


Abstract
Using FRET in bulk and on single molecules, we assessed the structural role of histone acetylation in nucleosomes reconstituted on the 170 bp long Widom 601 sequence. We followed salt-induced nucleosome disassembly, using donor–acceptor pairs on the ends or in the internal part of the nucleosomal DNA, and on H2B histone for measuring H2A/H2B dimer exchange. This allowed us to distinguish the influence of acetylation on salt-induced DNA unwrapping at the entry–exit site from its effect on nucleosome core dissociation. The effect of lysine acetylation is not simply cumulative, but showed distinct histone-specificity. Both H3- and H4-acetylation enhance DNA unwrapping above physiological ionic strength; however, while H3-acetylation renders the nucleosome core more sensitive to salt-induced dissociation and to dimer exchange, H4-acetylation counteracts these effects. Thus, our data suggest, that H3- and H4-acetylation have partially opposing roles in regulating nucleosome architecture and that distinct aspects of nucleosome dynamics might be independently controlled by individual histones.
INTRODUCTION
Eukaryotes employ various mechanisms to modulate local chromatin structure and provide on-demand access to specific DNA loci when needed. These mechanisms include posttranslational modifications (PTMs) on histones and DNA, such as histone acetylation or DNA methylation (1). Locally directed modifications allow specific gene activation; there is strong evidence that histone acetylation may influence the general level of chromatin compaction.
DNA is compacted into chromatin by first wrapping a stretch of ∼150 bp around a histone octamer core, composed of two copies each of histones H2A, H2B, H3 and H4, to form a nucleosome (2). Beyond a purely static means for compaction, the nucleosome is a highly dynamic entity whose complex conformational changes affect higher-order chromatin structure and DNA accessibility. These include reversible site exposure in the linker DNA region (3,4), nucleosome sliding (5) or ordered nucleosome disassembly (6).
About one-third of the total histone mass is located in N- and C-terminal histone tails, which are structurally disordered. Their general role in the regulation of nucleosome dynamics is uncontested, but the mechanism of this process is still a matter of debate. Among the different tails, salt-induced chromatin folding experiments identified the N-terminal regions to be more important than the respective C-terminal ends (7). Due to their high content of lysine and arginine residues, the N-terminal regions can be subjected to PTMs, such as acetylation or methylation. While these modifications can be recognized directly by specific enzymes—protein bromodomains, for example, recognize acetylated lysines (8)—the altered charge of the histones itself may influence electrostatic protein-DNA interactions, which affect nucleosome dynamics and the formation of higher-order chromatin structure.
A decompacting effect of hyperacetylation of isolated oligonucleosomes or chromatin fibers has been reported from gel mobility and ultracentrifugation measurements (9). Thermal and mechanical stabilities were reduced as well (10,11). Acetylation also inhibited the folding of reconstituted nucleosome arrays and increased the rate of transcription in cell extracts (12). While the in vivo effect of histone tail modifications has to be seen in the context of the overall chromatin structure, many studies showed an influence of such modifications on the structure of isolated mononucleosomes: for instance, histone hyperacetylation led to increased DNA accessibility (3) and to the opening of the external regions of nucleosomal DNA (13). Polach et al. (14) showed that clipping of histone H3 or H4 tail domains led to up to a 14-fold increased accessibility of mononucleosomal DNA.
The question whether the histone tails act independently or have synergistic effects is still disputed. In functional studies, acetylation of histone H4 is often found to be anticorrelated with acetylation of H3 or the other histones in binding of transcription factors, expressing genes or remodeling the chromatin (e.g. (15,16)). While earlier work suggested that N-terminal tails work independently and additive toward the folding of chromatin fibers (7), other studies identified H3 and H4 tails to be more important for folding than H2A and H2B tails (17). Truncation of the H3 and H2B N-terminal tails resulted in increased sliding and reduced thermal stability (18,19). Truncation of H3 also increased unwrapping of the nucleosome ends, while removal of half of the H4 tail only had a minor effect on the unwrapping rate (20). Interestingly, this study also proposed a synergistic effect of H3 and H4 tails on unwrapping of the DNA ends, where simultaneous truncation of both tails further enhanced DNA unwrapping. Our own results indicate that—in contrast to hyperacetylation—exclusive acetylation of histone H4 leads to the closing of the DNA arms 20 bp away from the exit (13). Acetylation-induced conformational changes in the structure of isolated histone H4 tails have also been observed in simulation studies (21,22), but their effect on the structure and stability of the full nucleosome is still unclear.
Thus, DNA–protein dynamics in the nucleosome entry–exit region have been studied rather thoroughly, but structural changes within internal nucleosome sites are relatively poorly understood. This information is essential, however, to derive a detailed picture of nucleosome architecture and how it is affected by posttranslational modifications. Here, we try to gain more detailed insight into the effect of histone tail acetylation from Förster Resonance Energy Transfer (FRET) measurements.
FRET (23) measures the distance-dependent energy transfer between a donor and an acceptor fluorophore on the length scale of a few nanometers, which compares well with the dimensions of the nucleosome. When the probes are attached to nucleosomal DNA and/or histone proteins, changes in nucleosome architecture can be followed via changes in the donor–acceptor distance. Existing FRET studies focused on the dynamics of DNA unwrapping in the entry–exit region (4,20,24–28), which was shown to be enhanced by specific acetylation of lysine 56 in histone H3 (29), and on nucleosome sliding (30,31). Global changes in nucleosome structure were studied in equilibrium and under disassembly conditions (6,32,33), as a result of DNA methylation (34) or specific acetylation in the histone-fold region (35). Changes in architecture were also analyzed near physiological concentrations if nucleosomes were enzymatically (36) or chemically (37) acetylated. These single molecule studies confirmed the general picture that histone acetylation alters nucleosome structure; yet a detailed picture of the role of individual histone acetylation, in particular at the N-terminal tails, on nucleosome structure and dynamics is still overdue.
Using FRET in bulk solution and on the single molecule level (single pair FRET, spFRET), we analyzed here changes in nucleosome structure and stability upon acetylation of one or more histones. We refer to a stable nucleosome as one either in its canonical form or in a (reversible) unwrapped state with increased opening of DNA ends but unaltered DNA topology in the core region. Combining data from the external and internal regions of nucleosomal DNA, we study the effect of selective histone acetylation on unwrapping of the DNA ends, and on nucleosome core disassembly. We present data on salt-dependent nucleosome stability, structural heterogeneity during disassembly and H2A/H2B dimer exchange to show that histones H3 and H4 have opposing roles in regulating these different aspects of nucleosome architecture. While acetylation of H3 dominates DNA end unwrapping and nucleosome dissociation, acetylation of histone H4 surprisingly counteracted the role of H3-acetylation during disassembly and dimer exchange but not its effect on DNA unwrapping in the entry–exit region. The lack of synergy between H3- and H4-acetylation demonstrates that eukaryotes can utilize modifications of individual histone tails to control DNA end dynamics and disassembly independently.
MATERIALS AND METHODS
Preparation of labeled mononucleosomes
All mononucleosomes were assembled on 170 bp long fragments of the Widom 601 nucleosome positioning DNA (38) or the naturally occurring 5S rDNA. Fluorescently labeled DNA was first prepared by PCR using labeled primers as described previously (13,37). Primer sequences for the 601 sequence are listed in the supplemental information, After PCR donor and acceptor labels were located either at positions +42 and −52 with respect to the center of the DNA fragment, denoted as ‘dl-I’, or at the ends of the DNA fragment, denoted as ‘dl-E’, see Figure 1A. For dimer exchange experiments additional DNA was prepared, which only carried the acceptor at position −52. The quality of primer labeling was checked on a denaturing 20% polyacrylamide/7M urea gel. After PCR, labeled DNA fragments were purified on an ion exchange column (Waters) using HPLC (Unicam); only the fraction where the ratio between Alexa 594 and Alexa 488 absorption was closest to the ratio of their extinction coefficients was used for nucleosome reconstitution. We estimated the fraction of non double-labeled DNA after purification to be <5%.
Figure 1.
