

Abstract
Identifying all essential genomic components is critical for the assembly of minimal artificial life. In the genome-reduced bacterium Mycoplasma pneumoniae, we found that small ORFs (smORFs; < 100 residues), accounting for 10% of all ORFs, are the most frequently essential genomic components (53%), followed by conventional ORFs (49%). Essentiality of smORFs may be explained by their function as members of protein and/or DNA/RNA complexes. In larger proteins, essentiality applied to individual domains and not entire proteins, a notion we could confirm by expression of truncated domains. The fraction of essential non-coding RNAs (ncRNAs) non-overlapping with essential genes is 5% higher than of non-transcribed regions (0.9%), pointing to the important functions of the former. We found that the minimal essential genome is comprised of 33% (269,410 bp) of the M. pneumoniae genome. Our data highlight an unexpected hidden layer of smORFs with essential functions, as well as non-coding regions, thus changing the focus when aiming to define the minimal essential genome.
Keywords: minimal genome, non-coding RNAs, small proteins
Introduction
Defining the minimal genome that is required for sustaining life is currently one of the major challenges in biology. The essential genome of an organism, aside from protein-coding regions (ORFs), comprises regulatory (5′-UTRs and non-coding RNAs (ncRNAs)) and structural elements (Gil et al, 2004; Christen et al, 2011). Most of the previous essentiality studies (Glass et al, 2006; Lluch-Senar et al, 2007; French et al, 2008) made use of the conventional genome annotations which are biased against small proteins (smORFs; < 100 aa) (Samayoa et al, 2011) and regulatory elements such as ncRNAs. However, an accurate essentiality study is limited by the completeness of the genome annotation. Therefore, M. pneumoniae is an ideal organism due to its reduced genome size (816 kb) (Guell et al, 2009; Kuhner et al, 2009; Yus et al, 2009, 2012; Schmidl et al, 2010; Maier et al, 2011; van Noort et al, 2012; Lluch-Senar et al, 2013) and its detailed genome annotation based on experimental data. The current annotation of the M. pneumoniae genome contains 694 ORFs (32 of which are smORFs), 311 ncRNAs and 43 conventional RNAs (tRNAs, rRNAs, etc.) (Supplementary Table S2); all genes are well supported by transcriptome data, or in combination with proteome data [Supplementary Materials and Methods or http://mycoplasma.crg.eu/ for details (Wodke et al, 2014)]. This fine annotation of M. pneumoniae has been facilitated by the vast “-omics” datasets collected over the years (Guell et al, 2009; Maier et al, 2011; Yus et al, 2012), providing a better chance to gain a biased view on all putative essential elements in a minimal cell.
Results and Discussion
To determine the essentiality map, we used two mini-transposon mutant libraries (differing in the antibiotic resistance) of M. pneumoniae (Fig1A and B, Materials and Methods) and high-throughput insertion tracking by deep sequencing (HITS) (Wong et al, 2011) of cells at different days and serial passages (Fig1C). We analyzed day 12 sample since the number of insertions for the essential ORF gold set is close to zero, while this number for non-essential genes remains approximately constant (Fig1C) (Supplementary Table S1). We found a small insertion bias against G/C-rich quadruplet base sequences, but this does not affect the essentiality of smORFs since they have a similar composition as ORFs (Supplementary Materials and Methods). Based on the number of reads per insertion in the essential and non-essential gold sets, we define two thresholds to decide whether an insertion was annotated or not (a relaxed one with seven reads per insertion, and a stringent one with 41 reads) (Supplementary Materials and Methods). In the following, unless specified, we used the stringent value.
Figure 1.
