5 | 2 | 0 |
| Mean numbers of Ub+ cells/slide | 67.4 | 34.4 | 10.9 | 4.6 | 0.3 | 0 |
| Scores* for: | ||||||
| Erosion | 1.8 | 1.6 | 1.1 | 0.9 | 0.01 | 0 |
| Intraluminal granulation tissue | 2.8 | 2.7 | 2.1 | 1.2 | 0.8 | 0 |
| Interstitial fibrosis | 2.0 | 2.8 | 2.2 | 1.1 | 1.0 | 0.1 |
DAD: diffuse alveolar damage; UIP: usual interstitial pneumonia; NSIP: non-specific interstitial pneumonia; OP: organizing pneumonia; AEF, airspace enlargement with fibrosis; CLE: centrilobular emphysema.
Scores 0–3 according to the severity or extent of the lesion.
Inclusions contain different ubiquitinated proteins. Major constituents of the inclusions include cytokeratins (CK8, 18), Ub and p62 protein/sequestosome 1 (SQSTM1).34 A marked increase of p62 is detected following autophagy inhibition. Similarly, the inclusions in our studies of diseased lungs were positive for several molecular types of cytokeratins (CK5, 6, 8, 18).43 To compare the precise H&E and Ub staining characteristics of the inclusions, we immunostained the inclusions for Ub after de-staining of H&E-stained sections, and all the inclusions were Ub+.43 Ultrastructural observation of the Ub+ inclusions demonstrated that they were composed of electron-dense, bundle-like structures and innumerable filamentous structures (15–20 nm in width),43 similar to the inclusions described in previous studies.18,23–25 No viral-associated inclusions in the IP specimens were studied, although viral particles have been found previously in pneumocytes from these patients.45
Inclusions in the pneumocytes of interstitial pneumonias
We performed histological and immunohistochemical studies on lung tissue specimens from 165 patients with ILD and other lung diseases. This cohort consisted of: 54 patients with acute interstitial pneumonia diagnosed as DAD;43 35 patients with IPF and subclinical IPF showing UIP in lobectomy specimens;46 17 primary NSIPs and 11 secondary NSIPs in surgical lung biopsy specimens, which were divided into 13 cases of fibrosing-NSIP (f-NSIP), nine cases of cellular-NSIP (c-NSIP) and six cases of fibrosing/cellular-NSIP (f/c-NSIP);47 34 patients with a secondary OP found at the periphery of lobectomized lung cancer;48 nine patients with AEF; and five patients with CLE.49 Each type of IP was diagnosed based on the criteria of the American Thoracic Society/European Respiratory Society (ATS/ERS),6–9 and the diagnosis of CLE was based on WHO criteria.50 Cases of cryptogenic organizing pneumonia were not included in this study.
Variable percentages of patients with DAD, UIP, NSIP, OP or AEF had inclusions in their pneumocytes, as determined by light microscopy. Inclusions were identified in pneumocytes in 40.7% of the DAD patients, 74.3% of the UIP patients, 17.9% of the NSIP patients and 5.9% of the OP patients. They were identified in only one case (11%) of AEF, and none were found among patients with CLE. The mean number of inclusions per slide that were identified by light microscopy was 27.0 cells for DAD, 11.8 for UIP, 2.3 for NSIP, 1.5 for OP and 0.1 for AEF (Table2). These results suggest that each type of IP and AEF has a different degree of pneumocyte injury. Pneumocyte injury from stimulants, bacteria and other causes may inhibit the UPS, and an abnormal amount of target proteins accumulate during the cell cycle. Poly-ubiquitinated proteins then form inclusions in variable sizes and shapes that can be identified microscopically.
Ub+ pneumocytes
All the inclusions evaluated in the IPs were Ub+ (Figures2 and 4), and they were identical to the inclusions identified by novel immunostaining. Ub+ granules were also found in the cytoplasm of pneumocytes in DAD.43 Ub+ granules could also comprise part of a fragmented inclusion, an inclusion precursor, or another form of accumulation (Table1). It was also found that Ub+ cells with a granular pattern (not representative of inclusions) were increased in the patients with IPF.42 The mean number of Ub+ pneumocytes per slide was 67.4 cells in cases of DAD; 34.4 in UIPs; 10.9 in NSIPs; 4.6 in OPs; and 0.3 in AEF cases (Table2). Ub+ pneumocytes were not found in cases of CLE. The number of Ub+ pneumocytes in each type of IP was higher than the number of inclusions detected by light microscopy. Ub immunostaining could detect most of the inclusions, as well as granules (non-inclusion-type), in the lung fields, demonstrating that this is a useful, sensitive assay for the detection of inclusions.
Figure 4.

Inclusion detected by H&E staining and Ub immunostaining. H&E staining (left) shows an inclusion (arrow) in a pneumocyte from a patient with DAD. Image (right) of the same pneumocyte with Ub immunostaining after removal of H&E staining shows a Ub+ inclusion in the pneumocyte cytoplasm (from Yamada et al.43). Bar, 5 μm.
The differential diagnosis of acute interstitial pneumonia (DAD histology), IPF (UIP histology), NSIP and OP is especially important for the determination of prognosis among IIPs.6 We studied the histology of these four major types of IPs. Ub+ inclusions were confirmed in the same pneumocytes that were Ub+ (Figure4). The patients with NSIP, and even the incidental secondary OP groups, had Ub+ inclusions in the pneumocytes, and characteristics of these inclusions were similar to those found in the DAD and UIP groups.43 The highest frequency of Ub+ cases was found in the UIP group, while the DAD group had the greatest number of Ub+ pneumocytes per slide among the IPs.
