

Abstract
The Pygo-BCL9 complex is a chromatin reader, facilitating β-catenin-mediated oncogenesis, and is thus emerging as a potential therapeutic target for cancer. Its function relies on two ligand-binding surfaces of Pygo’s PHD finger that anchor the histone H3 tail methylated at lysine 4 (H3K4me) with assistance from the BCL9 HD1 domain. Here, we report the first use of fragment-based screening by NMR to identify small molecules that block protein–protein interactions by a PHD finger. This led to the discovery of a set of benzothiazoles that bind to a cleft emanating from the PHD–HD1 interface, as defined by X-ray crystallography. Furthermore, we discovered a benzimidazole that docks into the H3K4me specificity pocket and displaces the native H3K4me peptide from the PHD finger. Our study demonstrates the ligandability of the Pygo–BCL9 complex and uncovers a privileged scaffold as a template for future development of lead inhibitors of oncogenesis.
β-catenin is a key effector of Wnt signaling, and also a potent oncogene, judging by the fact that activating mutations in β-catenin have been found in many types of cancer.1 β-catenin is also activated by disabling mutations in its immediate negative regulators, notably in Adenomatous polyposis coli (APC), a crucial tumor suppressor in the intestine that is mutated in >80% of all cases of colorectal cancers, but also in Axin, which, together with APC, promotes the proteasomal degradation of β-catenin in the absence of Wnt signaling.2 In normal development and adult tissues, Wnt signaling blocks β-catenin degradation; consequently, β-catenin accumulates and binds to TCF/LEF transcription factors to coactivate context-dependent transcriptional programmes that specify cell fates and differentiation, most notably in stem cell compartments.3 For example, in mammalian intestinal crypts, β-catenin is required for stem and progenitor cells, which can become the cells-of-origin for colorectal cancer.4
Despite its importance in cancer, there are no well-validated small molecule inhibitors of β-catenin.5 The reason for this is that β-catenin is a challenging target: there are no enzymes required for its activity that could be inhibited, and its interface with TCF factors involves most of its structured domain, the Armadillo Repeat domain (ARD), which is extensive and also constitutes the interface for its negative regulators, including APC and Axin, whose interaction with the ARD overlaps that of TCF.6 Unsurprisingly, attempts to block specifically the interaction between β-catenin and TCFs have met with little success and failed to uncover any promising leads.5
However, the N-terminus of the ARD harbors a separate interaction surface for the BCL9 adaptor proteins, which bind to β-catenin through a short α-helical domain (called HD2), simultaneously with TCF7 (Figure 1A). In turn, BCL9 adaptors use a separate domain (called HD1) to bind to the rear of the Pygo PHD fingers; they thus induce a subtle allosteric modulation of the PHD, which facilitates its binding to the histone H3 tail methylated at lysine 4 (H3K4me) through its frontal surface8−10 (Figure 1B,C). Humans have two closely related Pygo and BCL9 proteins (Pygo1 and Pygo2, BCL9 and BCL9–2/B9L, respectively), each of which are required for the elevated levels of TCF-dependent transcription in colorectal cancer cells due to the hyperactivated (“oncogenic”) β-catenin in these cells.11−14 Furthermore, Pygo and BCL9 orthologs behave as tumor promoters in murine intestinal and mammary tumor models.15,16 Thus, the Pygo-BCL9 complex emerges as a promising target for inhibiting oncogenic β-catenin, providing three unique and relatively small protein–protein interfaces that could be blocked.
Figure 1.
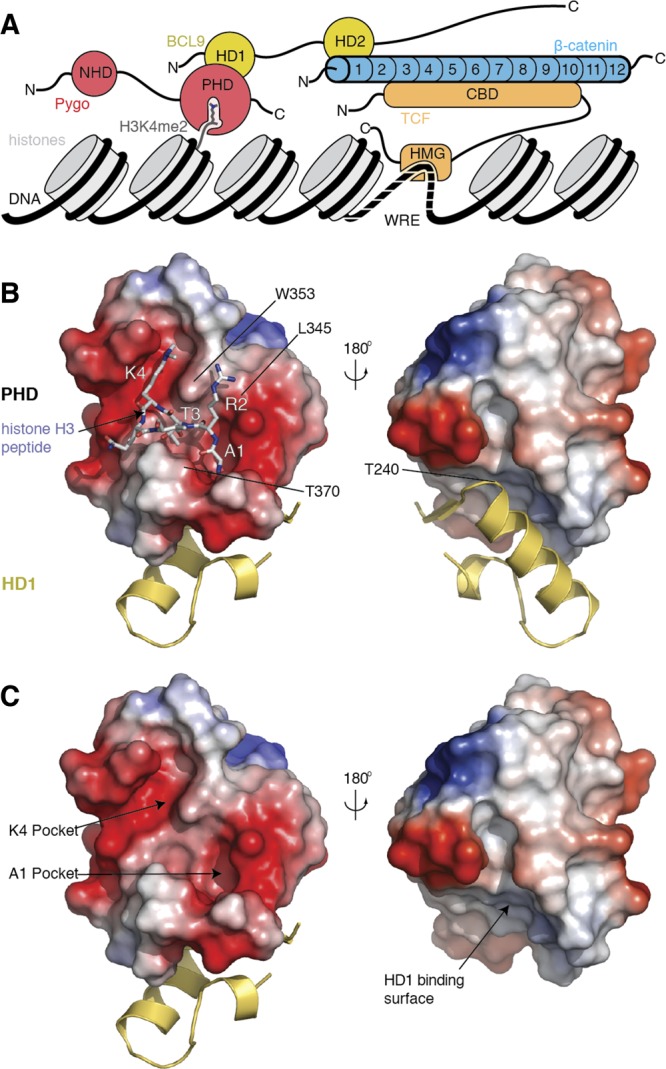
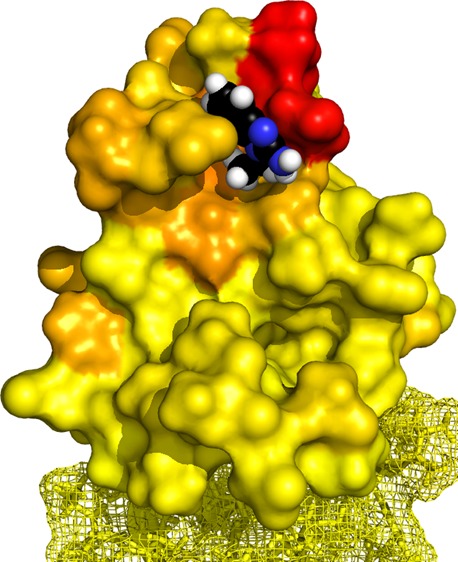
Ligand interfaces of the PHD–HD1 complex. (A) Schematic representation of the Pygo–BCL9 complex and its interaction surfaces with the β-catenin ARD N-terminus, and with methylated histone H3 tail (H3K4me2); recruitment of β-catenin to Wnt target genes requires its binding to TCF factors (bound to specific enhancer sequences through their HMG domain) but also its binding to Pygo–BCL9. (B, C) Molecular surface representations of the PHD finger from hPygo2, colored according to electrostatic potential (red, negative; blue, positive), in complex with HD1 from hB9L (yellow, in ribbon representation; omitted in lower right panel, to reveal the HD1-binding surface), (B) with H3K4me2K9ac (4UP0) or (C) without peptide (in stick representation; red, oxygen; blue, nitrogen), to visualize its deep K4me and A1 pockets (arrows), and its flat HD1-interacting surface on its rear (right-hand views, rotated by 180°). Key residues are labeled.
