

Abstract
Cycas multipinnata C.J. Chen & S.Y. Yang is a cycad endemic to the Red River drainage region that occurs under evergreen forest on steep limestone slopes in Southwest China and northern Vietnam. It is listed as endangered due to habitat loss and over-collecting for the ornamental plant trade, and only several populations remain. In this study, we assess the genetic variation, population structure, and phylogeography of C. multipinnata populations to help develop strategies for the conservation of the species. 60 individuals from six populations were used for chloroplast DNA (cpDNA) sequencing and 100 individuals from five populations were genotyped using 17 nuclear microsatellites. High genetic differentiation among populations was detected, suggesting that pollen or seed dispersal was restricted within populations. Two main genetic clusters were observed in both the cpDNA and microsatellite loci, corresponding to Yunnan China and northern Vietnam. These clusters indicated low levels of gene flow between the regions since their divergence in the late Pleistocene, which was inferred from both Bayesian and coalescent analysis. In addition, the result of a Bayesian skyline plot based on cpDNA portrayed a long history of constant population size followed by a decline in the last 50,000 years of C. multipinnata that was perhaps affected by the Quaternary glaciations, a finding that was also supported by the Garza-Williamson index calculated from the microsatellite data. The genetic consequences produced by climatic oscillations and anthropogenic disturbances are considered key pressures on C. multipinnata. To establish a conservation management plan, each population of C. multipinnata should be recognized as a Management Unit (MU). In situ and ex situ actions, such as controlling overexploitation and creating a germplasm bank with high genetic diversity, should be urgently implemented to preserve this species.
Introduction
The Cycad is considered an old lineage because of its ancient morphological characters and fossil records which could date to the Early Permian [1] or possibly even the late Carboniferous period (approximately 300 million years ago) [2]. Cycads became a dominant plant group during the Mesozoic, as shown by numerous fossils of megasporophylls and ovalate strobili, as well as vegetative shoots, leaves and trunks [3–9]. However, one recent fossil-calibrated molecular phylogenetic study based on multiple DNA sequence data proposed that extant Cycads originated no more than 12 million years ago; the Cycads underwent a recent synchronous global rediversification beginning in the late Miocene, followed by a slowdown towards the recent [10].
Cycas, as the basal lineage of the living cycads supported by both phylogenetic studies and the recent obtained genome size of nuclear DNA [11–14], is the sole genus of Cycadaceae, and one distinguishing character of Cycas from other Cycads in morphology is its leaflets with an obvious midrib, lacking lateral veins. The leaf of Cycas is pinnate or rarely bipinnate, and Cycas multipinnata C.J. Chen & S.Y. Yang is one of the four species characterized by the latter [15]. The somatic chromosome number of C. multipinnata is 2n = 2x = 22 [16], and the karyotype is classified as 3B according to Stebbin’s category.
Cycas multipinnata distributes in the Red River drainage zone under an evergreen forest canopy on the sharp limestone slopes of southwest China (Yunnan Province) and northern Vietnam (Yen Bai and Tuyen Quang Province) [17,18]. The Red River (also called the Yuanjiang River in China and Song Hong in Vietnam) flows for 692 kilometers in China, southeastward across Hekou County into northern Vietnam, before emptying into the sea. The area, through which the Red River flows, in geology terms, is known as the Red River fault zone. The collision of the India Plate with Asia caused the Indochina Plate to move approximately 700 km southeast relative to the South China Block along the Red River fault line [19,20]. At least 24 Cycas species occur in this small region, which ranges from the coast of Vietnam well into the interior of China along the Red River. Cycas multipinnata is one of the 17 Cycas species endemic to this area [21]. However, the wild individuals of C. multipinnata in both China and Vietnam declined dramatically in recent decades due to the anthropogenic disturbances, particularly habitat loss and over-exploitation for the ornamental plant trade. Cycas multipinnata was assessed as Endangered (EN) by the IUCN (the International Union for the Conservation of Nature) Categories and Criteria system [21,22].
Chloroplast DNA of Cycas is maternally inherited [23], so it is dispersed by seeds; its nuclear microsatellite is biparental inheritance, which is dispersed via both seeds and pollen. In this study, three intergenic spacers of chloroplast DNA and 17 microsatellite loci were employed to assess the genetic diversity, the structure and the population dynamics of C. multipinnata. Further, we discuss the causes of the genetic consequences, such as an environment with climatic oscillations or human impacts. These results show the urgency and significant implications of the management and recovery of this endangered species.
Materials and Methods
Ethics Statement
Cycas multipinnata is the First Grade conservation plant in China [24]. It was assessed as Endangered (EN) by the IUCN (the International Union for the Conservation of Nature) [22]. We got the permission of the Wildlife Protection and Administration Office under the Forestry Department of Yunnan, China and the permission of owners for some private cultivated plants. There is cooperation agreement between the Center for Plant Conservation (CPC) of Vietnam Union of Science and Technology Association and the Kunming Institute of Botany (KIB) of the Chinese Academy of Sciences on the joint exploration to the flora of globally threatened plant. We also got the permission of local forestry department in Vietnam when collecting the Cycas samples in the wild. The sampling process was under the guidance of local rangers. Our sampling will not affect the regular growth of C. multipinnata, and it was solely used for scientific research.
Population sampling and DNA extraction
A total of 105 individual samples were collected from six populations of C. multipinnata (four populations, including a cultivated SD population in a village because the nearby wild habitat was totally destroyed, were sampled in Yunnan Province, China and two populations were sampled in northern Vietnam). All six populations were located north of the Red River. Of the 105 samples, 60 individuals from the six populations were used for chloroplast DNA sequencing. The population known as SZD was eliminated from SSR analysis because there were only five individuals in the population. A total of 100 individuals from five populations were used for the microsatellite study. Information on each sampling location and the number of individuals from each population that were used in DNA sequences and SSR analysis is presented in Table 1 and Fig. 1, respectively.
Table 1. Information on sampling sites of Cycas multipinnata.
| Population(sampling site) | Code | Latitude (N) | Longitude (E) | Altitude (m) | N(for SSR) | Hd | π |
|---|---|---|---|---|---|---|---|
| Shi dong, Yunnan, China | SD | 22°59′ | 103°24′ | 254 | 10(20) | 0.6000 | 0.00072 |
| Shibanzhai, Yunnan, China | SBZ | 22°53′ | 103°37′ | 580 | 10(20) | 0.4667 | 0.00024 |
| Ganlongjing, Yunnan, China | GLJ | 22 °56′ | 103°31′ | 600 | 13(20) | 0.2821 | 0.00014 |
| Shazudi, Yunnan, China | SZD | 23°06′ | 103°23′ | 1000 | 5(0) | 0 | 0 |
| Ham Yen, Tuyen Quang, Vietnam | HY | 22°07′ | 105°02′ | 50 | 12(23) | 0 | 0 |
| Yen Binh, Yen Bai, Vietnam | YB | 21°56′ | 104°53′ | 225 | 10(17) | 0 | 0 |
N: the number of individuals for cpDNA sequencing, and the number for SSR genotyping
Fig 1. Geographic distribution and evolutionary relationships of the eight cpDNA haplotypes in Cycas multipinnata.