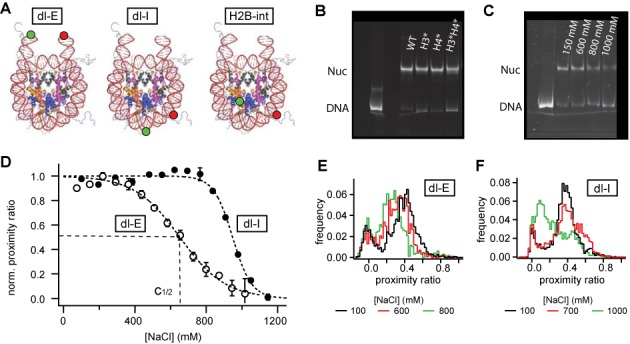
Bulk and spFRET experiments on mononucleosomes: (A) crystal structure (pdb file 1kx5, with hypothetical DNA extension taken from pdb file 1bna) and approximate dye positions for end-labeled nucleosomes (dl-E), internally labeled nucleosomes (dl-I) and protein–DNA labeled nucleosomes (H2B-int) that are formed in dimer exchange experimenys. (B) nucleosome positioning after reconstitution visualized in a native 6% polyacrylamide gel. All constructs showed homogeneous positioning. (C) electrophoretic mobility shift assay (EMSA) of nucleosomes after incubation at different ionic strength. No salt-induced repositioning of the octamer along DNA was observed. (D) salt-titration of non-acetylated nucleosomes measured with μpsFRET. Normalized bulk proximity ratio is plotted as a function of NaCl concentration. The decay in FRET is approximated by a sigmoidal function; the c1/2 value is used to quantify nucleosome stability. FRET between dyes at the DNA ends is lost at significantly lower ionic strength than between the dyes at internal DNA sites, showing that nucleosome opening in the entry–exit region precedes disassembly. (E) spFRET histograms of non-acetylated dl-E nucleosomes at 100, 600 and 800 mM NaCl. At higher ionic strength nucleosomes assume, an open structure. (F) spFRET histograms of non-acetylated dl-I nucleosomes at 100, 700 and 1000 mM NaCl. Prior to disassembly nucleosomes transit into an alternative conformation with increased proximity ratio.
Histone proteins were expressed and purified as described previously (24). Individual histones were chemically treated using acetyl phosphate, resulting in random acetylation of the lysines. MALDI-TOF mass spectrometry and Edman sequencing verified that acetylation occurred predominantly on the N termini of the histones (see Supplemental Table S1). Octamers with different composition of acetylated and non-acetylated proteins were refolded as described in (13). We prepared octamers with only histone H3 acetylated (H3*), only histone H4 acetylated (H4*), histones H3 and H4 acetylated simultaneously (H3*H4*), all histones acetylated (all*) or no histones acetylated (wt). For dimer exchange experiments we used nucleosomes where ∼5% of all histones H2B were labeled with Alexa 488. Threonine 112 in H2B was first replaced by cysteine (mutant H2B–T112C, obtained from Planet Protein (Colorado State University, Fort Collins) to introduce a site-specific SH-group to which Alexa 488-maleimide was attached via standard coupling chemistry. Further details on labeling and protein purification can be found in (6). Labeled and unlabeled H2B were mixed at a ratio of 1:20, added to unlabeled H2A, H3 and H4 and refolded into octamers. Approximately 10% of all octamers carried one donor-labeled H2B, while a negligible amount (<1%) of octamers contained two donor fluorophores. Purified DNA and octamers were mixed at 2 M NaCl–TE buffer and reconstituted into nucleosomes by gradual salt dialysis down to 5 mM NaCl. The molar ratio between DNA and octamer was optimized between 1:1.6 and 1:2 to avoid aggregation and to minimize excess free DNA. Where needed, nucleosomes were centrifuged at 10 000 rpm (Eppendorf Centrifuge 5417R, corresponding to a RCF of 10 600 g) for 10 min to remove residual aggregates. The quality of nucleosomes was checked on a native 6% polyacrylamide gel (acrylamide:bisacrylamide = 60:1), see Figure 1B. Nucleosomes were stored in stock solution at 4°C for up to 3 weeks.
Single pair FRET experiments
Quasi-bulk spFRET experiments were performed in a device of our own construction (39). A schematic view is shown in Supplemental Figure S1. All experiments were performed in 384-well microplates (SensoPlate Plus, Greiner Bio-One) that were passivated with Sigmacote before each use. Nucleosomes were freshly diluted into the experimental buffer (10 mM Tris, 0.1 mM EDTA, pH 7.5, supplemented with 0.01% Nonidet P40 (Roche Diagnostics), 0.5 mM ascorbic acid (Sigma–Aldrich) to minimize photobleaching, and NaCl as noted). 40–50 pM of labeled nucleosomes were mixed with an appropriate excess of unlabeled nucleosomes that were reconstituted with unlabeled, 170 bp long DNA fragments. Samples were incubated for 60 min, spFRET data were then taken for 20 min, during which ∼2000–3000 single molecule bursts were detected.
spFRET data were analyzed by our own software that filtered the raw data stream of a time-correlated single photon counting (TCSPC) board (TimeHarp200, PicoQuant) and selected single molecule bursts. A burst was defined as a group of at least 60 detected photons with a mutual separation of <120 μs. For each burst several parameters were recorded, including proximity ratio, burst duration and photon intensity per time. Single molecule distributions of the proximity ratio and other parameters were built and further analyzed with IGOR Pro software (WaveMetrics). Calculation of the moments of spFRET distributions was performed in IGOR Pro as described in supplemental information, section S2.
Microplate-scanning FRET analysis
Salt-dependent dissociation of nucleosomes was quantified using microplate-scanning FRET (μpsFRET) as described in (39,40). Samples were incubated in the same 384-well microplates used for spFRET; for each set of samples three fluorescence images were taken on a variable mode scanner (Typhoon 9400, GE Healthcare) corresponding to donor channel (excitation at 488 nm, detection at 500–540 nm), acceptor channel (excitation at 532 nm, detection at 595–625 nm) and transfer channel (excitation at 488 nm, detection at 595–625 nm). Pixel resolution was 100 μm and voltages on the photomultiplier tubes (PMT) were set to 625 V for the donor channel and 675 V for the transfer and acceptor channel. Images were analyzed with ImageQuant™ software and proximity ratios were calculated for each well as described below.
Estimation of FRET efficiencies via the proximity ratio
Salt-induced changes in energy transfer were measured by the sensitized emission of the acceptor upon selective donor excitation (41). Fluorescence was detected in two spectral windows, yielding signal intensities 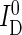 and
and 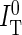 for the donor and transfer channel, respectively. These intensities either represent the intensity within a microplate well (μpsFRET) or the number of donor and acceptor photons per single molecule burst (spFRET).
for the donor and transfer channel, respectively. These intensities either represent the intensity within a microplate well (μpsFRET) or the number of donor and acceptor photons per single molecule burst (spFRET). 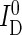 and
and 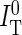 were corrected for background from buffer (BD and BT), spectral crosstalk from the donor into the transfer channel (αDT) and direct excitation of the acceptor dye (fdir), to yield corrected intensities
were corrected for background from buffer (BD and BT), spectral crosstalk from the donor into the transfer channel (αDT) and direct excitation of the acceptor dye (fdir), to yield corrected intensities
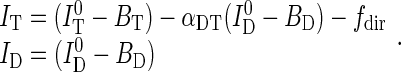 |
(1) |
All correction factors were determined in independent control experiments with donor-only samples, acceptor-only samples and pure buffer solution, see SI. The proximity ratio P was calculated as a measure of energy transfer:
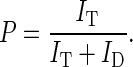 |
(2) |
Salt-induced destabilization of nucleosomes
To measure salt-induced changes in stability 300 pM nucleosomes were incubated in TE buffer containing 1 mM ascorbic acid, 0.01% (v/v) Nonidet P40 and NaCl concentrations between 100 and 1300 mM. For microplate-scanning FRET experiments all 300 pM nucleosomes were labeled, while the samples for spFRET contained 50 pM labeled and 250 pM unlabeled nucleosomes. The proximity ratio was calculated from bulk or spFRET data and plotted against salt concentration. A sigmoidal function (Equation (3)) was then fitted to the experimental curves; stability was quantified by the c1/2 parameter, which is the salt concentration at which P has dropped to half its initial value at low salt.
