

Abstract
Background
Mycobacterium tuberculosis is one of the most dangerous human pathogens, the causative agent of tuberculosis. While this pathogen is considered as extremely clonal and resistant to horizontal gene exchange, there are many facts supporting the hypothesis that on the early stages of evolution the development of pathogenicity of ancestral Mtb has started with a horizontal acquisition of virulence factors. Episodes of infections caused by non-tuberculosis Mycobacteria reported worldwide may suggest a potential for new pathogens to appear. If so, what is the role of horizontal gene transfer in this process?
Results
Availing of accessibility of complete genomes sequences of multiple pathogenic, conditionally pathogenic and saprophytic Mycobacteria, a genome comparative study was performed to investigate the distribution of genomic islands among bacteria and identify ontological links between these mobile elements. It was shown that the ancient genomic islands from M. tuberculosis still may be rooted to the pool of mobile genetic vectors distributed among Mycobacteria. A frequent exchange of genes was observed between M. marinum and several saprophytic and conditionally pathogenic species. Among them M. avium was the most promiscuous species acquiring genetic materials from diverse origins.
Conclusions
Recent activation of genetic vectors circulating among Mycobacteria potentially may lead to emergence of new pathogens from environmental and conditionally pathogenic Mycobacteria. The species which require monitoring are M. marinum and M. avium as they eagerly acquire genes from different sources and may become donors of virulence gene cassettes to other micro-organisms.
Keywords: Mycobacterium, horizontal gene transfer, virulence
Background
Mycobacterium tuberculosis is one of the most dangerous human bacterial pathogens, causing a potentially deadly disease that has been around for a long time. [1]. Despite of an optimistic report that tuberculosis and AIDS death rates are steadily declining around the world over recent several years [2], they remain the main killers. Moreover, HIV patients are much more vulnerable to tuberculosis and often become carriers for other non-tuberculous mycobacterial pathogens, such as M. avium [3-5], M. kansasii [4,5], M. abscessus [5], M. timonense [6] and M. genavense [7]. In the future these new pathogens may undergo the same evolutionary process of pathogenicity formation that was assumed for M. tuberculosis [8]. According to this hypothesis the evolution has started with an expansion phase involving active horizontal acquisition of virulence factors and gene duplication.
In the work by Reva & Bezuidt [9] a new channel of transfer of virulence genes from pathogenic Enterobacteria to Brucella, Mycobacterium and Nocardia was reported, which may pose a serious impact on emergence of new pathogens. Genomic islands found in Mycobacterium were most likely originated from alpha-Proteobacteria [10] and gamma-Proteobacteria [11]. An unexpectedly high frequency of mercury-resistant strains showing also an increased tolerance to gentamicin, streptomycin and D-cycloserine has been reported among the clinical non-tuberculous mycobacteria isolates of species M. avium, M. intracellulare and M. scrofulaceum [12]. The genome of a fish pathogen M. marinum, which sometimes causes opportunistic infections in humans, comprises a 23 kb mercury-resistance plasmid pMM2329. BLASTn and oligonucleotide composition comparison showed that this plasmid comprising a mercury resistance operon had originated from either Nocardia or Pseudonocardia [9]. It looked like that this plasmid has been acquired by M. marinum quite recently as it still shows a strong sequence and oligonucleotide pattern similarities to Nocardia. These newly acquired genes may be behind the increased drug resistance reported for M. marinu isolates [13]. Mercury resistance plasmid similar to that of M. marinum together with multiple GIs of Pseudomonas and Actinobacteria origin were identified in M. abscessus, a pseudotuberculous lung disease causing microbe [14]; and in a frog pathogen M. ulcerans, which sometimes cause skin ulcers in human [15].
In this work we analysed acquired genes and patterns of distribution of genomic islands in available genomes of Mycobacteria by comparing complete genome sequences and sequences of genomic islands previously identified in these organisms. The aim was to study the possibility of emergence of new mycobacterial pathogens in result of acquiring of virulence genomic islands.