Indeed, the interaction between BCL9–HD2 and β-catenin has been targeted successfully with a small-molecule inhibitor that destabilizes oncogenic β-catenin in human colorectal cancer cell lines17 and in the murine intestine.18 Furthermore, a stapled HD2-like α-helix caused dissociation of β-catenin from BCL9 and showed potent tumor-suppressive effects in mouse xenograft models.19 However, the druggability or ligandability20 of the Pygo PHD finger has not yet been assessed. In fact, there is no systematic study of the ligandability of any PHD finger by small molecules as yet, although chromatin reader domains are generally considered attractive targets for small-molecule inhibition, in light of recent successes.21−24 There is one recent report of small molecules attenuating the histone binding of a PHD finger (from ARID1A), identified by alpha screening involving HaloTag technology;25 however there is only limited information on how these compounds interact with their cognate PHD finger.
As mentioned above, native H3K4me peptides bind to the Pygo PHD finger, whose “face” contains two deep pockets—an anchoring pocket that buries its N-terminal alanine (A1) and a specificity pocket that embeds methylated lysine 4 (K4me)—connected by a short channel that accommodates threonine 3 (T3 channel;8 Figure 1C). The A1 pocket and T3 channel are allosterically linked to the HD1-interacting surface at the “rear” surface of the PHD finger, whereby the PHD signature residue (an invariant tryptophan, W377 in hPygo2) plays a pivotal role in relaying the allosteric communication through the PHD structural core.10 Given that minimal alterations of the histone H3 tail peptide drastically reduce its affinity for PHD–HD1 complexes,8,26 we surmised that we might be able to identify small molecules that bind to these histone pockets and interfere with their binding to the histone H3 tail.
We thus conducted a screen by two-dimensional nuclear magnetic resonance (NMR) for chemical fragments (CF) binding to the PHD–HD1 complex. This identified two closely related benzothiazole compounds binding to its rear surface. Structure–activity relationship (SAR) analysis defined the functionally relevant groups within their scaffold, including a crucial amine which binds to its cognate cleft extending from the PHD–HD1 interface, as defined by X-ray crystallography. In addition, SAR also uncovered a second binding site in the histone-binding surface recognized by a set of benzimidazoles. One of these (CF16) docks into the distal portion of the K4me pocket, as revealed by an NMR-based structural model, and displaces its natural ligand, the methylated H3 tail, from the PHD finger. We also used de novo virtual screening to identify four sets of larger compounds, each with a distinct chemical scaffold, whose binding poses span both K4me and A1 pockets. This is the first systematic study of the ligandability of a PHD finger, uncovering small chemical scaffolds that bind to its pockets with high specificity and efficiency. These could provide templates for subsequent chemical development toward lead inhibitors of the Pygo–BCL9 complex.
Results and Discussion
Targeting the PHD Histone-Binding Surface of Pygo
The HD1-interacting surface of the Pygo PHD finger is hydrophobic8 and unstable in aqueous solution, causing undesirable self-aggregation.27 We thus decided to use the PHD finger in a complex with HD1 for small-molecule screening, initially by taking an in silico approach, to see whether we could identify small compounds (up to 500 Da) that recognize the histone-binding surface of this complex. This surface is essentially the same in the PHD–HD1 complexes from Pygo1–BCL98 and its paralog Pygo2–B9L,10 so both crystal structures were used.
The first pilot screen of 225 000 commercially available chemicals identified 313 hits, which were subsequently tested for their binding to PHD–HD1 by NMR spectroscopy. We thus incubated a purified 15N-labeled complex with pools of five hit compounds (each at 1 mM) and recorded heteronuclear single-quantum correlation (HSQC) spectra for each pool. Most pools turned out to be negative for binding, judging by their spectra that superimposed perfectly on a reference HSQC (whose resonances had been assigned previously;10 see also below). Only three compounds (IS1–3) proved to be positive, reflecting a very low hit rate (0.001%). Each of these hits elicited several weak chemical shift perturbations (CSPs) of the same PHD residues (Figure 2A), consistent with their near-identical chemical scaffold (Figure 2B). Projecting these CSPs onto the crystal structure of PHD–HD110 allowed us to generate “heat-maps,” which confirmed that IS1–3 bind to its histone pockets: in silico docking predicts a hydrogen bond between their central amide nitrogen and the main-chain carbonyl oxygen of the highly conserved A343 at the T3 channel floor (Figure 2C). These docking poses further predict that their bicyclic core reaches into the A1 pocket, while their phenyl ring could stack against the indole ring of the tryptophan (W353) that separates this pocket from the adjacent K4me pocket.
Figure 2.

In silico hits binding to PHD–HD1. (A) Overlay of HSQC spectra of 50 μM 15N-labeled PHD–HD1 + 1 mM IS1 (cyan) onto PHD–HD1 alone (maroon), with residues experiencing CSPs labeled. (B) Structure of IS1–3 and IS hits used in D–F. (C) Heat-maps of CSPs elicited by IS1 projected onto the structure of PHD–HD1 (2XB1; coloring thresholds: yellow <0.04 ppm; orange <0.1 ppm; red <0.15 ppm) and calculated docking pose (in stick representation; red, oxygen; blue, nitrogen; yellow, sulfur; green, chlorine); front and rear views as in Figure 1B (HD1, mesh representation) and zoomed-in view at the right, with key interacting residues labeled (note L345 whose side-chain fills the R2 cavity found in other PHD fingers in this position, see text). (D–F) Docking poses of representatives of each IS group (Figures S1 and S2), as indicated in the panels.
An undesirable property of IS1–3 is their poor solubility. We therefore conducted another three consecutive virtual screens, each increasingly refined and constrained based on the preceding one (see Supporting Information, Supplementary Methods), and also applied a filter to exclude compounds with predicted low solubility. Collectively, these screens identified 32 additional hits whose binding to the PHD–HD1 complex was confirmed by NMR. Of these, 28 can be classified into three groups, based on a common substructure; common to each group is an amide-containing linker fragment (except IS4–6), typically with substitutions at each end (Figure S1), similar to the arrangement in IS1–3.
On the basis of the heat-maps of these 28 hits and their docking poses, we were able to build a picture of how these compounds might recognize the histone-binding surface (Figure 2D–F; Figure S2): representative poses suggest that their central amide fits into the T3 channel while their terminal ring systems project into the A1 and K4me pockets, as described for IS1–3. They predict charge interactions between the IS basic group and the aspartate (D339) at the lip of the K4me pocket and a hydrogen bond between a carbonyl oxygen in their termini with the main-chain amide of a conserved leucine (L345) which lines the side-wall of the T3 channel at the opening of the A1 pocket (Figure 2C–F), somewhat reminiscent of the binding of histone H3 tail to this wall8 (Figure 1B). Note that this region is structurally variable in different PHD fingers: some of these exhibit a cavity at this position, which envelops unmodified arginine 2 (R2)28,29 or methylated R2,26 but this R2 cavity is obliterated in the PHD fingers of typical Pygo orthologs, e.g. by the side-chain of L345 in human Pygo2 (Figure 1B).
The degree to which the A1 cavity is occupied by the IS hits varies between the groups and appears maximal for group 3 (e.g., IS19; Figure 2F), while members of group 2 penetrate less far into the A1 cavity (Figure 2E), and those of group 1 merely interact with L345 at the pocket opening (Figure 2D). We classed IS19 as our top hit, based on the magnitude of induced CSPs as a rough guide (0.15 ppm), which was confirmed by titration experiments that led us to estimate the affinity of this hit for PHD–HD1 (3.5 ± 1.8 mM). This indicates a relatively poor LE for the IS hits (an estimated 0.12 kcal mol–1 per heavy atom for IS19).