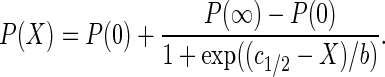 |
(3) |
Here, X is the salt concentration in mM and b describes the slope of the curve at X = c1/2. P(0) and P(∞) are amplitude (at 0 mM NaCl) and offset (at maximum [NaCl]) of the salt-titration curve.
Heterodimer exchange experiments
Dimer exchange between nucleosomes was monitored by μpsFRET. Nucleosomes were mixed at a molar ratio of 1:2 dimer-labeled(donor)/DNA-labeled(acceptor) and incubated in buffer containing 100–900 mM NaCl. During incubation at higher salt nucleosomes will start to exchange dimers, reaching equilibrium between unlabeled, single-labeled and double-labeled nucleosomes. After incubation for 70 min, samples were diluted with twice the volume of low salt buffer (TE + 10 mM NaCl) to stop heterodimer exchange. After incubation for another 45 min, the amount of double labeled nucleosomes was quantified by the increase in FRET.
Electrophoretic mobility shift assay (EMSA)
EMSA was performed on a native 6% polyacrylamide gel (acrylamide:bisacrylamide = 60:1). If not otherwise stated, nucleosomes were incubated for 60 min in TE buffer at NaCl concentrations as denoted. Samples were then threefold diluted into Tris–borate–EDTA (TBE) buffer supplemented with 12% glycerol. About 60 fmol nucleosomes were then loaded onto the gel, and run at 100 V (E = 16 V/cm) for 55 min at room temperature. Nucleosome and DNA bands were visualized on a Typhoon multimode imager using the same excitation and detection settings as in µpsFRET.
RESULTS
Nucleosome unwrapping precedes nucleosome disassembly
Variation of buffer salinity is a convenient way to study changes in nucleosome conformation in vitro. At elevated ionic strength and sub-nM sample concentration nucleosomes gradually dissociate and undergo characteristic changes in their architecture. These can be followed by changes in FRET between fluorophores that are attached to the sites of interest.
We first performed salt titration experiments on non-acetylated nucleosomes to analyze their general dissociation behavior. Bulk FRET experiments were done with our microplate-scanning FRET (μpsFRET) assay (39) at a nucleosome concentration of 300 pM, which was easily detectable on the Typhoon scanner, while not too much valuable sample was required per experiment. As shown in Supplemental Figure S2, this concentration was still an order of magnitude above the regime where spontaneous nucleosome dissociation due to mass action was observed at physiological ionic strength. Nucleosomes were incubated at NaCl concentrations between 10 and 1200 mM for 60 min, after which the array of samples was imaged on a Typhoon scanner. Each [NaCl] was measured in triplicate and the proximity ratio averaged between wells. Average proximity ratios were then plotted against salt concentration. Figure 1D shows the salt-dependence of P for end-labeled (dl-E) and internally labeled (dl-I) unmodified nucleosomes. For visualization, all data were normalized between 1 at low salt and 0 at high salt, when all nucleosomes were dissociated.
The bulk FRET of end-labeled nucleosomes initially increases between 10 and 200 mM NaCl, followed by a decrease in FRET at higher ionic strength (∼500–700 mM NaCl). The initial decrease in DNA end-to-end distance up to physiological salt has been observed in our own and others’ previous work (13,20) and is probably due to reduced electrostatic repulsion between the DNA arms. spFRET experiments revealed that the underlying distribution of nucleosomal FRET was rather narrow, indicating that almost all nucleosomes assume a compact structure where the DNA ends come closer together (Figure 1E). At higher salt, the distribution shifts toward an open structure with lower proximity ratio. Distribution broadening at 600 mM NaCl revealed the co-existence of open and closed conformation at intermediate salt concentrations, at which loss in FRET occurred. The open conformation probably stems from DNA unpeeling at the nucleosome ends (4,27) rather than salt-induced sliding of the octamer on the DNA. To exclude the latter we performed an electrophoretic mobility shift assay (EMSA). As shown in Figure 1C, incubation at higher salt did not change positioning of the octamer on the DNA and the amount of free DNA only increased above 800 mM NaCl.
Supplemental Figure S3A compares salt titration curves for 300 pM and 3 nM dl-E nucleosomes. We did not observe a notable dependence of the loss of FRET on nucleosome concentration, supporting our conclusion that reversible, concentration-independent DNA unwrapping at the entry–exit site is responsible for the decrease in FRET. Importantly, this transition occurs at similar ionic strength as the ‘butterfly’ transition described earlier (6), where the nucleosome opens up before H2A/H2B dissociation.
Internally labeled nucleosomes show fairly constant FRET at lower ionic strength, followed by a slight increase in P in the salt range where loss in FRET was observed for end-labeled samples. This increase in FRET is related to an increase in sample heterogeneity, where spFRET shows a second nucleosome population emerging at higher P (Figure 1F). A steep decrease in FRET above 800 mM NaCl indicates gross unpeeling of the DNA and nucleosome disassembly, consistent with the increase in free DNA observed in EMSA. FRET between internal labels reports on nucleosome stability rather than reversible DNA breathing at the entry–exit site. Supplemental Figure S3B shows a ≈10% increase in c1/2 (dl-I) at 10-fold increased nucleosome concentration, consistent with reduced dissociation and increased stability at higher nucleosome concentration (42). The onset of a second population prior to dissociation shows that disassembly occurs through one or more intermediate states (6,39).
Acetylation of histone H4 stabilizes nucleosomes against salt-induced dissociation
We next investigated the effect of histone acetylation on salt-dependent nucleosome architecture. Nucleosomes were reconstituted on octamers with only histone H3 acetylated (H3*), only histone H4 acetylated (H4*), histones H3 and H4 acetylated (H3*H4*) and no histones acetylated (wt). Samples were diluted to 300 pM and incubated at 75–1200 mM NaCl for 60 min. To minimize errors due to day-to-day variations in experimental procedure, each acetylated sample was measured in parallel to a non-acetylated reference sample. Representative salt-titration curves of μpsFRET for end- and internally labeled nucleosomes are shown in Figure 2A and B. The ratio c1/2(X)/c1/2(wt) was computed to quantify the change in stability associated with acetylation of histone(s) X (X = H3*, H3*H4*, H4*, wt). c1/2 values were calculated with respect to the maximum in P around 200–300 mM NaCl (dl-E) and 600–700 mM NaCl (dl-I). Salt-titrations were repeated at least three times per construct, the average values of c1/2(X)/c1/2(wt) are shown in Figure 2C.
Figure 2.
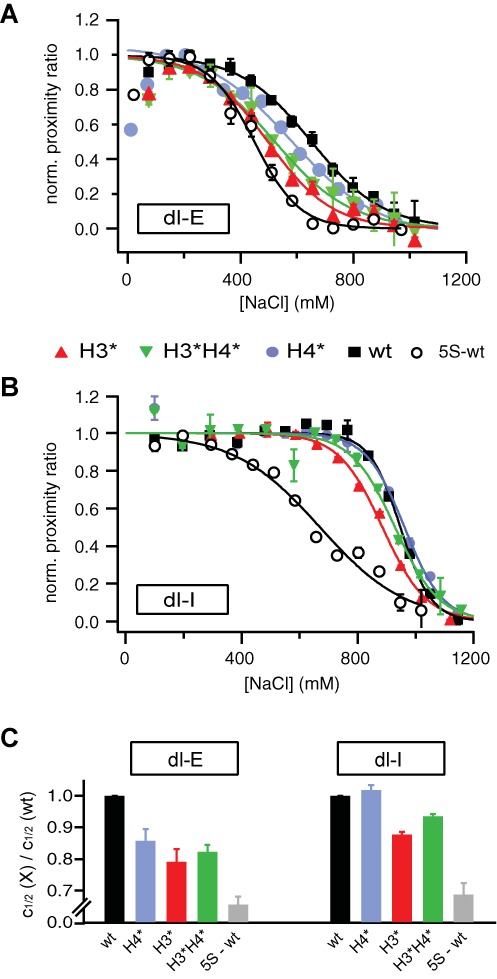
Effect of histone acetylation on DNA unwrapping and nucleosome core stability: (A and B) exemplary μpsFRET curves for selectively acetylated nucleosomes (dl-E (A) and dl-I (B)). For visualization, all curves were normalized between 1 at the maximum value and 0 at high ionic strength. Salt-titration curves of nucleosomes containing 5S rDNA and non-acetylated histones are shown for comparison (open circles) (C) ratio c1/2(X)/c1/2(wt) as a function of histone acetylation (X = H3*, H3*H4*, H4* or wt). Unwrapping of the DNA ends is enhanced for all acetylated samples, while H4-acetylation counteracted the role of H3-acetylation in disassembly. The dynamic range of the y-axes is rescaled for easy comparison of data with the 5S reference sample.