The presence of Ub+ inclusions (i.e. the Ub+ pneumocytes) revealed pneumocyte injury with greater sensitivity than did the light microscopy detection of inclusions (Table2). Ub immunostaining revealed all the inclusions in the lung fields; not only inclusions with typical forms, but also those with indistinct forms in H&E-stained sections (Figure3). Our data also showed that the number of Ub+ inclusions was correlated with the severity of pneumocyte injury (erosions and granulation tissue; Table2), suggesting that the presence of inclusions is a histological marker for pneumocyte injury. Additionally, the variable intensity and/or positivity of Ub+ pneumocytes may be of relevance for the evaluation of the progression of ILD.
Erosions and granulation tissue subtypes in interstitial pneumonias
In H&E sections, erosion is defined as a complete loss of pneumocytes next to the alveolar interstitium with inflammatory cell infiltration. Epithelial membrane antigen (EMA) immunostaining confirms the presence of erosions in tissue samples (Figure5). The pattern of intraluminal granulation tissue can be used to classify the severity of pneumocyte injury; the classification system developed by Basset et al.51 (Figure6) consists of an intraluminal bud (or polypoid) type, a mural incorporation (or mural) type, and obliterative changes (or occluded type).
Figure 5.
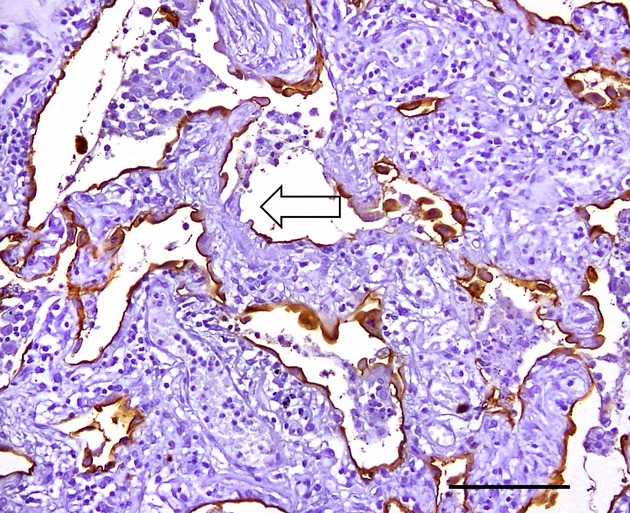
Erosion of pneumocytes (arrow) in the alveolar wall: defect of pneumocytes and inflammatory cell infiltration (EMA immunostaining). No intracytoplasmic inclusions are noted. The tissue sample is from a patient with non-specific interstitial pneumonia. Bar, 100 μm.
Figure 6.
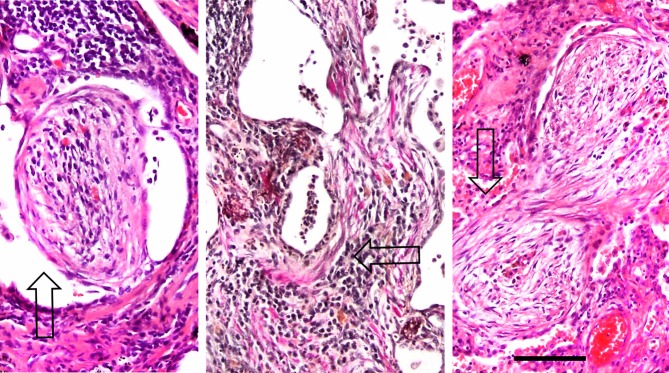
Granulation tissue subtypes. These images show representative histological characteristics of intraluminal granulation tissue subtypes, at the same magnification. The polypoid type is on the left (arrow, H&E staining), the mural type is in the middle (arrow, Weigert-Van Gieson staining), and the occluded type is on the right (arrow, H&E staining). The subtypes are related to the extent and severity of pneumocyte injury. Bar, 100 μm.
The Ub+ NSIP subgroup showed significant increases in the amount of pneumocyte erosion and granulation tissue compared to the Ub– NSIP subgroup, and these findings were unique to the histopathological features in the NSIP group.47 The Ub+ OP subgroup also had a greater amount of granulation tissue in comparison to that of the Ub– OP subgroups.48 The Ub+ pneumocytes tended to be located adjacent to the granulation tissue in the lung fields.47 The histology of DAD, UIP or NSIP cases was marked by severe pneumocyte injury (erosion or granulation tissue), in contrast to the histology of OP and AEF. Each type of IP has different localizations and distributions of inflammation and fibrosis in the lung fields, and these differences among the pulmonary diseases may determine the clinical manifestation of each type of IP. Pneumocyte injury was detected histologically by evaluating the erosions, granulation tissue subtypes and inclusions by light microscopy, and Ub+ pneumocytes were identified by immunohistochemistry. These features, which result from pneumocyte injury, were observed in some of the patients with OP, while cases of AEF had the least pneumocyte injury among ILDs.49
The UPS plays a critical role in cellular damage during the developmental course of IPs52–54 and disorders in other organs.55,56 In addition to our studies, there have been several other reports of the immunohistochemical detection of Ub in pulmonary diseases,54 including discussions of the pathogenesis of IPF (UIP histology) and NSIP.57,58 We found that the amount of granulation tissue in the Ub+ NSIP subgroup was greater than that observed in the Ub– NSIP subgroup, with significant increases in the mural type and the occluded type found in f-NSIP compared to c-NSIP. It has been suggested that these granulation tissue subtypes play an important role in pulmonary remodelling.51 The mural and occluded subtypes of granulation tissue reflected pneumocyte injury that was more severe than the injury seen in the polypoid type. Furthermore, even in the Ub+ OP subgroup, the degree of mural and occluded subtypes was greater than that of the Ub– OP subgroup. Increases of these subtypes in the UIP, NSIP and OP groups may correlate with the localization of Ub+ pneumocytes that were found frequently in front of the granulation tissue (Figure1). These findings suggested that the severity of the pneumocyte injury may affect the development of NSIP and OP, similar to its effect in cases of DAD or UIP.