Co-crystallization of the PHD–HD1 complex with several of the hits, including IS19, was unsuccessful, likely due to the combination of the low solubility and affinity of these compounds. Also, PHD–HD1 has a strong tendency to engage in pseudoligand interactions through its histone-binding surface, e.g., with lysine-containing peptides from symmetry-related proteins,8 which could hinder the access of small molecules to this surface. We thus used a slightly modified PHD for our subsequent NMR screens (see Methods), to minimize pseudoligand blocking.
NMR-Based Fragment Screening
The two main problems of our IS hits are their low solubility and LE, as mentioned above. To identify soluble compounds that bind to the PHD–HD1 complex with increased LE, we adopted a fragment-based approach,30−32 but using protein-observed NMR spectroscopy as our primary screen. We chose the Maybridge “rule of three” (Ro3) library of 1000 chemical fragments, divided into pools of five compounds (each at 1 mM, and selected to avoid 1H resonance overlap), which we incubated with 50 μM 15N-labeled PHD–HD1, to monitor binding by recording HSQC spectra.
Numerous pools produced multiple CSPs, from which we selected the top 7 (eliciting the most pronounced CSPs) for further analysis. Using both ligand-observed and protein-observed NMR techniques, we succeeded in unambiguous deconvolution for 2/7 pools: we identified a single compound in each of the two pools (CF1, CF2) that was responsible for the CSPs initially recorded for the whole pool (Figure 3A). Strikingly, CF1 and CF2 are almost identical, differing only in the atom attached to position C6 of their benzothiazole ring (fluorine or chlorine; Figure 3B), which explains the high similarity of their HSQC spectra and indicates that they bind to the same site. Titration experiments led us to estimate the affinity of CF1 for the complex to be low (3.1 mM ± 1.3; Figure 3C), as expected for a small chemical fragment (168 Da). The calculated LE of 0.31 kcal mol–1 per heavy atom for CF1 suggests an excellent fit with its cognate binding site on PHD–HD1.
Figure 3.
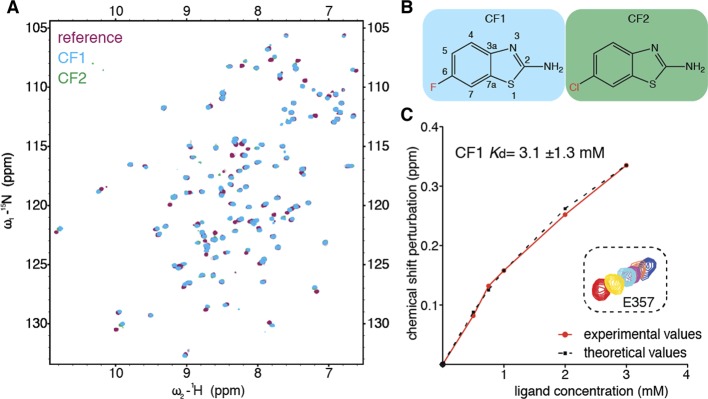
Benzothiazoles binding to PHD–HD1. (A) Overlay of HSQC spectra of 50 μM 15N-labeled PHD–HD1 + 1 mM CF1 (cyan) or CF2 (green) onto PHD–HD1 alone (maroon). (B) Structure of CF1 (with positions labeled in benzothiazole scaffold) and CF2. (C) Graphical plot based on NMR titration of a single PHD residue (E357) by increasing fragment concentrations of CF1, as indicated (red, no fragment; yellow, 600 μM; cyan, 900 μM; purple, 2.2. mM; blue, 3.2 mM); red, experimentally observed CSPs plotted against ligand concentration; black dotted, model curve. A mean Kd of 3.1 ± 1.3 (standard error) was derived from CSPs of seven different correlation peaks (labeled in A).
None of the other positive pools were deconvoluted successfully, suggesting that the CSPs observed with these pools may have resulted from aggregation between individual compounds. Our fragment screen thus yielded a rate of 0.2% confirmed hits, 200 fold higher than that from our initial in silico screen, and close to the average hit rate (0.24%) reported for similar protein-observed fragment screens.33 This indicates an average ligandability of the PHD–HD1 complex.
To identify the features of these CF hits that determine their interaction with PHD–HD1, we conducted a first round of SAR, screening analogues with chemical alterations at either end of their bicyclic core. This identified two hits with different pendant groups at C6, confirming that this position can be altered without a loss of binding. Testing additional modifications at this position, we found that binding is compatible with surprisingly bulky C6 pendants (Figure 4A). This suggests that C6 is solvent-exposed and suitable for further chemical development. Conversely, modifying the amine attached to C2 (2-amine) caused a loss of binding, judging by the lack of CSPs in NMR tests of two compounds with 2-amine pendants. This indicates the functional importance of the 2-amine of the benzothiazole hits.
Figure 4.
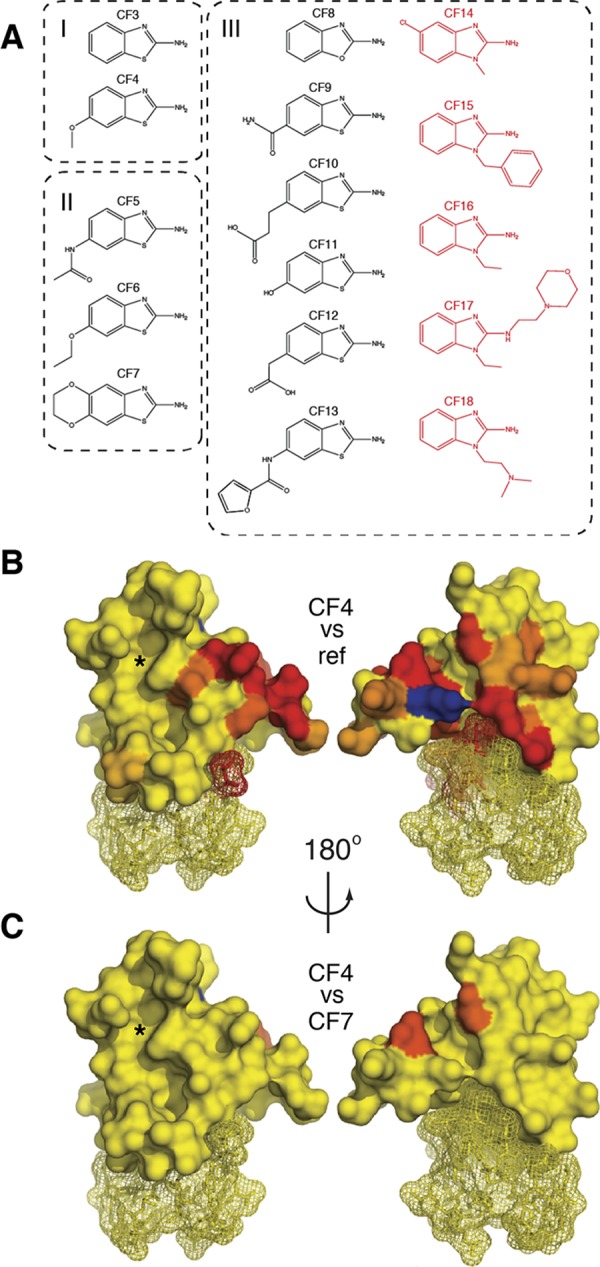
Identifying the benzothiazole binding site. (A) SAR defining three groups of chemical relatives of CF1 that bind to PHD–HD1. In red is the subset of compounds (extended at N1) that acquired a distinct binding site on PHD–HD1 (see also Figures 6 and 7). (B) Heat-maps of CSPs induced by CF4 (coloring and views as in Figure 2C; blue, peak exchange broadening); left, front surface, with histone-binding pockets (indicated by asterisk); right, rear surface. (C) Differential heat-map (coloring and views as in B), representing the ratios of CSPs induced by CF7 versus CF4, which identifies the benzothiazole binding site at the rear surface of PHD–HD1 (see also Figure 5).
