

Abstract
Besides single-nucleotide variants in the human genome, large-scale genomic variants, such as copy number variations (CNVs), are being increasingly discovered as a genetic source of human diversity and the pathogenic factors of diseases. Recent experimental findings have shed light on the links between different genome architectures and CNV mutagenesis. In this review, we summarize various genomic features and discuss their contributions to CNV formation. Genomic repeats, including both low-copy and high-copy repeats, play important roles in CNV instability, which was initially known as DNA recombination events. Furthermore, it has been found that human genomic repeats can also induce DNA replication errors and consequently result in CNV mutations. Some recent studies showed that DNA replication timing, which reflects the high-order information of genomic organization, is involved in human CNV mutations. Our review highlights that genome architecture, from DNA sequence to high-order genomic organization, is an important molecular factor in CNV mutagenesis and human genomic instability.
Keywords: DNA copy number variations, DNA replication, genetic recombination, genomic instability
Introduction
Genetic mutations have been known as one of the key factors in the pathogenesis of human diseases. Besides the well-known single-nucleotide variants, it has been shown that the large-scale genomic variants also make a great contribution to human health. 'Genomic disorders' are human diseases caused by relatively large genomic rearrangements [1]. Such large-scale genomic variants (named copy number variation [CNV]) can also be frequent in human populations [2, 3]. CNV involves DNA segments larger than 1 kb and exhibits variable copy numbers among individuals, comprising deletions and duplications/insertions [4, 5]. In the past 10 years, CNV has been found to play an important role in both sporadic Mendelian disorders and complex diseases. Previous studies have reported that CNV can be mediated by multiple molecular mechanisms involving various genomic features. Here, we focus on CNV mutagenesis and review the involvement of human genome architecture in CNV instability and the underlying molecular mechanisms.
Non-allelic Homologous Recombination between Human Genomic Repeats
Genomic disorders and low-copy repeats
Large-scale genomic changes in the human genome can be associated with human diseases. Such clinical conditions resulting from human genome architecture are termed 'genomic disorders' [1]. The structural features, such as genomic repeats, can provide substrates for homologous recombination and induce genomic rearrangements and genomic disorders.
Stankiewicz and Lupski [6] defined region-specific lowcopy repeats (LCRs) as paralogous genomic segments spanning 10-400 kb of genomic DNA and sharing ≥95%-97% sequence identity. The non-allelic homologous recombination (NAHR) between directly oriented LCRs can generate microdeletions and microduplications of megabases in size, which are frequently associated with genomic disorders (Fig. 1). For example, the 22q11.2 deletion syndrome is a well-investigated disorder caused by microdeletions between the paired LCRs in human 22q11.2, which deletes one copy of TBX1, CRKL, MAPK1, and several additional genes [7, 8, 9, 10]. In addition to microdeletions, microduplications can also manifest as genomic disorders. The 1.4-Mb microduplication involving the PMP22 gene in human 17p12 can lead to CMT1A, which is a classical model for disease resulting from gene dosage effects [11, 12].
Fig. 1.
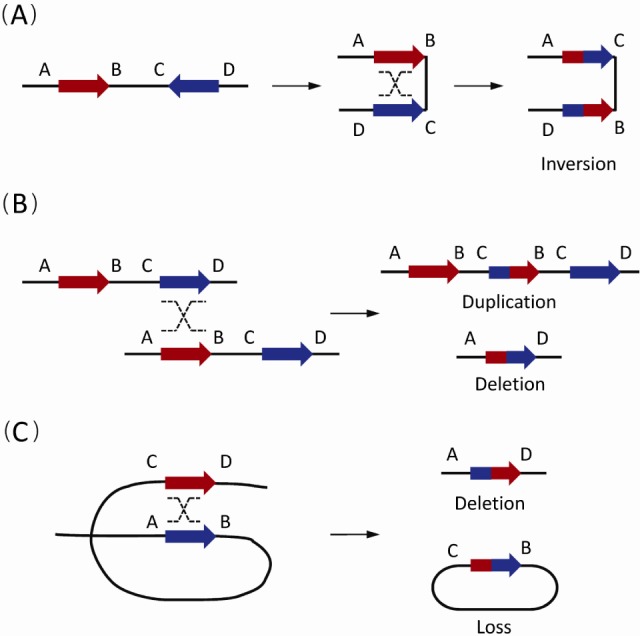
The non-allelic homologous recombination (NAHR) events between paired low-copy repeats (LCRs)/segmental duplications (SDs) [1]. Paired LCRs/SDs are depicted as bold arrows (red and blue) with the orientation indicated by arrowheads. Capital letters near the LCRs/SDs refer to the flanking unique sequences, while the same letter on different lines indicates the homologues on the other strand. Dashed crossed lines represent a homologous recombination event. (A) The NAHR event between reversely oriented LCRs/SDs can cause inversion, a copy-neutral structural variation. (B) The inter-chromatid NAHR events between directly oriented LCRs/SDs result in deletions and duplications. (C) The intra-chromatid NAHR events between directly oriented LCRs/SDs can generate deletions and ring-shaped DNA segments that will be lost in subsequent cell divisions.
Segmental duplication and NAHR
Genomic repeats play a significant role in human evolution and have a strong association with genomic CNVs [6, 13, 14, 15]. In 2001, Eichler [16] initially conducted a systematically bioinformatic analysis for such low-copy genomic repeats and defined them as segmental duplications (SDs), which have a high degree of sequence identity (>90%-95%) and large genomic sizes (1-100 kb). After that, Bailey et al. [17] further performed a whole-genome assembly comparison to detect SDs with pair-wise alignments ≥ 90% and ≥ 1 kb in the human genome. In addition to human SDs, the subsequent analyses also identified the SD architecture in the genomes of other primates, including chimpanzee, gorilla, and orangutan, and even in the mouse genome [17, 18, 19, 20], all of which have been archived in the online database of the University of California Santa Cruz (UCSC) Genome Browser (http://genome.ucsc.edu/).
