

Abstract
There is a need for low cost, sensitive and chemical specific detectors for routine characterization of biomolecules. In this study, we utilize sheath-flow surface-enhanced Raman scattering (SERS) to analyze a mixture of eight biologically-active peptides separated by capillary zone electrophoresis (CZE). Analysis of the SERS electropherogram resulting from online detection resolves the characteristic Raman bands attributed to the amino acid constituents of each peptide, which enables identification. The detection limit by SERS was found to be 10−8 M. Our results suggest that the structural information obtained from the detected vibrational modes provides complementary characterization to other chemically specific detectors like mass spectrometry and improved chemical identification over other commonly used optical-based post-chromatographic detection methods. In addition, the sheath-flow SERS detection results in band narrowing in the observed electropherogram that enables distinction of closely migrating species. The results presented here indicate that online SERS detection can provide fast, robust, reproducible, and chemical specific detection to facilitate the characterization of peptides.
Introduction
The identification of biomolecules has broad application in areas such as clinical diagnostics, environmental monitoring, and pharmaceutical production. In the last few decades, progress has been made toward improving separation and developing better characterization methods for a variety of complex biomolecules. Recent improvements in microcolumn separation science and more specifically the incorporation of high sensitivity detectors have allowed analysis of minute amounts of sample with reduced processing time.1
Improving separations has been a key factor for identifying biomolecules such as DNA, lipids, metabolites, amino acids, peptides and proteins in complex biological mixtures.2 The most common detection method for characterizing these biomolecules is mass spectrometry (MS) due to its universality, sensitivity, and selectivity.3 While mass spectrometry can provide exquisite analyte identification, the cost of high-resolution mass spectrometers typically limits this analysis to core facilities. Additionally, certain classes of molecules require complex sample derivatization, while others exhibit poor ionization and are difficult to detect.4–6 Furthermore, the interface between the column and the mass spectrometer can limit the breadth of applications available for analysis.7,8
A low cost, chemical specific detector could facilitate analysis of biomolecular samples. Optical-based detection techniques provide an appealing alternative as they are typically nondestructive and relatively inexpensive. It is quite common to use optical detectors such as UV-visible absorption or laser-induced fluorescence (LIF) to detect analytes post-separation. However, LIF requires incorporation of a fluorescent label to achieve high sensitivity.9–12 And whiole UV-visible absorption offers a low cost and flexible alternative, it suffers from modest sensitivity.13, 14 More importantly, LIF and UV-visible absorption both lack molecular specificity which precludes their use for direct analyte identification.
Raman spectroscopy is an appealing optical-based detection method for post-separation analysis of biomolecules. Raman detection is readily incorporated to liquid-phased separation and provides label-free structural information through signals arising from vibrational modes.15–17 Previous studies have shown that post-chromatographic Raman detection suffers from poor sensitivity without sample concentration or resonance enhancement.18, 19 Recently, we demonstrated surface-enhanced Raman scattering (SERS) detection of rhodamine isomers and of amino acids following a capillary zone electrophoresis separation.20, 21 The SERS enhancement enabled us to detect concentrations ranging from 10−5 – 10−10 M without resonant enhancement. The structural information provided by the SERS spectra offers a chemical specific alternative for routine analysis of biomolecules.
In this report, we demonstrate the ability of our sheath-flow SERS detector to characterize and identify eight biologically-active peptides separated by capillary zone electrophoresis (CZE). Peptides are a class of biomolecules well characterized in bioanalysis. As a result, they provide an established test system to assess the sensitivity and robustness of our SERS detection method. Our results demonstrate the ability of our sheath-flow SERS detector to identify peptides from changes in the observed vibrational bands that correlate with the peptides’ amino acid composition.
Experimental Methods
Materials and Reagents
Lyophilized peptides were purchased from Peptides International (Louisville, KY). The peptides were dissolved in water to a concentration of 500 μM, aliquoted and stored at −20°C. Ammonium bicarbonate was purchased from Sigma-Aldrich (St. Louis, MO). Ultrapure water (18.2 MΩ cm) was obtained from a Barnstead Nanopure filtration system. All other chemicals were of analytical grade and used without any further purification.
SERS Substrate Fabrication
Silver (Ag, Sigma-Aldrich, 99.999%) was vapor deposited onto a commercial anodized aluminum oxide filter (Anodisc 13, Whatman) with 0.1 μm pores as previously described.22 Prior to deposition, the Anodisc filters were cleaned for 5 minutes in an Ar+ plasma (Model PDC-32G, Harrick Plasma, Ithaca, NY) to remove surface contamination and mounted on a substrate holder in the deposition chamber. Silver was deposited at a constant rate of 1.0–1.5 Å/s until a quartz crystal microbalance (QCM) registered a final nominal thickness of 500 nm. Following deposition, the substrates were allowed to cool to room temperature under vacuum inside the deposition chamber for 30 minutes. Prior to use, the SERS-active substrates were stored under vacuum to prevent oxidation and surface contamination.