Results and discussion
Comparison of 22 mycobacterial genomes (Table 1) revealed 2,337 clusters of orthologous genes (COGs) shared by all these organisms. Concatenated alignment of these proteins was 657,505 amino acid residues long. A species tree inferred from the concatenated alignment is shown in Figure 1.
Table 1.
Micobacterial genomes and genomic islands used in this study.
| Genome and NCBI accession | Number of genomic islands | Total number of genes in genomic islands |
|---|---|---|
| M. abscessus [NC_010397] | 10 | 300 |
| M. avium 104 [NC_008595] | 20 | 538 |
| M. avium ssp. paratuberculosis K-10 [NC_002944] | 11 | 230 |
| M. bovis AF2122/97 [NC_002945] | 11 | 212 |
| M. bovis BCG str. Pasteur 1173P2 [NC_008769] | 10 | 187 |
| M. bovis BCG str. Tokyo 172 [NC_012207] | 11 | 209 |
| M. canettii CIPT 140010059 [NC_015848] | 11 | 194 |
| M. leprae Br4923 [NC_011896] | 22* | Not used |
| M. leprae TN [NC_002677] | 23* | Not used |
| M. marinum M [NC_010612] | 23 | 387 |
| M. smegmatis MC2 155 [NC_008596] | 12 | 329 |
| M. tuberculosis CDC1551 [NC_002755] | 10 | 203 |
| M. tuberculosis F11 [NC_009565] | 12 | 217 |
| M. tuberculosis H37Ra [NC_009525] | 12 | 225 |
| M. tuberculosis H37Rv [NC_000962] | 9 | 170 |
| M. ulcerans Agy99 [NC_008611] | 3 | 44 |
| M. vanbaalenii PYR-1 [NC_008726] | 16 | 360 |
| Mycobacterium sp. JDM601 [NC_015576] | 6 | 172 |
| Mycobacterium sp. JLS [NC_009077] | 14 | 430 |
| Mycobacterium sp. KMS [NC_008705] | 15 | 445 |
| Mycobacterium sp. MCS [NC_008146] | 12 | 356 |
| Mycobacterium sp. Spyr1 [NC_014814] | 14 | 419 |
* These genomic islands were excluded from this study.
Figure 1.

Species phylogenetic tree. The tree was constructed by Neighbour-Joining algorithm on concatenated alignments of 2,337 COGs.
242 Genomic islands identified in 20 genomes (excluding two M. leprae genomes, see discussion in the 'Methods' section) comprised 5,627 genes, which formed 1,563 COGs. A binary data table representing the presence and absence of orthologous accessory genes associated with different genomic islands was created for inferring a parsimony tree shown in Figure 2.
Figure 2.
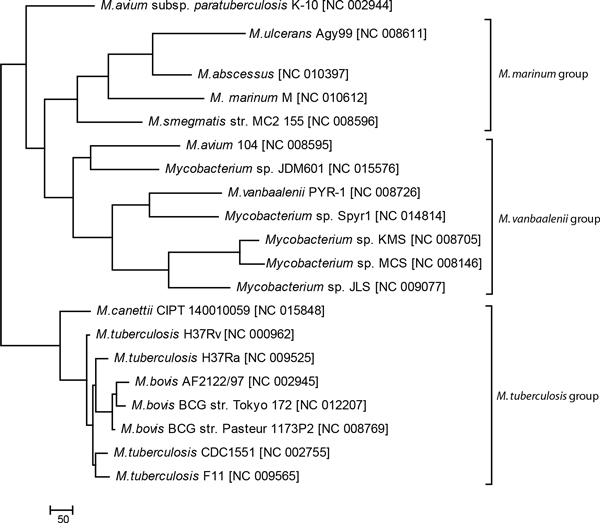
Accessory gene tree. Clustering of Mycobacterial genomes by sharing the accessory genes associated with genomic islands was performed by Wagner parsimony algorithm.