LCR/SD is a very important category of DNA architecture in the human genome. It has been found that they are associated with duplicated genes and pseudogenes [21], co-localize and overlap with Alu elements and CNV [22, 23], and play an important role in genome evolution [24, 25, 26]. LCR/SD pairs, acting as substrates, are thought to be a key factor in triggering NAHR events and causing CNV mutations [4, 27, 28, 29, 30, 31, 32].
Generally, reversely oriented SDs can align and subsequently crossover with each other via NAHR, resulting in copy-neutral inversions of the flanking DNA fragments (Fig. 1A). Similarly, NAHR events between direct SD pairs can cause CNVs. Based on the different positions of SD pairs, different types of CNVs occur. Duplications or deletions take place in NAHR events between different chromatids (interchromatid) (Fig. 1B), while only deletions occur in NAHR on the same chromatid (intra-chromatid) (Fig. 1C).
Based on the observations in specific pathogenic loci, SD properties (including homology length, distance, and sequence similarity) were shown to affect the incidence of NAHR [33]. In a recent study on common CNVs in human populations, it was found that SD length and inter-SD distance were the major SD properties involved in NAHR frequency [34]. A model of chromosomal compression/extension/looping has also been proposed for homology mispairing in NAHR [34].
High-copy repeats in CNV instability
The genomic repeats representing DNA primary structures can be divided into LCR/SD and high-copy repeats. Compared to LCR/SD, high-copy repeats constitute a great portion of the human genome. Interspersed repeats are the most common type of high-copy repeats, which cover over 44% of the human genome [35]. One of the major classes of interspersed repeats is the retrotransposon, including short interspersed elements (SINEs), long interspersed elements (LINEs), and endogenous retroviruses (ERVs).
SINEs are short DNA sequences (100-400 bp in length) with an internal (mobile) polymerase III promoter [15], making up about 11% of the human genome [36]. The most common SINEs are Alu elements, which burst out in the evolution of primates [37]. Moreover, Alu elements play an important role in disease, such as breast cancer, Ewing's sarcoma, familial hypercholesterolemia, and so on [38]. In 2008, Kim et al. [22] found a strong association between Alu elements and old SDs. By means of NAHR, Alu elements contribute to the formation of CNVs, especially deletions [39, 40, 41, 42, 43].
The most classic repeats in LINEs are the LINE-1 (L1) elements, which are 6-8 kb in length [44]. Covering about 17% of the human genome, L1 elements can elevate genomic instability, provide resources for NAHR [39, 40, 41, 45, 46], and cause human diseases [47, 48, 49, 50].
In the human genome, there exist at least 50,000 copies of ERVs [51], which are defined as human endogenous retroviruses (HERVs), covering about 4.9% of human DNA sequences [21]. Via the NAHR mechanism, HERVs were found to induce large deletions and cause hypotonia and motor, language, and cognitive delays [52, 53]. Intriguingly, a series of studies show that there is a strong association between HERVs and CNVs in the region of AZFa, a well-known locus related to male infertility [54, 55, 56, 57].
Repeat-Induced DNA Replication Error and CNV Mutation
As discussed above, repeat-mediated NAHR is one of the major mutational mechanisms for CNV formations. In these recombination-based events, paired repeats in direct orientation contribute to CNV instability and disease traits. However, is it the only way for the genomic repeats to induce CNV mutations? The recent investigation of DNA replication-based mechanisms provides novel insights into repeat-mediated CNV instability.
Inverted repeats involved in CNV instability
Genomic repeats, especially inverted repeats (IRs), can cause DNA replication error and induce CNV formation. IRs, sharing high sequence similarity in adjacent loci, are found to align or crossover with each other and form specific DNA secondary structures, such as cruciform structures, during DNA replication [58]. Formation of such secondary structures can cause DNA replication fork stalling, and later, jumping into the wrong locus to continue replicating (Fig. 2). This mechanism of triggering replication errors subsequently results in genomic rearrangements and CNV mutations [24].
Fig. 2.
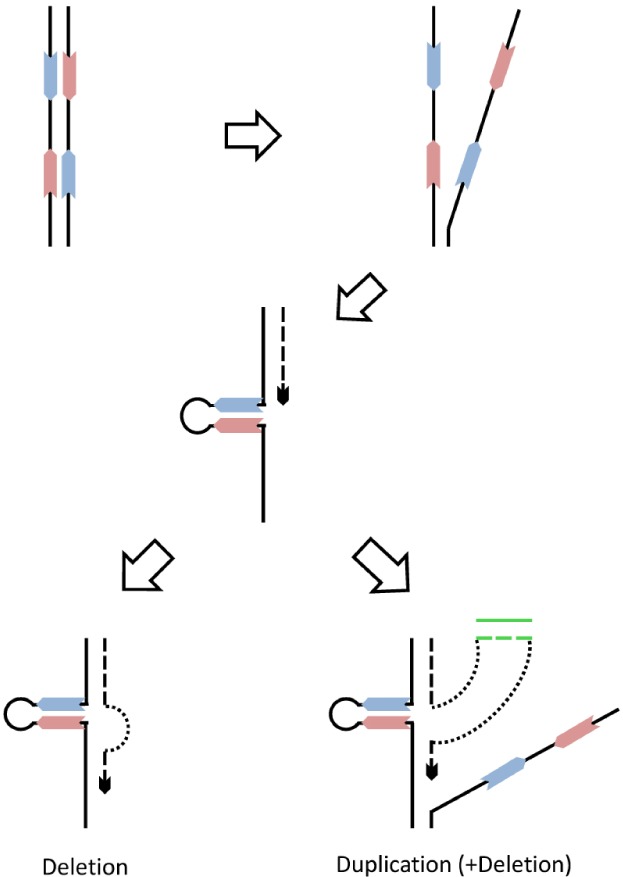
Repeat-induced DNA replication errors and copy number variation (CNV) formation. The straight lines depict single DNA strands, and the solid arrows (red and blue) represent genomic repeats. The dashed lines indicate newly synthesized DNA strands. During DNA replication, adjacent repeats could form DNA secondary structures (such as hairpin) that consequently result in replication fork stalling. Then, CNVs are generated via DNA template switching. For example, (1) jumping over the secondary structures and restarting DNA replication lead to deletions and (2) switching to a new template (shown in green lines) and switching back result in duplications of the green DNA segment.