A: General position of map B; B: Pie charts show the proportions of haplotypes within each population. C: The network of the eight cpDNA haplotypes. The numbers on the branches represent the number of mutations between two connected haplotypes and black dots (mv) represent missing haplotypes (not sampled or extinct).
Healthy leaves were dried and preserved in silica gel immediately after being collected. Genomic DNA was extracted from the leaves, which was ground into a powder after being frozen with liquid nitrogen, following the cetyltrimethyl ammonium bromide (CTAB) method [25]. The total DNA was dissolved in TE (Tris-EDTA) buffer and used as the template in the polymerase chain reaction (PCR).
Chloroplast DNA analysis
Three cpDNA regions atpB-rbcL, psbA-trnH and psbB-psbH were selected. The PCR amplifying conditions and primers of atpB-rbcL and psbA-trnH were identical to those described by Zhan [26]. The primers and cycles of psbB-psbH fragment were as follows: psbB (F) 5′-TCCAAAAACTGGGAGATCCAAC-3′, psbH (R) 5′-TCAATGGTCTGTGTAGCCAT-3′ [27]; one cycle of four min at 80°C, 30 cycles of 50 s at 94°C, 50 s at 52°C, and 50 s at 72°C, with a final extension step for eight min at 72°C.
Purified PCR productions were sequenced in both directions using the same primers for the amplification reactions, using an ABI 3730 automated sequencer at Shanghai Majorbio Bio-pharm Technology Co., Ltd.
Microsatellite genotyping
Microsatellite primers of Cycas have been developed by several research groups. 17 variable microsatellite loci were selected from the published 65 microsatellite loci [28–34] that developed in other species of Cycas to investigate the genetic diversity of C. multipinnata (Table A in S1 File). The PCR amplification was performed in a 25 μL reaction, with the forward primers labeled with fluorescent dye (FAM, TEMRA or HEX) and visualized on an ABI 3730xl Capillary DNA Analyzer by Sangon Biotech (Shanghai) Co., Ltd. Fragment sizes were assessed using GeneMapper version 4.0.
Data analysis of cpDNA
After being edited and jointed in the SeqMan of software package (DNA Star), sequences were aligned using Clustal X version 1.81 [35], and fine adjustments were performed manually using the BioEdit version 7.0.1 [36] software. The incongruence length difference test was conducted using the partition homogeneity test in PAUP version 4.0b10 [37] to examine the congruence between datasets. Insertions and/or deletions were coded as binary characters. All character states were specified as unordered and equally weighted.
DnaSP version 5.10 [38] was used to calculate the number of haplotypes, variable sites and nucleotide diversity per site (π). A map of the geographic distribution of haplotypes was drawn via ArcGIS 10.2 [39]. Permut 1.0 (http://www.pierroton.inra.fr/genetics/labo/Software/Permut) was used to calculate the within-population diversity (HS), the total diversity (HT) and two measures of population differentiation, GST and NST [40]. Arlequin version 3.11 [41] was used for the molecular variance (AMOVA) analysis [42] to estimate genetic variations within and among populations.
We calculated the genetic distance (GD) among the six populations following the formula GD = FST/ (1-FST) [43] using Arlequin version 3.11 [41]. Geography distance (GGD) was calculated via GenAlEx version 6.4.1 [44]. A Mantel test was designed to test the relationship of the elements of two matrices in GenAlEx version 6.4.1 [44].
The genealogical haplotype network was constructed using Network version 4.6.1.2 (http://www.fluxus-engineering.com/sharenet.htm) following the media-joining calculation [45]. Phylogenetic relationships among the cpDNA haplotypes were reconstructed by Maximum Parsimony (MP), Neighbor Joining (NJ) and Maximum Likelihood (ML) analyses using PAUP version 4.0b10 [37] and Bayesian methods implemented in MrBayes version 3.1.2 [46], with C. panzhihuaensis serving as the outgroup. BEAST version 1.7 [47] was used to estimate the ages of the most recent common ancestor (TMRCA). The analysis was run for 107 iterations with a burn-in of 106 under the HKY (Hasegawa-Kishino-Yano) nucleotide substitution model, which was determined to be the most suitable model by Modeltest in Mega 6.06 [48] and a strict molecular clock. A well-documented evolutionary rate for cpDNA, 1.01×10-9 substitution per site per year [49,50] for synonymous sites, was used to estimate the coalescent time between lineages across haplotypes.
A Bayesian skyline plot was constructed by BEAST version 1.7 [47] and TRACER version 1.5 [51] to infer the past population dynamics. To infer the possible demographic expansion of C. multipinnata, mismatch distribution analysis based on the sudden population expansion model using the observed number of differences between pairs of haplotypes were conducted with DnaSP version 5.10 [38]. The sum of squared deviations (SSD) between the observed and expected mismatch distributions, the raggedness index (HRag) and their significance [52] were calculated in Arlequin version 3.11 [41]. We also conducted neutrality tests, with Tajima’s D [53], Fu and Li’s F*& D* [54], as well as Fu’s F S [55], using Arlequin version 3.11 [41], to detect departures from the population equilibrium.
Microsatellite fingerprinting analysis
The dataset was edited and transformed to other formats in GenAlEx version 6.4.1 [44]. We tested for evidence of selection on each locus using LOSITAN [56], which can detect excessively high or low FST compared with neutral expectations. Deviations from Hardy-Weinberg equilibrium (HWE) could indicate the presence of population structure or inbreeding [57]. HWE was tested for each locus and each population with default parameters using Genepop version 4.1.4 [58]. Linkage disequilibrium was investigated at the 5% statistical significance level among loci pairs with 1000 permutations using Arlequin version 3.11 [41].
The indices of genetic diversity within populations, such as the number of alleles (NT), the number of private alleles (AP), the mean number of alleles (NA), the effective number of alleles (NE), the observed heterozygosity (HO), the expected heterozygosity (HE), the information index (I), the fixation index (F) and the percentage of polymorphic loci (PPB) were calculated using GenAlEx version 6.4.1 [44]. Differentiation between pairs of populations was computed using FST and tested with GenAlEx version 6.4.1. Allelic richness (AR) was estimated with FSTAT, version 2.9.3) was estimated with FSTAT, version 2.9.3 [59].
As in the analysis of cpDNA, genetic distance (GD) and geography distance (GGD) among the five populations was calculated using the Arlequin version 3.11 [41] and GenAlEx version 6.4.1 [44], respectively. A Mantel test was designed to test whether this species was isolated by distance (IBD) using GenAlEx version 6.4.1 [44]. Gene flow between pairs of populations was estimated based on Wright’s principles [60], Nm = (1-FST)/4 FST.