The heat-map of the CSPs of our strongest binder (CF4; Table S1) identified multiple residues, mostly located at the rear PHD surface (Figure 4B, right), whereas the histone-binding surface (Figure 4B, left, asterisk) was largely unaffected. Other CF hits (e.g., CF1, CF7) produced similar heat-maps, and pairwise overlays allowed us to identify residues that are differentially affected because of their different C6 pendants: for example, if we overlay the HSQC spectra of CF4 and CF7, this highlights two residues at the rear of PHD–HD1 (Figure 4C). Collectively, these differential heat-maps point to a narrow cleft emanating from the HD1-binding surface of PHD as the binding site for these benzothiazoles.
The Benzothiazole Cleft
The high solubility of the CF hits made them suitable for cocrystallization with PHD–HD1 at high compound excess (20 mM). Diffracting crystals were obtained under multiple conditions, and the subsequent structure determination revealed that one of the crystals (solved at 1.65 Å resolution; 4UP5; Table S2) contained CF4 in the narrow cleft emanating from the PHD–HD1 interface (Figure 5A), as shown by NMR (Figure 4C). This demonstrates the validity of our approach of generating differential heat-maps from chemically related hits in predicting their binding sites. Below, we shall call this binding site the benzothiazole cleft.
Figure 5.

Structure of the benzothiazole cleft. (A) Representation of PHD–HD1 rear surface (as in Figure 1B, right), with CF4 bound to its cleft above the PHD–HD1 interface (4UP5; conserved HD1 T240 side-chain in stick representation). (B) Detailed view of CF4 (electron density contoured to 1.2 σ) interacting with the benzothiazole cleft (orange, interacting residues; yellow, noninteracting residues); dotted lines, hydrogen bonds (with PHD D380 and HD1 T240). (C, D) Ligplots of benzothiazoles binding to their cognate clefts of (C) PHD–HD1 and (D) p53_Y220C (as specified in key).
Consistent with our SAR results, the 2-amine is crucial for the binding of CF4, forming a hydrogen bond with the side-chain of T240 of B9L HD1 (Figure 5B,C). A second hydrogen bond is formed between the thiazole nitrogen (N3) and the main-chain amide of D380 at the cleft gate. Multiple hydrophobic interactions between the CF4 benzene ring and F354 and A332 and between its thiazole and T359 of Pygo2 PHD are likely to strengthen the binding between the compound and protein complex (Figure 5C). Intriguingly, three of these key interacting residues of the PHD finger, including its two gatekeepers (D380 and T359) which separate the cleft from the HD1-binding surface, are highly conserved among all Pygo orthologs, from placozoa to humans,26 and the CF-interacting residue of B9L (T240 whose side-chain points into the cleft gate) is invariant among B9L and BCL9 orthologs. This striking conservation suggests that the benzothiazole cleft may constitute the binding site for an unknown natural ligand.
Conversely, the sulfur (S1) of CF4 faces the solvent (Figure 5B), like the methoxy group at C6 (as predicted from our SAR studies), which faces away from the HD1-binding surface of PHD. Indeed, the latter is the most exposed group of the ligand, and the only one that does not contact PHD at all, which explains why bulky groups can be attached to C6 without a loss of binding. For example, this position in CF7 is replaced by an additional ring system (a dioxane ring; Figure 4A), but its estimated affinity for PHD–HD1 (Table S1) is comparable to that of CF1 and CF2. CF4 appears to have the highest affinity for PHD–HD1 of all CF compounds (2.5 mM ± 0.5; Table S1), which corresponds to a moderately high LE (0.29 kcal mol–1 per heavy atom).
Intriguingly, CF7 is identical to compound 15,34 one of four related fragment hits that bind to a narrow pocket in the p53 tumor suppressor formed by the Y220C cancer mutation. This p53–Y220C pocket does not show any obvious structural or chemical similarity to the benzothiazole cleft (Figure 5C,D), although the 2-amine of the benzothiazole is also crucial for the binding of compound 15 to p53–Y220C34 (Figure 5D). Notably, benzothiazole resembles benzoxazole, a relatively simple chemical scaffold considered to be a privileged substructure, due to its intrinsic versatility in forming interactions with a range of different protein environments.35,36 Privileged structures have emerged as useful starting points for rational drug design.37,38
Benzimidazoles Binding to the K4me Pocket
Given the apparent versatility of the benzothiazole scaffold, we attempted to improve its affinity for its PHD–HD1 target, by screening additional derivatives. Specifically, we asked whether the 2-amine could be extended through the cleft gate (between D380 and T359) toward the PHD–HD1 interface without losing compound binding, ultimately to develop compounds that interfere with PHD–HD1 binding. We thus tested another 51 chemical derivatives with variations at S1, C6, or 2-amine for binding to PHD–HD1. Of 25 compounds in the 2-amine test group, only one (CF17) retained binding, but this hit produced a distinct CSP pattern (see below), indicating that it binds to a distinct site. This reconfirms that the 2-amine is crucial for the binding of the benzothiazoles to their cognate cleft.
Among the eight tested compounds with C6 pendants, we identified five additional hits that produced CSP patterns similar to those of the original CF hits (Figure 3A), increasing the number of hits in this group to 9 (Figure 4A). Substitutions of S1 proved to be permissive for binding, as expected from the cocrystal structure: one of the new hits in this group is a benzoxazole (CF8), but each of the remaining five hits is a benzimidazole, with a nitrogen at position 1 to which various side groups are attached (CF14–18; Figure 4A).
Interestingly, when comparing the CSPs of these benzimidazoles with those of the benzothiazoles, we noticed that two of them (CF15 and CF18) produced additional CSPs, while CF14, CF16, and CF17 produced altogether distinct CSP patterns. Heat-maps of the latter indicate that they interact predominantly with the K4me pocket, while only retaining a residual interaction with the benzothiazole cleft, and no significant binding elsewhere in the complex (Figure S3). Notably, the benzimidazole scaffold is another known privileged structure,35,36 providing an explanation for why CF15 and CF18 bind to two distinct sites in PHD–HD1—to its benzothiazole cleft and its K4me pocket.
We attempted cocrystallization for CF16 and CF18, the top two K4me binders (based on their CSPs of K4me pocket residues), but were unable to obtain crystals with the compound bound. We thus used NMR, to determine their binding mode in the K4me pocket, recording half-filtered NOESY spectra of 13C–15N double-labeled PHD–HD1 incubated with the compound, which allowed us to observe numerous intermolecular 1H(12C)–1H(13C) NOEs in each case. Assignments of these NOEs confirmed our results from the 15N-HSQC shift-maps that CF16 binds almost exclusively to the K4me pocket: 33/35 NOEs were assigned to residues flanking this pocket (Figures S4 and S5), while CF18 also produced numerous strong NOEs assigned to residues flanking the benzothiazole cleft (Figure S3).