IRs induce complex CNVs by replication errors
During DNA replication, IRs could form DNA secondary structures, which will induce replication fork stalling. Template switching and replication resumption further result in CNV mutations (Fig. 2). Notably, replication-associated events usually lead to complex CNVs, which include the combined segments of deletions and duplications. As was reported, Chen et al. [59] identified three complex CNVs that could be explained by a model of serial replication slippage (SRS). In this model, IRs have the potential to induce SRS and cause CNV mutations.
IRs can induce complex CNVs, as observed in the MECP2 locus in chromosome Xq28 and the PLP1 locus in chromosome Xq22 [60]. To elucidate the mechanisms of complex CNVs in the PLP1 locus, Hastings et al. [61] found both microhomology and IRs at the breakpoints. They proposed that both breakage of replication forks and the IR-mediated aberrant repair process can result in complex CNVs. This model was termed 'microhomology-mediated break-induced replication,' which was used to explain the formation of the complex CNVs involving individual genes or even single exons [62].
Self-chains in CNV formation
The aforementioned SDs are long (>1 kb) and LCRs in the human genome. Besides SDs, self-chains (SCs) are another type of short LCRs, which were previously analyzed and mapped via self-alignment in the human genome utilizing BLASTZ [63, 64]. SCs are short in length (91% of which range from 150 bp to 1 kb in size) [14]. Furthermore, SCs have a limited number of matched alignments in the human genome. Thus, SCs represent a distinct category of human short LCRs.
In 2013, Chen et al. [65] studied deletion CNVs in the NRXN1 gene and its flanking regions. After mapping and analyzing the breakpoints of 32 deletions, they found a significant bias that minus SCs (i.e., paired SCs in the inverted orientation) were overrepresented in the vicinity of deletion breakpoints in the NRXN1 region. Furthermore, they claimed that the SCs can increase genomic instability and cause deletions via DNA replication errors. Their work contributes to the exploration on SC-mediated CNVs.
To perform a genomewide analysis on the contribution of SCs to human CNV instability, Zhou et al. [14] plotted the numbers of SC regions with different orientations in the entire human genome. After masking the SDs and gaps in the human genome, utilizing the germline CNVs in human populations and the somatic CNVs in various cancer genomes, they observed a significant biased distribution of CNV breakpoints to SC regions, which indicated that SC-mediated secondary structures may induce DNA replication errors and potentially generate different types of CNVs, such as deletions and duplications. In this case, SCs represent a new genomic architecture for the underlying regional susceptibility to genomic instability, further giving rise to CNVs.
DNA repair and nonhomologous end-joining
While DNA double-strand breaks (DSBs) occur, nonhomologous end-joining (NHEJ) is one of the molecular mechanisms for repairing DSBs and maintaining genome integrity. Once a DSB is detected, the broken DNA ends are bridged and modified by the enzyme machinery. After that, the final ligation is needed for DNA repair. Unlike NAHR, NHEJ can take place without any homology as the substrate. Notably, deletions or insertions of several base pairs are usually brought to the joint point. More mechanistic details of NHEJ are provided in some previous works [66, 67, 68].
DNA Replication Dynamics in CNV Mutagenesis
In addition to the aforementioned genomic features, some high-order genome organizations might contribute to genome instability. New observations in the human genome showed that the DNA mutation rate is associated with DNA replication timing. Stamatoyannopoulos et al. [69] found that the human point mutation rate is markedly increased in genomic regions of late replication. This correlation indicates that DNA replication timing, as an important feature of replication dynamics, is involved in genomic instability and enlightens the investigation on the relationship of replication timing and CNV instability.
Replication timing as a high-order genomic feature
DNA replication takes place at replication forks following a fixed way [70]. In the human genome, the segments of chromosomes replicate in a temporal order [71], and the whole genome is spatially segregated by replication zones of different organizations. With some replicons in one spatial compartmentalization of chromatin fired synchronously, this chromosomal unit shares the same replication timing, termed the 'replication domain.' Therefore, the genome consists of several replication domains with different replication timing and the timing transition regions.
Replication timing can be measured by two distinct methods, based on current genome technologies [72, 73, 74]. One method is to label the newly replicated DNA with chemically tagged nucleotides. Then, the DNA will be isolated from cells at various times during S phase by immune-precipitation or density fractionation. In the other method, since DNA segments that replicate earlier accumulate more copies than those that replicate late in most cells-the DNA content of a region simply reflects the replication timing. After being classified by florescence-activated cell sorting, the DNAs extracted from S phase and G1 phase cells, respectively, are compared by next-generation sequencing or microarray technologies. By either way, a replication timing profile can be generated (Fig. 3) [75].
Fig. 3.