Near physiological ionic strength, end-labeled nucleosomes show an initial increase in bulk FRET regardless of the state of acetylation. The maximum proximity ratio decreased in the order wt ≈ H4* > H3*H4* > H3* (see Supplemental Figure S4), indicating an opening of the nucleosome ends upon H3-acetylation, while acetylation of H4 did not affect DNA geometry at the entry–exit region around 200 mM NaCl. The salt concentration at which loss in FRET is observed strongly varied between non-acetylated and acetylated samples; c1/2(H3*):c1/2(H3*H4*):c1/2(H4*):c1/2(wt) = 0.79:0.82:0.86:1. The most pronounced nucleosome opening (lowest c1/2) was observed for H3-acetylation, in H4-acetylated samples this occurred at higher NaCl concentration. c1/2(H4*) was still significantly smaller than c1/2(wt), however, suggesting that acetylation of H4 enhances DNA breathing at higher ionic strength as well. This increase in DNA opening upon H4-acetylation was not observed in previous work which used monovalent salt concentrations in the physiological range (<300 mM) only (13,20). Interestingly, nucleosomes where both H3 and H4 were acetylated were less susceptible to salt-induced unwrapping than H3-acetylated nucleosomes. These data suggest that H3-acetylation dominates reversible unwrapping of the DNA ends and that H4-acetylation can partially counteract this effect. Supplemental gel shift assays verified that the loss in FRET around 600 mM NaCl was indeed caused by opening of the DNA ends rather than octamer sliding on the DNA (see Supplemental Figure S5).
Variations in the stability of acetylated nucleosomes were analysed with dl-I samples, where the loss of FRET reports on actual nucleosome disassembly. Compared to non-acetylated nucleosomes, acetylation of H3 significantly destabilized the nucleosome with c1/2 being reduced by 120–130 mM, while acetylation of H4 did not affect nucleosome stability at all. More striking was the result from nucleosomes where both H3 and H4 were acetylated. The decrease in c1/2 was only half the change observed for H3-acetylated samples (7% versus 13%). This difference is intriguing, since intuitively one would expect that electrostatic protein–DNA interactions would decrease if more charges in the histone tails were neutralized. Apparently, the effect of lysine acetylation is not simply cumulative, but exhibits a histone-specific component.
To put the differences in c1/2 into better perspective we measured the salt-dependent unwrapping of the DNA ends in and stability of non-acetylated nucleosomes reconstituted onto the naturally occurring 5S rDNA. Previous work from the Widom lab had shown that the 5S sequence has a roughly 150-fold lower positioning strength than the artificial 601 sequence (43). c1/2 values for 5S nucleosomes were about 35% lower than those for wt 601 samples. The effect of histone acetylation on the opening of the nucleosomal ends is of comparable magnitude to the change in DNA sequence. The effect of histone acetylation on nucleosome stability, however, is more subtle than the sequence-dependence.
We finally analyzed nucleosomes where H2A and H2B were acetylated in addition to H3 and H4; these samples showed a stability similar to H3*H4* nucleosomes, but slightly enhanced unwrapping of the ends of nucleosomal DNA (Supplemental Figure S6). This suggests that acetylation of H2A and H2B does not influence nucleosome stability, but may well affect the nucleosome entry–exit region.
In summary, μpsFRET data show that acetylation of histone H3 promotes nucleosome opening and disassembly, whereas acetylation of H4 alone leads to an increase of unwrapping of the DNA ends, but does not enhance disassembly compared to unmodified histones. More importantly, H4-acetylation significantly counteracts H3-acetylation in nucleosome disassembly.
Histone acetylation does not change the dissociation pathway
Microplate-scanning FRET is a powerful tool to quantify overall nucleosome stability accurately and quickly, but does not provide further insight into the disassembly mechanism, such as potential heterogeneity in nucleosome architecture. This information, however, is important to understand the structure-defining properties of histone acetylation in more detail. We thus performed spFRET salt-titration experiments on internally labeled nucleosomes (dl-I). The total nucleosome concentration was adjusted to 300 pM by adding 250 pM unlabeled nucleosomes to 50 pM labeled complexes. We had shown in earlier work that this ‘quasi-bulk spFRET’ approach yields comparable estimates of nucleosome stability with the benefits of subspecies resolution at ensemble concentrations (39).
We first analyzed nucleosome heterogeneity near physiological ionic strength. Single pair FRET histograms at 100 mM NaCl are shown in Figure 3A and B. Detection settings were deliberately varied between dl-E and dl-I samples, hence both showed similar proximity ratios despite the differences in interdye distance (37). Non-acetylated dl-E nucleosomes showed a narrow distribution peaking around P = 0.4. Acetylation of histone H4 did not notably alter the distribution, while acetylation of histone H3 or all histones induced a significant shift toward smaller P ratios, indicating a more open structure at the entry–exit region. These results confirm the trend in bulk P that was observed between 100 and 200 mM NaCl (Supplemental Figure S4). Internally labeled nucleosomes showed a fairly homogeneous distribution at 100 mM NaCl for all samples. These observations affirm that at physiological ionic strength the effect of histone acetylation is confined to the entry-exit-region of the nucleosome and dominated by histone H3.
Figure 3.
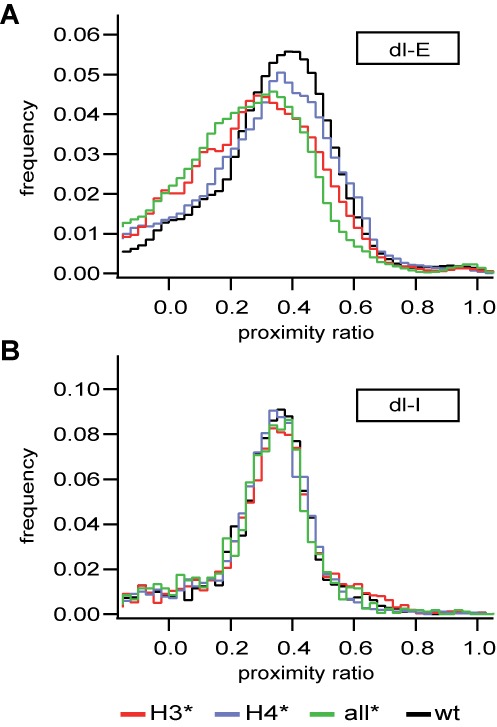
Effect of acetylation on nucleosome structure at physiological ionic strength: spFRET histograms of dl-E (A) and dl-I nucleosomes (B). Acetylation of H3 only or all histones leads to opening of the DNA ends but has no effect on DNA position in the histone-fold region. An excess of 10 nM unlabeled nucleosomes was added to each sample.
The structural heterogeneity during disassembly is shown in Figure 4A. P distributions of dl-I nucleosomes are shown for 500–1000 mM NaCl (top to bottom). At higher salt some nucleosomes progressively transited into an additional state with higher proximity ratio (P ≈ 0.55), while the amount of free DNA at first did not increase. At 1 M NaCl, most nucleosomes were dissociated, leaving a major DNA peak.
Figure 4.