Given that CF16 appears to bind to a single (new) site of PHD–HD1, we chose this compound for docking simulations with the HADDOCK software package,39 aiming to model its interaction with PHD. As inputs into this model, we used the 33 NOEs from CF16 to obtain unambiguous restraints (Figure S5), as well as ambiguous restraints derived from the 15N-HSQC CSPs (leading to the heat-map shown in Figure 6A). Of the 200 models generated by HADDOCK, each one showed CF16 occupying a single binding site in the distal part of the K4me pocket, with a buried surface area of 371 ± 16 Å2, and with 199/200 ligands in the same orientation. The five highest-ranked models of the cluster, based on the HADDOCK score, show only minor violations (>0.5 Å) in 2/33 NOEs (Figure 6B), and no close H–H contacts (<4 Å) without a corresponding NOE. Therefore, this model defines the contact interface of the K4me pocket with CF16 with high confidence, and it also predicts the binding pose of CF16 in its cognate pocket (see also Figure S5, for a further appraisal of the model).
Figure 6.

A benzimidazole binding to the K4me pocket. (A) Cluster of top five poses of CF16 (in stick representation; blue, nitrogen) docked into the distal K4me pocket of PHD–HD1 (4UP0, in surface representation), as calculated by HADDOCK, based on unambiguous restraints derived from 33 intermolecular NOEs between the compound and protein (see B), and ambiguous restraints derived from CSPs (colored as in Figure 4B). Key interacting residues of hPygo2 are labeled. (B) Top pose of CF16 (as in A) docked into the K4me pocket (in stick overlaid by surface representation), with protein 1H showing intermolecular NOEs (orange) and CSPs (blue) indicated (see also Figures S4 and S5). (C) PHD–HD1 with bound histone H3 tail (salmon, with nitrogen in cyan) and CF16 (colored as in A), visualizing the clash between the two ligands in the methyl–lysine binding site of the K4me pocket (underneath its pocket lip, D339).
Our model predicts that the imidazole ring of CF16 and its C1 pendant undergo three crucial interactions with the K4me pocket (Figure 6A): a π stacking interaction between this ring and the benzene ring of tyrosine 328 (Y328) which forms the lid of this pocket, a cation−π stacking interaction between the delocalized proton of its guanidinium group and the electronegative surface of the phenyl ring of W353 (the side wall of the pocket), and a series of hydrophobic interactions between its ethyl group and the K4me pocket floor formed by the hydrophobic side-chains of V337 and A343. Conversely, the benzene ring of CF16 is partially solvent-exposed, and although it does not appear to contribute majorly to the interactions of CF16 with PHD, it may nevertheless serve to “wedge” CF16 into its cognate pocket. The model suggests that compounds with appropriate substitutions at C4 or C5 of the benzene ring might exist that provide additional interactions with D339 (Figure 6A) and thus increase the affinity of the compound to PHD–HD1. Likewise, an extension of its ethyl group might allow it to form additional contacts with the T3 channel, similarly to the IS compounds (Figure 2C), and thus anchor it more firmly in the histone-binding surface of PHD–HD1.
Competitive Binding between CF16 and Histone H3 Tail
The estimated affinities of CF16 and CF18 for the K4me pocket appear to be lower than those of the benzothiazole cleft-binding compounds (7.3 mM ± 2.0 and 14.7 mM ± 5.3, respectively, based on CSP titrations for K4me pocket residues; Table S1). Nevertheless, we asked whether CF16 could compete with a native histone H3 tail peptide for binding to the PHD–HD1 complex. However, to conduct these competition experiments, we first wished to define the minimal histone H3 peptide that exhibits the full range of interactions with the histone-binding surface of PHD–HD1.
If the histone H3 tail is broken down into tripeptides (ART, or TKme2Q), we cannot detect any binding by NMR (Figure S6), although these tripeptides are larger than a typical chemical fragment. This indicates that a native histone H3 peptide can only bind to PHD–HD1 if it interacts with both the K4me and A1 pockets. Indeed, a minimal peptide capable of doing this is the penta-peptide ARTKme2Q: if we titrate 15N-labeled PHD–HD1 with increasing concentrations of this pentamer, we observe numerous CSPs of residues from both K4me and A1 pockets (Figure S7), allowing us to estimate a Kd of 528 ± 32 μM. Heat-maps of these titrations highlight the T3 channel at the lowest peptide concentration, with perturbation of the distal part of the K4me pocket appearing at 2× higher concentration, followed by additional interactions in the A1 pocket at a peptide concentration 3× below Kd (Figure 7A).
Figure 7.

Competitive binding between CF16 and histone H3 peptide. (A,B) Heat-maps of CSPs induced by increasing concentrations of (A) ARTKme2Q or (B) CF16, projected onto PHD–HD1 (in sphere representation, to visualize amino acid side-chains; coloring as in Figure 4B). (C) CSPs of selected residues differentially affected by the two ligands (see text), following simultaneous incubation of PHD–HD1 (50 μM) with ARTKme2Q (515 μM) and increasing concentrations of CF16 (as indicated in key), revealing gradual CF16-dependent displacement of ARTKme2Q from PHD–HD1.
We note that the affinity of ARTKme2Q to PHD–HD1 is considerably lower than that of longer histone H3 tail peptides (e.g., a 15-mer8,10), but this is likely to be due to the lack of the intramolecular hydrogen bond between T3 and T6 seen with extended histone H3 peptides bound to PHD fingers (e.g., ref (40)), which might rigidify the peptide and thus increase its binding affinity to PHD. Indeed, titration of 15N-PHD–HD1 with 15-mer H3K4me2 revealed that the extended peptide produces predominantly peak broadening, even at low (limiting) concentrations, in contrast to the pentamer which produces mostly CSPs (Figure S6), consistent with the notion of a reduced off-rate of 15-mer binding, possibly due to a scaffolding effect provided by the T3–T6 interaction within the extended peptide. Importantly, the two heat-maps obtained by these titrations are strikingly similar (Figure S7), implying that the two peptides form essentially the same set of interactions with their cognate histone-binding surface in PHD, consistent with the cocrystal structures which reveal no interactions between extended histone H3 peptides and PHD beyond H3Q58,10 (4UP0; Table S3).
By contrast, the interactions of CF16 with PHD are far more localized than those of the histone H3 peptide, being limited to the distal part of the K4me pocket at low compound concentration, and extending into the T3 channel at a higher compound concentration (Figure 7B), consistent with the structural model (Figure 6A). In particular, V376 and L369 (which line the A1 pocket) interact exclusively with the histone pentamer but not with CF16, and the same is true for Y366 (interacting with H3Q5; Figure 6C). Therefore, the CSPs from these three residues are suitable for monitoring specifically the binding of the histone peptide (versus compound) to PHD–HD1.
To test whether CF16 could compete with a histone pentamer in binding to PHD–HD1, we incubated 50 μM 15N-labeled PHD–HD1 with 515 μM ARTKme2Q, plus increasing concentrations of CF16 (0, 2, or 5 mM). We thus found that the magnitude of the CSPs of V376, L369, and Y366 was decreased by 2 mM CF16, and further still by 5 mM CF16 (Figure 7C), indicating that this compound reduces the fraction of PHD–HD1 bound to histone peptide. Competition for binding is even more apparent if the CSPs of A343 are recorded: this residue interacts with both ARTKme2Q and CF16, but the CSPs induced by these two ligands individually are distinct. Histone peptide causes a downfield shift of the A343 HN resonance, whereas CF16 causes an upfield shift. Simultaneous incubation with both ligands reverses the direction of the histone-specific CSP toward that of CF16, again in a concentration-dependent manner (Figure 7C). This observed displacement of ARTKme2Q from PHD–HD1 by CF16 is fully consistent with calculations of the fractions of peptide or compound bound to PHD–HD1, taking into account their respective affinities to the complex (i.e., theoretically, for a two-ligand equilibrium, 49%, 43%, and 37% of PHD–HD1 is bound to ARTKme2Q, contrasting with 0%, 12%, and 25% of PHD–HD1 bound to CF16, at the three concentrations tested in the competition assays; these proportional changes are reflected by the observed CSPs). We conclude that the small benzimidazole compound CF16, by docking into the distal portion of the K4me pocket, is capable of displacing the much larger histone H3 peptide from PHD–HD1 at a relatively low molar excess. This is explained by our structural model (Figure 6A), which indicates a tight fit between CF16 and its interacting residues in the K4me pocket—namely its lid (Y328), side-wall (W353), and floor (V337 and A343).