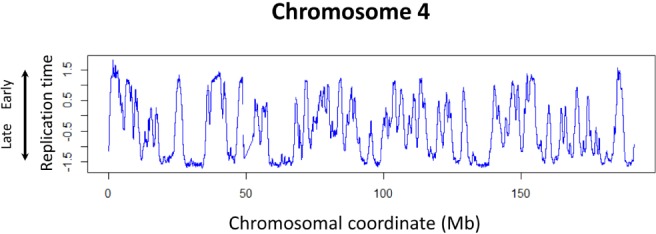
The DNA replication timing profile of human lymphoblastoid cells. The data of human chromosome 4, which were obtained from Koren et al. [75], are shown. The blue lines show the replication timing, high values of which indicate that DNA replicates early in these regions, and vice versa.
Based on the timing profiles, a lot of progress has been made on understanding the replication program and its relationship with other genome architectures. Recent findings indicate significant links between replication timing and the features of primary genomic structures [76]. The genomic regions where DNA replicates earlier usually have more genes, fewer LINEs, and higher GC content [77, 78]. Moreover, it is noticed that DNA replication timing correlates with transcription [79, 80, 81]. Expressed genes replicate earlier, while repressed genes replicate late. Although this correlation shows a discrepancy between multicellular and single-celled organisms, it is worth noting that such works indeed reveal the striking association of replication timing and transcriptional activity in humans [77, 82, 83]. Moreover, recent findings show that replication timing strongly correlates with three-dimensional chromatin structures [84]. In Hi-C data, it has been observed that chromatin is organized into two separate compartments. Remarkably, DNA that resides in close spatial structures replicates in near time, and chromatin that interacts between two compartments is exactly at the timing transition regions. This observation suggests replication timing as an independent advanced genomic feature.
Replication timing and CNV instability in human populations and cancers
The relationship of DNA replication timing and genomic instability, which is involved in genomic mutation and human disease, is what people are most concerned about. As mentioned above, human mutation rates, based on evolutionary divergence and single-nucleotide polymorphism frequency, are increased in late-replicated regions [69]. Koren et al. [75] generated a high-resolution timing profile of the human genome and investigated the relationship between DNA replication timing and point mutations. In accordance with the previous discovery, this association was also observed and proved to be much stronger.
How is CNV related to replication timing? Recent studies showed some distinct but multi-dimensional relationships between CNVs and replication timing. Based on the duplication hotspots conserved between two species of Drosophila, Cardoso-Moreira et al. [85] explored the roles of replication timing in genomic instability. They found that Drosophila duplication hotspots were enriched in late-replicated regions, unlike the aforementioned sequences of high sequence identity in the human genome. However, in spite of the association observed in Drosophila, the situation seems to be more complicated in mammalian genomes. In the study of Koren et al. [75], the relationship between early/late replication timing and CNV mutation was also investigated. The CNVs, mediated by different mechanisms, showed divergent patterns, suggesting a multi-dimensional interaction between CNVs and replication timing.
In addition to the observations in human populations, recent findings have also discovered the relationship of genomic reorganization and the subsequently generated genetic variation during cell fate changes. Lu et al. [86] have investigated the impact of altered replication timing on the CNV landscape during reprogramming. Approximately 40% of the human genome changes with regard to replication timing between human induced pluripotent stem cells (iPSCs) and their parent fibroblasts. Intriguingly, the CNV distribution shows a correlation with the changed timing profile. In particular, CNV gains tend to be located in the genomic regions that switch to replicate earlier. This correlation is conserved among different reprogramming methods.
Compared with cell fate changes, replication timing is disrupted in many disease states, including cancer [87]. It has been noticed that numerous alterations to the replication program take place during carcinogenesis. One of the changes is the aberrant asynchronous replication of loci that replicate synchronously in normal cells. This phenomenon exists in not only cancer but also noncancerous cells [88, 89, 90]. This abnormal replication program apparently has a notable impact on genomic stability and thus increases the frequency of chromosomal rearrangements and CNVs. Recent findings have indicated that aberrant DNA replication timing is involved in changes in gene expression, epigenetic modifications, and an increased CNV mutation frequency [91, 92]. An analysis of 331,724 somatic copy number alterations (SCNAs) has shown that SCNAs increase in late-replicating regions among cells of different cancer types. Like the findings in iPSCs, the SCNA distribution is related to replication timing in tumor cells. In particular, amplification boundaries tend to be located in early-replicated regions, whereas deletion boundaries are more likely to reside in late-replicated regions [93].
Integrated replication dynamic related with CNV instability
In the study of Koren et al. [75], point mutations and CNVs showed different patterns in their correlations with replication timing. These observations may reflect the distinct mutational mechanisms between these two types of genomic variants and suggest complex effects of DNA replication on CNV instability. We hypothesized that integrated replication dynamics, which are not just early/late replication timing, contribute to CNV mutation. It has been reported that dividing the genome into early/late replication timing alone does not give the entire characteristics of DNA replication fork dynamics [72]. Actually, the timing transitional regions represent the interactions of two spatially dependent chromatin compartments, which are DNA segments with low rates of replication fork progression. Notably, slower fork speed and increased fork stalling have been found to be associated with cancer cells and result in CNV mutations [94]. Actually, Chen et al. [95] have conducted a statistical method, estimating replication dynamics, and observed its significant association with CNV instability. Replication dynamics may be used as a measure of the progress of genome replication and regional replication stress and provides novel insights into the roles of DNA replication in CNV mutagenesis.