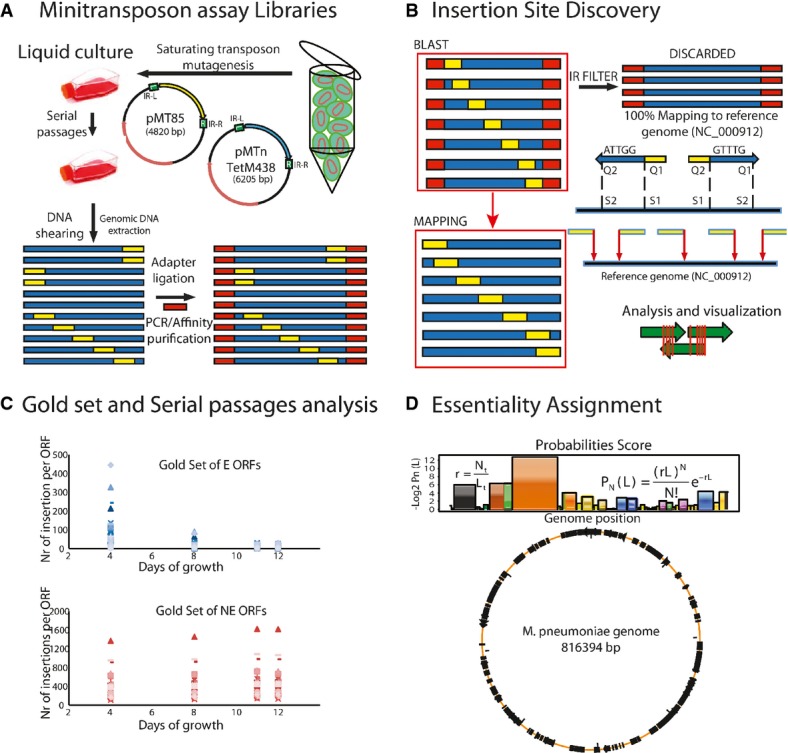
- Mini-transposon assay libraries. Map of pMT85 and pMTnTetM438 vectors and schematic representation of the procedure to obtain the mini-transposon libraries. Cells were grown in liquid culture (two serial passages) after transformation. Then, genomic DNAs were isolated and libraries were prepared for sequencing by HITS. Blue indicates regions of the M. pneumoniae genome, yellow represents transposon insertion sites, and red adaptor sequences.
- Insertion site discovery. Reads were first filtered by inverted repeat sequences and then mapped to the reference genome of M. pneumoniae. Insertion sites were defined by BLAST. Green arrows indicate ORFs and red lines transposon insertion sites.
- Determination of the optimal number of cell passages and culture days required for analyzing essentiality. Blue dots, in the upper graph, indicate the number of insertions for each gene in the essential gold set. Pink dots, in the lower graph, indicate the number of insertions for genes in the non-essential gold set. Growth of 11 to 12 days allows the best separation in terms of number of insertions for the essential and non-essential gold sets.
- Essentiality assignment. Upper panel shows the equations used to calculate the essentiality probabilities PN(L) with an example of a genome region (Supplementary Materials and Methods for the description of the formula). Lower circle represents essential regions in M. pneumoniae genome (in black); orange line represents non-essential regions.
The resulting integrated essentiality map (Supplementary File S1) after 12 days of growth consists of 69,994 unique mini-transposon insertions with a resolution of ∽4 bp for non-essential genes. Based on the analysis of the gold sets of essential and non-essential ORFs (Supplementary Table S1), we developed an essentiality probability criterion (Supplementary Materials and Methods; Fig1D) (Christen et al, 2011). Using this criterion, the 694 annotated ORFs were assigned to three distinct categories: essential (E; 342 ORFs), non-essential (NE; 259 ORFs) and fitness (F; 93 ORFs) (Supplementary Table S2) (Christen et al, 2011). The robustness of the classification was validated by the ability to isolate 92% of randomly selected F (12 genes) and NE (24 genes) clones, and the lack of success for 90% of E ORFs (28 out of 31, Supplementary Table S2). The 3 isolated E clones come out as fitness with the relaxed seven reads per insertion threshold, suggesting that they are severely affected in their growth. Moreover, when comparing with the predicted set of the minimal protein machinery in mollicutes, including 129 genes (Grosjean et al, 2014), we find 92% of them essential and 7% fitness. The dependency on the number of reads per insertion cutoff on fitness genes indicates that some of them could be incorrectly classified as essential when it is too strict. On the other hand, relaxing this cutoff results in some gold set essential genes being classified as fitness. This illustrates the limitation of transposon essentiality studies using deep sequencing for fitness genes.