UPS and autophagy in interstitial pneumonias
Recent studies have shown that autophagy plays an important regulatory role in cellular senescence and differentiation, helps to maintain homeostatic balance, and also plays a regulatory role in IPF pathogenesis.22 Araya et al. reported that insufficient autophagy was an underlying mechanism of both accelerated cellular senescence and myofibroblast differentiation in a cell type-specific manner and was a promising potential component in providing a better understanding of the pathogenesis of IPF.42,59 Although proteasome inhibition has an antifibrotic effect,59 autophagy inhibition appears to play a larger role in fibrosis in IPF, as well as in the accumulation of ubiquitinated proteins in pneumocyte compared to proteasome inhibition.60 Both proteasomes and autophagy degrade ubiquitinated protein chains; however, while proteasomes only degrade soluble proteins, autophagy degrades protein aggregates and micro-organelles, including mitochondria. Poly-ubiquitinated chains include different types of proteins. Lys-48-linked poly-ubiquitin chains are targeted for proteasomal degradation, whereas Lys-63-linked chains provide a non-degradative signal in UPS. Autophagy inhibition was reported to induce the accumulation of ubiquitinated proteins not only in the CNS but also in the respiratory epithelium.60 In our analyses, UPS appears to be more important than autophagy for pneumocyte injury in ILDs. However, further studies are needed to elucidate whether proteasome or autophagy predominantly degrades the accumulated ubiquitinated proteins.
Ub+ pneumocyte and prognosis in interstitial pneumonias
The severity of hyaline membrane formation (as determined by histological evaluation) correlates with an increased number of Ub+ inclusions in patients with acute interstitial pneumonia (DAD histology). Patients with UIP have a poor prognosis and strict pathological diagnosis, and surgical lung biopsy is required to differentiate UIP from other types of IPs.61 Additionally, many patients with acute clinical deterioration of an unknown aetiology experience subsequent acute deteriorations in respiratory function.62 However, an increase in the number of fibroblastic foci (i.e., mural type granulation tissue in UIP) could be an important histological marker for acceleration of UIP, or for predicting acute exacerbation.46,61,63 IP is complicated by superimposed acute lung injury,64,65 and some reports have suggested that viral infection may cause an acute exacerbation of UIP. We performed a background study of acute exacerbation cases in patients with UIP and found that Ub+ pneumocytes tended to be increased in the acute exacerbation subgroup compared to the stable subgroup of UIP cases. However, there was no statistically significant correlation between acute exacerbation and Ub+ pneumocytes as a background factor.48
Although patients with NSIP have a relatively good prognosis,8,47,66 some patients are resistant to therapy.67–69 In our analyses, patients with NSIP were designated as therapy-resistant after a mean follow-up period of 4.4 years following surgical lung biopsy. Follow-up included patients with secondary NSIP, and NSIP was classified as f-NSIP or c-NSIP based on the proportions of interstitial inflammation and fibrosis.66 Some patients with NSIP had fibroblastic foci (mural-type granulation tissue) attached to the interstitium, confirmed by elastic Weigert-Van Gieson staining, but no histological markers were clearly associated with therapy resistance in patients with NSIP. The Ub+ NSIP subgroup, as well as the f-NSIP and c-NSIP subtypes, contained both responding and resistant cases (61% of NSIP). Ub+ pneumocytes were found in the patients with f-, c- or f/c-NSIP, with variable proportions. Subsequently, the presence of Ub+ pneumocytes did not correlate with NSIP subtypes or clinical features. The Ub+ pneumocyte was not a histological indicator of resistance to therapy in the NSIP group.47
The OP group had fewer Ub+ pneumocytes in comparison to the NSIP group (Figure7). The histological characteristics of OP are heterogeneous with various degrees of lung injury. Although cases of cryptogenic organizing pneumonia (COP) were not included in this study, the histological features of secondary OP were identical with those of COP (mainly intraluminal polypoid granulation tissue with mild alveolitis, unpublished data). In fact, fibrin exudation (an occasional feature in the alveolar spaces) and/or prominent reactive pneumocytes are seen in lung biopsy specimens from patients clinically diagnosed as COP who showed amelioration with or without steroid therapy. In the present study, no fibrosing variant of an OP-like case was observed; by the ATS criteria, COP is considered to be a benign disease without severe damage or death,9 and we speculate that this finding is consistent with the better prognosis of COP among the IPs.
Figure 7.