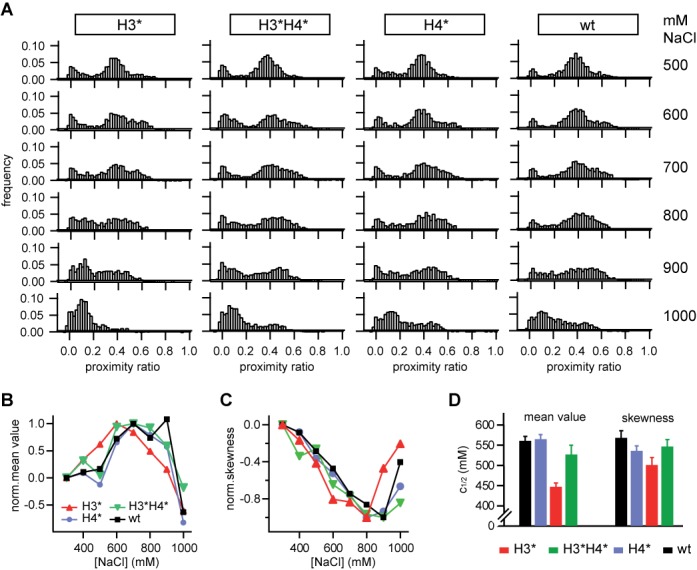
Effect of histone acetylation on nucleosome disassembly: (A) (from left to right) Single molecule distributions of H3-acetylated, H3/H4-acetylated, H4-acetylated and non-acetylated nucleosomes at 500, 600, 700, 800, 900 and 1000 mM NaCl (from top to bottom). (B and C) Normalized mean value and skewness of the nucleosome subpopulation as a function of NaCl concentration. (D) Salt-dependence of the change in distribution moments. c1/2 values were extracted from a sigmoidal fit to the data shown in Supplemental Figure S8.
The same transition was observed for all samples, suggesting that histone acetylation does not alter the mechanism of disassembly as such. Since acetylation does affect nucleosome stability, the salt concentration at which the transition was observed was histone-specific. To quantify this dependence in more detail, we analyzed the salt-induced changes in position and form of the nucleosome subpopulation as a function of [NaCl] and histone acetylation. To do so, we first subtracted the contribution from donor-only and free DNA in the histogram, PFRET = Pall − PD-only − PDNA (see Supplemental Figure S8), and analyzed position and shape of the resulting ‘net’ distribution PFRET.
As described in SI section S2, this information is parameterized by the various statistical moments of the distribution, such as its mean value or its standard deviation. To decide which moments are most promising for analysis we first simulated a heterogeneous mixture of two model species with PA = 0.4 and PB = 0.55 and equal standard deviation σA = σB = 0.08, which resembled the heterogeneity observed in nucleosome conformation. The ratio between both species was varied from 100% species A to 100% species B to mimic a transition from state A to state B. We then computed the first four distribution moments (mean value, variance, skewness and kurtosis) as a function of mixing ratio for N = 2000 bursts, a typical value in our single molecule experiments. As shown in Supplemental Figure S7, changes in mean value and skewness are most suitable for analysis, as they provide a linear readout over an extended range of species ratios.
Salt-dependent mean value and skewness of the nucleosome samples are shown in Figure 4B and C. For visualization, data were normalized to 0 at 100 mM NaCl and ±1 at the respective maximum in mean value (minimum in skewness). The corresponding non-normalized data are shown in Supplemental Figure S8. As expected, the mean value increases, while the asymmetry-related skewness decreases. For quantification, data were smoothed by a box filter, averaging each data point over its first adjacent neighbors and approximated by a sigmoidal function; c1/2 values are compared in Figure 4D. Evidently, H3-acetylated nucleosomes transit into the high FRET state at lower ionic strength than nucleosomes where H3 and H4 were acetylated, non-acetylated and H4-acetylated nucleosomes retained their initial structure up to higher ionic strength, with similar overall changes in distribution moments. These findings are in agreement with the bulk FRET data and strongly support the opposing role of H4 acetylation to salt-induced dissociation.
Histone acetylation affects dimer exchange between nucleosomes
Based on our previous work we hypothesize that the conformational transition might arise from H2A/H2B eviction following disruption of the dimer-tetramer interface, the first step during nucleosome disassembly (6,39). If this assumption was correct we expect heterodimer exchange between nucleosomes to show a dependence on histone acetylation that is similar to our results from spFRET. We thus performed heterodimer exchange experiments using our μpsFRET assay. Acceptor-only labeled, acetylated nucleosomes were mixed with H2B-labeled donor-only wt nucleosomes and incubated at various NaCl concentrations. During incubation at higher ionic strength, nucleosomes will start to exchange dimers, reaching an equilibrium where a subpopulation of nucleosomes is formed that contains both fluorophores, resulting in an increase in FRET signal. The exchange process is stopped by adding low salt buffer and the relative increase in FRET is analyzed as a function of histone acetylation.
A typical salt-dependence of the proximity ratio is shown in Figure 5A. Incubation at low salt did not result in an increase in FRET, indicating that spontaneous dimer exchange is negligible at physiological ionic strength. Significant dimer exchange was observed above 600 mM NaCl only, in agreement with our current model of nucleosome disassembly. Heterodimer exchange did not result in a change of nucleosome positioning as verified by gel electrophoresis, see Figure 5B. The effect of histone acetylation on heterodimer exchange is shown in Figure 5C. Exchange experiments were repeated at least 4 times for each sample; the average and standard deviation of the difference ΔP = P(X mM NaCl) − P(100 mM NaCl) is shown for X = 600, 700 and 800 mM NaCl. At all three salt concentrations H3-acetylated nucleosomes showed the largest amount of dimer exchange (red bars), whereas acetylation of H4 only did not result in an increase in dimer exchange compared to wt samples. Noteworthy, H3/H4-acetylated samples (green bars) consistently showed less dimer exchange than H3-acetylated samples, again supporting our conclusion that H4-acetylation can counteract the effect of acetylated H3.
Figure 5.
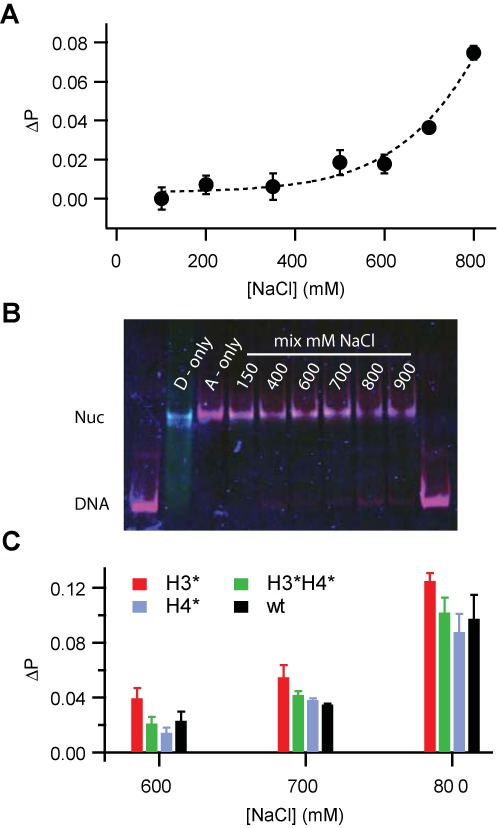
Histone acetylation affects dimer exchange between nucleosomes: (A) Difference in proximity ratio, ΔP = P(X mM) – P(100 mM) as a function of incubation salt concentration (X) for H4-acetylated nucleosomes. Significant dimer exchange between nucleosomes results in an increase in FRET signal above 600 mM NaCl. A polynomial fit is shown as visual guidance. (B) Electrophoretic shift assay of nucleosome positioning after incubation at 150–900 mM NaCl. Intensities from donor, acceptor and transfer channel are overlaid. Dimer exchange did not alter nucleosome positioning but caused an increase in FRET in the nucleosome band at higher ionic strength. (C) Bar chart showing average ΔP values for all samples at 600, 700 and 800 mM NaCl (n > 3 experiments for each construct). H3-acetylated nucleosomes showed the largest dimer exchange, whereas less exchange was observed for H3/H4-acetylated and H4-acetylated nucleosomes.
DISCUSSION
Histone tails and their modifications control nucleosome structure and genetic activity—but how they regulate DNA accessibility, whether through direct action on the nucleosome or through associated protein factors, whether they act independently of each other or in a concerted fashion, is still controversial. Several PTMs have been shown to constitute binding epitopes for protein factors that in turn can modify DNA accessibility, e.g. (44), but direct effects of histone tail modifications on nucleosome architecture have been reported as well (29). Our work addresses the ongoing debate of synergy between histone tails in regulating nucleosome architecture.