CZE-SERS Setup
SERS-active substrates were integrated into a custom-built flow cell, as previously described.23 A 50 cm (72 μm i.d., 143 μm o.d.) uncoated fused silica capillary (Polymicro Technologies, Phoenix, AZ) was used. When selecting the capillary, our previous work shows that a thin capillary wall thickness is important for efficient hydrodynamic focusing. The distal end of the separation capillary was placed in a sheath-flow channel that was defined by a 250 μm thick silicone gasket with a 2 mm slit, covered with a standard cover glass that was held in place by the stainless steel top plate. The proximal end of the capillary was inserted into a custom-made injection block.24 A syringe pump (Model NE-500 OEM, New Era Pump Systems Inc., Farmingdale, NY) controlled by LabView (National Instruments, Austin, TX) was used to pump the sheath liquid (10 mM ammonium bicarbonate buffer, pH 8) at a rate of 10 μL/min, which provided hydrodynamic focusing of the sample into the detection region.
CZE was performed in positive mode by applying a constant potential of 15 kV to the Pt electrode embedded in the custom-built injection block using a Spellman, CZE 1000R power supply (Spellman High Voltage Electronics Corp., Hauppauge, NY). The system was grounded directly from the SERS substrate during the CZE separations. Sample injection was performed using a 2 s pressure injection, which introduced ~34 nL of sample into the capillary. Following injection, the sample was replaced with 10 mM ammonium bicarbonate buffer (pH 8) in the injection block and 15 kV (~15 μA, 300 V cm−1) was applied to the Pt electrode at the sample end of the capillary.
Raman spectra were collected using a previously described home-built Raman microscope.22 Excitation was provided by a 632.8 nm HeNe laser beam that was directed into the flow chamber using a 40× water-immersion objective (Olympus, NA = 0.8), resulting in a spot size of approximately 0.4 μm2. The laser power at the sample was attenuated to 1 mW. Raman back-scattering was collected by the same objective lens and directed to the Czerny-Turner spectrograph (Andor) and EMCCD (Newton 970, Andor). SERS spectra between 2000 and 500 cm−1 were recorded in kinetic series with 200 ms acquisition times. The spectral resolution of the home-built Raman instrument is 3 cm−1 based on the grating (600 groves/mm), entrance slit (25 μm), monochromator pathlength (320 mm), and CCD pixel size.
SERS Data Analysis
Band height and peak frequency determination were performed using MATLAB (R2012a, The Mathworks Inc., Natwick, MA). Baseline correction (Weighted Least Squares, basic filter, 2nd order) and data smoothing (Savitsky-Golay, 3 points, 0th order) were performed on the SERS electropherograms using PLS Toolbox version 6.2 (Eigenvector Research Inc., Wenatchee, WA) in MATLAB. The SERS electropherogram was created by plotting the Raman intensity on the z-axis as a function of Raman shift on the y-axis and migration time along the x-axis. The total photon electropherogram (TPE) was plotted by adding all the photons detected at all Raman shifts during each acquisition as a function of migration time.
Results and Discussion
Eight biologically-active peptides were chosen to assess the detection capability and sensitivity of our sheath-flow SERS detector. To study the amino acid contribution in the observed SERS signal, the peptides were chosen to have different combinations of aromatic and sulfur-containing amino acids, including two peptides containing a single amino acid polymorphism (Angiotensin II and Angiotensin III). The peptides range in mass from MW=593.68 (Laminin Pentapeptide) to 1637.9 (Somatostatin). The isoelectric points vary from 7.19 (Angiotensin II) to 11.12 (Substance P). The peptide mixture was prepared in 10 mM ammonium bicarbonate buffer (pH 8), where the concentration of each peptide is 50 μM. Table 1 summarizes the properties of the peptides used in this study.
Table 1.
Peptide name (listed in alphabetical order), molecular weight, one letter amino acid sequence, and pI for the eight biologically-active peptides used in this study. The red letters in the amino acid sequence indicate aromatic and sulfur-containing amino acids.