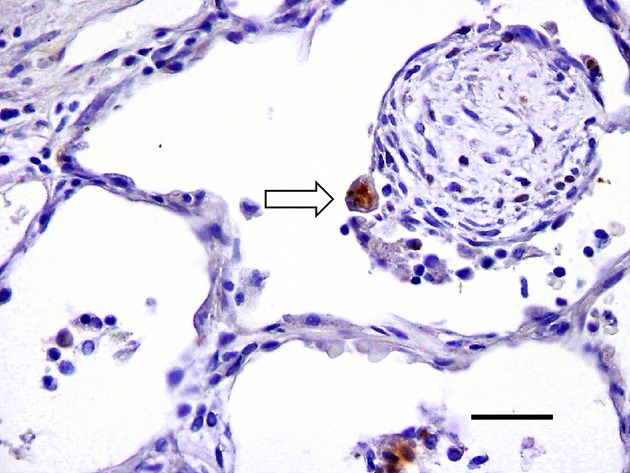
Ub+ pneumocyte in organizing pneumonia. Ub+ pneumocyte (fragmented form, arrow) is shown on the surface of intraluminal granulation tissue (polypoid type) from a patients with organizing pneumonia (Ub immunostaining). Bar, 50 μm.
Airspace enlargement with fibrosis, centrilobular emphysema and asbestosis
Asbestosis (another ILD) is characterised typically by Mallory bodies, but we found very few inclusions in our cases (unpublished data). AEF is characterized by subpleural cystic lesions with a thickened fibrous wall, and Kawabata et al.44 reported a close relationship between this condition and smoking-related lung disease. AEF has been included in the update of the international multidisciplinary classification of IPs by ATS/ERS,9 and other groups have categorized it similarly.19,70,71 When compared to IPs, AEF is characterized by less inflammatory cell infiltration, minimal intraluminal granulation tissues, few Ub+ pneumocytes and low erosion scores. It has the least pneumocyte injury among the ILDs, whereas CLE shows neither intraluminal granulation tissue formation nor erosion. Additionally, alphaB crystallin has been detected immunohistochemically in the AEF group, and a protective effect against pneumocyte injury was suggested along with UPS.37 AEF has a different pathogenesis from that of UIP and NSIP, and AEF frequently accompanies CLE. AEF is suggested to be an intermediate condition between CLE and IPF (unpublished data). The relationship between Ub positivity and pneumocyte injury among ILDs is summarized in Figure8.
Figure 8.
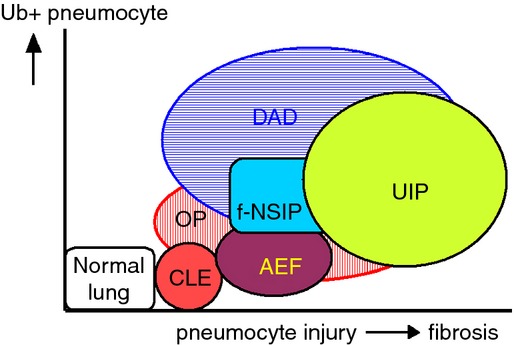
Schematic relationship of interstitial lung disease based on the degree of pneumocyte injury and numbers of Ub+ pneumocytes. Ub+ pneumocytes are most numerous in the DAD group, and interstitial fibrosis is most severe in the UIP group, (CLE: centrilobular emphysema; AEF: airspace enlargement with fibrosis; UIP: usual interstitial pneumonia; f-NSIP: fibrosing non-specific interstitial pneumonia; DAD: diffuse alveolar damage; OP: organizing pneumonia).
Conclusion
IPs develop through an inflammatory process accompanied by pneumocyte injury with disordered UPS, leading to the accumulation of ubiquitinated target proteins in pneumocytes. Pneumocyte injury might induce necrosis or erosion of pneumocytes, exudation and the formation of intraluminal granulation tissue, and the inclusions that are features of pneumocyte degeneration due to the accumulation of ubiquitinated proteins in the pneumocyte cytoplasm. These histological features were found in each type of IP. The Ub+ pneumocytes can be a marker of cellular injury in a variety of organs, including the lung. Additionally, pneumocyte injury caused by the disordered UPS was observed adjacent to granulation tissues during IP development. The presence of Ub+ pneumocytes is not an essential diagnostic criteria for IPs; however, Ub immunostaining is more useful than H&E staining for the detection of inclusions as a result of pneumocyte injury. Our findings may help to differentiate each type of IP by determining the severity of the pneumocyte injury based on microscopy of surgical lung biopsy specimens.
Acknowledgments
We wish to thank Dr Thomas V. Colby (Department of Laboratory Medicine and Pathology, Mayo Clinic Arizona, Scottsdale, Arizona, 85259, USA) for his revision of the manuscript and for his contribution to the interstitial lung diseases, including samples with airspace enlargement with fibrosis. We would also like to thank Dr Eishin Hoshi (Department of Thoracic Surgery, Saitama Prefectural Cardiovascular and Respiratory Center, Kumagaya City, Japan) for generously providing surgical lung materials, Dr Yoko Nakanishi for her statistical analysis and excellent research on lung cancer, Dr Taku Homma for his research support and helpful suggestion to consider central nervous system diseases, Dr Norimichi Nemoto (Department of Pathology, Nihon University School of Medicine, Tokyo, Japan) for his general support and for providing lung materials from Nihon University Itabashi Hospital, Dr Tomohiko Mizutani (University Research Center, Nihon University School of Medicine) for his contribution to the research on the ubiquitin-proteasome system in the central nervous system, Dr Jun Araya (Division of Respiratory Diseases, Department of Internal Medicine, Jikei University School of Medicine, Tokyo, Japan) for his revision of the autophagy section, and Mr Kenji Uehara (Division of Neurology, Department of Medicine, Nihon University School of Medicine) for his technical assistance, including immunostaining.