An individual-based principal coordinate analysis (PCO) was conducted in the MVSP version 3.12 software [61] using the genetic distances among SSR genotypes. The PCO could visualize genetic relationships among these 100 individuals from the five populations. We also conducted a Bayesian analysis of population structure on the SSR data using STRUCTURE version 2.3 [62]. The admixture model was used and the posterior probability of the grouping number (K = 1~8) was estimated by the Markov chain Monte Carlo (MCMC) method with 10 separate runs to evaluate the consistency of the results. Each run was estimated as 100,000 steps, with a 100,000-step burn-in. The best fit number of grouping [63] was evaluated using ΔK in the STRUCTURE HARVESTER v. 0.6.93 tool [64]. Finally, we identified geographical locations where major genetic barriers among populations might occur with a barrier boundary analysis using BARRIER version 2.2 [65], based on genetic distance matrices.
A heterozygosity excess test at the population level from BOTTLENECK 1.2.02 was used to detect the recent population bottleneck [66]. The computation was performed under the two phased model (TPM) [67]. Two methods, the Sign test and the Wilcoxon test, which are powerful and robust statistics when using less than 20 polymorphic loci, were executed in the model. Second, we used Bottleneck to examine the distribution of the alleles’ frequencies for a so-called mode-shift that discriminates recently bottlenecked from stable populations. The method implemented in the bottleneck has low power [68] unless the decline is greater than 90%, so we computed the Garza-Williamson index, a statistic that can detect population bottlenecks using Arlequin version 3.11. The Garza—Williamson index [69] is the mean ratio of the number of alleles at a given locus to the range in allele size, i.e., M = (k/r), where k is the number of alleles and r is the allelic range (i.e., the difference in repeat units between the shortest and the longest alleles at a locus). This measure is based on the assumption that in a bottleneck event, the number of alleles decreases faster than the allelic range because the latter is only reduced if the shortest and/or longest allele is lost, whereas the loss of any allele reduces the former. The Garza and Williamson found critical values for M < 0.68, which indicated a bottleneck, and M > 0.80, which indicated no reduction of effective population size.
Effective population sizes (Ne) are among the most important parameters in wildlife management and conservation because they can inform management and help predict the extinction risk of populations. We estimated effective population sizes (Ne) using the linkage disequilibrium (LD) method in LDNe [70] at three levels of lowest allele frequency (0.01, 0.02, 0.05) for a 95% confidence interval.
Results
Sequence variation and genetic diversity of cpDNA
The three cpDNA fragments, atpB-rbcL (delete the poly T), psbA-trnH and psbB-psbH, are 719, 597 and 644 bp in length, respectively (GenBank accession numbers: KP335666—KP335683). The partition-homogeneity test indicated that data sets of these cpDNA fragments are significantly congruent (P = 1). The 1960 bp combined cpDNA data set had 14 parsimony-informative polymorphic sites and 8 haplotypes (H1–H8) (Table B in S1 File). The haplotype frequencies in each population and geographical distribution are presented in Fig. 1. H2 is the most abundant haplotype. The two populations in Vietnam have only one unique haplotype, respectively. The haplotype diversity (Hd) is 0.7718, and the nucleotide diversity per site (π) is 0.00149. The total diversity (HT) is 0.896, and the within-population diversity (HS) is 0.225.
Significant population differentiation was observed, with a GST = 0.749 and an NST = 0.922, and the permutation test showed that GST and NST were not significantly different from each other (NST > GST, P > 0.05). The results of the AMOVA analysis indicated that 92.3% variation occurs among the populations (Table 2), with FST = 0.92304.
Table 2. Results of Analysis of Molecular Variance (AMOVA, cpDNA/nSSR) of C. multipinnata.
| Source of variation | d.f. | Sum of squares | Variance components | Percent of variation (%) |
|---|---|---|---|---|
| Among populations | 5/4 | 144.774/228.593 | 2.90808/1.35128 Va | 92.304/29.57 |
| Within population | 54/195 | 13.092/627.642 | 0.24254/3.21868 Vb | 7.696/70.43 |
d.f.: degree of freedom
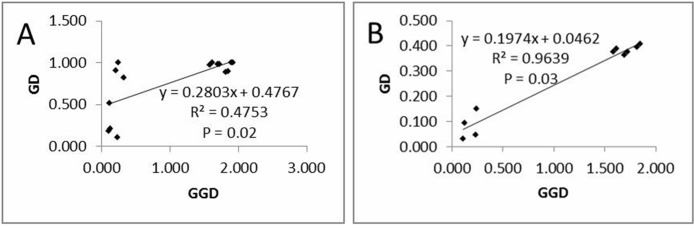
The Mantel test (Fig. 2A) showed that there was a significant positive correlation (P < 0.05) between the genetic distance (GD) and the geographic distance (GGD), suggesting that C. multipinnata was isolated by distance.
Fig 2. Scatterplots showing the relationship between genetic distance (GD) and geographic distance (GGD) based on cpDNA (A) and SSR data (B).
Phylogenetic inferences based on the combined cpDNA sequence data
The genealogical haplotype network is shown in Fig. 1(C). In this figure, we can observe that each of the two populations in Vietnam has a unique haplotype. Additionally, there were at least eight mutations between the haplotypes (H7 or H8) in Vietnam and haplotypes (H1–H6) in China. The topology of the Bayesian tree of the eight haplotypes detected from the combined cpDNA of C. multipinnata, with C. panzhihuaensis as outgroup, is shown in Fig. 3, and the major clades were the same as the topology of MP and ML trees. Two clades were identified in the tree and the network. Clade I contained two haplotypes (H7, H8) from the northern Vietnam, and clade II included the remaining haplotypes that occurred in Yunnan provinces, China. The date of the most recent common ancestor (TMRCA) of the China clade and Vietnam clade was approximately 1.0307 million years ago (MYA).
Fig 3. Phylogenetic relationships of the cpDNA haplotypes based on the Bayesian analysis.

Demographic analysis of the cpDNA
The mismatch distributions under the sudden expansion model for all the populations were multimodal (Fig. 4), with an SSD of 0.07017 (P = 0.08833) and a HRag of 0.14270 (P = 0.14333), indicating no recent population expansion. This conclusion was also supported by the results of the Neutrality Test, Tajima’s D 3.09754 (P < 0.01), Fu and Li’s D* 1.16049, Fu and Li’s F* 2.09321 (P < 0.01), Fu’ Fs 5.672, Tajima’s D and Fu and Li’s F* were all positive values. According to the variation pattern in cpDNA, the Bayesian skyline plot was reconstructed. It showed that the demographic scenario for population of C. multipinnata was a long history of constant population size, followed by a decline over the last 50,000 years with no subsequent expansion (Fig. 5).