| Name | M.W | One Letter Sequence | pI |
|---|---|---|---|
| Amyloid β-Protein | 1060.3 | GSNKGAIIGLM | 8.88 |
| Angiotensin I | 1296.5 | DRVYIHPFHL | 7.38 |
| Angiotensin II | 1046.2 | DRVYIHPF | 7.19 |
| Angiotensin III | 931.09 | RVYIHPF | 9.06 |
| Bombesin | 1619.9 | Pyr-QRLGNQWAVGHLM | 10 |
| Laminin Pentapeptide | 593.68 | YIGSR-NH2 | 8.88 |
| Somatostatin | 1637.9 | AGCKNFFWKTFTSC (Disulfide bridge between Cys3-Cys14) | 8.59 |
| Substance P | 1347.6 | RPKPQQFFGLM | 11.12 |
Figure 1A shows the SERS electropherogram resulting from the separation and online detection of the peptide mixture. The electropherogram shows signals associated with the peptides at tm1 = 274 ± 6 s, tm2 = 300 ± 11 s, tm3 = 328 ± 9 s, tm4 = 332 ± 8 s, tm5 = 380 ± 13s, tm6 = 389 ± 6 s, tm7 = 395 ± 13 s, and tm8 = 430 ± 15 s. Analysis of Figure 1A shows that each analyte is very well resolved. The observed migration peak widths vary from 0.5 to 1.5 s, as seen in to the total photon electropherogram (TPE) shown in Figure 1B. Similar to the total ion count commonly plotted in separations with MS detection, the total photon count helps characterize peptide migration.
Figure 1.
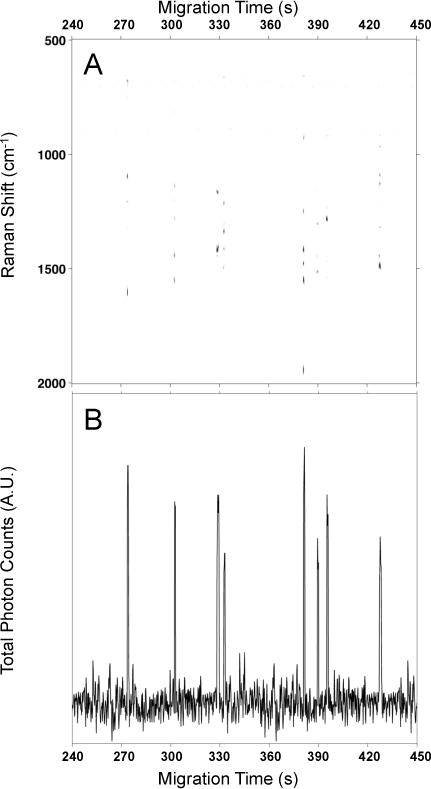
(A) The electropherogram shows the observed SERS intensity at each Raman shift as a function of migration time for the electrophoretic separation of the eight biologically-active peptides. The SERS signals indicate that Laminin Pentapeptide migrates at tm1 = 273 ± 6 s, Bombesin at tm2 = 302 ± 11 s, Angiotensin III at tm3 = 328 ± 9 s, Somatostatin at tm4 = 332 ± 8 s, Amyloid β -Protein at tm5 = 381 ± 13s, Angiotensin I at tm6 = 389 ± 6 s, Angiotensin II at tm7 = 395 ± 13 s, and Substance P at tm8 = 427 ± 15 s. (B) Total photon electropherogram (TPE) extracted from (A) showing the photons detected at all Raman shifts as a function of migration time.
We performed CZE-ESI-MS to confirm the identity, the elution order and the migration times of the peptides observed in the CZE-SERS experiments. CZE-ESI-MS has been used extensively for the study of peptides and proteins.25–28 Figure S1 in the ESI shows the total (A) and extracted (B) ion (MS) electropherograms of the same peptide mixture resulting from the CZE-ESI-MS experiments following a 1:10 dilution such that the concentration of each peptide is 5.0×10−6 M. The capillary dimensions and separation buffer composition were kept identical to those used in the optimized SERS experiments to provide a direct comparison. As seen in Figure S1, the CZE-ESI-MS experiments detected Laminin Pentapeptide (m/z = 594.3367) at tm1 = 318 s, Bombesin (m/z = 810.4175) at tm2 = 324 s, Angiotensin III (m/z = 466.2615) at tm3 = 361 s, Amyloid β-Protein (m/z = 530.7956) at tm4 = 367 s, Somatostatin (m/z = 819.3678) at tm5 = 378 s, Angiotensin I (m/z = 648.8478) at tm6 = 388 s, Angiotensin II (m/z = 523.775) at tm7 = 391 s and Substance P (m/z = 674.3733) at tm8 = 695 s. The advantage of MS detection is that it provides unambiguous identification of the known peptides based on accurate mass to charge ratios. Figure S2 in the ESI shows the MS spectra of the eight detected peptides. Table S1 in the ESI summarizes the results of the CZE-ESI-MS experiment and lists each peptide detected, along with the observed m/z, predicted m/z, and sample concentration.