In the tree in Figure 2 the clusters represent groups of organisms, which share the same pool of interchangeable mobile genetic elements. Species of the M. tuberculosis group were clustered separately from other mycobacteria in both trees in Figure 1 and 2. Finding of genomic islands in these micro-organisms contributed to the hypothesis by Veyrier et al. [8] that the pathogen evolution might be triggered out by the acquisition of horizontally transferred genes. However, all genomic islands in M. tuberculosis most likely are ancient acquisitions. The identification of relative time of insertion is grounded in the assumption that the process of amelioration alters the island nucleotide composition from the time of insertion to reconcile with that of the host in which it occurs [9]. In an interactive Web-based network of genomic islands prepared for our previous publication [16] the relative age of acquisition is depicted by grey gradient where the darker colour means recent acquisitions and lighter colours indicates ancient islands.
In total 48 genes were found which were associated exclusively with these genomic islands of the Mtb cluster and which were not present in genomic islands of other Mycobacteria. Among them there were argFGHR arginine biosynthesis operon; PE-PGRS family genes; lpqD lipoprotein; idsB and grcC2 genes involved in terpenoid biosynthesis; mscL osmotic pressure regulator; moaB2 stress response regulator and several hypotheticals.
Another group of micro-organisms clustered around M. marinum comprised phylogenetically related M. ulcerans and more distant M. abscessus and M. smegmatis. Hypothesis of sharing of common mobile genetic elements by these bacteria is supported by the fact that M. marinum, M. ulcerans and M. abscessus genomes contained almost identical plasmids with several virulence genes [13-15]. There were 5 hypothetical genes (17 genes when M. smegmatis is excluded), which were unique for the genomic islands of this group of micro-organisms. The third group clustered around M. vanbaalenii consisted of multiple environmental Mycobacteria. They shared 20 hypothetical genes unique for this group.
The strain M. avium subsp. paratuberculosis was located apart from other groups (Figure 2) and far away on the tree from its closest relative M. avium 104 (compare to Figure 1). Presence of multiple genomic islands in M. avium subsp. paratuberculosis was confirmed by alternative genomic island prediction methods, as it was shown in Pre_GI database. Genetic content of the genomic islands and their ontological links to genomic islands from other micro-organisms were summarized in Additional file 1 supplementary Table S1.
From Additional file 1 Table S1 it was seen that the genomic islands of M. avium K-10 shared sequences with those from M. avium 104, but according to Figure 2 the genetic content is rather different. Several genomic islands showed sequence similarity to rather distant genomic islands from Mycobacterium canettii and Alicycliphilus denitrificans. M. avium subsp. paratuberculosis is a causative agent of Johne's disease in cattle and other ruminants [17]. The non-paratuberculosis strain M. avium 104 also was isolated from an adult AIDS patient, but this micro-organism was considered as an opportunist rather than an established pathogen [18]. It looks that the horizontal gene transfer was the driving force of evolution of the paratuberculosis lineage of M. avium, as regarding to other proteins both these subspecies are very much similar (Figure 1).
An interesting finding was that all these mycobacterial genomes possessed several common genes present in all species, which nevertheless were associated with horizontally transferred mobile genetic elements. These genes were fadD22-fadE, which are important for virulence and mycobactin synthesis [19,20], and also the O-succinylbenzoic acid-CoA ligase menE involved in menaquinone biosynthesis and considered as a potential target for antibiotics [21]. To ensure that these genes were associated with the horizontal gene transfer and were not falsely predicted due to some peculiarities in their sequences, a tree was constructed based on an alignment of the FadD22 proteins (Figure 3).
Figure 3.