Fig 4. Mismatch distributions of cpDNA haplotypes based on pairwise sequence difference plotted against the frequency of occurrence.

Fig 5. Bayesian skyline plot based on cpDNA for the effective population size fluctuation throughout time.

black line, median estimation; area between gray lines, 95% confidence interval.
Genetic diversity of microsatellite loci
Only the locus Cha5 was in the positive selection and it did not reach the significant level of the Fst-outlier. The other sixteen loci fell under the neutral selection. Some of the loci in populations deviated from the Hardy-Weinberg expectations (Table C in S1 File) due to the deficiency of the heterozygotes. All 17 microsatellite loci used for the further analysis.
All 17 microsatellite loci were polymorphic in C. multipinnata, and a total of 199 alleles were identified. The total number of alleles (NT) of each population varied from 71 (HY) to 88 (GLJ) (Table 3). The two populations in Vietnam (YB, HY) have more private alleles (AP) than that in the three populations in China (Table 3). The mean number of alleles (NA) and the effective number of alleles (NE) for each population ranged from 4.176 to 5.176 and 2.314 to 3.197, respectively (Table 3). The observed heterozygosity (Ho) and expected heterozygosity (HE) were 0.367 to 0.410 and 0.449 to 0.551, respectively (Table 3). The percentage of polymorphic loci (PPB) had higher values that varied from 88.24% to 100% (Table 3).
Table 3. Genetic variability of microsatellites within C. multipinnata populations.
| Pop. | NT | NP | AR | NA | NE | HO | HE | I | F | PPB (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| SD | 80 | 15 | 3.703 | 4.706 | 2.314 | 0.367 | 0.449 | 0.910 | 0.274 | 100 |
| SBZ | 82 | 8 | 3.724 | 4.824 | 2.580 | 0.375 | 0.462 | 0.943 | 0.243 | 94.12 |
| GLJ | 88 | 18 | 4.070 | 5.176 | 2.885 | 0.407 | 0.500 | 1.031 | 0.199 | 94.12 |
| HY | 71 | 26 | 3.382 | 4.176 | 2.572 | 0.378 | 0.523 | 0.966 | 0.315 | 94.12 |
| YB | 82 | 30 | 4.253 | 4.824 | 3.197 | 0.410 | 0.551 | 1.122 | 0.207 | 88.24 |
| Mean | 80.6 | 19.4 | 3.826 | 4.741 | 2.709 | 0.387 | 0.497 | 0.994 | 0.248 | 94.12 |
Differing from the result of the cpDNA sequences analysis, the AMOVA analysis (Table 2) of the 17 microsatellite loci indicated that 70.43% genetic variation occurs within populations. The mean FST across all loci and populations was 0.29569 (P < 0.0001), and the FST values for each population (all P < 0.0001) ranged from 0.29386 to 0.29761. The Mantel test (Fig. 2B) based on the SSR data also showed that there was a significant positive correlation (P < 0.05) between the genetic distance (GD) and the geographic distance (GGD), suggesting that C. multipinnata was isolated by distance.
Gene flow between each pair of the five populations is shown in Table 4, and that between the populations GLJ and SBZ was the highest. The gene flow between populations from China and Vietnam was smaller than 1.
Table 4. Estimates of gene flow between each pair of the five populations of C. multipinnata.
| SD | SBZ | GLJ | HY | |
|---|---|---|---|---|
| SBZ | 4.9779 | |||
| GLJ | 2.4589 | 7.8722 | ||
| HY | 0.3628 | 0.3916 | 0.4160 | |
| YB | 0.4389 | 0.4116 | 0.4379 | 1.4188 |
The two-dimensional PCO (Fig. 6) indicated that these individuals could be approximately divided into two groups. One consisted of the three populations (SD, SBZ and GLJ) in China, and the other consisted of the two populations (HY and YB) in the northern Vietnam. The best and second fit numbers of grouping is inferred as two and four based on the ΔK evaluation (ΔK = 506.453 when K = 2 and ΔK = 3.357 when K = 4) in the Bayesian clustering analysis, when using the 17 microsatellite loci (Fig. 7). At K = 2, as in the PCO analysis, the C. multipinnata was divided into two major groups. The BARRIER indicated that there was only one barrier, with a 57.65% mean bootstrap value, between the populations in China and Vietnam (Fig. A in S2 File), which means that two clusters came to being among all the individuals of the five populations.
Fig 6. Principal coordinates analysis (PCO) of microsatellites from five populations and 100 individuals of C. multipinnata.
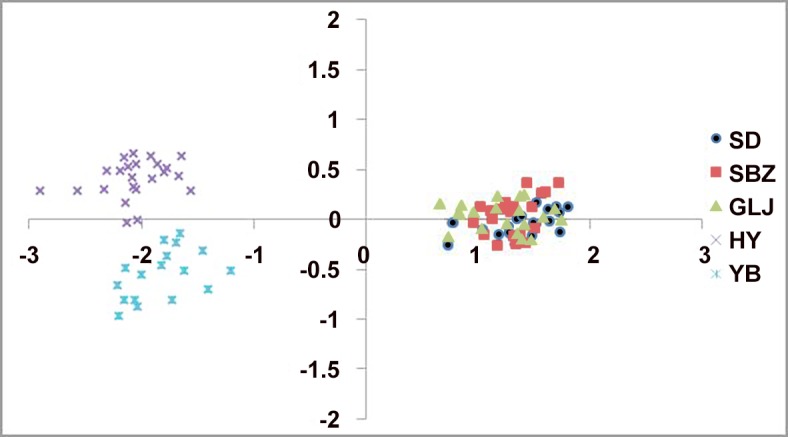
Fig 7. Structure analysis results using the admixture and correlated allele frequencies among populations model.

Population bottleneck analysis of the microsatellite loci
The probabilities of the Wilcoxon test and the sign test in the TPM model showed no significant difference (P > 0.05) except for the YB population (P < 0.01) (Table 5). The allele-distribution mode-shift of all the populations is a normal L-shaped distribution (Table 5). Taken together, bottleneck analysis did not yield evidence of any significant reduction of population size.
Table 5. Bottleneck analysis for the five populations of C. multipinnata based on SSR data.
| POP. | T.P.M. | Mode-Shift | G-W index | |
|---|---|---|---|---|
| Sign Test | Wilcoxon Test | |||
| SD | 0.09676 | 0.10889 | L-shaped | 0.40903 |
| SBZ | 0.40522 | 0.85364 | L-shaped | 0.34567 |
| GLJ | 0.38307 | 0.43068 | L-shaped | 0.33602 |
| HY | 0.41521 | 0.14543 | L-shaped | 0.31695 |
| YB | 0.00866** | 0.00025** | L-shaped | 0.33259 |
**: P<0.01, most significant difference; G-W index: Garza-Williamson index, the ratio of number of alleles to range in allele size
However, the Garza-Williamson indices (Table 5) were low, ranging from 0.31695 to 0.40903 across populations, which were lower than the critical value of 0.68 proposed by Garza and Williamson, suggesting past population size reductions.