The structural response of nucleosomes to salt-induced destabilization is often used to probe the influence of histone and DNA modifications on nucleosome architecture. It is generally accepted that increasing salt concentrations induce changes in nucleosome architecture that mimic transiently populated conformations observed near physiological conditions (45). Using FRET labels at the DNA ends and at an internal DNA site near its H2A/H2B binding region, we performed ensemble and single molecule FRET experiments to assess the effect of histone acetylation on two important mechanisms by which nucleosome architecture controls DNA accessibility; (transient) unwrapping of the DNA ends and nucleosome core disassembly.
To probe the effect of selective acetylation of the different histone tails many sample conditions have to be analyzed. This is very time consuming if samples were measured one-at-a-time like in conventional fluorimetry. Therefore, we employed our recently developed ‘microplate-scanning FRET’ (μpsFRET) assay, in which bulk FRET efficiencies of many samples are determined in parallel (39). After quantification of nucleosome stability, we then used single molecule FRET to follow the structural heterogeneity during nucleosome disassembly. We introduced a phenomenological, computationally very efficient analysis of our spFRET data, where the statistical moments of the measured distribution are used to locate changes within the nucleosome subpopulation. This analysis is model-independent and does not need a priori information about the number of subspecies, whose determination can be non-trivial in a complex spFRET histogram.
Loss of FRET between the DNA ends is caused by concentration-independent DNA unwrapping (4,46,47) and occurs at lower salt (around 600 mM NaCl) than loss of FRET in the internal region of nucleosomal DNA, which reports on nucleosome disassembly (∼900 mM NaCl). Together with the onset of heterodimer exchange above 600 mM NaCl (Figure 5) the data confirm our current picture of nucleosome disassembly; the DNA ends unwrap first, followed by breakage of the dimer/tetramer interface, eviction of dimers and gross unpeeling of DNA (6,48).
At physiological ionic strength acetylation of H3, but not of H4, enhanced the opening of the DNA ends, while the internal nucleosome architecture did not change significantly after acetylation. These findings suggest that at low salt DNA binding is not significantly affected by histone tail acetylation. Under similar conditions, Lee et al. (36) only found minor rearrangements of internal DNA in immobilized, enzymatically acetylated nucleosomes as well. Consistent with existing literature the observed changes were more pronounced in the entry-exit region of the nucleosome.
At higher ionic strength, we observed an increase in opening of the DNA ends regardless of the state of acetylation. Apparently, screening of the charges in the tails leads to an opening of the nucleosome structure, which suggests that electrostatic charges in the tails dominate their interaction with the DNA ends. Using tail truncation instead of acetylation, Nurse et al. recently proposed a cooperative effect of H3 and H4 tails in nucleosome unwrapping (20). Our data challenge this idea of cooperativity when the tails are acetylated rather than clipped. Nucleosomes where H3 and H4 were acetylated did not show an increased opening compared to H3-acetylated samples, and H4-acetylation resulted in more compact DNA ends than H3- or H3/H4-acetylation. Thus, the effects of lysine acetylation seem not to be cumulative but show strong histone specificity. The dominating effect of H3 tails on the DNA ends is not surprising, since H3 tails exit the octamer near the DNA entry-exit region (49). H4 tails exit the nucleosome at a more internal region which could explain their smaller role in the unwrapping of the DNA ends. Our data demonstrate that nucleosomes will behave differently when the histone tails are acetylated rather than truncated, so that truncated tails are not a good mimic for acetylation.
More distinct roles of histones were observed during nucleosome core disassembly. H3-acetylated nucleosomes were least stable and dissociated at lower ionic strength than nucleosomes where H3 and H4 were acetylated. More stable nucleosomes were formed with H4-only acetylated or non-acetylated octamers. Importantly, during disassembly all nucleosomes transited into a conformation with increased proximity ratio before complete disruption. This intermediate state is suppressed at higher nucleosome concentration and might represent a (sub-)nucleosomal particle with either a transiently disrupted octamer (‘butterfly state’) or with one or both dimers missing (39). The ordered disassembly mechanism of nucleosomes does not seem to be altered by acetylation; the salt-dependence of this transition, however, correlated with the differences in bulk stability; H3* < H3*H4* < H4* ≤ wt. This corroborates our findings of H4 opposing H3 in altering nucleosome stability and demonstrates the special role that H4 assumes in regulating nucleosome architecture. In functional studies such as transcription factor binding assays or gene expression analysis, acetylation of histone H4 is also often found to be anticorrelated with acetylation of H3 or the other histones (15,16).
Comparing the stability of the 601 sequence to another known nucleosome-positioning sequence, the well-studied 5S rDNA which binds about 150-fold less strongly to octamers than the 601 sequence, shows that the effect of acetylation on the opening of the nucleosomal DNA ends is comparable to the differences associated with DNA sequence. In contrast, the effect of acetylation on nucleosome core stability was more subtle than the sequence-dependence. We might speculate that in the context of chromatin, histone tail acetylation—besides modulating local compaction for increased accessibility —can also fine-tune nucleosome stability for subsequent enzymatic processing where needed.
The N-terminal tails of histone H4 emerge from the octamer near the dimer:tetramer interface (49); their modification could significantly affect disassembly initiated by the loss of dimer:tetramer contacts (6). Using FRET as a probe for heterodimer exchange we found that sole acetylation of H3 caused more dimers to exchange between nucleosomes than combined acetylation of H3 and H4. This raises the idea that H4-acetylation can stabilize the dimer:tetramer interface. Computer simulations have shown that upon acetylation, residues 15–20 in H4 adopt a more helical structure, which causes shortening of the tails and disruption of internucleosome interactions in chromatin fibers (50). This shortening may facilitate additional intranucleosome interactions of the H4 tail, for instance with the acidic patch of H2A in the same nucleosome, similar to internucleosome tail bridging (51,52), which would hinder disruption of the dimer:tetramer interface and/or dimer eviction. An increase in dimer dynamics upon H3 modification was also observed by Ferreira et al., who reported increased thermal dimer exchange (not salt-induced) upon deletion of the H3 tail (18); however, their study did not analyze the combined effect of H3 and H4 acetylation.
From the current data we cannot unambiguously infer a difference in nucleosome core stability between non-acetylated and H4-acetylated nucleosomes. We attribute this to our choice of DNA sequence, one of the strongest nucleosome positioning sequences known (38). It is conceivable that the effect of H4-acetylation might be too subtle in relation to the strong binding of the 601 sequence. Additional destabilization by acetylated H3 might have lowered nucleosome stability to a point where the effect of H4-acetylation became visible.
The role of individual histone-fold residues in nucleosome dynamics has recently been studied using a chemical ligation protocol (35). In this study, the authors showed that acetylation of lysines in distinct regions of the histone-fold selectively affect unwrapping or disassembly. Our chemical acetylation method predominantly targets lysines in the histone tail regions (see Supplemental Table S1), but we cannot exclude the existence of acetylated lysines in the histone-fold region. In histone H4 we found by mass spectrometry (SI Table S1) that H4K91, located in the C-terminal region, was acetylated in all our samples. Since we detected no other acetylated lysines in that region, we can conclude that H4K77 and H4K79, two other lysines that reduce nucleosome stability when being acetylated (35), were not modified. H4K91 is located in the dimer:tetramer interface; its acetylation has been implicated in nucleosome destabilization (53). The absence of any detectable destabilization of H4-acetylated nucleosomes suggests that a potentially destabilization by H4K91ac is compensated by the presence of other acetylation sites in the tail region of H4. This further strengthens our hypothesis that acetylation of lysines in the histone H4 tail does not mediate nucleosome destabilization but can exert the opposite effect.
The central part of histone H3 which was not amenable to T3 sequencing includes two lysines, H3K56 and H3K64, which are known to affect nucleosome unwrapping and stability (29,54). Assuming random acetylation we estimate that both residues are acetylated in ∼30% of all histone H3. This would seem to be insufficient to dominate the strong effects observed for H3-acetylation. On the other hand, we cannot exclude that either one is acetylated to a higher degree as in the case of H4K91, but even then the observation that H4K91ac alone does not dominate the role of H4-acetylation suggests a similar interplay between acetylated lysines in the tail of H3 and H3K56 or H3K64, albeit with a synergistic outcome as opposed to H4.