Glossary
- AEF
airspace enlargement with fibrosis
- AIP
acute interstitial pneumonia
- ATS/ERS
American Thoracic Society/European Respiratory Society
- CLE
centrilobular emphysema
- CNS
central nervous system
- c-NSIP
cellular NSIP
- COP
cryptogenic organizing pneumonia
- DAD
diffuse alveolar damage pattern
- DIP
desquamative interstitial pneumonia
- EMA
epithelial membrane antigen
- f/c-NSIP
fibrosing/cellular NSIP
- FF
fibroblastic foci
- f-NSIP
fibrosing NSIP
- HE
hematoxylin-eosin
- IIP
idiopathic interstitial pneumonia
- ILD
interstitial lung disease
- inclusion
intracytoplasmic eosinophilic inclusion
- IPF
idiopathic pulmonary fibrosis
- IP
interstitial pneumonia
- NSIP
nonspecific interstitial pneumonia pattern
- OP
organizing pneumonia pattern
- RB-ILD
respiratory bronchiolitis-interstitial lung disease
- SLB
surgical lung biopsy
- S-NSIP
secondary NSIP
- TGF-β1
transforming growth factor beta 1
- Ub+
Ub-positive
- Ub
ubiquitin
- UIP
usual interstitial pneumonia pattern
- UPS
ubiquitin–proteasome system
- WHO
World Health Organization
Conflict of interest
The authors declare no potential conflicts of interest.
References
- Prata LO, Oliveira FM, Ribeiro TM, et al. Exercise attenuates pulmonary injury in mice with bleomycin-induced pulmonary fibrosis. Exp. Biol. Med. 2012;23:873–883. doi: 10.1258/ebm.2012.011334. [DOI] [PubMed] [Google Scholar]
- Citrin DE, Shankavaram U, Horton JA, et al. Role of type II pneumocyte senescence in radiation-induced lung fibrosis. J. Natl Cancer Inst. 2013;105:1474–1484. doi: 10.1093/jnci/djt212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs CA, Montgomery DW, Merkle CJ. Age-related differences in cigarette smoke extract-induced H2O2 production by lung endothelial cells. Microvasc. Res. 2011;82:311–317. doi: 10.1016/j.mvr.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araya J, Maruyama M, Inoue A, et al. Inhibition of proteasome activity is involved in cobalt-induced apoptosis of human alveolar macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002;283:L849–L858. doi: 10.1152/ajplung.00422.2001. [DOI] [PubMed] [Google Scholar]
- Matute-Bello G, Winn RK, Jonas M, Chi EY, Martin TR, Liles WC. Fas (CD95) induces alveolar epithelial cell apoptosis in vivo: implications for acute pulmonary inflammation. Am. J. Pathol. 2001;158:153–161. doi: 10.1016/S0002-9440(10)63953-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Thoracic Society/European Respiratory Society. International Multidisciplinary Consensus Classification of the Interstitial Pneumonia. Am. J. Respir. Crit. Care Med. 2002;165:277–304. doi: 10.1164/ajrccm.165.2.ats01. [DOI] [PubMed] [Google Scholar]
- Raghu G, Collard HR, Egan JJ, et al. ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis WD, Hunninghake G, King TE, Jr, et al. Idiopathic nonspecific interstitial pneumonia: report of an American Thoracic Society project. Am. J. Respir. Crit. Care Med. 2008;177:1338–1347. doi: 10.1164/rccm.200611-1685OC. [DOI] [PubMed] [Google Scholar]
- Travis WD, Costabel U, Hansell DM, et al. Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 2013;188:733–748. doi: 10.1164/rccm.201308-1483ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata Y, Takemura T, Hebisawa A, et al. Desquamative interstitial pneumonia may progress to lung fibrosis as characterized radiologically. Respirology. 2012;17:1214–1221. doi: 10.1111/j.1440-1843.2012.02226.x. [DOI] [PubMed] [Google Scholar]
- Goto K, Kodama T, Sekine I, et al. Serum levels of KL-6 are useful biomarkers for severe radiation pneumonitis. Lung Cancer. 2001;34:141–148. doi: 10.1016/s0169-5002(01)00215-x. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Fujishima T, Koba H, et al. Serum surfactant proteins A and D as prognostic factors in idiopathic pulmonary fibrosis and their relationship to disease extent. Am. J. Respir. Crit. Care Med. 2000;162:1109–1114. doi: 10.1164/ajrccm.162.3.9910080. [DOI] [PubMed] [Google Scholar]
- Xu L, Drachenberg C, Tavora F, Burke A. Histologic findings in lung biopsies in patients with suspected graft-versus-host disease. Hum. Pathol. 2013;44:1233–1240. doi: 10.1016/j.humpath.2012.11.012. [DOI] [PubMed] [Google Scholar]
- Kuhn C, III, Kuo TT. Cytoplasmic hyalin in asbestosis. A reaction of injured alveolar epithelium. Arch. Pathol. 1973;95:190–194. [PubMed] [Google Scholar]
- Gould VE, Miller J. Sclerosing alveolitis induced by cyclophosphamide. Ultrastructural observations on alveolar injury and repair. Am. J. Pathol. 1975;81:513–530. [PMC free article] [PubMed] [Google Scholar]