These results also imply that the histone H3 peptide is readily displaced from its cognate surface in PHD–HD1. For example, loss of the single methyl group from H3A1 reduces the affinity of histone H3 tail to PHD–HD1 by 2 orders of magnitude.8 Furthermore, a loss of the methyl groups from H3K4me eliminates histone binding to PHD–HD1,8,26 emphasizing the crucial role of the K4me pocket in determining the specificity of the Pygo PHD finger for the K4-methylated state of the histone H3 tail. Notably, the ethyl group of CF16 occupies the methyl-lysine binding site of this pocket, clashing with histone peptide in this crucial region (Figure 6C), which could explain why this compound is capable of displacing this peptide. Our competition data demonstrate that small compounds with a good fit to the methyl-binding site of the K4me pocket (such as CF16) can be excellent tools for displacing the histone H3 tail from PHD fingers.
Conclusions
Our study describes the first systematic screening effort to identify small molecules that bind to a PHD finger—namely the PHD finger from human Pygo, a chromatin reader module that recognizes the histone H3K4me mark associated with active transcription. We thus discovered two sets of privileged substructures, with tight fits to the distal portion of the K4me pocket and to a highly conserved narrow cleft with unknown physiological function at the rear of PHD abutting its HD1-binding surface. This confirms the usefulness of the fragment-based screening approach to determine the ligandability of the PHD–HD1 complex and to identify compounds that bind to this complex with high ligand efficiency and complementarity to their cognate pockets. The success of our approach is consistent with the rationales for fragment screening layed out by others (e.g., refs (30−32)). Using protein-observed NMR as a primary screen allowed us to minimize the rate of false positives that are inherent to ligand-observed NMR and other biophysical methods.32 Our hits include a benzimidazole scaffold that displays competitive binding to the K4me pocket, which could serve as an attractive template for further chemical development. This paves the way toward the discovery of lead inhibitors of the Pygo–BCL9 complex that block its binding to methylated histone H3 tail or that destabilize the PHD–HD1 interaction itself, which should prevent it from enabling oncogenic β-catenin to operate transcriptional switches that drive cancer.
Methods
In Silico Screening
Four successive rounds of virtual screening based on the PHD–HD1 crystal structures of both human paralog complexes8,10 were conducted, with various strategies for docking and shortlisting as detailed in the Supporting Information Supplementary Methods.
Protein Purification
For crystallography, hPygo2 PHD (amino acids 327–387) linked by GSGSGSGS to hB9L HD1 (amino acids 235–263) was fused to a 6xHis-tag separated by a TEV cleavage site. Expression was done in E. coli BL21-CodonPlus(DE3)-RIL cells (Stratagene), and the PHD–HD1 complex was purified by Ni-NTA resin and TEV cleavage, followed by size exclusion chromatography. For some of the NMR validation experiments, a modified version of PHD–HD1 was used (PHD–HD1ATAE, bearing two mutations K384A and K386A, to avoid intermolecular pseudoligand interactions by these residues),8,10 but this behaved indistinguishably from wild-type PHD–HD1.
NMR Spectroscopy
All NMR samples were prepared in aqueous buffer containing 25 mM phosphate, pH 6.7, and 150 mM NaCl. Spectra were recorded on one of several Bruker spectrometers operating at 500–800 MHz 1H frequency and equipped with cryogenic inverse probes. Spectra were processed with TopSpin (Bruker) and analyzed using Sparky version 3.110 (Goddard and Kneller, UCSF).
Fragment Screening by NMR
{1H,15N}-Fast-HSQC spectra41 were obtained for 30 μM protein and five ligands at 1 mM each, with a digital resolution of 3.7 and 4.6 Hz/point in f2 and f1, respectively. Compound pools were selected to avoid 1H resonance overlap between the different components, to allow validation and deconvolution by ligand-observed methods. Compounds were dissolved in DMSO-d6 at 100 mM (resulting in a final DMSO concentration of 5% v/v in the NMR sample). Ligand-observed NMR spectra were recorded on a Bruker 500 MHz DRX spectrometer, with a sample temperature of 25 °C and a protein concentration of 10 μM. WaterLOGSY spectra42 were acquired with 4096 points, a 6 kHz spectral width, 25 ms 3-Gaussian 180° water selection pulse, 0.9 s NOE mixing time, 2.5 s relaxation delay, 512 scans, and a T1ρ filter (50 ms square pulse with 2.2 kHz B1 field) to suppress signals from the protein. Saturation transfer difference spectra were acquired using a pseudo-2D pulse sequence (unmodified Bruker pulse program stddiffesgp.3), 16k points, 8 kHz spectral width, 96 scans, interleaving on-resonance (−0.2 ppm) or off-resonance (25.2 ppm) presaturation (repeating 50 ms 1% truncated Gaussian pulses with 105 Hz B1 field) throughout the 7.0 s recycle delay, and a 15 ms T1ρ trim pulse (square pulse, 5.8 kHz B1). Ligand-observed deconvolution of pots was attempted with MBP-tagged PHD–HD1 (with increased molecular weight), but these proved unsuccessful, yielding results that were not reliably validated by subsequent protein-observed experiments.
Recording and Assignments of NOEs
Complete backbone and side-chain resonance assignments were obtained for a complex containing 500 μM 13C–15N double-labeled PHD–HD1 and 5 mM CF16, from standard triple resonance correlation spectra (HNCACB, CBCA(CO)NH, HN(CA)CO, HNCO, (H)CC(CO)NH, H(CCCO)NH, using unmodified Bruker pulse programs), and were used to assign a 2D {1H,13C}-HSQC of a separate sample in D2O buffer. Aromatic side-chain 1H resonances were assigned from 2D (HB)CB(CGCD)HD and (HB)CB(CGCDCE)HE spectra, and the tryptophan side-chain 1H resonances were assigned from an unfiltered 2D H–H NOESY (150 ms NOE mix, 800 MHz 1H). 2D ω1-13C-filtered-ω2-13C-edited H–H NOESY spectra, with X half-filters set to accept only cross-peaks between 12C-coupled 1H- and 13C-coupled 1H, were acquired at 800 MHz for the sample in a D2O buffer, with an NOE mixing time of 250 ms. A total of 128 complex points were recorded in t1, expanded to 256 by linear prediction, to yield a digital resolution of 8.6 and 2.7 Hz/point in f1 and f2, respectively.
HADDOCK Calculations
Docking simulations were performed with HADDOCK version 2.139 linked to CNS version 1.3.43 CNS topology parameters for CF16 were generated using the PRODRG server,44 and partial charges were assigned from MOPAC semiempirical calculations using the PM7 Hamiltonian. The starting coordinates for PHD–HD1 were taken from 4UP0, with hydrogen atoms added (using PyMol version 1.6). Unambiguous NOE distance restraints were each applied as a symmetric biharmonic potential without penalty in the distance range 1.8–3.8 Å, 1.8–4.6 Å, or 1.8–5.7 Å, according to the intensity of the NOE correlation (note that the tightening of these ranges by 1 Å each resulted in essentially the same models as shown in Figure 6A). For the final models, 200 structures were refined with explicit water, all of which occupied a single cluster.