We investigated the sensitivity of the SERS detector by performing CZE-SERS using varying concentrations of Angiotensin I. The separation and detection conditions were kept identical to those previously described. Figure 2 shows the averaged SERS spectra resulting from the detection of Angiotensin I at concentrations ranging from 50 μM to 50 nM. The high degree of spectral similarity of the SERS spectra of Angiotensin I shown in Figure 2 demonstrates the reproducibility and robustness of our sheath-flow SERS detector. As expected, the Raman signal decreases with decreasing analyte concentration. Figure 2 shows that Angiotensin I was detected at 50 nM (orange trace). The observed signal at 50 nM is just above the noise level (green trace), suggesting this concentration is near the detection limit.
Figure 2.
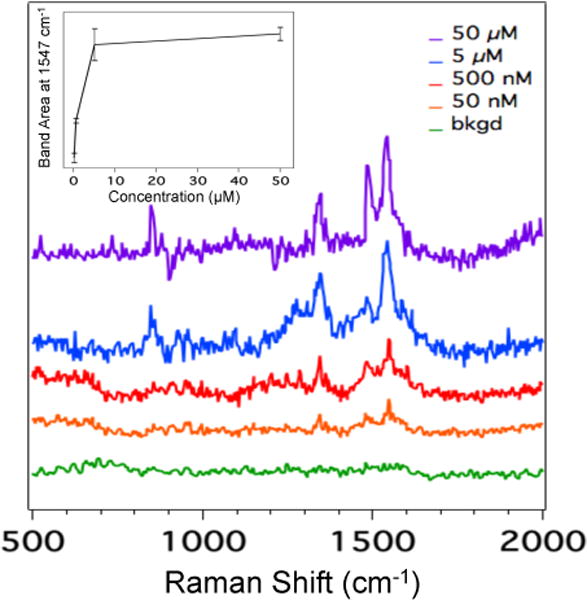
Averaged SERS spectra of Angiotensin I at concentrations ranging from 50 μM to 50 nM. The inset (plotted in a linear scale) is a plot of the area of the band at 1547 cm−1 assigned to the amide II vibration as a function of Angiotensin I concentration. Error bars in the inset represent the standard deviation.
The SERS electropherogram shown in Figure 1A resolves all eight peptides. The total ion count electropherogram in Figure S1A shows co-migration of many of the peptides in the CZE-ESI-MS experiments. The inset in Figure 2 shows the area of the Angiotensin I amide II band at 1547 cm−1 as a function of increasing concentration. The plot exhibits the Langmuir-type behavior typical of SERS. This observation is consistent with our previous findings showing that hydrodynamic focusing promotes molecule confinement at the silver surface, which enhances the physical interaction with the SERS substrate.23 As a result, the SERS signal is observed from the portion of the migrating analyte band with sufficient concentration to promote adsorption,29 which narrows the observed width of the migration peak. Figure 3 shows the change in migration peak width determined from the 1547 cm−1 band of Angiotensin I with increasing concentration. As the concentration exceeds the critical value for adsorption, the width of the peaks is observed to increase. This increase in migration peak width corresponds to the concentration of analyte in the migration band sufficient to promote adsorption. This agrees with other results where SERS signals in solution were correlated with adsorbed molecules.30 The dependence on Langmuir-type adsorption implies a nonlinear response that results in peak narrowing, which provides improved separation resolution. This peak-narrowing is valuable for resolving co-migrating species. Furthermore, sample pre-concentration techniques, such as isotachophoresis that have been used with MS detection,31 may also improve SERS detection.
Figure 3.
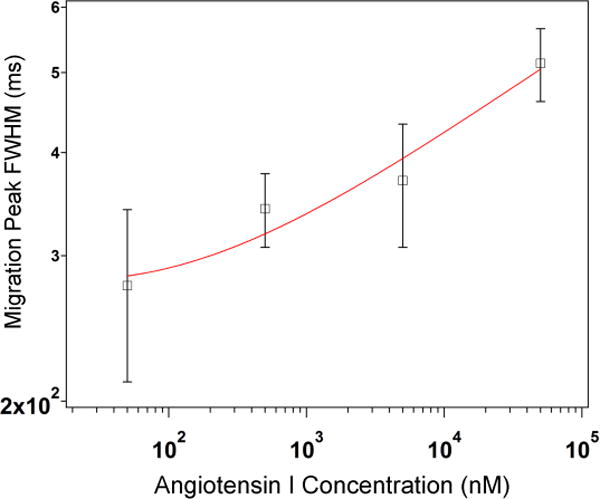
Log-log plot of the FWHM of the migration peak determined from the 1547 cm−1 band of Angiotensin I as a function of concentration in the range from 50 to 50,000 nM. The red trace represents the log fit. The FWHM values were extracted from the SERS electropherograms for each concentration from the data shown in Fig. 2. Error bars represent the standard deviation.