- Shimizu S, Kobayashi H, Watanabe H, Ohnishi Y. Mallory body-like structures in the lung. Acta Pathol. Jpn. 1986;36:105–112. doi: 10.1111/j.1440-1827.1986.tb01464.x. [DOI] [PubMed] [Google Scholar]
- Fukuda Y, Basset F, Ferrans VJ, Yamanaka N. Significance of early intra-alveolar fibrotic lesions and integrin expression in lung biopsy specimens from patients with idiopathic pulmonary fibrosis. Hum. Pathol. 1995;26:53–61. doi: 10.1016/0046-8177(95)90114-0. [DOI] [PubMed] [Google Scholar]
- Michel RP, Limacher JJ, Kimoff RJ. Mallory bodies in scar adenocarcinoma of the lung. Hum. Pathol. 1982;13:81–85. doi: 10.1016/s0046-8177(82)80143-3. [DOI] [PubMed] [Google Scholar]
- Katzenstein AL. Smoking-related interstitial fibrosis (SRIF), pathogenesis and treatment of usual interstitial pneumonia (UIP), and transbronchial biopsy in UIP. Mod. Pathol. 2012;25(Suppl. 1):S68–S78. doi: 10.1038/modpathol.2011.154. [DOI] [PubMed] [Google Scholar]
- Ciehanover A, Hod Y, Hershko A. A heat-stable polypeptide component of an ATP-dependent proteolytic system from reticulocytes. Biochem. Biophys. Res. Commun. 1978;81:1100–1105. doi: 10.1016/0006-291x(78)91249-4. [DOI] [PubMed] [Google Scholar]
- Reinstein E, Ciechanover A. Narrative review: protein degradation and human diseases: the ubiquitin connection. Ann. Intern. Med. 2006;145:676–684. doi: 10.7326/0003-4819-145-9-200611070-00010. [DOI] [PubMed] [Google Scholar]
- Weiss CH, Budinger GR, Mutlu GM, Jain M. Proteasomal regulation of pulmonary fibrosis. Proc. Am. Thorac. Soc. 2010;7:77–83. doi: 10.1513/pats.200906-055JS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry G, Friedman R, Shaw G, Chau V. Ubiquitin is detected in neurofibrillary tangles and senile plaque neurites of Alzheimer disease brains. Proc. Natl Acad. Sci. USA. 1987;84:3033–3036. doi: 10.1073/pnas.84.9.3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel MB, Majetschak M. Distribution and interrelationship of ubiquitin proteasome pathway component activities and ubiquitin pools in various porcine tissues. Physiol. Res. 2007;56:341–350. doi: 10.33549/physiolres.931005. [DOI] [PubMed] [Google Scholar]
- Driscoll J. The role of the proteasome in cellular protein degradation. Histol. Histopathol. 1994;9:197–202. [PubMed] [Google Scholar]
- Murayama S, Mori H, Ihara Y, et al. Immunocytochemical and ultrastructural studies of lower motor neurons in amyotrophic lateral sclerosis. Ann. Neurol. 1990;27:137–148. doi: 10.1002/ana.410270208. [DOI] [PubMed] [Google Scholar]
- Kawanishi R, Mizutani T, Yamada H, et al. Ubiquitin-positive inclusions in ependymal cells. Acta Neuropathol. 2003;106:129–136. doi: 10.1007/s00401-003-0712-3. [DOI] [PubMed] [Google Scholar]
- Lennox G, Lowe J, Morrell K, Landon M, Mayer RJ. Anti-ubiquitin immunocytochemistry is more sensitive than conventional techniques in the detection of diffuse Lewy body disease. J. Neurol. Neurosurg. Psychiatry. 1989;52:67–71. doi: 10.1136/jnnp.52.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love S, Saitoh T, Quijada S, Cole GM, Terry RD. Alz-50, ubiquitin and tau immunoreactivity of neurofibrillary tangles, Pick bodies and Lewy bodies. J. Neuropathol. Exp. Neurol. 1988;47:393–405. doi: 10.1097/00005072-198807000-00001. [DOI] [PubMed] [Google Scholar]
- Shimura H, Schlossmacher MG, Hattori N, et al. Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson's disease. Science. 2001;293:263–269. doi: 10.1126/science.1060627. [DOI] [PubMed] [Google Scholar]
- Ardley HC, Scott GB, Rose SA, Tan NG, Markham AF, Robinson PA. Inhibition of proteasomal activity causes inclusion formation in neuronal and non-neuronal cells overexpressing Parkin. Mol. Biol. Cell. 2003;14:4541–4556. doi: 10.1091/mbc.E03-02-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilo K, Colby TV, Travis WD, Franks TJ. Exuberant type 2 pneumocyte hyperplasia associated with spontaneous pneumothorax: secondary reactive change mimicking adenocarcinoma. Mod. Pathol. 2007;20:352–356. doi: 10.1038/modpathol.3800744. [DOI] [PubMed] [Google Scholar]
- Marques LJ, Teschler H, Guzman J, Costabel U. Smoker's lung transplanted to a nonsmoker. Long-term detection of smoker's macrophages. Am. J. Respir. Crit. Care Med. 1997;156:1700–1702. doi: 10.1164/ajrccm.156.5.9611052. [DOI] [PubMed] [Google Scholar]
- Zatloukal K, French SW, Stumptner C, et al. From Mallory to Mallory-Denk bodies: what, how and why? Exp. Cell Res. 2007;313:2033–2049. doi: 10.1016/j.yexcr.2007.04.024. [DOI] [PubMed] [Google Scholar]