The effective population sizes (Ne) calculated using the linkage disequilibrium (LD) method with LDNe at the three lowest allele frequency levels with a 95% confidence interval are shown in Table 6. All of the effective population sizes (Ne) in wild (SBZ, GLJ, HY, YB) are smaller than 100, and some are even smaller than 50.
Table 6. The effective population size of each population of C. multipinnata.
| population | Ne (Fl = 0.05) | Ne (Fl = 0.02) | Ne (Fl = 0.01) |
|---|---|---|---|
| SD | 39.9 | 151.7 | 151.7 |
| SBZ | 12.9 | 48.6 | 48.6 |
| GLJ | 7.4 | 16.7 | 16.7 |
| HY | 67.9 | 46.6 | 46.6 |
| YB | 21.7 | 80.5 | 80.5 |
Ne: Effective population; Fl: Lowest Alleles Frequency used
Discussion
The survival of a species relies on adequate genetic diversity to enable adaptation to a changing environment. We used two molecular markers, three chloroplast DNA and 17 nuclear microsatellite loci, to detect the exact genetic variation, differentiation, structure and demographic scenarios of C. multipinnata. The AMOVA analysis revealed that the variation partition of the three cpDNA intergenic spacers is 92.3% variation among populations and 7.7% within populations, whereas the 17 microsatellite loci indicated that 70.43% genetic variation occurs within populations and 29.57% among populations (Table 2). The cause of this discordance is that the microsatellite mutation rate (3.0×10-5 ~ 7.1×10-4 substitutions per site per year for synonymous sites [71,72]) is several orders of magnitude greater than that of cpDNA sequence (approximately 1.01×10-9 substitutions per site per year for synonymous sites [49,50]), and the microsatellite mutation could be transmitted to the next generation via pollen and seed, so during the same time in a population, the microsatellite DNA could accumulate more variations than cpDNA sequence. For microsatellite DNA evolving neutrally, the amount of polymorphism is expected to be directly proportional to the underlying mutation rate [73]. The two molecular markers have different mutation rate and dispersed mechanism, hence, they could reflect respective genetic events, and it is necessary to choose these two markers to study on the C. multipinnata.
Genetic variation and genetic differentiation
Generally speaking, the genetic diversity of C. multipinnata is moderate in cpDNA sequence. It is higher than that of C. debaoensis (atpB-rbcL and psbA-trnH, Hd = 0.49160, HS = 0.179, HT = 0.564, π = 0.00132) [26] but much lower than C. revoluta (atpB-rbcL, π = 0.0581) and C. taitungensis (atpB-rbcL, π = 0.0181) [74]. For C. simplicipinna, a unique cpDNA haplotype was detected in all seven populations from both China and Laos [75], so the haplotype diversity (Hd = 0.864) and the total diversity (HT = 1.000) is higher than C. multipinnata, whereas the within-population diversity (HS = 0.076) is lower. The genetic variation within populations for C. multipinnata, C. debaoensis, and C. simplicipinna is low.
In the cpDNA analysis of C. multipinnata, H2 is the predominant chloroplast haplotype among the eight haplotypes detected in the 60 individuals from six populations. The other five haplotypes in the four populations (SD, SBZ, GLJ and SZD) in China with short branches to H2 represent recently evolved haplotypes. A unique cpDNA haplotype is detected in each of the two Vietnamese populations. The cpDNA genetic diversity in China tends to be higher than that in Vietnam because there are more populations of C. multipinnata in Yunnan, China, and greater gene flow among them.
Cycas multipinnata was characterized by isolation by distance (IBD), i.e., a significant positive correlation (P < 0.05) was detected between genetic distance (GD) and geographic distance (GGD), which was addressed by the accordant results of the Mantel test based on the cpDNA and SSR data.
As with most Cycads, the dioecious C. multipinnata plants are pollinated by insects, primarily by beetles [76]. Seeds disperse by their own gravity beside the maternal individuals or by rodents that are attracted by the fleshy sarcotesta of cycads [77]. The current gene flow between extant populations is restricted due to the geographical isolation and the low seed dispersal. Thus, the levels of gene flow (Nm, Table 4) between populations cannot be interpreted as current gene flow between populations; they represent either ancient migratory events or shared ancestral polymorphism. So C. multipinnata has a high genetic differentiation (FST = 0.92304 based on the cpDNA data; FST = 0.29569 based on 17 microsatellite loci) according to the Wright’s criterion [78], which an FST value greater than 0.25 would indicate that there is significant genetic differentiation among populations. Moreover, high genetic differentiation has been detected in many cycads, such as C. debaoensis (FST = 0.80102, based on the data of atpB-rbcL and psbA-trnH) [26] and C. simplicipinna (FST = 0.987, based on the data of psbA-trnH and trnL-trnF, FST = 0.26 based on 16 microsatellite loci) [75]. In contrast with the three inland Cycas species above, C. revoluta and C. taitungensis, which are coastal or island distributed, possess high genetic variation and low genetic differentiation between populations [74]. One possible reason for this is that the glaciation had different effects on the Cycas species occurring inland and on islands. In detail, during the glacial period, migration corridors across islands may have formed that enhanced the gene flow, whereas on land, the environmental niche of Cycas plants became restricted to isolated refuges, resulting in limited gene flow. Another reason may be that different Cycas species have different lengths of evolution history; for example, the most recent common ancestor (TMRCA) of C. revolute and C. taitungensisis is estimated at 327.3 MYA in mtDNA and 204.0 MYA in cpDNA [74]. In a longer evolutionary history with more glaciations, C. revoluta and C. taitungensis experienced more complex in-range expansion and contraction fluctuations in population size.
Late Pleistocene divergence and population contraction
The genealogical haplotype network and topology of the Bayesian tree based on the data of the combined cpDNA sequences showed that C. multipinnata can be divided into two groups: one included the populations (SD, GLJ, SBZ and SZD) distributed in China and the other included the two populations (HY and YB) that occurred in northern Vietnam. The same result was obtained from three different analyses of the 17 microsatellite loci. First, in the principal coordinate analyses (PCO), 100 individuals from the five populations (SD, GLJ, SBZ, HY and YB) were spilt into two parts: one included the three populations (SD, GLJ and SBZ) in China, and the other contained the remaining two populations in Vietnam. Second, in the Bayesian clustering analysis using the STRUCTURE version 2.3 software, it was strongly recommended that the five populations should be divided into the same two groups suggested in the PCO analysis. Finally, the BARRIER analysis also showed that only one barrier existed between the populations in China and Vietnam. Overall, these analysis based on two different markers reached the same conclusion: C. multipinnata has a distinct structure, and it contains the China clade and Vietnam clade. The date of the most recent common ancestor (TMRCA) between the clades, based on the cpDNA data is approximately 1.0307 million years ago (MYA) i.e., during the Pleistocene.