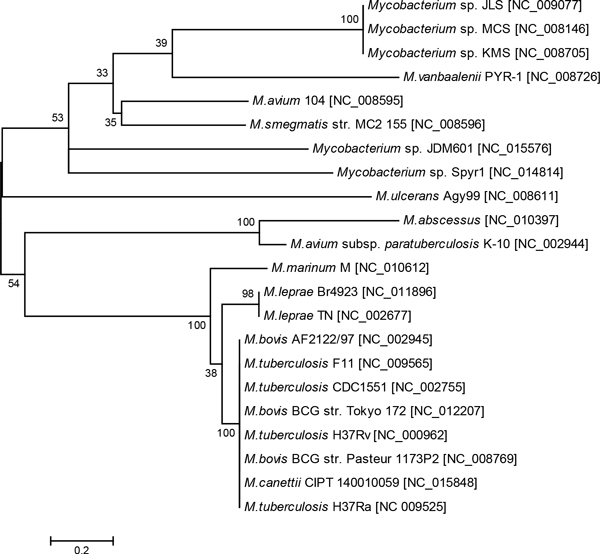
FadD22 gene tree. This dendrogram was inferred by Neighbour-Joining algorithm based on alignment of protein sequences of FadD22 found in different Mycobacterial genomes.
A combination of high level variability of FadD22 across Mycobacteria with its conservation within taxonomic units indicates crucial importance of this protein for bacteria. FadD22 proteins showed much higher conservation in M. tuberculosis and M. leprae than that observed for other orthologous proteins (compare Figure 1 and 3) implying an indispensability of this protein for the pathogenesis. FadD22 of M. marinum also belonged to the Mtb group despite that this organism phylogenetically is quite distant from M. tuberculosis. Two strains of M. avium K-10 and 104 were separated in the FadD22 tree the same like in the genomic island gene tree (see Figure 2). In M. avium subsp. paratuberculosis K-10 this protein was similar to that from the pathogenic strain M. abscessus, while in M. avium 104 the protein FadD22 was similar to orthologs in environmental strains. In general the topology of the FadD22 tree was not exactly congruent to either the species tree (Figure 1) or the accessory gene tree (Figure 2). It implies existing of a complex network of gene exchange between Mycobacteria. Two reticulate networks were designed by using the program SplitsTree: the first was based on incongruences of 2,337 individual core COG alignments (Figure 4A); and the second was based on the matrix of shared 1,563 COGs of horizontally transferred genes (Figure 4B).
Figure 4.
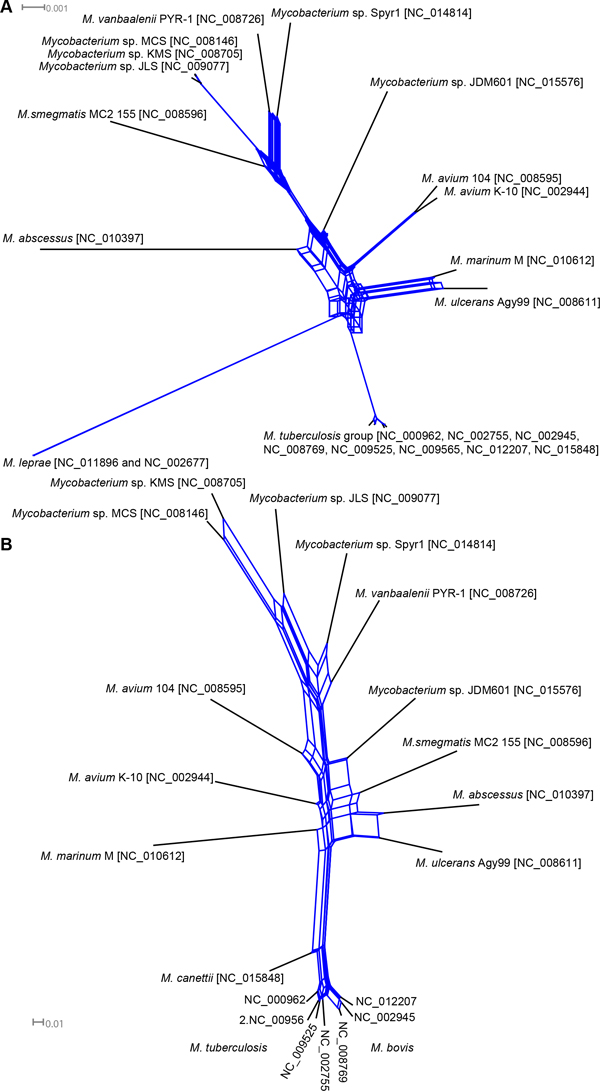
Reticulate networks. These networks demonstrate the events of exchange of A) core genes and B) accessory horizontally transferred genes between Mycobacteria. Reticulate events are depicted by blue lines.