Figure 4 shows the averaged SERS spectra of each of the eight peptides extracted from the SERS electropherogram shown in Figure 1A. Although SERS spectra provide structural information in the form of bands arising from bond vibrations, it is difficult to unambiguously identify the peptides on the basis of the SERS spectra alone. As expected based on the peptides’ composition, the SERS spectra shown in Figure 4 share a high degree of similarity. Notably, characteristic vibrations of the amine and carboxyl groups are present in each spectrum, suggesting that charged groups are interacting with the silver SERS surface. However, the amino acid constituents in each peptide give rise to unique spectral features that enable differentiation.
Figure 4.
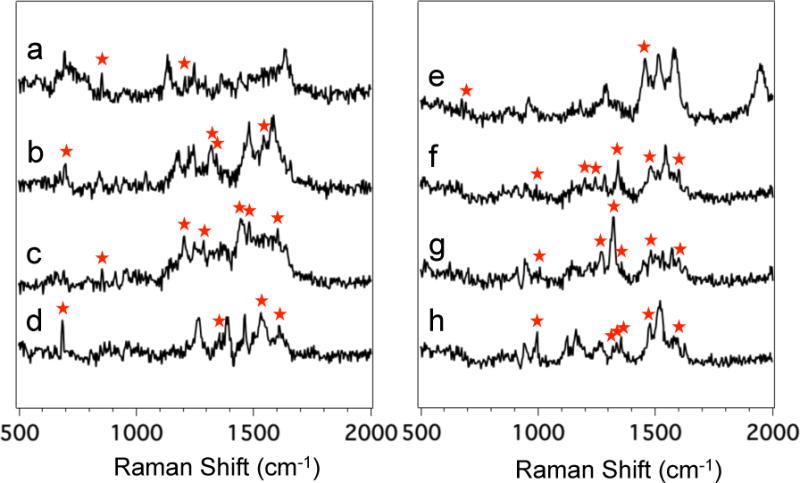
Averaged SERS spectra of (a) Laminin Pentapeptide extracted from Figure 1A between t = 273.6 and 274.4 s, (b) Bombesin between t = 302.4 and 303.0 s, (c) Angiotensin III between t = 328.2 and 330.0 s, (d) Somatostatin between t = 332.6 and 333.4 s, (e) Amyloid β-Protein between 381.0 and 381.8 s, (f) Angiotensin I between t = 389.4 and 390.4 s, (g) Angiotensin II between t = 395.2 and 396.0 s, and (h) Substance P between t = 427.2 and 428.6 s. Red asteriks mark the bands indicative of aromatic, sulfur-containing, or side chain vibrations that enabled differentiation and identification of the eight peptides.
Spectral analysis of the SERS data provides chemical insight into the detected peptides. Figure 4a shows the averaged SERS spectrum of Laminin Pentapeptide extracted from the SERS electropherogram shown in Figure 1A between t = 273.6 and 274.4 s. Table S2 in the ESI summarizes the observed bands and their assignments in the SERS spectrum of Laminin Pentapeptide. The presence of the two distinct vibrations intrinsic to Tyr and the absence of other aromatic features (Phe or Trp) support the identification of this peptide as Laminin Pentapeptide. Other peptides containing tyrosine also contain other aromatic amino acids, which tend to have distinct SERS signals. Other vibrational bands common to peptides are also observed.
Figure 4b shows the SERS spectrum of Bombesin extracted from the SERS electropherogram (Figure 1A) between t = 302.4 and 303.0 s. Table S3 in the ESI summarizes the observed bands in the SERS spectrum of Bombesin shown in Figure 4b along with their literature assignments. The SERS spectrum is characterized by unique bands that can be directly assigned to the vibrations of aromatic and sulfur-containing amino acids. We assign the band at 674 cm−1 to the characteristic C-S stretching vibration of Met.32, 33 The band at 1547 cm−1 is attributed to the indole vibration of Trp.34–37 These two distinct bands are unique to Bombesin, the only peptide in the mixture to contain Met and Trp. In addition, the CH3 symmetric and asymmetric bending modes observed at 1344 and 1481 cm−1 can be assigned to the methyl group in pyruvic acid at the N-terminus of Bombesin.38
The SERS spectrum of Angiotensin III is shown in Figure 4c, extracted from Figure 1A between t = 328.2 and 330.0 s. The main bands present in the SERS spectrum of Angiotensin III are tabulated in Table S4 in the ESI, along with band assignments to particular vibrational modes. The SERS spectrum shows distinct bands characteristic of Tyr and Phe found in the sequence of all the Angiotensins. These features include the bands at 854 (Fermi resonance between ring breathing and out-of-plane ring bending overtone of Tyr),37, 39 1204 (combined ring breathing mode of Phe and Cβ-Cγ stretching mode of Tyr),34, 36, 40 1288 (imidazole C-H in-plane bending),41 1482 (combined imidazole ring stretching and imidazole C1-H in plane bending),42 and 1602 cm−1 (ring C-C stretching of Phe).43
Figure 3d shows the SERS spectrum of Somatostatin extracted from the SERS electropherogram (Figure 1A) between t = 332.6 and 333.4 s. Table S5 in the ESI summarizes the observed bands in the SERS spectrum of Somatostatin, along with their literature assignments. The presence of the C-S stretching mode of cysteine and bands attributed to the aromatic constituents (Phe and Trp) are observed. Somatostatin is reported to form a disulfide bond (Cys3-Cys14). While sulfur is known to have a strong affinity for silver, the S-S vibration typically present around 530 cm−1 is not observed. This could indicate the bond is cleaved or oriented parallel to the SERS substrate where it would not be observed. The only spectral contribution from the disulfide bond is the C-S stretching mode observed as a sharp band at 674 cm−1.