The Bayesian skyline plot of cpDNA showed that the C. multipinnata population size experienced a significant reduction approximately 50,000 years ago (Fig. 5), and this result was supported by the microsatellite-based Garza-Williamson index (Table 5). It is likely that the conditions during the Quaternary glacial periods substantially affected the distribution and genetic structure of C. multipinnata populations as the temperature fluctuated [79]. Comparing the annual temperature at the fossil locality in northern China (Liaoning, 42° North) with that of the present distributions of cycads, we find that the current distribution of cycads is strongly affected by climate changes. Conversely, the evidence from the molecular analyses supported that both the time of the two clade divergence and the population contraction of C. multipinnata are associated with the severe climatic oscillation during the Quaternary glaciations. Similarly, that Quaternary glaciations played an important role in the population demographic histories has also been indicated in other Cycas species. For example, it was estimated that the TMRCA of different chloroplast haplotypes in C. debaoensis is approximately 2.66 MYA [26], for C. simplicipinna, it is 2.682 MYA in cpDNA, and 1.429 MYA according to the ITS4-ITS5 of nrDNA data [75]. The estimated divergence time of the living C. multipinnata, C. debaoensis and C. simplicipinna is could thus date to the Pleistocene. Additionally, a population contraction event of all three species has been detected in the last 50,000 years. Thus, the extant C. multipinnata populations in China and Vietnam represent the glacial refuges owning the high levels of nucleotide diversity in the cpDNA and the highest number of private haplotypes. Anthropogenic disturbances in the last three decades, such as large scale deforestation, road construction and over exploiting, merely accelerated the decrease in population size.
Conservation suggestions
There are only six small and fragmented populations of C. multipinnata remaining; the population SZD has only five individuals. During the fieldwork, we found there are few seedlings and coning plants (particularly for female individuals) due to overexploitation and habitat destruction. Population size is the most important factor among the five criteria to identify threatened species in IUCN [80]. It is believed that effective population size (Ne) of 50 individuals is the minimum to maintain sufficient allelic richness, and the effective population size of 500 individuals is barely sufficient to maintain the genetic variation of quantitative characteristics within populations and the adaptive ability for future environmental change [81]. When 0.02 is used as the lowest allele’s frequency used, the Ne of the YB population is only 80.5, which is greater than the minimum effective population size of 50. The Ne of population SBZ, GLJ and HY is 48.6, 16.7 and 46.6, respectively (Table 6). What is worse, both the Bayesian skyline plot of cpDNA and the microsatellite-based Garza-Williamson indices suggest a reduction in population size. Thus, it is rather urgent to take measures to protect this species.
In situ conservation is one important measure to take because the entire gene pools are preserved in their native habitat. Considering the limitation of the wild C. multipinnata resource and the apportionment of genetic diversity, we should regard all six populations as Management Units (MUs). In addition to public education and legal constraints to curtail overexploitation and habitat destruction, a long-term recovery of C. multipinnata package should be launched with the active participation of local people.
Ex situ conservation is an insurance policy that can be carried out with living plants cultivated in nature reserves or botanic gardens. Because Cycas could propagate via vegetative techniques [77], the germplasm collection objects could be basal offsets (suckers) or seeds of C. multipinnata from as many populations as possible to increase the genetic diversity.
Considering the main impact due to the population contraction is the low genetic variation and low gene flow between populations for both the ex situ and in situ conservation of C. multipinnata, we should increase the genetic diversity of the plant material during forest management. Of course, scientific research on C. multipinnata’s environment and pollination biology should also be carried out in time.
Conclusions
The historical divergence between the main genetic clusters of C. multipinnata took place approximately 1.0307 MYA, namely in the Pleistocene. The reconstruction of the population demographic history of C. multipinnata indicates that over the last 50,000 years, this species underwent a population contraction, with no subsequent expansion. The severe climate oscillation during the Pleistocene is a factor that contributed to the currently isolated geographical distribution of C. multipinnata. Our study revealed that the extant C. multipinnata displays low genetic diversity within populations and high genetic differentiation among populations in cpDNA sequence. Overall, the pattern of genetic variation within and among populations in C. multipinnata was related to the geographic distance. Practical measures should be launched immediately to protect the Endangered C. multipinnata.
Supporting Information
Table A. Information of 17 microsatellite loci used to study the population genetics. Table B. Variable sites from the three cpDNA combined sequence in Cycas multipinnata. Table C. P-value of Hardy-Weinberg equilibrium test for the five populations of C. multipinnata.
(DOCX)
The boundaries detected using the BARRIER program based on matrices of Nei’s (1983) unbiased genetic distance.
(DOCX)
Acknowledgments
We thank Dr. Yu-Juan Zhao for comments on the manuscript.
Data Availability
New DNA sequences were deposited in Genbank (NCBI) with accession numbers KP335666-KP335683.