Chemical acetylation results in considerable heterogeneity between acetylated histones, which averages out in bulk stability experiments but can contribute to broadening in spFRET histograms. Although random, acetylation was highly enriched in the N-terminal tails. Heterogeneity between nucleosomes was thus limited to the number of acetylated lysines per tail and did not affect tail-specificity; this still allowed us to target our study toward the structure-defining properties of histone tail modification. Our findings thus demonstrate that eukaryotes can additionally employ acetylation of the flexible histone tails to de-couple unwrapping and nucleosome disassembly. Additional broadening related to instrument noise and photon counting statistics prevented direct mapping of heterogeneity in acetylation using spFRET. The similarity of histograms of dl-I nucleosomes suggests that the impact of heterogeneous tail acetylation on structure is minimal. At this time, we do not have access to methods that allow us to acetylate specific lysines in a preparative manner and with 100% efficiency (to prepare defined samples for physical studies). A more detailed study using a histone specifically acetylated by peptide ligation is underway and will be the subject of a later publication.
Taken together, our analysis of entry–exit DNA unwrapping, bulk stability, structural heterogeneity during disassembly and dimer exchange between nucleosomes convincingly show that histones H3 and H4 can assume distinct roles in controlling nucleosome architecture. In combination with acetylated H3, H4-acetylation opposes destabilization and reduces dimer exchange. Tail-selective histone modifications thus provide another modus operandi to spatially control structural properties in different regions of the nucleosome.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We gratefully thank C.A.M. Seidel for helpful discussions.
FUNDING
DFG [La 500/18-1, La 500/18-2 to J.L.]. Funding for open access charge: DFG [La 500/18-2].
Conflict of interest statement. None declared.
REFERENCES
- 1.Zentner G.E., Henikoff S. Regulation of nucleosome dynamics by histone modifications. Nat. Struct. Mol. Biol. 2013;20:259–266. doi: 10.1038/nsmb.2470. [DOI] [PubMed] [Google Scholar]
- 2.Olins A.L., Olins D.E. Spheroid chromatin units (v bodies) Science. 1974;183:330–332. doi: 10.1126/science.183.4122.330. [DOI] [PubMed] [Google Scholar]
- 3.Anderson J.D., Lowary P.T., Widom J. Effects of histone acetylation on the equilibrium accessibility of nucleosomal DNA target sites. J. Mol. Biol. 2001;307:977–985. doi: 10.1006/jmbi.2001.4528. [DOI] [PubMed] [Google Scholar]
- 4.Li G., Levitus M., Bustamante C., Widom J. Rapid spontaneous accessibility of nucleosomal DNA. Nat. Struct. Mol. Biol. 2005;12:46–53. doi: 10.1038/nsmb869. [DOI] [PubMed] [Google Scholar]
- 5.Flaus A., Owen-Hughes T. Mechanisms for nucleosome mobilization. Biopolymers. 2003;68:563–578. doi: 10.1002/bip.10323. [DOI] [PubMed] [Google Scholar]
- 6.Böhm V., Hieb A.R., Andrews A.J., Gansen A., Rocker A., Toth K., Luger K., Langowski J. Nucleosome accessibility governed by the dimer/tetramer interface. Nucleic Acids Res. 2011;39:3093–3102. doi: 10.1093/nar/gkq1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gordon F., Luger K., Hansen J.C. The core histone N-terminal tail domains function independently and additively during salt-dependent oligomerization of nucleosomal arrays. J. Biol. Chem. 2005;280:33 701–33 706. doi: 10.1074/jbc.M507048200. [DOI] [PubMed] [Google Scholar]
- 8.Owen D.J., Ornaghi P., Yang J.C., Lowe N., Evans P.R., Ballario P., Neuhaus D., Filetici P., Travers A.A. The structural basis for the recognition of acetylated histone H4 by the bromodomain of histone acetyltransferase gcn5p. EMBO J. 2000;19:6141–6149. doi: 10.1093/emboj/19.22.6141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garcia-Ramirez M., Rocchini C., Ausio J. Modulation of chromatin folding by histone acetylation. J. Biol. Chem. 1995;270:17 923–17 928. doi: 10.1074/jbc.270.30.17923. [DOI] [PubMed] [Google Scholar]
- 10.Wang X.Y., He C., Moore S.C., Ausio J. Effects of histone acetylation on the solubility and folding of the chromatin fiber. J. Biol. Chem. 2001;276:12 764–12 768. doi: 10.1074/jbc.M100501200. [DOI] [PubMed] [Google Scholar]
- 11.Brower-Toland B., Wacker D.A., Fulbright R.M., Lis J.T., Kraus W.L., Wang M.D. Specific contributions of histone tails and their acetylation to the mechanical stability of nucleosomes. J. Mol. Biol. 2005;346:135–146. doi: 10.1016/j.jmb.2004.11.056. [DOI] [PubMed] [Google Scholar]
- 12.Tse C., Fletcher T.M., Hansen J.C. Enhanced transcription factor access to arrays of histone H3/H4 tetramer.DNA complexes in vitro: implications for replication and transcription. Proc. Natl. Acad. Sci. U.S.A. 1998;95:12 169–12 173. doi: 10.1073/pnas.95.21.12169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toth K., Brun N., Langowski J. Chromatin compaction at the mononucleosome level. Biochemistry. 2006;45:1591–1598. doi: 10.1021/bi052110u. [DOI] [PubMed] [Google Scholar]
- 14.Polach K.J., Lowary P.T., Widom J. Effects of core histone tail domains on the equilibrium constants for dynamic DNA site accessibility in nucleosomes. J. Mol. Biol. 2000;298:211–223. doi: 10.1006/jmbi.2000.3644. [DOI] [PubMed] [Google Scholar]
- 15.Kurdistani S.K., Tavazoie S., Grunstein M. Mapping global histone acetylation patterns to gene expression. Cell. 2004;117:721–733. doi: 10.1016/j.cell.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 16.Agricola E., Verdone L., Di Mauro E., Caserta M. H4 acetylation does not replace H3 acetylation in chromatin remodelling and transcription activation of Adr1-dependent genes. Mol. Microbiol. 2006;62:1433–1446. doi: 10.1111/j.1365-2958.2006.05451.x. [DOI] [PubMed] [Google Scholar]
- 17.Allahverdi A., Yang R., Korolev N., Fan Y., Davey C.A., Liu C.F., Nordenskiold L. The effects of histone H4 tail acetylations on cation-induced chromatin folding and self-association. Nucleic Acids Res. 2011;39:1680–1691. doi: 10.1093/nar/gkq900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferreira H., Somers J., Webster R., Flaus A., Owen-Hughes T. Histone tails and the H3 alphaN helix regulate nucleosome mobility and stability. Mol. Cell. Biol. 2007;27:4037–4048. doi: 10.1128/MCB.02229-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamiche A., Kang J.G., Dennis C., Xiao H., Wu C. Histone tails modulate nucleosome mobility and regulate ATP-dependent nucleosome sliding by NURF. Proc. Natl. Acad. Sci. U.S.A. 2001;98:14316–14321. doi: 10.1073/pnas.251421398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nurse N.P., Jimenez-Useche I., Smith I.T., Yuan C. Clipping of flexible tails of histones H3 and H4 affects the structure and dynamics of the nucleosome. Biophys. J. 2013;104:1081–1088. doi: 10.1016/j.bpj.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Potoyan D.A., Papoian G.A. Energy landscape analyses of disordered histone tails reveal special organization of their conformational dynamics. J. Am. Chem. Soc. 2011;133:7405–7415. doi: 10.1021/ja1111964. [DOI] [PubMed] [Google Scholar]
- 22.Potoyan D.A., Papoian G.A. Regulation of the H4 tail binding and folding landscapes via Lys-16 acetylation. Proc. Natl. Acad. Sci. U.S.A. 2012;109:17 857–17 862. doi: 10.1073/pnas.1201805109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Förster T. Energiewanderung und Fluoreszenz. Naturwissenschaften. 1946;6:166–175. [Google Scholar]