- French BA, van Leeuwen F, Riley NE, et al. Aggresome formation in liver cells in response to different toxic mechanisms: role of the ubiquitin–proteasome pathway and the frameshift mutant of ubiquitin. Exp. Mol. Pathol. 2001;71:241–246. doi: 10.1006/exmp.2001.2401. [DOI] [PubMed] [Google Scholar]
- Mayer RJ, Landon M, Lowe J. Ubiquitin and the molecular pathology of human disease. In: Peters J-M, Harris JR, Finley D, editors. Ubiquitin and the biology of the cell. New York/London: Plenum Press; 1998. pp. 436–452. [Google Scholar]
- Lowe J, Mayer RJ, Landon M. Ubiquitin in neurodegenerative diseases. Brain Pathol. 1993;3:55–65. doi: 10.1111/j.1750-3639.1993.tb00726.x. [DOI] [PubMed] [Google Scholar]
- Oda Y, Okada Y, Katsuda S, Nakanishi I. Immunohistochemical study on the infection of herpes simplex virus, human cytomegalovirus, and Epstein-Barr virus in secondary diffuse interstitial pneumonia. Hum. Pathol. 1994;25:1057–1062. doi: 10.1016/0046-8177(94)90065-5. [DOI] [PubMed] [Google Scholar]
- Grant RM, Weitzman SS, Sherman CG, Sirkin WL, Petric M, Tellier R. Fulminant disseminated Varicella Zoster virus infection without skin involvement. J. Clin. Virol. 2002;24:7–12. doi: 10.1016/s1386-6532(01)00217-7. [DOI] [PubMed] [Google Scholar]
- Yamada T, Kawanishi R, Yamada H, et al. An autopsy case of ubiquitin-positive eosinophilic intracytoplasmic inclusion bodies in alveolar epithelium. Kokyu To Junkan. 2004;52:759–763. [Google Scholar]
- Herrmann J, Ciechanover A, Lerman LO, Lerman A. The ubiquitin–proteasome system in cardiovascular diseases – a hypothesis extended. Cardiovasc. Res. 2004;61:11–21. doi: 10.1016/j.cardiores.2003.09.033. [DOI] [PubMed] [Google Scholar]
- Araya J, Kojima J, Takasaka N, et al. Insufficient autophagy in idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013;304:L56–L69. doi: 10.1152/ajplung.00213.2012. [DOI] [PubMed] [Google Scholar]
- Yamada T, Uehara K, Kawanishi R, et al. Immunohistochemical detection of ubiquitin-positive intracytoplasmic eosinophilic inclusion bodies in diffuse alveolar damage. Histopathology. 2006;48:846–854. doi: 10.1111/j.1365-2559.2006.02445.x. [DOI] [PubMed] [Google Scholar]
- Kawabata Y, Hoshi E, Murai K, et al. Smoking-related changes in the background lung of specimens resected for lung cancer: a semiquantitative study with correlation to postoperative course. Histopathology. 2008;53:707–714. doi: 10.1111/j.1365-2559.2008.03183.x. [DOI] [PubMed] [Google Scholar]
- Browning JD, More IA, Boyd JF. Adult pulmonary cytomegalic inclusion disease: report of a case. J. Clin. Pathol. 1980;33:11–18. doi: 10.1136/jcp.33.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata Y, Hoshi E, Ubukata M, et al. Histological changes are background factors in acute exacerbation of usual interstitial pneumonia pattern/lesions following lung resection. Jpn J. Lung Cancer. 2006;46:329–336. [Google Scholar]
- Yamada T, Uehara K, Mizutani T, et al. Ubiquitin-positive pneumocytes are present in nonspecific interstitial pneumonia with immunohistochemical analyses. Histopathology. 2008;53:742–744. doi: 10.1111/j.1365-2559.2008.03149.x. [DOI] [PubMed] [Google Scholar]
- Yamada T, Sunagawa K, Homma T, Uehara K, Mizutani T, Kawabata Y. Ubiquitin-positive pneumocytes and inclusion bodies are present in secondary organizing pneumonia. Intern. Med. 2011;50:277–283. doi: 10.2169/internalmedicine.50.4156. [DOI] [PubMed] [Google Scholar]
- Yamada T, Nakanishi Y, Homma T, et al. Airspace enlargement with fibrosis shows characteristic histology and immunohistology different from usual interstitial pneumonia, nonspecific interstitial pneumonia, and centrilobular emphysema. Pathol. Int. 2013;63:206–213. doi: 10.1111/pin.12054. [DOI] [PubMed] [Google Scholar]
- Snider GL. Emphysema: the first two centuries – and beyond. A historical overview, with suggestions for future research: Part 2. Am. Rev. Respir. Dis. 1992;146:1615–1622. doi: 10.1164/ajrccm/146.6.1615. [DOI] [PubMed] [Google Scholar]
- Basset F, Ferrans VJ, Soler P, Takemura T, Fukuda Y, Crystal RG. Intraluminal fibrosis in interstitial lung disorders. Am. J. Pathol. 1986;122:443–461. [PMC free article] [PubMed] [Google Scholar]
- Nakashima N, Kuwano K, Maeyama T, et al. The p53–Mdm2 association in epithelial cells in idiopathic pulmonary fibrosis and non-specific interstitial pneumonia. J. Clin. Pathol. 2005;58:583–589. doi: 10.1136/jcp.2004.022632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Boggaram V. Proteasome dysfunction inhibits surfactant protein gene expression in lung epithelial cells: mechanism of inhibition of SP-B gene expression. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007;292:L74–L84. doi: 10.1152/ajplung.00103.2006. [DOI] [PubMed] [Google Scholar]