The network of core genes (Figure 4A) showed that the species of Mycobacterium were quite isolated from each other and the individual gene trees in most cases were congruent to the species tree shown in Figure 1. Exceptions were M. marinum clustered with M. ulcerans in one case, and M. vanbaalenii clustered with Mycobacterium sp. Spyr1 in another case, which apparently have exchanged the core genes frequently. The turnover of genomic islands, which usually is associated with sharing the same pool of mobile genetic vectors such as conjugative plasmids and phages, was more intensive than the core gene exchange, especially between environmental M. vanbaalenii and Mycobacterium sp. Surprisingly, another common pool of mobile genes was shared by conditionally pathogenic M. abscessus and M. ulcerans with saprophytic soil M. smegmatis and Mycobacterium sp. JDM601. Although bacteria of the M. tuberculosis group were believed to be resistant to horizontal gene transfer and comprised only ancient genomic islands [9,22], it was still possible to root them to the common pool of mycobacterial horizontally transferred genes. Contrary, M. avium and M. marinum genomes were placed apart in the reticulate network. The reason for this might be that they acquired genes from different sources including those which were not common to other Mycobacteria (see Additional file 1 Table S1).
Conclusions
Veyrier et al. [8] hypothesized that M. tuberculosis had undergone a biphasic evolutionary process involving genome expansion (gene acquisition and duplication) and reductive evolution (deletions). Nowadays the evolution of this pathogen including the development of drug resistance fully relies on selective mutations, genome recombination and gene duplication [22], but the evolution towards pathogenicity initially might be triggered by an acquisition of several virulence factors [9]. Over the recent decades the humankind has witnessed a drastic emergence of outbreaks of new pathogens. A question of an acute medicinal importance is where, when and which pathogens may cause new outbreaks in the near future? Drawing the strongest attention to control on M. tuberculosis, we have not to forget that other non-tuberculosis Mycobacteria have a potential to put humankind under risk of new invasions. This is why it is very important to study in detail the development of pathogenicity of M. tuberculosis so that an emergence of new mycobacterial pathogens will not catch us unaware.
Genomic islands found in Mycobacteria share DNA composition and sequence similarities with a big group of genomic islands originated from Actinobacteria, alpha-, beta- and gamma-Proteobacteria, Deinococcus/Thermus and some other bacteria [23] (see also the online interactive network of genomic islands at [16]). In the same paper it was shown that the genomic islands of M. tuberculosis most likely have originated from alpha-Proteobacterial intracellular parasites and symbionts of Agrobacterium, Rhizobium and Brucella genera. An activation of genetic vectors of this group was reported and it was hypothesized that it might be resulted from up-growing ocean water pollution with heavy metal ions and other industrial pollutants [9,24]. According to Bezuidt et al. [24], the recent outbreak of the enterohemorrhagic E. coli O104:H4 in 2011 was associated with an activation of this pool of mobile virulence genes. The same virulence vectors may affect in future the environmental and conditionally pathogenic Mycobacteria. Potentially the most risky species in this regard are M. marinum (fish pathogen) and M. avium ssp. paratuberculosis (cattle pathogen) as i) they were promiscuous in acquiring mobile genetic elements from different sources including taxonomically distant organisms; and ii) M. marinum might actively exchange genes with other environmental and conditionally pathogenic species of Mycobacterium. The latter capability is potentially dangerous as the exchange of readily available virulence genes between compatible potentially pathogenic bacteria may lead to spontaneous stochastic outbreaks of new diseases.