Conclusion
Human genomic repeats play an important role in CNV mutation, genomic disorders, and genome evolution. Both low-copy genomic repeats (including LCRs and SDs) and high-copy repeats (including Alu, LINEs, and HERVs) can induce CNV formation via classical DNA recombination-based mechanisms, such as NAHR. Furthermore, paired repeats (especially those in the inverted orientation) are even more crucial as substrates to form DNA secondary structures and cause DNA replication fork stalling and replication stress. This will induce DNA replication errors and subsequently generate CNV mutations. Besides the primary structural features (e.g., the organization of repeat sequences in the human genome) and repeat-mediated secondary DNA structures, higher-order genomic architecture (such as replication timing) is also involved in CNV instability. Further investigation of the role of DNA replication dynamics in CNV mutagenesis will reveal more mutational mechanisms underlying genomic disorders and genome evolution.
Acknowledgments
This work was supported by the National Basic Research Program of China (2012CB944600 and 2011CBA00401), National Natural Science Foundation of China (81222014, 31171210 and 31000552), Shu Guang Project (12SG08), Shanghai Pujiang Program (10PJ1400300), and Recruitment Program of Global Experts.
References
- 1.Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–422. doi: 10.1016/s0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- 2.Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, et al. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 3.Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 4.Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. 2006;7:85–97. doi: 10.1038/nrg1767. [DOI] [PubMed] [Google Scholar]
- 5.Lee C, Scherer SW. The clinical context of copy number variation in the human genome. Expert Rev Mol Med. 2010;12:e8. doi: 10.1017/S1462399410001390. [DOI] [PubMed] [Google Scholar]
- 6.Stankiewicz P, Lupski JR. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;18:74–82. doi: 10.1016/s0168-9525(02)02592-1. [DOI] [PubMed] [Google Scholar]
- 7.Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, et al. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362:1366–1373. doi: 10.1016/s0140-6736(03)14632-6. [DOI] [PubMed] [Google Scholar]
- 8.Paylor R, Glaser B, Mupo A, Ataliotis P, Spencer C, Sobotka A, et al. Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: implications for 22q11 deletion syndrome. Proc Natl Acad Sci U S A. 2006;103:7729–7734. doi: 10.1073/pnas.0600206103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McDonald-McGinn DM, Sullivan KE. Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome) Medicine (Baltimore) 2011;90:1–18. doi: 10.1097/MD.0b013e3182060469. [DOI] [PubMed] [Google Scholar]
- 10.Breckpot J, Thienpont B, Bauters M, Tranchevent LC, Gewillig M, Allegaert K, et al. Congenital heart defects in a novel recurrent 22q11.2 deletion harboring the genes CRKL and MAPK1. Am J Med Genet A. 2012;158A:574–580. doi: 10.1002/ajmg.a.35217. [DOI] [PubMed] [Google Scholar]
- 11.Lupski JR, de Oca-Luna RM, Slaugenhaupt S, Pentao L, Guzzetta V, Trask BJ, et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell. 1991;66:219–232. doi: 10.1016/0092-8674(91)90613-4. [DOI] [PubMed] [Google Scholar]
- 12.Zhang F, Seeman P, Liu P, Weterman MA, Gonzaga-Jauregui C, Towne CF, et al. Mechanisms for nonrecurrent genomic rearrangements associated with CMT1A or HNPP: rare CNVs as a cause for missing heritability. Am J Hum Genet. 2010;86:892–903. doi: 10.1016/j.ajhg.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smit AF. The origin of interspersed repeats in the human genome. Curr Opin Genet Dev. 1996;6:743–748. doi: 10.1016/s0959-437x(96)80030-x. [DOI] [PubMed] [Google Scholar]
- 14.Zhou W, Zhang F, Chen X, Shen Y, Lupski JR, Jin L. Increased genome instability in human DNA segments with self-chains: homology-induced structural variations via replicative mechanisms. Hum Mol Genet. 2013;22:2642–2651. doi: 10.1093/hmg/ddt113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smit AF. Interspersed repeats and other mementos of transposable elements in mammalian genomes. Curr Opin Genet Dev. 1999;9:657–663. doi: 10.1016/s0959-437x(99)00031-3. [DOI] [PubMed] [Google Scholar]
- 16.Eichler EE. Recent duplication, domain accretion and the dynamic mutation of the human genome. Trends Genet. 2001;17:661–669. doi: 10.1016/s0168-9525(01)02492-1. [DOI] [PubMed] [Google Scholar]
- 17.Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, et al. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 18.Bailey JA, Eichler EE. Primate segmental duplications: crucibles of evolution, diversity and disease. Nat Rev Genet. 2006;7:552–564. doi: 10.1038/nrg1895. [DOI] [PubMed] [Google Scholar]
- 19.Marques-Bonet T, Kidd JM, Ventura M, Graves TA, Cheng Z, Hillier LW, et al. A burst of segmental duplications in the genome of the African great ape ancestor. Nature. 2009;457:877–881. doi: 10.1038/nature07744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olson MV, Varki A. Sequencing the chimpanzee genome: insights into human evolution and disease. Nat Rev Genet. 2003;4:20–28. doi: 10.1038/nrg981. [DOI] [PubMed] [Google Scholar]