- Renzoni EA, Abraham DJ, Howat S, et al. Gene expression profiling reveals novel TGFbeta targets in adult lung fibroblasts. Respir. Res. 2004;5:24–35. doi: 10.1186/1465-9921-5-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Chen D, Shiloh A, et al. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature. 2002;416:648–653. doi: 10.1038/nature737. [DOI] [PubMed] [Google Scholar]
- Mortenson MM, Schlieman MG, Virudachalam S, Bold RJ. Effects of the proteasome inhibitor bortezomib alone and in combination with chemotherapy in the A549 non-small-cell lung cancer cell line. Cancer Chemother. Pharmacol. 2004;54:343–353. doi: 10.1007/s00280-004-0811-4. [DOI] [PubMed] [Google Scholar]
- Riha RL, Duhig EE, Clarke BE, Steele RH, Slaughter RE, Zimmerman PV. Survival of patients with biopsy-proven usual interstitial pneumonia and nonspecific interstitial pneumonia. Eur. Respir. J. 2002;19:1114–1118. doi: 10.1183/09031936.02.00244002. [DOI] [PubMed] [Google Scholar]
- Katzenstein AL, Zisman DA, Litzky LA, Nguyen BT, Kotloff RM. Usual interstitial pneumonia: histologic study of biopsy and explant specimens. Am. J. Surg. Pathol. 2002;26:1567–1577. doi: 10.1097/00000478-200212000-00004. [DOI] [PubMed] [Google Scholar]
- Araya J, Hara H, Kuwano K. Autophagy in the pathogenesis of pulmonary disease. Intern. Med. 2013;52:2295–2303. doi: 10.2169/internalmedicine.52.1118. [DOI] [PubMed] [Google Scholar]
- Fujii S, Hara H, Araya J, et al. Insufficient autophagy promotes bronchial epithelial cell senescence in chronic obstructive pulmonary disease. Oncoimmunology. 2012;1:630–641. doi: 10.4161/onci.20297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzenstein A-LA, Mukhopadhyay S, Myers JL. Diagnosis of usual interstitial pneumonia and distinction from other fibrosing interstitial lung diseases. Hum. Pathol. 2008;39:1275–1294. doi: 10.1016/j.humpath.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Collard HR, Moore BB, Flaherty KR, et al. Acute exacerbations of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2007;176:636–643. doi: 10.1164/rccm.200703-463PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KR, Colby TV, Travis WD, et al. Fibroblastic foci in usual interstitial pneumonia: idiopathic versus collagen vascular disease. Am. J. Respir. Crit. Care Med. 2003;167:1410–1415. doi: 10.1164/rccm.200204-373OC. [DOI] [PubMed] [Google Scholar]
- Churg A, Müller NL, Silva CI, Wright JL. Acute exacerbation (acute lung injury of unknown cause) in UIP and other forms of fibrotic interstitial pneumonias. Am. J. Surg. Pathol. 2007;31:277–284. doi: 10.1097/01.pas.0000213341.70852.9d. [DOI] [PubMed] [Google Scholar]
- Park IN, Kim DS, Shim TS, et al. Acute exacerbation of interstitial pneumonia other than idiopathic pulmonary fibrosis. Chest. 2007;132:214–220. doi: 10.1378/chest.07-0323. [DOI] [PubMed] [Google Scholar]
- Katzenstein AL, Fiorelli RF. Nonspecific interstitial pneumonia/fibrosis. Histologic features and clinical significance. Am. J. Surg. Pathol. 1994;18:136–147. [PubMed] [Google Scholar]
- Park IN, Jegal Y, Kim DS, et al. Clinical course and lung function change of idiopathic nonspecific interstitial pneumonia. Eur. Respir. J. 2009;33:68–76. doi: 10.1183/09031936.00158507. [DOI] [PubMed] [Google Scholar]
- Sakamoto N, Mukae H, Fujii T, et al. Nonspecific interstitial pneumonia with poor prognosis associated with amyopathic dermatomyositis. Intern. Med. 2004;43:838–842. doi: 10.2169/internalmedicine.43.838. [DOI] [PubMed] [Google Scholar]
- Nicholson AG, Colby TV, du Bois RM, Hansell DM, Wells AU. The prognostic significance of the histologic pattern of interstitial pneumonia in patients presenting with the clinical entity of cryptogenic fibrosing alveolitis. Am. J. Respir. Crit. Care Med. 2000;162:2213–2217. doi: 10.1164/ajrccm.162.6.2003049. [DOI] [PubMed] [Google Scholar]
- Yousem SA. Respiratory bronchiolitis-associated interstitial lung disease with fibrosis is a lesion distinct from fibrotic nonspecific interstitial pneumonia: a proposal. Mod. Pathol. 2006;19:1474–1479. doi: 10.1038/modpathol.3800671. [DOI] [PubMed] [Google Scholar]
- Reddy TL, Mayo J, Churg A. Respiratory bronchiolitis with fibrosis. High-resolution computed tomography findings and correlation with pathology. Ann. Am. Thorac. Soc. 2013;10:590–601. doi: 10.1513/AnnalsATS.201304-088OC. [DOI] [PubMed] [Google Scholar]