The SERS spectrum of Amyloid β is shown in Figure 4e, extracted from Figure 1A between 381.0 and 381.8 s. A complete table of the observed bands in the SERS spectrum of Amyloid β-Protein and their assignments is provided in Table S6 in the ESI. The absence of bands attributed to characteristic aromatic vibrations and the presence of the band attributed to the C-S stretching vibration at 674 cm−1 provide spectral evidence for identification of this peptide as Amyloid β-Protein since it is the only peptide in the mixture that does not contain any aromatic constituents and that possesses a Met group at the end of the peptide chain.
Figure 4f and 4g show the averaged SERS spectra of Angiotensin I and Angiotensin II, respectively, which migrate closely. The Raman signals from these two peptides were extracted from the SERS electropherogram in Figure 1A between t = 389.4 and 390.4 s and t = 395.2 and 396.0 s, for Angiotensin I and Angiotensin II, respectively. Tables S7 and S8 in the ESI summarize the observed bands in the SERS spectra along with their literature assignments. The SERS spectra show a high degree of similarity, as expected by their structural similarity. However, as seen in Figure 4f and 4g, many bands at the same frequency are observed to vary in relative intensity, suggesting that identical molecular constituents provide different scattering intensities based on minor differences in molecular composition. However, the spectra present some spectral variations that allow differentiation and further identification of the peptides.
Substance P is the last peptide to migrate off the capillary. Although near the noise in the MS results, the averaged SERS spectrum is readily detected as shown in Figure 4h. The SERS spectrum was extracted from the heatmap shown in Figure 1A between t = 427.2 and 428.6 s. The SERS spectrum shows distinct bands characteristic of aromatic vibrations. The main bands present in the SERS spectrum of Substance P shown in Figure 3h are tabulated and assigned in Table S9 in the ESI. The observed bands arise from the expected aromatic and hydrocarbon amino acid chains based on the peptide’s sequence.
The CZE-SERS and CZE-ESI-MS experiments generated identical elution order and equivalent migration times using identical capillary conditions and buffer composition. The advantage of MS detection is that an accurate mass provides unambiguous identification of the known peptides. However, little additional information about the chemical nature of the peptide is provided by the MS spectrum. As demonstrated in this study, the SERS spectra of the analytes can be used for identification. A significant advantage of the sheath-flow SERS detector is the reproducible SERS spectra obtained from analytes, something that has been problematic in other SERS based detection schemes. Figure 5 illustrates the hierarchical cluster analysis (HCA) dendrogram based on the thirty-five 200 ms SERS spectra extracted from the SERS electropherogram shown in Figure 1A. The spectra of the peptides were clustered based on Ward’s linkage method for minimizing variance.44 The vertical bars in the dendrogram specify which spectra and classes are linked, while the horizontal bars represent the distance between the linked classes. The dendrogram shown in Figure 5 demonstrates that HCA classified the SERS spectra of the eight peptides into eight distinct clusters with 100% accuracy, showing the robustness of HCA classification for differentiation of SERS spectra of different peptides. Interestingly, Angiotensin I, Angiotensin II and Angiotensin III, which are derived from the same protein, combine to form one higher order cluster. This observation demonstrates that cluster analysis is able to distinguish the spectral differences between SERS spectra of closely-related peptides, including single amino acid polymorphism (Angiotensin II and III). This result suggests that biochemical variations, such as post-translational modifications, may be detectable by SERS and linked to known, previously identified sequences.
Figure 5.

Dendrogram produced by hierarchical cluster analysis (HCA) of the SERS spectra extracted from the electropherogram shown in Figure 1A using Ward’s method. A total of 35 SERS spectra extracted from the electropherogram shown in Figure 1A were used to generate this dendrogram.