- 21.Nelson PN, Hooley P, Roden D, Davari Ejtehadi, H, Rylance P, Warren P, et al. Human endogenous retroviruses: transposable elements with potential. Clin Exp Immunol. 2004;138:1–9. doi: 10.1111/j.1365-2249.2004.02592.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim PM, Lam HY, Urban AE, Korbel JO, Affourtit J, Grubert F, et al. Analysis of copy number variants and segmental duplications in the human genome: Evidence for a change in the process of formation in recent evolutionary history. Genome Res. 2008;18:1865–1874. doi: 10.1101/gr.081422.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang Z, Tang H, Ventura M, Cardone MF, Marques-Bonet T, She X, et al. Ancestral reconstruction of segmental duplications reveals punctuated cores of human genome evolution. Nat Genet. 2007;39:1361–1368. doi: 10.1038/ng.2007.9. [DOI] [PubMed] [Google Scholar]
- 24.Dittwald P, Gambin T, Gonzaga-Jauregui C, Carvalho CM, Lupski JR, Stankiewicz P, et al. Inverted low-copy repeats and genome instability: a genome-wide analysis. Hum Mutat. 2013;34:210–220. doi: 10.1002/humu.22217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu W, Zhang F, Wang Y, Gu X, Jin L. Identification of copy number variation hotspots in human populations. Am J Hum Genet. 2010;87:494–504. doi: 10.1016/j.ajhg.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Itsara A, Cooper GM, Baker C, Girirajan S, Li J, Absher D, et al. Population analysis of large copy number variants and hotspots of human genetic disease. Am J Hum Genet. 2009;84:148–161. doi: 10.1016/j.ajhg.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nat Rev Genet. 2011;12:363–376. doi: 10.1038/nrg2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dittwald P, Gambin T, Szafranski P, Li J, Amato S, Divon MY, et al. NAHR-mediated copy-number variants in a clinical population: mechanistic insights into both genomic disorders and Mendelizing traits. Genome Res. 2013;23:1395–1409. doi: 10.1101/gr.152454.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, et al. Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470:59–65. doi: 10.1038/nature09708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med. 2010;61:437–455. doi: 10.1146/annurev-med-100708-204735. [DOI] [PubMed] [Google Scholar]
- 32.Sharp AJ, Locke DP, McGrath SD, Cheng Z, Bailey JA, Vallente RU, et al. Segmental duplications and copy-number variation in the human genome. Am J Hum Genet. 2005;77:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu P, Lacaria M, Zhang F, Withers M, Hastings PJ, Lupski JR. Frequency of nonallelic homologous recombination is correlated with length of homology: evidence that ectopic synapsis precedes ectopic crossing-over. Am J Hum Genet. 2011;89:580–588. doi: 10.1016/j.ajhg.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peng Z, Zhou W, Fu W, Du R, Jin L, Zhang F. Correlation between frequency of non-allelic homologous recombination and homology properties: evidence from homology-mediated CNV mutations in the human genome. Hum Mol Genet. 2014 Oct 16; doi: 10.1093/hmg/ddu533. [Epub]. http://dx.doi.org/10.1093/hmg/ddu533. [DOI] [PubMed] [Google Scholar]
- 35.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 36.Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet. 2009;10:691–703. doi: 10.1038/nrg2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kriegs JO, Churakov G, Jurka J, Brosius J, Schmitz J. Evolutionary history of 7SL RNA-derived SINEs in Supraprimates. Trends Genet. 2007;23:158–161. doi: 10.1016/j.tig.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 38.Batzer MA, Deininger PL. Alu repeats and human genomic diversity. Nat Rev Genet. 2002;3:370–379. doi: 10.1038/nrg798. [DOI] [PubMed] [Google Scholar]
- 39.Sasaki M, Lange J, Keeney S. Genome destabilization by homologous recombination in the germ line. Nat Rev Mol Cell Biol. 2010;11:182–195. doi: 10.1038/nrm2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Smith AJ, Walters RG, Coin LJ, Steinfeld I, Yakhini Z, Sladek R, et al. Small deletion variants have stable breakpoints commonly associated with alu elements. PLoS One. 2008;3:e3104. doi: 10.1371/journal.pone.0003104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gu W, Zhang F, Lupski JR. Mechanisms for human genomic rearrangements. Pathogenetics. 2008;1:4. doi: 10.1186/1755-8417-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Erez A, Patel AJ, Wang X, Xia Z, Bhatt SS, Craigen W, et al. Alu-specific microhomology-mediated deletions in CDKL5 in females with early-onset seizure disorder. Neurogenetics. 2009;10:363–369. doi: 10.1007/s10048-009-0195-z. [DOI] [PubMed] [Google Scholar]
- 43.Matejas V, Huehne K, Thiel C, Sommer C, Jakubiczka S, Rautenstrauss B. Identification of Alu elements mediating a partial PMP22 deletion. Neurogenetics. 2006;7:119–126. doi: 10.1007/s10048-006-0030-8. [DOI] [PubMed] [Google Scholar]
- 44.Warren WC, Hillier LW, Marshall Graves JA, Birney E, Ponting CP, Grützner F, et al. Genome analysis of the platypus reveals unique signatures of evolution. Nature. 2008;453:175–183. doi: 10.1038/nature06936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Higashimoto K, Maeda T, Okada J, Ohtsuka Y, Sasaki K, Hirose A, et al. Homozygous deletion of DIS3L2 exon 9 due to non-allelic homologous recombination between LINE-1s in a Japanese patient with Perlman syndrome. Eur J Hum Genet. 2013;21:1316–1319. doi: 10.1038/ejhg.2013.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Janoušek V, Karn RC, Laukaitis CM. The role of retrotransposons in gene family expansions: insights from the mouse Abp gene family. BMC Evol Biol. 2013;13:107. doi: 10.1186/1471-2148-13-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang N, Kazazian HH., Jr L1 retrotransposition is suppressed by endogenously encoded small interfering RNAs in human cultured cells. Nat Struct Mol Biol. 2006;13:763–771. doi: 10.1038/nsmb1141. [DOI] [PubMed] [Google Scholar]