X-ray Crystallography
Concentrated protein was mixed with CF4 (100 mM in DMSO), or with 15-mer H3K4me2K9ac, to obtain final solutions containing 9.1 mg mL–1 PHD–HD1, 20% (v/v) DMSO and 20 mM CF4, or 9 mg mL–1 PHD–HD1 and 5 mM 15-mer, respectively. Solutions were cleared by centrifugation at 100 000 g for 20 min prior to crystallization as described8 (initial screen of >1500 different crystallization conditions in 100 nL drops in a 96-well sitting-drop format). Crystals emerged under multiple conditions after several days at 19 °C using the vapor diffusion method and were cryoprotected by perfluoropolyether (PHD–HD1-CF4) or 30% (w/v) glucose in the mother liquor (PHD–HD1–H3K4me2K9ac) before flash-cooling in liquid nitrogen. X-ray diffraction data were collected at the Diamond synchrotron I04 or ESRF synchrotron ID29 beamlines, from crystals grown in 60% (w/v) tacsimate (pH 7.0; PHD–HD1–CF4) or in 1 M sodium citrate, 0.1 M Tris (pH 7), 0.2 M NaCl (PHD–HD1–H3K4me2K9ac), and the data were processed as described in the Supporting Information Supplementary Methods, using molecular replacement with Phaser45 based on 2XB18,10 (see Table S3, for refinement statistics). Structural images were drawn with PyMol.
Acknowledgments
We thank J. Sutherland for helpful discussions, R. Williams and R. Wilcken for technical advice and critical comments on the manuscript, and the staff at Diamond Light Source and at the ESRF for support during data collection at the beamlines I04 (Diamond) and ID29 (ESRF). This work was supported by the Medical Research Council (U105192713) and by Cancer Research UK (grant no. C7379/A8709 to M.B.).
Supporting Information Available
Supplementary methods, Figures S1–S7, Tables S1–S3, and supplementary references. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
Coordinates and structure factors have been deposited with the Protein Data Bank under accession codes 4UP0 (PHD–HD1-H3K4me2K9ac) and 4UP5 (PHD–HD1-CF4).
Author Present Address
§ EMBL, Meyerhofstraße 1, 69117 Heidelberg, Germany
Author Contributions
Experiments were designed, performed, and interpreted by T.C.R.M. (crystallography, NMR spectroscopy, SAR analysis), M.F. (NMR screening), T.J.R. (NMR screening and spectroscopy, HADDOCK modeling), and K.B. and J.C. (virtual screening); study design, supervision, and conclusions were done by M.B. and M.F.; M.B. drafted the manuscript.
Author Contributions
∥ These authors contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Polakis P. (2012) Wnt signaling in cancer. Cold Spring Harbor Perspect. Biol. 4, a008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienz M.; Clevers H. (2000) Linking colorectal cancer to Wnt signaling. Cell 103, 311–320. [DOI] [PubMed] [Google Scholar]
- Clevers H.; Nusse R. (2012) Wnt/β-Catenin Signaling and Disease. Cell 149, 1192–1205. [DOI] [PubMed] [Google Scholar]
- Barker N.; Ridgway R. A.; van Es J. H.; van de Wetering M.; Begthel H.; van den Born M.; Danenberg E.; Clarke A. R.; Sansom O. J.; Clevers H. (2009) Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 457, 608–611. [DOI] [PubMed] [Google Scholar]
- Polakis P. (2012) Drugging Wnt signalling in cancer. EMBO J. 31, 2737–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels D. L.; Eklof Spink K.; Weis W. I. (2001) β-catenin: molecular plasticity and drug design. Trends Biochem. Sci. 26, 672–678. [DOI] [PubMed] [Google Scholar]
- Sampietro J.; Dahlberg C. L.; Cho U. S.; Hinds T. R.; Kimelman D.; Xu W. (2006) Crystal structure of a β-catenin/BCL9/Tcf4 complex. Mol. Cell 24, 293–300. [DOI] [PubMed] [Google Scholar]
- Fiedler M.; Sanchez-Barrena M.; Nekrasov M.; Mieszczanek J.; Rybin V.; Muller J.; Evans P.; Bienz M. (2008) Decoding of methylated histone H3 tail by the Pygo-BCL9 Wnt signaling complex. Mol. Cell 30, 507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu B.; Sun P.; Yuan Y.; Moraes R. C.; Li A.; Teng A.; Agrawal A.; Rheaume C.; Bilanchone V.; Veltmaat J. M.; Takemaru K.; Millar S.; Lee E. Y.; Lewis M. T.; Li B.; Dai X. (2009) Pygo2 expands mammary progenitor cells by facilitating histone H3 K4 methylation. J. Cell Biol. 185, 811–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller T. C.; Rutherford T. J.; Johnson C. M.; Fiedler M.; Bienz M. (2010) Allosteric remodelling of the histone H3 binding pocket in the Pygo2 PHD finger triggered by its binding to the B9L/BCL9 co-factor. J. Mol. Biol. 401, 969–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson B.; Townsley F.; Rosin-Arbesfeld R.; Musisi H.; Bienz M. (2002) A new nuclear component of the Wnt signalling pathway. Nat. Cell Biol. 4, 367–373. [DOI] [PubMed] [Google Scholar]
- Adachi S.; Jigami T.; Yasui T.; Nakano T.; Ohwada S.; Omori Y.; Sugano S.; Ohkawara B.; Shibuya H.; Nakamura T.; Akiyama T. (2004) Role of a BCL9-related β-catenin-binding protein, B9L, in tumorigenesis induced by aberrant activation of Wnt signaling. Cancer Res. 64, 8496–8501. [DOI] [PubMed] [Google Scholar]
- Brembeck F. H.; Schwarz-Romond T.; Bakkers J.; Wilhelm S.; Hammerschmidt M.; Birchmeier W. (2004) Essential role of BCL9–2 in the switch between β-catenin’s adhesive and transcriptional functions. Genes Dev. 18, 2225–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Roche M.; Worm J.; Bienz M. (2008) The function of BCL9 in Wnt/β-catenin signaling and colorectal cancer cells. BMC Cancer 8, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deka J.; Wiedemann N.; Anderle P.; Murphy-Seiler F.; Bultinck J.; Eyckerman S.; Stehle J. C.; Andre S.; Vilain N.; Zilian O.; Robine S.; Delorenzi M.; Basler K.; Aguet M. (2010) Bcl9/Bcl9l are critical for Wnt-mediated regulation of stem cell traits in colon epithelium and adenocarcinomas. Cancer Res. 70, 6619–6628. [DOI] [PubMed] [Google Scholar]