Methods
Genomes and annotation data used in this research
Complete genome sequences of 22 strains of Mycobacteria were obtained from NCBI database [25] in genbank format. Genomic island data including the annotation of all associated genes were obtained from Pre_GI database [26]. Numbers of genomic islands and horizontally acquired genes per genome are summarized in Table 1.
Genomic islands stored in Pre_GI were identified by SeqWord Gene Island Sniffer (SWGIS) program [27,28]. The analysis of gene content of genomic islands showed that the predicted genomic loci in M. leprae contained many unexpected conserved core genes like dnaA replication helicase and gyrase sub-units. These genomic islands most likely were false predicted resulting from a degeneration of the genome specific pattern of biased frequencies of oligonucleotides probably due to a higher rate of mutations [27]. Extremely high level of compositional variability of M. leprae genomes was confirmed by genome visualization using SWGIS [29,30]. According to Pre_GI data, the multiple genomic islands identified by SWGIS in the M. leprae genomes were not confirmed by other programs (IslandViewer and PAGIDB). To avoid further false predictions, the genomic islands of M. leprae were excluded from consideration in this research.
Phylogenomic inferences
Clusters of orthologous genes (COG) were identified by BLASTp alignment of all genes from different genomes against each other. Pairs of genes in two genomes where considered as orthologs if they reciprocally returned the best BLASTp hits. On the next step the MUSCLE alignment [31] was used to filter out those BLASTp predictions where the alignment coverage was less than 70% of the length of aligned proteins. Resulting alignment files were used for designing gene trees, but prior to phylogenetic analysis every alignment file was edited by the program Gblocks to remove ambiguous blocks [32]. For phylogenetic inferences based on alignments of orthologous sequences the super-matrix and super-tree approaches were used. In the former case all alignments were concatenated sequentially into an artificial super-alignment that was then analysed by the Neighbour-Joining algorithm implemented in MEGA6 [33]. In the latter case the phylogenetic trees were inferred individually for every COG alignment by the Neighbour-Joining algorithm implemented in neighbor.exe executable file of the PHYLIP package and then all the gene trees were reconciled into a reticulate phylogenetic network by the program SplitsTree [34].
A phylogenetic tree based on the presence and absence of accessory genes associated with the genomic islands was inferred by using the Wagner parsimony algorithm in pars.exe executable file of the PHYLIP package.
List of abbreviations
COGs - clusters of orthologous genes;
SWGIS - SeqWord Genomic Island Sniffer;
Mtb - Mycobacterium tuberculosis.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
OR - bioinformatics support, programming, manuscript preparation;
IK and AI contributed equally to data validating and manuscript preparation.
Supplementary Material
Genomic islands of M. avium subsp. paratuberculosis K-10
Declarations
Publication of this article has been funded by the program "Study on the reversion of antibiotic resistance in pathogenic microorganisms" funded in Kazakhstan (#0113PК00831). Programming and bioinformatics research was funded by the National Research Foundation of South Africa (#86941).
This article has been published as part of BMC Evolutionary Biology Volume 15 Supplement 1, 2015: Selected articles from the IX International Conference on the Bioinformatics of Genome Regulation and Structure\Systems Biology (BGRS\SB-2014): Evolutionary Biology. The full contents of the supplement are available online at http://www.biomedcentral.com/bmcevolbiol/supplements/15/S1.