Funding Statement
This work was supported by the National Natural Science Foundation of China and Yunnan Natural Science Foundation (U1136602 to XG). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Gao Z, Barry AT (1989) A review of fossil cycad megasporophylls, with new evidence of Crossozamia pomel and its associated leaves from the lower permian of Taiyuan, China. Review of Palaeobotany and Palynology 60: 205–223. [Google Scholar]
- 2. Leary RL (1990) Possible early Pennsylvanian ancestor of the Cycadales. Science 249: 1152–1154. [DOI] [PubMed] [Google Scholar]
- 3. Cantrill D (2000) A petrified cycad trunk from the Late Cretaceous of the Larsen Basin, Antarctica. Alcheringa 24: 307–318. [Google Scholar]
- 4. Li N, Fu X, Zhang W, Zheng S, Cao Y (2005) A New Genus of Cycadalean Plants from the Early Triassic of Western Liaoning, China-Mediocycas gen. nov. and Its Evolutionary Significance. Acta Palaeontologica Sinica 44: 423. [Google Scholar]
- 5. Hermsen EJ, Taylor TN, Taylor EL, Stevenson DW (2006) Cataphylls of the Middle Triassic cycad Antarcticycas schopfii and new insights into cycad evolution. American Journal of Botany 93: 724–738. 10.3732/ajb.93.5.724 [DOI] [PubMed] [Google Scholar]
- 6. Wang X, Li N, Wang Y, Zheng S (2009) The discovery of whole-plant fossil cycad from the Upper Triassic in western Liaoning and its significance. Chinese Science Bulletin 54: 3116–3119. [Google Scholar]
- 7. Wang X, Li N, Cui J (2006) Jurastrobus chenii gen. et sp. nov., a cycadalean pollen cone connected with vegetative parts from Inner Mongolia, China. Progress in Natural Science 16: 213–221. [Google Scholar]
- 8. Zhang W, Yang X-J, Fu X-P, Zheng S-L, Wang Y-D (2012) A polyxylic Cycad trunk from the Middle Jurassic of western Liaoning, China, and its evolutionary implications. Review of Palaeobotany and Palynology 183: 50–60. [Google Scholar]
- 9. Zhang JW, Yao JX, Chen JR, Li CS (2010) A new species of Leptocycas (Zamiaceae) from the Upper Triassic sediments of Liaoning Province, China. Journal of Systematics and Evolution 48: 286–301. [Google Scholar]
- 10. Nagalingum N, Marshall C, Quental T, Rai H, Little D, et al. (2011) Recent synchronous radiation of a living fossil. Science 334: 796–799. 10.1126/science.1209926 [DOI] [PubMed] [Google Scholar]
- 11. Chaw S-M, Walters TW, Chang C-C, Hu S-H, Chen S-H (2005) A phylogeny of cycads (Cycadales) inferred from chloroplast matK gene, trnK intron, and nuclear rDNA ITS region. Molecular phylogenetics and evolution 37: 214–234. [DOI] [PubMed] [Google Scholar]
- 12. Zgurski JM, Rai HS, Fai QM, Bogler DJ, Francisco-Ortega J, et al. (2008) How well do we understand the overall backbone of cycad phylogeny? New insights from a large, multigene plastid data set. Molecular phylogenetics and evolution 47: 1232–1237. 10.1016/j.ympev.2008.03.002 [DOI] [PubMed] [Google Scholar]
- 13. Rai HS, O’Brien HE, Reeves PA, Olmstead RG, Graham SW (2003) Inference of higher-order relationships in the cycads from a large chloroplast data set. Molecular phylogenetics and evolution 29: 350–359. [DOI] [PubMed] [Google Scholar]
- 14. Zonneveld B (2012) Genome sizes for all genera of Cycadales. Plant Biology 14: 253–256. 10.1111/j.1438-8677.2011.00522.x [DOI] [PubMed] [Google Scholar]
- 15. Chen J, Yang S (1994) Cycas multipinnata CJ Chen & SY Yang: A remarkable new cycad from China. Acta Phytotax Sin 32: 239. [Google Scholar]
- 16. Tian B, Gong X, Zhang Q (2002) Karyotypes of five species in Cycas. Acta Botanica Yunnanica 24: 370–376. [Google Scholar]
- 17. Hill KD, Nguyen HT, Loc PK (2004) The genusCycas (Cycadaceae) in Vietnam. The botanical review 70: 134–193. [Google Scholar]
- 18. Hill K (2008) The genus Cycas (Cycadaceae) in China. Telopea 12: 71–118. [Google Scholar]
- 19. Hall R (1996) Reconstructing Cenozoic SE Asia. Geological Society, London, Special Publications 106: 153–184. [Google Scholar]
- 20. Tang W (2004) Continental drift and the evolution of Asian cycas. Encephalartos. pp. 23–28. [Google Scholar]
- 21. Osborne R, Calonje MA, Hill KD, et al. (2012) The World List of Cycads. Memoirs New York Botanical Garden Vol. 106: 480–510. [Google Scholar]
- 22. Donaldson JS (2003) Cycads: status survey and conservation action plan: IUCN—the World Conservation Union. [Google Scholar]
- 23. Zhong Z-R, Li N, Qian D, Jin J-H, Chen T (2011) Maternal inheritance of plastids and mitochondria in Cycas L.(Cycadaceae). Molecular Genetics and Genomics 286: 411–416. 10.1007/s00438-011-0653-9 [DOI] [PubMed] [Google Scholar]
- 24. Fu L, Jin J (1992) China plant red data book: rare and endangerd plants. Science Press, Beijing. [Google Scholar]
- 25. Doyle J (1991) DNA protocols for plants—CTAB total DNA isolation, Molecular Techniques in Taxonomy Hewitt GM, Johnston A(Eds), Molecular Techniques in Taxonomy: 283–293. [Google Scholar]
- 26. Zhan Q-Q, Wang J-F, Gong X, Peng H (2011) Patterns of chloroplast DNA variation in Cycas debaoensis (Cycadaceae): conservation implications. Conservation Genetics 12: 959–970. [Google Scholar]
- 27. Shaw J, Lickey EB, Beck JT, Farmer SB, Liu W, et al. (2005) The tortoise and the hare II: relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. American Journal of Botany 92: 142–166. 10.3732/ajb.92.1.142 [DOI] [PubMed] [Google Scholar]
- 28. Wang Z-F, Ye W-H, Cao H-L, Li Z-C, Peng S-L (2008) Identification and characterization of EST-SSRs and cpSSRs in endangered Cycas hainanensis. Conservation genetics 9: 1079–1081. 10.1002/ar.20719 [DOI] [PubMed] [Google Scholar]
- 29. Yang Y, Li Y, LI LF, GE XJ, Gong X (2008) Isolation and characterization of microsatellite markers for Cycas debaoensis YC Zhong et CJ Chen (Cycadaceae). Molecular ecology resources 8: 913–915. 10.1111/j.1755-0998.2008.02114.x [DOI] [PubMed] [Google Scholar]
- 30. Cibrián-Jaramillo A, Marler TE, DeSalle R, Brenner ED (2008) Development of EST-microsatellites from the cycad Cycas rumphii, and their use in the recently endangered Cycas micronesica. Conservation Genetics 9: 1051–1054. [Google Scholar]
- 31. Li L, Wang Z-F, Jian S-G, Zhu P, Zhang M, et al. (2009) Isolation and characterization of microsatellite loci in endangered Cycas changjiangensis (Cycadaceae). Conservation genetics 10: 793–795. [Google Scholar]
- 32. Zhang M, Wang Z-F, Jian S-G, Ye W-H, Cao H-L, et al. (2009) Isolation and characterization of microsatellite markers for Cycas hainanensis CJ Chen (Cycadaceae). Conservation genetics 10: 1175–1176. [Google Scholar]
- 33. Zhang F, Su T, Yang Y, Zhai Y, Ji Y, et al. (2010) Development of seven novel EST—SSR markers from Cycas panzhihuaensis (Cycadaceae). American journal of botany 97: e159–e161. 10.3732/ajb.1000377 [DOI] [PubMed] [Google Scholar]
- 34. Ju L-P, Kuo C-C, Chao Y-S, Cheng Y-P, Gong X, et al. (2011) Microsatellite primers in the native perennial cycad Cycas taitungensis (Cycadaceae). American journal of botany 98: e84–e86. 10.3732/ajb.1000504 [DOI] [PubMed] [Google Scholar]
- 35. Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic acids research 25: 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic acids symposium series 41: 95–98. [Google Scholar]
- 37.Swofford D (2003) PAUP*: phylogenetic analysis using parsimony, version 4.0 b10.