- Watanabe K.; Fallahi M.; Dai X. (2014) Chromatin effector Pygo2 regulates mammary tumor initiation and heterogeneity in MMTV-Wnt1 mice. Oncogene 33, 632–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Roche M.; Rutherford T. J.; Gupta D.; Veprintsev D. B.; Saxty B.; Freund S. M.; Bienz M. (2012) An intrinsically labile α-helix abutting the BCL9-binding site of β-catenin is required for its inhibition by carnosic acid. Nat. Commun. 3, 680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Roche M.; Ibrahim A. E.; Mieszczanek J.; Bienz M. (2014) LEF1 and B9L shield β-catenin from inactivation by Axin, desensitizing colorectal cancer cells to tankyrase inhibitors. Cancer Res. 74, 1495–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada K.; Zhu D.; Bird G. H.; Sukhdeo K.; Zhao J. J.; Mani M.; Lemieux M.; Carrasco D. E.; Ryan J.; Horst D.; Fulciniti M.; Munshi N. C.; Xu W.; Kung A. L.; Shivdasani R. A.; Walensky L. D.; Carrasco D. R. (2012) Targeted disruption of the BCL9/β-catenin complex inhibits oncogenic Wnt signaling. Sci. Transl. Med. 4, 148ra117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edfeldt F. N.; Folmer R. H.; Breeze A. L. (2011) Fragment screening to predict druggability (ligandability) and lead discovery success. Drug Discovery Today 16, 284–287. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Knapp S. (2014) Targeting bromodomains: epigenetic readers of lysine acetylation. Nat. Rev. Drug Discovery 13, 337–356. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Qi J.; Picaud S.; Shen Y.; Smith W. B.; Fedorov O.; Morse E. M.; Keates T.; Hickman T. T.; Felletar I.; Philpott M.; Munro S.; McKeown M. R.; Wang Y.; Christie A. L.; West N.; Cameron M. J.; Schwartz B.; Heightman T. D.; La Thangue N.; French C. A.; Wiest O.; Kung A. L.; Knapp S.; Bradner J. E. (2010) Selective inhibition of BET bromodomains. Nature 468, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James L. I.; Barsyte-Lovejoy D.; Zhong N.; Krichevsky L.; Korboukh V. K.; Herold J. M.; MacNevin C. J.; Norris J. L.; Sagum C. A.; Tempel W.; Marcon E.; Guo H.; Gao C.; Huang X. P.; Duan S.; Emili A.; Greenblatt J. F.; Kireev D. B.; Jin J.; Janzen W. P.; Brown P. J.; Bedford M. T.; Arrowsmith C. H.; Frye S. V. (2013) Discovery of a chemical probe for the L3MBTL3 methyllysine reader domain. Nat. Chem. Biol. 9, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simhadri C.; Daze K. D.; Douglas S. F.; Quon T. T.; Dev A.; Gignac M. C.; Peng F.; Heller M.; Boulanger M. J.; Wulff J. E.; Hof F. (2014) Chromodomain antagonists that target the Polycomb-Group methyllysine reader protein Chromobox Homolog 7 (CBX7). J. Med. Chem. 57, 2874–2883. [DOI] [PubMed] [Google Scholar]
- Wagner E. K.; Nath N.; Flemming R.; Feltenberger J. B.; Denu J. M. (2012) Identification and characterization of small molecule inhibitors of a plant homeodomain finger. Biochemistry 51, 8293–8306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller T. C.; Mieszczanek J.; Sanchez-Barrena M.; Rutherford T. J.; Fiedler M.; Bienz M. (2013) Evolutionary adaptation of the fly Pygo PHD finger towards recognizing histone H3 tail methylated at arginine 2. Structure 21, 2208–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y.; Umehara T.; Hamana H.; Hayashizaki Y.; Inoue M.; Kigawa T.; Shirouzu M.; Terada T.; Tanaka A.; Padmanabhan B.; Yokoyama S. (2007) Crystal structure analysis of the PHD domain of the transcription co-activator Pygopus. J. Mol. Biol. 370, 80–92. [DOI] [PubMed] [Google Scholar]
- Pena P. V.; Davrazou F.; Shi X.; Walter K. L.; Verkhusha V. V.; Gozani O.; Zhao R.; Kutateladze T. G. (2006) Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature 442, 100–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna S. D.; Li H.; Ruthenburg A. J.; Allis C. D.; Patel D. J. (2007) How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 14, 1025–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajduk P. J.; Greer J. (2007) A decade of fragment-based drug design: strategic advances and lessons learned. Nat. Rev. Drug Discovery 6, 211–219. [DOI] [PubMed] [Google Scholar]
- Murray C. W.; Rees D. C. (2009) The rise of fragment-based drug discovery. Nat. Chem. 1, 187–192. [DOI] [PubMed] [Google Scholar]
- Silvestre H. L.; Blundell T. L.; Abell C.; Ciulli A. (2013) Integrated biophysical approach to fragment screening and validation for fragment-based lead discovery. Proc. Natl. Acad. Sci. U. S. A. 110, 12984–12989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajduk P. J.; Huth J. R.; Fesik S. W. (2005) Druggability indices for protein targets derived from NMR-based screening data. J. Med. Chem. 48, 2518–2525. [DOI] [PubMed] [Google Scholar]
- Basse N.; Kaar J. L.; Settanni G.; Joerger A. C.; Rutherford T. J.; Fersht A. R. (2010) Toward the rational design of p53-stabilizing drugs: probing the surface of the oncogenic Y220C mutant. Chem. Biol. 17, 46–56. [DOI] [PubMed] [Google Scholar]
- Welsch M. E.; Snyder S. A.; Stockwell B. R. (2010) Privileged scaffolds for library design and drug discovery. Curr. Opin. Chem. Biol. 14, 347–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barelier S.; Krimm I. (2011) Ligand specificity, privileged substructures and protein druggability from fragment-based screening. Curr. Opin. Chem. Biol. 15, 469–474. [DOI] [PubMed] [Google Scholar]
- Duarte C. D.; Barreiro E. J.; Fraga C. A. (2007) Privileged structures: a useful concept for the rational design of new lead drug candidates. Mini Rev. Med. Chem. 7, 1108–1119. [DOI] [PubMed] [Google Scholar]
- Smith S. G.; Sanchez R.; Zhou M. M. (2014) Privileged Diazepine Compounds and Their Emergence as Bromodomain Inhibitors. Chem. Biol. 21, 573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez C.; Boelens R.; Bonvin A. M. (2003) HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125, 1731–1737. [DOI] [PubMed] [Google Scholar]
- Ramon-Maiques S.; Kuo A. J.; Carney D.; Matthews A. G.; Oettinger M. A.; Gozani O.; Yang W. (2007) The plant homeodomain finger of RAG2 recognizes histone H3 methylated at both lysine-4 and arginine-2. Proc. Natl. Acad. Sci. U. S. A. 104, 18993–18998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori S.; Abeygunawardana C.; Johnson M. O.; van Zijl P. C. (1995) Improved sensitivity of HSQC spectra of exchanging protons at short interscan delays using a new fast HSQC (FHSQC) detection scheme that avoids water saturation. J. Magn. Reson. B 108, 94–98. [DOI] [PubMed] [Google Scholar]
- Dalvit C.; Fogliatto G.; Stewart A.; Veronesi M.; Stockman B. (2001) WaterLOGSY as a method for primary NMR screening: practical aspects and range of applicability. J. Biomol. NMR 21, 349–359. [DOI] [PubMed] [Google Scholar]
- Brunger A. T.; Adams P. D.; Clore G. M.; DeLano W. L.; Gros P.; Grosse-Kunstleve R. W.; Jiang J. S.; Kuszewski J.; Nilges M.; Pannu N. S.; Read R. J.; Rice L. M.; Simonson T.; Warren G. L. (1998) Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr., Sect. D: Biol. Crystallogr. 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Schuttelkopf A. W.; van Aalten D. M. (2004) PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 1355–1363. [DOI] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Storoni L. C.; Read R. J. (2005) Likelihood-enhanced fast translation functions. Acta Crystallogr., Sect. D: Biol. Crystallogr. 61, 458–464. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.