Conclusion
We have demonstrated the ability of our sheath-flow SERS detector to characterize and identify eight biologically-active peptides separated by CZE. CZE-ESI-MS was used to confirm the identity, the elution order and the migration times of the peptides observed in the CZE-SERS experiments. The reproducible SERS results obtained provide a chemical specific signature that, once defined, can also be used for identification. More specifically, our sheath-flow SERS detector appears to be sensitive to the characteristic functional groups of aromatic and sulfur-containing amino acids, as well as the amine, carboxyl, and side chain constituents located at the N- and C-terminus of the peptides. The SERS assay has a limit of detection of 10−8 M. This limit of detection appears to be limited by the Langmuir adsorption behavior. Hierarchical clustering analysis of the SERS spectra can be used to identify differences in the molecular composition of closely-related analytes.
Classic molecular characterization consists of combining orthogonal detection methods. Here we present the use of our sheath-flow SERS detector with capillary zone electrophoresis (CZE) for trace molecule characterization in solution. The implementation of this robust, sensitive and high throughput sheath-flow SERS detector is shown to provide complementary characterization to mass spectrometry and improved chemical identification of complex biomolecular mixtures over other commonly used post-chromatographic detection methods for analytes with high biochemical relevance.
Supplementary Material
Acknowledgments
The University of Notre Dame, NIH Award R21 GM107893 (ZDS), and the Cottrell Scholar Award from the Research Corporation for Science Advancement (ZDS) supported this work. We thank Liangliang Sun for helpful suggestions with the CZE-ESI-MS experiments.
Footnotes
Electronic Supporting Information (ESI) available: Supporting figures S1 and S2 show the total and extracted ion electropherograms as well as the MS spectra of the eight peptides resulting from the CZE-ESI-MS experiments. Supporting tables S1–S9 summarize the results from the CZE-ESI-MS experiments as well as spectral assignments observed in the SERS spectra of the peptides.
References
- 1.Legido-Quigley C, Marlin ND, Melin V, Manz A, Smith NW. Electrophoresis. 2003;24:917–944. doi: 10.1002/elps.200390136. [DOI] [PubMed] [Google Scholar]
- 2.Albertsson P-Å. Partition of cell particles and macromolecules : seperation and purification of biomolecules, cell organelles, membranes, and cells in aqueous polymer two-phase systems and their use in biochemical analysis and biotechnology. 3. New York: 1986. [Google Scholar]
- 3.Kaltashov IA, Eyles SJ. Mass Spectrometry Reviews. 2002;21:37–71. doi: 10.1002/mas.10017. [DOI] [PubMed] [Google Scholar]
- 4.Banks JF. Electrophoresis. 1997;18:2255–2266. doi: 10.1002/elps.1150181216. [DOI] [PubMed] [Google Scholar]
- 5.Figeys D, Aebersold R. Electrophoresis. 1998;19:885–892. doi: 10.1002/elps.1150190603. [DOI] [PubMed] [Google Scholar]
- 6.von Brocke A, Nicholson G, Bayer E. Electrophoresis. 2001;22:1251–1266. doi: 10.1002/1522-2683(200105)22:7<1251::AID-ELPS1251>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 7.Smith RD, Olivares JA, Nguyen NT, Udseth HR. Analytical Chemistry. 1988;60:436–441. [Google Scholar]
- 8.Kebarle P, Tang L. Analytical Chemistry. 1993;65:A972–A986. [Google Scholar]
- 9.Chen DY, Dovichi NJ. Journal of Chromatography B-Biomedical Applications. 1994;657:265–269. doi: 10.1016/0378-4347(94)00014-x. [DOI] [PubMed] [Google Scholar]
- 10.MacTaylor CE, Ewing AG. Electrophoresis. 1997;18:2279–2290. doi: 10.1002/elps.1150181218. [DOI] [PubMed] [Google Scholar]
- 11.Santesson S, Andersson M, Degerman E, Johansson T, Nilsson J, Nilsson S. Analytical Chemistry. 2000;72:3412–3418. doi: 10.1021/ac000140r. [DOI] [PubMed] [Google Scholar]
- 12.Otsuka K, Karuhaka K, Higashimori M, Terabe S. Journal of Chromatography A. 1994;680:317–320. doi: 10.1016/0021-9673(94)80082-0. [DOI] [PubMed] [Google Scholar]
- 13.Beck W, Engelhardt H. Chromatographia. 1992;33:313–316. [Google Scholar]