References
- Brosch R, Pym AS, Gordon SV, Cole ST. The evolution of mycobacterial pathogenicity: clues from comparative genomics. Trends Microbiol. 2001;9:452–458. doi: 10.1016/S0966-842X(01)02131-X. [DOI] [PubMed] [Google Scholar]
- Murray CJ, Ortblad KF, Guinovart C, Lim SS, Wolock TM, Roberts DA, Dansereau EA, Graetz N, Barber RM, Brown JC. et al. Global, regional, and national incidence and mortality for HIV, tuberculosis, and malaria during 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2014;S0140-6736:60844–60848. doi: 10.1016/S0140-6736(14)60844-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams IB, Schafer JJ, Roberts AL, Short WR. Mycobacterium avium complex (MAC) immune reconstitution syndrome (IRIS) with reduced susceptibility to ethambutol in an HIV-infected patient: case report and review of the literature. Ann Pharmacother. 2014;48:1219–1224. doi: 10.1177/1060028014536879. [DOI] [PubMed] [Google Scholar]
- Sheu LC, Tran TM, Jarlsberg LG, Marras TK, Daley CL, Nahid P. Non-tuberculous mycobacterial infections at San Francisco General Hospital. Clin Respir J. in press . [DOI] [PubMed]
- Johnson MM, Odell JA. Nontuberculous mycobacterial pulmonary infections. J Thorac Dis. 2014;6:210–220. doi: 10.3978/j.issn.2072-1439.2013.12.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurita J, Ortega-Paredes D, Mora M, Espinel N, Parra H, Febres L, Zurita-Salinas C. Characterization of the first report of Mycobacterium timonense infecting an HIV patient in an Ecuadorian hospital. Clin Microbiol Infect. in press . [DOI] [PubMed]
- Santos M, Gil-Brusola A, Escandell A, Blanes M, Gobernado M. Mycobacterium genavense infections in a Tertiary Hospital and reviewed cases in non-HIV patients. Patholog Res Int. 2014;2014:371370. doi: 10.1155/2014/371370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veyrier FJ, Dufort A, Behr MA. The rise and fall of the Mycobacterium tuberculosis genome. Trends Microbiol. 2011;19:156–161. doi: 10.1016/j.tim.2010.12.008. [DOI] [PubMed] [Google Scholar]
- Reva O, Bezuidt O. Distribution of horizontally transferred heavy metal resistance operons in recent outbreak bacteria. Mob Genet Elements. 2012;2:96–100. doi: 10.4161/mge.19963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsella RJ, Fitzpatrick DA, Creevey CJ, McInerney JO. Fatty acid biosynthesis in Mycobacterium tuberculosis: lateral gene transfer, adaptive evolution, and gene duplication. Proc Natl Acad Sci USA. 2003;100:10320–10325. doi: 10.1073/pnas.1737230100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas-Magallanes V, Deschavanne P, Quintana-Murci L, Brosch R, Gicquel B, Neyrolles O. Horizontal transfer of a virulence operon to the ancestor of Mycobacterium tuberculosis. Mol Biol Evol. 2006;23:1129–1135. doi: 10.1093/molbev/msj120. [DOI] [PubMed] [Google Scholar]
- Fry KL, Meissner PS, Falkinham JO III. Epidemiology of infection by nontuberculous mycobacteria. VI. Identification and use of epidemiologic markers for studies of Mycobacterium avium, M. intracellulare, and M. scrofulaceum. Am Rev Respir Dis. 1986;134:39–43. doi: 10.1164/arrd.1986.134.1.39. [DOI] [PubMed] [Google Scholar]
- Schué M, Dover LG, Besra GS, Parkhill J, Brown NL. Sequence and analysis of a plasmid-encoded mercury resistance operon from Mycobacterium marinum identifies MerH, a new mercuric ion transporter. J Bacteriol. 2009;191:439–444. doi: 10.1128/JB.01063-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripoll F, Pasek S, Schenowitz C, Dossat C, Barbe V, Rottman M, Macheras E, Heym B, Herrmann JL, Daffé M, Brosch R, Risler JL, Gaillard JL. Non mycobacterial virulence genes in the genome of the emerging pathogen Mycobacterium abscessus. PLoS One. 2009;4:e5660. doi: 10.1371/journal.pone.0005660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobias NJ, Doig KD, Medema MH, Chen H, Haring V, Moore R, Seemann T, Stinear TP. Complete genome sequence of the frog pathogen Mycobacterium ulcerans ecovar Liflandii. J Bacteriol. 2013;195:556–564. doi: 10.1128/JB.02132-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An online interactive network of distribution of genomic islands. http://www.bi.up.ac.za/SeqWord/maps/map.html