- 48.Miki Y, Nishisho I, Horii A, Miyoshi Y, Utsunomiya J, Kinzler KW, et al. Disruption of the APC gene by a retrotransposal insertion of L1 sequence in a colon cancer. Cancer Res. 1992;52:643–645. [PubMed] [Google Scholar]
- 49.Kazazian HH, Jr, Wong C, Youssoufian H, Scott AF, Phillips DG, Antonarakis SE. Haemophilia A resulting from de novo insertion of L1 sequences represents a novel mechanism for mutation in man. Nature. 1988;332:164–166. doi: 10.1038/332164a0. [DOI] [PubMed] [Google Scholar]
- 50.Belancio VP, Deininger PL, Roy-Engel AM. LINE dancing in the human genome: transposable elements and disease. Genome Med. 2009;1:97. doi: 10.1186/gm97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dangel AW, Mendoza AR, Baker BJ, Daniel CM, Carroll MC, Wu LC, et al. The dichotomous size variation of human complement C4 genes is mediated by a novel family of endogenous retroviruses, which also establishes species-specific genomic patterns among Old World primates. Immunogenetics. 1994;40:425–436. doi: 10.1007/BF00177825. [DOI] [PubMed] [Google Scholar]
- 52.Shuvarikov A, Campbell IM, Dittwald P, Neill NJ, Bialer MG, Moore C, et al. Recurrent HERV-H-mediated 3q132-q1331 deletions cause a syndrome of hypotonia and motor, language, and cognitive delays. Hum Mutat. 2013;34:1415–1423. doi: 10.1002/humu.22384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hermetz KE, Surti U, Cody JD, Rudd MK. A recurrent translocation is mediated by homologous recombination between HERV-H elements. Mol Cytogenet. 2012;5:6. doi: 10.1186/1755-8166-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kamp C, Ditton H, Huellen K, Vogt PH. Complex human Y-chromosomal HERV sequence structure in the AZFa region: new candidate genes for the control of early germ cell proliferation? Eur J Hum Genet. 2001;9(Suppl 1):C044. [Google Scholar]
- 55.Bosch E, Jobling MA. Duplications of the AZFa region of the human Y chromosome are mediated by homologous recombination between HERVs and are compatible with male fertility. Hum Mol Genet. 2003;12:341–347. doi: 10.1093/hmg/ddg031. [DOI] [PubMed] [Google Scholar]
- 56.Arruda JT, Silva DM, Silva CC, Moura KK, da Cruz AD. Homologous recombination between HERVs causes duplications in the AZFa region of men accidentally exposed to cesium-137 in Goiania. Genet Mol Res. 2008;7:1063–1069. doi: 10.4238/vol7-4gmr492. [DOI] [PubMed] [Google Scholar]
- 57.Koh E. Male infertility and genome disease -mechanism of microdeletions in azoospermia factor (AZF) regions and genomic diversification of the Y chromosome through analysis of human endogenous retrovirus. Genes Genet Syst. 2010;85:445. [Google Scholar]
- 58.Carvalho CM, Zhang F, Lupski JR. Structural variation of the human genome: mechanisms, assays, and role in male infertility. Syst Biol Reprod Med. 2011;57:3–16. doi: 10.3109/19396368.2010.527427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen JM, Chuzhanova N, Stenson PD, Férec C, Cooper DN. Complex gene rearrangements caused by serial replication slippage. Hum Mutat. 2005;26:125–134. doi: 10.1002/humu.20202. [DOI] [PubMed] [Google Scholar]
- 60.Carvalho CM, Ramocki MB, Pehlivan D, Franco LM, Gonzaga-Jauregui C, Fang P, et al. Inverted genomic segments and complex triplication rearrangements are mediated by inverted repeats in the human genome. Nat Genet. 2011;43:1074–1081. doi: 10.1038/ng.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hastings PJ, Ira G, Lupski JR. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5:e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang F, Khajavi M, Connolly AM, Towne CF, Batish SD, Lupski JR. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat Genet. 2009;41:849–853. doi: 10.1038/ng.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kent WJ, Baertsch R, Hinrichs A, Miller W, Haussler D. Evolution's cauldron: duplication, deletion, and rearrangement in the mouse and human genomes. Proc Natl Acad Sci U S A. 2003;100:11484–11489. doi: 10.1073/pnas.1932072100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schwartz S, Kent WJ, Smit A, Zhang Z, Baertsch R, Hardison RC, et al. Human-mouse alignments with BLASTZ. Genome Res. 2003;13:103–107. doi: 10.1101/gr.809403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen X, Shen Y, Zhang F, Chiang C, Pillalamarri V, Blumenthal I, et al. Molecular analysis of a deletion hotspot in the NRXN1 region reveals the involvement of short inverted repeats in deletion CNVs. Am J Hum Genet. 2013;92:375–386. doi: 10.1016/j.ajhg.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moore JK, Haber JE. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:2164–2173. doi: 10.1128/mcb.16.5.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Conrad DF, Hurles ME. The population genetics of structural variation. Nat Genet. 2007;39(7 Suppl):S30–S36. doi: 10.1038/ng2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stamatoyannopoulos JA, Adzhubei I, Thurman RE, Kryukov GV, Mirkin SM, Sunyaev SR. Human mutation rate associated with DNA replication timing. Nat Genet. 2009;41:393–395. doi: 10.1038/ng.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yao NY, O'Donnell M. SnapShot: the replisome. Cell. 2010;141:1088. doi: 10.1016/j.cell.2010.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rhind N, Gilbert DM. DNA replication timing. Cold Spring Harb Perspect Biol. 2013;5:a010132. doi: 10.1101/cshperspect.a010132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Farkash-Amar S, Simon I. Genome-wide analysis of the replication program in mammals. Chromosome Res. 2010;18:115–125. doi: 10.1007/s10577-009-9091-5. [DOI] [PubMed] [Google Scholar]