- 14.Song JZ, Huang DP, Tian SJ, Sun ZP. Electrophoresis. 1999;20:1850–1855. doi: 10.1002/(SICI)1522-2683(19990701)20:9<1850::AID-ELPS1850>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 15.Chen CY, Morris MD. Applied Spectroscopy. 1988;42:515–518. [Google Scholar]
- 16.Kneipp K, Kneipp H, I I, Dasari RR, Feld MS. Chemical Reviews. 1999;99:2957–2976. doi: 10.1021/cr980133r. [DOI] [PubMed] [Google Scholar]
- 17.Moskovits M. Reviews of Modern Physics. 1985;57:783–826. [Google Scholar]
- 18.Swinney K, Bornhop DJ. Electrophoresis. 2000;21:1239–1250. doi: 10.1002/(SICI)1522-2683(20000401)21:7<1239::AID-ELPS1239>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 19.Chen CY, Morris MD. Journal of Chromatography. 1991;540:355–363. doi: 10.1016/s0021-9673(01)88822-1. [DOI] [PubMed] [Google Scholar]
- 20.Negri P, Flaherty RJ, Dada OO, Schultz ZD. Chemical Communications. 2014;50:2707–2710. doi: 10.1039/c3cc49030k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Negri P, Schultz ZD. Analyst. 2014;139:5990–5999. doi: 10.1039/c4an01177e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Asiala SM, Schultz ZD. Analyst. 2011;136:4472–4479. doi: 10.1039/c1an15432j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Negri P, Jacobs KT, Dada OO, Schultz ZD. Anal Chem. 2013;85:10159–10166. doi: 10.1021/ac401537k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krylov SN, Starke DA, Arriaga EA, Zhang Z, Chan NW, Palcic MM, Dovichi NJ. Anal Chem. 2000;72:872–877. doi: 10.1021/ac991096m. [DOI] [PubMed] [Google Scholar]
- 25.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Mass Spectrometry Reviews. 1990;9:37–70. [Google Scholar]
- 26.Issaq HJ. Electrophoresis. 2001;22:3629–3638. doi: 10.1002/1522-2683(200109)22:17<3629::AID-ELPS3629>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 27.Aebersold R, Goodlett DR. Chemical Reviews. 2001;101:269–295. doi: 10.1021/cr990076h. [DOI] [PubMed] [Google Scholar]
- 28.Aebersold R, Mann M. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 29.Lee HJ, Li Y, Wark AW, Corn RM. Analytical Chemistry. 2005;77:5096–5100. doi: 10.1021/ac050815w. [DOI] [PubMed] [Google Scholar]
- 30.Asiala SM, Schultz ZD. Analytical Chemistry. 2014;86:2625–2632. doi: 10.1021/ac403882h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Larsson M, Lutz ESM. Electrophoresis. 2000;21:2859–2865. doi: 10.1002/1522-2683(20000801)21:14<2859::AID-ELPS2859>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 32.Rycenga M, McLellan JM, Xia YN. Chemical Physics Letters. 2008;463:166–171. doi: 10.1016/j.cplett.2008.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watanabe T, Maeda H. Journal of Physical Chemistry. 1989;93:3258–3260. [Google Scholar]
- 34.Rava RP, Spiro TG. Journal of the American Chemical Society. 1984;106:4062–4064. [Google Scholar]
- 35.Miura T, Takeuchi H, Harada I. Biochemistry. 1991;30:6074–6080. doi: 10.1021/bi00238a035. [DOI] [PubMed] [Google Scholar]
- 36.Wei F, Zhang DM, Halas NJ, Hartgerink JD. Journal of Physical Chemistry B. 2008;112:9158–9164. doi: 10.1021/jp8025732. [DOI] [PubMed] [Google Scholar]
- 37.Smith E, Dent G. Modern Raman Spectroscopy: A Practical Approach. Wiley; 2005. [Google Scholar]
- 38.Stewart S, Fredericks PM. Spectrochimica Acta Part a-Molecular and Biomolecular Spectroscopy. 1999;55:1615–1640. [Google Scholar]
- 39.Siamwiza MN, Lord RC, Chen MC, Takamatsu T, Harada I, Matsuura H, Shimanouchi T. Biochemistry. 1975;14:4870–4876. doi: 10.1021/bi00693a014. [DOI] [PubMed] [Google Scholar]
- 40.Nabiev IR, Trakhanov SD, Efremov ES, Marinyuk VV, Lasorenkomanevich RM. Bioorganicheskaya Khimiya. 1981;7:941–945. [Google Scholar]
- 41.Takeuchi H, Watanabe N, Satoh Y, Harada I. Journal of Raman Spectroscopy. 1989;20:233–237. [Google Scholar]
- 42.Martusevicius S, Niaura G, Talaikyte Z, Razumas V. Vibrational Spectroscopy. 1996;10:271–280. [Google Scholar]
- 43.Naumann D. Applied Spectroscopy Reviews. 2001;36:239–298. [Google Scholar]
- 44.Beebe KR, Pell RJ, Seasholtz MB. Chemometrics: A Practical Guide. New York: 1998. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.