- Li L, Bannantine JP, Zhang Q, Amonsin A, May BJ, Alt D, Banerji N, Kanjilal S, Kapur V. The complete genome sequence of Mycobacterium avium subspecies paratuberculosis. Proc Natl Acad Sci USA. 2005;102:12344–12349. doi: 10.1073/pnas.0505662102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horan KL, Freeman R, Weigel K, Semret M, Pfaller S, Covert TC, van Soolingen D, Leão SC, Behr MA, Cangelosi GA. Isolation of the genome sequence strain Mycobacterium avium 104 from multiple patients over a 17-year period. J Clin Microbiol. 2006;44:783–789. doi: 10.1128/JCM.44.3.783-789.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rindi L, Bonanni D, Lari N, Garzelli C. Requirement of gene fadD33 for the growth of Mycobacterium tuberculosis in a hepatocyte cell line. New Microbiol. 2004;27:125–131. [PubMed] [Google Scholar]
- Babbette B, LaMarca D, Zhu W, Arceneaux JE, Byers BR, Lundrigan MD. Participation of fad and mbt genes in synthesis of mycobactin in Mycobacterium smegmatis. J Bacteriol. 2004;186:374–382. doi: 10.1128/JB.186.2.374-382.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Zhang H, Tonge PJ, Tan DS. Mechanism-based inhibitors of MenE, an acyl-CoA synthetase involved in bacterial menaquinone biosynthesis. Bioorg Med Chem Lett. 2008;18:5963–5966. doi: 10.1016/j.bmcl.2008.07.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namouchi A, Didelot X, Schöck U, Gicquel B, Rocha EP. After the bottleneck: Genome-wide diversification of the Mycobacterium tuberculosis complex by mutation, recombination, and natural selection. Genome Res. 2012;22:721–734. doi: 10.1101/gr.129544.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezuidt O, Ganesan H, Labuschange P, Emmett W, Pierneef R, Reva O. In: Systems and computational biology - molecular and cellular experimental systems. Yang NS, editor. Croatia: Intech; 2011. Linguistic approaches for annotation, visualization and comparison of prokaryotic genomes and environmental sequences; pp. 27–52. [Google Scholar]
- Bezuidt O, Pierneef R, Mncube K, Lima-Mendez G, Reva ON. Mainstreams of horizontal gene exchange in enterobacteria: consideration of the outbreak of enterohemorrhagic E. coli O104:H4 in Germany in 2011. PLoS One. 2011;6:e25702. doi: 10.1371/journal.pone.0025702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCBI FTP database. ftp://ftp.ncbi.nih.gov/genomes/Bacteria/
- Pre_GI database. http://pregi.bi.up.ac.za/index.php
- Bezuidt O, Lima-Mendez G, Reva ON. SeqWord Gene Island Sniffer: a program to study the lateral genetic exchange among bacteria. World Academy of Science, Engineering and Technology. 2009;58:1169–1274. [Google Scholar]
- SeqWord Gene Island Sniffer home page. http://www.bi.up.ac.za/SeqWord/sniffer/index.html
- Ganesan H, Rakitianskaia AS, Davenport CF, Tümmler B, Reva ON. The SeqWord Genome Browser: an online tool for the identification and visualization of atypical regions of bacterial genomes through oligonucleotide usage. BMC Bioinformatics. 2008;9:333. doi: 10.1186/1471-2105-9-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SeqWord Genome Browser. http://www.bi.up.ac.za/SeqWord/mhhapplet.php
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56:564–577. doi: 10.1080/10635150701472164. [DOI] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Genomic islands of M. avium subsp. paratuberculosis K-10