- 38. Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452. 10.1093/bioinformatics/btp187 [DOI] [PubMed] [Google Scholar]
- 39. Bader E (2005) ArcGIS Server Administrator and Developer Guide: ArcGIS 9: ESRI Press. [Google Scholar]
- 40. Pons O, Petit R (1996) Measwring and testing genetic differentiation with ordered versus unordered alleles. Genetics 144: 1237–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evolutionary bioinformatics online 1: 47. [PMC free article] [PubMed] [Google Scholar]
- 42. Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131: 479–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Slatkin M (1993) Isolation by distance in equilibrium and non-equilibrium populations. Evolution: 264–279. [DOI] [PubMed] [Google Scholar]
- 44. Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Molecular ecology notes 6: 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bandelt HJ, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Molecular Biology and Evolution 16: 37–48. [DOI] [PubMed] [Google Scholar]
- 46. Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19: 1572–1574. [DOI] [PubMed] [Google Scholar]
- 47. Drummond AJ, Suchard MA, Xie D, Rambaut A (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Molecular biology and evolution 29: 1969–1973. 10.1093/molbev/mss075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Molecular biology and evolution 30: 2725–2729. 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wolfe KH, Li W-H, Sharp PM (1987) Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proceedings of the National Academy of Sciences 84: 9054–9058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li W-H, Graur D (1991) Fundamentals of molecular evolution: Sinauer Associates Sunderland, MA. [Google Scholar]
- 51.Rambaut A, Drummond A (2009) TRACER: MCMC Trace Analysis Tool Version v1. 5.0. Available from: http://tree.bio.ed.ac.uk/software/tracer/ University of Oxford.
- 52. Harpending H (1994) Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Human biology: 591–600. [PubMed] [Google Scholar]
- 53. Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fu Y-X, Li W-H (1993) Statistical tests of neutrality of mutations. Genetics 133: 693–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fu Y-X (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147: 915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Antao T, Lopes A, Lopes RJ, Beja-Pereira A, Luikart G (2008) LOSITAN: a workbench to detect molecular adaptation based on a Fst-outlier method. BMC bioinformatics 9: 323 10.1186/1471-2105-9-323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hartl DL, Clark AG (1997) Principles of population genetics: Sinauer associates Sunderland. [Google Scholar]
- 58. Raymond M, Rousset F (1995) GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. Journal of heredity 86: 248–249. [Google Scholar]
- 59.Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9. 3).
- 60. Wright S (1931) Evolution in Mendelian populations. Genetics 16: 97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kovach W (1999) MVSP-A multivariate statistical Package for Windows, ver. 3.1 Kovach Computing Services, Pentraeth, Wales, UK: 137. [Google Scholar]
- 62. Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155: 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molecular ecology 14: 2611–2620. [DOI] [PubMed] [Google Scholar]
- 64. Earl DA (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation Genetics Resources 4: 359–361. [Google Scholar]
- 65. Manni F, Guerard E, Heyer E (2004) Geographic patterns of (genetic, morphologic, linguistic) variation: how barriers can be detected by using Monmonier’s algorithm. Human biology 76: 173–190. [DOI] [PubMed] [Google Scholar]
- 66. Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144: 2001–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Di Rienzo A, Peterson A, Garza J, Valdes A, Slatkin M, et al. (1994) Mutational processes of simple-sequence repeat loci in human populations. Proceedings of the National Academy of Sciences 91: 3166–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Williamson-Natesan EG (2005) Comparison of methods for detecting bottlenecks from microsatellite loci. Conservation Genetics 6: 551–562. [Google Scholar]
- 69. Garza J, Williamson E (2001) Detection of reduction in population size using data from microsatellite loci. Molecular ecology 10: 305–318. [DOI] [PubMed] [Google Scholar]
- 70. Waples RS, Do C (2008) LDNE: a program for estimating effective population size from data on linkage disequilibrium. Molecular Ecology Resources 8: 753–756. 10.1111/j.1755-0998.2007.02061.x [DOI] [PubMed] [Google Scholar]
- 71. Kuchma O, Vornam B, Finkeldey R (2011) Mutation rates in Scots pine (Pinus sylvestris L.) from the Chernobyl exclusion zone evaluated with amplified fragment-length polymorphisms (AFLPs) and microsatellite markers. Mutation Research/Genetic Toxicology and Environmental Mutagenesis 725: 29–35. [DOI] [PubMed] [Google Scholar]
- 72. O’Connell L, Ritland K (2004) Somatic mutations at microsatellite loci in western redcedar (Thuja plicata: Cupressaceae). Journal of Heredity 95: 172–176. [DOI] [PubMed] [Google Scholar]
- 73. Kimura M (1984) The neutral theory of molecular evolution: Cambridge University Press. [Google Scholar]
- 74. Chiang Y-C, Hung K-H, Moore S-J, Ge X-J, Huang S, et al. (2009) Paraphyly of organelle DNAs in Cycas Sect. Asiorientales due to ancient ancestral polymorphisms. BMC evolutionary biology 9: 161 10.1186/1471-2148-9-161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Feng X, Wang Y, Gong X (2014) Genetic diversity, genetic structure and demographic history of Cycas simplicipinna (Cycadaceae) assessed by DNA sequences and SSR markers. BMC plant biology 14: 187 10.1186/1471-2229-14-187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jolivet P (2005) Cycads and beetles: recent views on pollination. The Cycad Newsletter 28: 3. [Google Scholar]
- 77. Jones D (2002) Cycads of the World-Ancient Plant in Today’s Landscape. Washington, DC: Smithsonian Institution Press; 10.1002/bdrc.21052 [DOI] [Google Scholar]
- 78. Wright S (1978) Evolution and the Genetics of Populations: A Treatise in Four Volumes: Vol. 4: Variability Within and Among Natural Populations: University of Chicago Press. [Google Scholar]
- 79. Hewitt G (2000) The genetic legacy of the Quaternary ice ages. Nature 405: 907–913. [DOI] [PubMed] [Google Scholar]
- 80. Frankham R, Briscoe DA, Ballou JD (2002) Introduction to conservation genetics: Cambridge University Press. [Google Scholar]
- 81. Frankel OH (1995) The conservation of plant biodiversity: Cambridge University Press. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table A. Information of 17 microsatellite loci used to study the population genetics. Table B. Variable sites from the three cpDNA combined sequence in Cycas multipinnata. Table C. P-value of Hardy-Weinberg equilibrium test for the five populations of C. multipinnata.
(DOCX)
The boundaries detected using the BARRIER program based on matrices of Nei’s (1983) unbiased genetic distance.
(DOCX)
Data Availability Statement
New DNA sequences were deposited in Genbank (NCBI) with accession numbers KP335666-KP335683.