- 73.Ryba T, Battaglia D, Pope BD, Hiratani I, Gilbert DM. Genome-scale analysis of replication timing: from bench to bioinformatics. Nat Protoc. 2011;6:870–895. doi: 10.1038/nprot.2011.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Raghuraman MK, Brewer BJ. Molecular analysis of the replication program in unicellular model organisms. Chromosome Res. 2010;18:19–34. doi: 10.1007/s10577-009-9099-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Koren A, Polak P, Nemesh J, Michaelson JJ, Sebat J, Sunyaev SR, et al. Differential relationship of DNA replication timing to different forms of human mutation and variation. Am J Hum Genet. 2012;91:1033–1040. doi: 10.1016/j.ajhg.2012.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bechhoefer J, Rhind N. Replication timing and its emergence from stochastic processes. Trends Genet. 2012;28:374–381. doi: 10.1016/j.tig.2012.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Desprat R, Thierry-Mieg D, Lailler N, Lajugie J, Schildkraut C, Thierry-Mieg J, et al. Predictable dynamic program of timing of DNA replication in human cells. Genome Res. 2009;19:2288–2299. doi: 10.1101/gr.094060.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Farkash-Amar S, Lipson D, Polten A, Goren A, Helmstetter C, Yakhini Z, et al. Global organization of replication time zones of the mouse genome. Genome Res. 2008;18:1562–1570. doi: 10.1101/gr.079566.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hansen RS, Canfield TK, Lamb MM, Gartler SM, Laird CD. Association of fragile X syndrome with delayed replication of the FMR1 gene. Cell. 1993;73:1403–1409. doi: 10.1016/0092-8674(93)90365-w. [DOI] [PubMed] [Google Scholar]
- 80.MacAlpine DM, Rodríguez HK, Bell SP. Coordination of replication and transcription along a Drosophila chromosome. Genes Dev. 2004;18:3094–3105. doi: 10.1101/gad.1246404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hiratani I, Ryba T, Itoh M, Yokochi T, Schwaiger M, Chang CW, et al. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 2008;6:e245. doi: 10.1371/journal.pbio.0060245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Woodfine K, Fiegler H, Beare DM, Collins JE, McCann OT, Young BD, et al. Replication timing of the human genome. Hum Mol Genet. 2004;13:191–202. doi: 10.1093/hmg/ddh016. [DOI] [PubMed] [Google Scholar]
- 83.Aran D, Toperoff G, Rosenberg M, Hellman A. Replication timing-related and gene body-specific methylation of active human genes. Hum Mol Genet. 2011;20:670–680. doi: 10.1093/hmg/ddq513. [DOI] [PubMed] [Google Scholar]
- 84.Ryba T, Hiratani I, Lu J, Itoh M, Kulik M, Zhang J, et al. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res. 2010;20:761–770. doi: 10.1101/gr.099655.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cardoso-Moreira M, Emerson JJ, Clark AG, Long M. Drosophila duplication hotspots are associated with late-replicating regions of the genome. PLoS Genet. 2011;7:e1002340. doi: 10.1371/journal.pgen.1002340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lu J, Li H, Hu M, Sasaki T, Baccei A, Gilbert DM, et al. The distribution of genomic variations in human iPSCs is related to replication-timing reorganization during reprogramming. Cell Rep. 2014;7:70–78. doi: 10.1016/j.celrep.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Watanabe Y, Maekawa M. Spatiotemporal regulation of DNA replication in the human genome and its association with genomic instability and disease. Curr Med Chem. 2010;17:222–233. doi: 10.2174/092986710790149756. [DOI] [PubMed] [Google Scholar]
- 88.Litmanovitch T, Altaras MM, Dotan A, Avivi L. Asynchronous replication of homologous alpha-satellite DNA loci in man is associated with nondisjunction. Cytogenet Cell Genet. 1998;81:26–35. doi: 10.1159/000015003. [DOI] [PubMed] [Google Scholar]
- 89.Grinberg-Rashi H, Cytron S, Gelman-Kohan Z, Litmanovitch T, Avivi L. Replication timing aberrations and aneuploidy in peripheral blood lymphocytes of breast cancer patients. Neoplasia. 2010;12:668–674. doi: 10.1593/neo.10568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fritz A, Sinha S, Marella N, Berezney R. Alterations in replication timing of cancer-related genes in malignant human breast cancer cells. J Cell Biochem. 2013;114:1074–1083. doi: 10.1002/jcb.24447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Donley N, Thayer MJ. DNA replication timing, genome stability and cancer: late and/or delayed DNA replication timing is associated with increased genomic instability. Semin Cancer Biol. 2013;23:80–89. doi: 10.1016/j.semcancer.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Woo YH, Li WH. DNA replication timing and selection shape the landscape of nucleotide variation in cancer genomes. Nat Commun. 2012;3:1004. doi: 10.1038/ncomms1982. [DOI] [PubMed] [Google Scholar]
- 93.De S, Michor F. DNA replication timing and long-range DNA interactions predict mutational landscapes of cancer genomes. Nat Biotechnol. 2011;29:1103–1108. doi: 10.1038/nbt.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501:338–345. doi: 10.1038/nature12625. [DOI] [PubMed] [Google Scholar]
- 95.Chen L, Zhou W, Zhang C, Lupski JR, Jin L, Zhang F. CNV instability associated with DNA replication dynamics: evidence for replicative mechanisms in CNV mutagenesis. Hum Mol Genet. 2014 Nov 14; doi: 10.1093/hmg/ddu572. [Epub]. http://dx.doi.org/10.1093/hmg/ddu572. [DOI] [PMC free article] [PubMed] [Google Scholar]