- 24.Toth K., Brun N., Langowski J. Trajectory of nucleosomal linker DNA studied by fluorescence resonance energy transfer. Biochemistry. 2001;40:6921–6928. doi: 10.1021/bi002695m. [DOI] [PubMed] [Google Scholar]
- 25.Tomschik M., Zheng H., van Holde K., Zlatanova J., Leuba S.H. Fast, long-range, reversible conformational fluctuations in nucleosomes revealed by single-pair fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. U.S.A. 2005;102:3278–3283. doi: 10.1073/pnas.0500189102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Torres T., Levitus M. Measuring conformational dynamics: a new FCS-FRET approach. J. Phys. Chem. B. 2007;111:7392–7400. doi: 10.1021/jp070659s. [DOI] [PubMed] [Google Scholar]
- 27.Koopmans W.J., Brehm A., Logie C., Schmidt T., van Noort J. Single-pair FRET microscopy reveals mononucleosome dynamics. J. Fluoresc. 2007;17:785–795. doi: 10.1007/s10895-007-0218-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jimenez-Useche I., Yuan C. The effect of DNA CpG methylation on the dynamic conformation of a nucleosome. Biophys. J. 2012;103:2502–2512. doi: 10.1016/j.bpj.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neumann H., Hancock S.M., Buning R., Routh A., Chapman L., Somers J., Owen-Hughes T., van Noort J., Rhodes D., Chin J.W. A method for genetically installing site-specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Mol. Cell. 2009;36:153–163. doi: 10.1016/j.molcel.2009.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang J.G., Narlikar G.J. FRET-based methods to study ATP-dependent changes in chromatin structure. Methods. 2007;41:291–295. doi: 10.1016/j.ymeth.2006.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deindl S., Hwang W.L., Hota S.K., Blosser T.R., Prasad P., Bartholomew B., Zhuang X. ISWI remodelers slide nucleosomes with coordinated multi-base-pair entry steps and single-base-pair exit steps. Cell. 2013;152:442–452. doi: 10.1016/j.cell.2012.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gansen A., Valeri A., Hauger F., Felekyan S., Kalinin S., Toth K., Langowski J., Seidel C.A. Nucleosome disassembly intermediates characterized by single-molecule FRET. Proc. Natl. Acad. Sci. U.S.A. 2009;106:15 308–15 313. doi: 10.1073/pnas.0903005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kelbauskas L., Chan N., Bash R., DeBartolo P., Sun J., Woodbury N., Lohr D. Sequence-dependent variations associated with H2A/H2B depletion of nucleosomes. Biophys. J. 2008;94:147–158. doi: 10.1529/biophysj.107.111906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee J.Y., Lee T.H. Effects of DNA methylation on the structure of nucleosomes. J. Am. Chem. Soc. 2012;134:173–175. doi: 10.1021/ja210273w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simon M., North J.A., Shimko J.C., Forties R.A., Ferdinand M.B., Manohar M., Zhang M., Fishel R., Ottesen J.J., Poirier M.G. Histone fold modifications control nucleosome unwrapping and disassembly. Proc. Natl. Acad. Sci. U.S.A. 2011;108:12 711–12 716. doi: 10.1073/pnas.1106264108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee J.Y., Wei S., Lee T.H. Effects of histone acetylation by Piccolo NuA4 on the structure of a nucleosome and the interactions between two nucleosomes. J. Biol. Chem. 2011;286:11 099–11 109. doi: 10.1074/jbc.M110.192047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gansen A., Toth K., Schwarz N., Langowski J. Structural variability of nucleosomes detected by single-pair Förster resonance energy transfer: histone acetylation, sequence variation, and salt effects. J. Phys. Chem. B. 2009;113:2604–2613. doi: 10.1021/jp7114737. [DOI] [PubMed] [Google Scholar]
- 38.Lowary P.T., Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- 39.Gansen A., Hieb A.R., Bohm V., Toth K., Langowski J. Closing the Gap between Single Molecule and Bulk FRET Analysis of Nucleosomes. PLoS One. 2013;8:e57018. doi: 10.1371/journal.pone.0057018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hieb A.R., D'Arcy S., Kramer M.A., White A.E., Luger K. Fluorescence strategies for high-throughput quantification of protein interactions. Nucleic Acids Res. 2012;40:e33. doi: 10.1093/nar/gkr1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clegg R.M. Fluorescence resonance energy transfer and nucleic acids. Methods Enzymol. 1992;211:353–388. doi: 10.1016/0076-6879(92)11020-j. [DOI] [PubMed] [Google Scholar]
- 42.Wolffe A. Chromatin: Structure and Function. 2nd edn. London: Academic Press; 1995. [Google Scholar]
- 43.Lowary P.T., Widom J. Nucleosome packaging and nucleosome positioning of genomic DNA. Proc. Natl. Acad. Sci. U.S.A. 1997;94:1183–1188. doi: 10.1073/pnas.94.4.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Musselman C.A., Gibson M.D., Hartwick E.W., North J.A., Gatchalian J., Poirier M.G., Kutateladze T.G. Binding of PHF1 Tudor to H3K36me3 enhances nucleosome accessibility. Nat. Commun. 2013;4:2969. doi: 10.1038/ncomms3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen Y., Tokuda J.M., Topping T., Sutton J.L., Meisburger S.P., Pabit S.A., Gloss L.M., Pollack L. Revealing transient structures of nucleosomes as DNA unwinds. Nucleic Acids Res. 2014;42:8767–8776. doi: 10.1093/nar/gku562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gurunathan K., Levitus M. Single-molecule fluorescence studies of nucleosome dynamics. Curr. Pharmaceut. Biotechnol. 2009;10:559–568. doi: 10.2174/138920109788922074. [DOI] [PubMed] [Google Scholar]
- 47.Koopmans W.J., Buning R., Schmidt T., van Noort J. spFRET using alternating excitation and FCS reveals progressive DNA unwrapping in nucleosomes. Biophys. J. 2009;97:195–204. doi: 10.1016/j.bpj.2009.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tóth K., Böhm V., Sellmann C., Danner M., Hanne J., Berg M., Barz I., Gansen A., Langowski J. Histone- and DNA sequence-dependent stability of nucleosomes studied by single-pair FRET. Cytometry A. 2013;83:839–846. doi: 10.1002/cyto.a.22320. [DOI] [PubMed] [Google Scholar]
- 49.Davey C.A., Sargent D.F., Luger K., Maeder A.W., Richmond T.J. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 a resolution. J. Mol. Biol. 2002;319:1097–1113. doi: 10.1016/S0022-2836(02)00386-8. [DOI] [PubMed] [Google Scholar]
- 50.Korolev N., Zhao Y., Allahverdi A., Eom K.D., Tam J.P., Nordenskiold L. The effect of salt on oligocation-induced chromatin condensation. Biochem. Biophys. Res. Commun. 2012;418:205–210. doi: 10.1016/j.bbrc.2011.12.112. [DOI] [PubMed] [Google Scholar]
- 51.Mangenot S., Raspaud E., Tribet C., Belloni L., Livolant F. Interactions between isolated nucleosome core particles: a tail-bridging effect. Eur. Phys. J. E. 2002;7:221–231. [Google Scholar]
- 52.Schalch T., Duda S., Sargent D.F., Richmond T.J. X-ray structure of a tetranucleosome and its implications for the chromatin fibre. Nature. 2005;436:138–141. doi: 10.1038/nature03686. [DOI] [PubMed] [Google Scholar]
- 53.Ye J., Ai X., Eugeni E.E., Zhang L., Carpenter L.R., Jelinek M.A., Freitas M.A., Parthun M.R. Histone H4 lysine 91 acetylation a core domain modification associated with. Mol. Cell. 2005;18:123–130. doi: 10.1016/j.molcel.2005.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Di Cerbo V., Mohn F., Ryan D.P., Montellier E., Kacem S., Tropberger P., Kallis E., Holzner M., Hoerner L., Feldmann A., et al. Acetylation of histone H3 at lysine 64 regulates nucleosome dynamics and facilitates transcription. eLife. 2014;3:e01632. doi: 10.7554/eLife.01632. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.