Notably, we found that the insertions were not evenly distributed along the entire ORFs as previously observed in Caulobacter crescentus (Christen et al, 2011). In this respect, it is important to note that our mini-transposon has an internal promoter that could allow expression of downstream genes or domains if there is a start codon for translation. This hints at the existence of individual domains that mediate the interactions within sub-complexes. Indeed, we found that multi-domain proteins involved in protein complexes are frequently more essential than proteins with a single domain and they are involved in important cellular processes such as transcription and DNA replication (Supplementary Fig S1). Analyzing the essentiality of individual protein domains revealed that in 81 multi-domain proteins, the essentiality status of individual structural domains differs (Fig2, Supplementary Table S2, Supplementary Materials and Methods). Furthermore, cloning and expression of some of these structural domains (C-terminus of MPN241, Fig2A and N-terminus of MPN683, Fig2B) showed autonomous folding since they can be expressed in a soluble manner (Supplementary Fig S2). These results indicate that identification of a transposon insertion as criterion for protein essentiality should be revised and domain essentiality analysis should be routinely applied instead.
Figure 2.
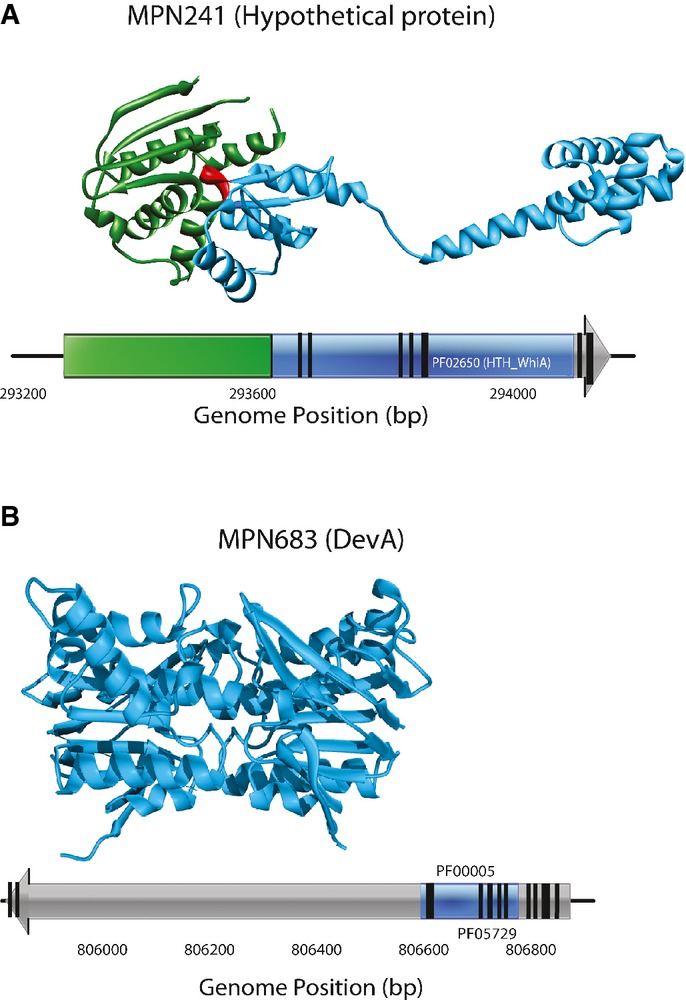
Essentiality at the protein domain level
Within non-transcriptionally active sequences of the M. pneumoniae genome, we detected 0.9% of essential intergenic regions (> 100 bp), which may function as structural elements including the origin of replication (oriC) (Fig3A, Supplementary Fig S3, Supplementary Table S3). In addition, we found that the percentages of essential transcriptionally active 5′-UTRs and ncRNAs (intergenic and overlapping with non-essential genes; for those overlapping with essential genes, no essentiality could be assigned) are 26 and 5%, respectively, and for conventional RNAs 82% (Fig3, Supplementary Table S2, Supplementary Materials and Methods). Strikingly, a large number of the ncRNAs (∽95%) overlap with coding genes on the opposite strand, which suggests that they have regulatory roles in gene expression. To gain insight into their functionality, we studied the correlation of expression of ncRNAs with their overlapping ORFs along 10 different time points of the growth curve by RNAseq. Interestingly, ncRNAs that anti-correlate with the overlapping ORF have higher essentiality coefficients than those that correlate (Fig3B, Supplementary Table S4). More importantly, the percentage of essential anti-correlated ORFs is higher than that of correlated ones (Fig3C; means of percentages: 50% versus 37%, respectively; P = 5.63e-10 applying Welch's two sample t-test), suggesting that essential ORFs are down-regulated by ncRNAs.
Figure 3.
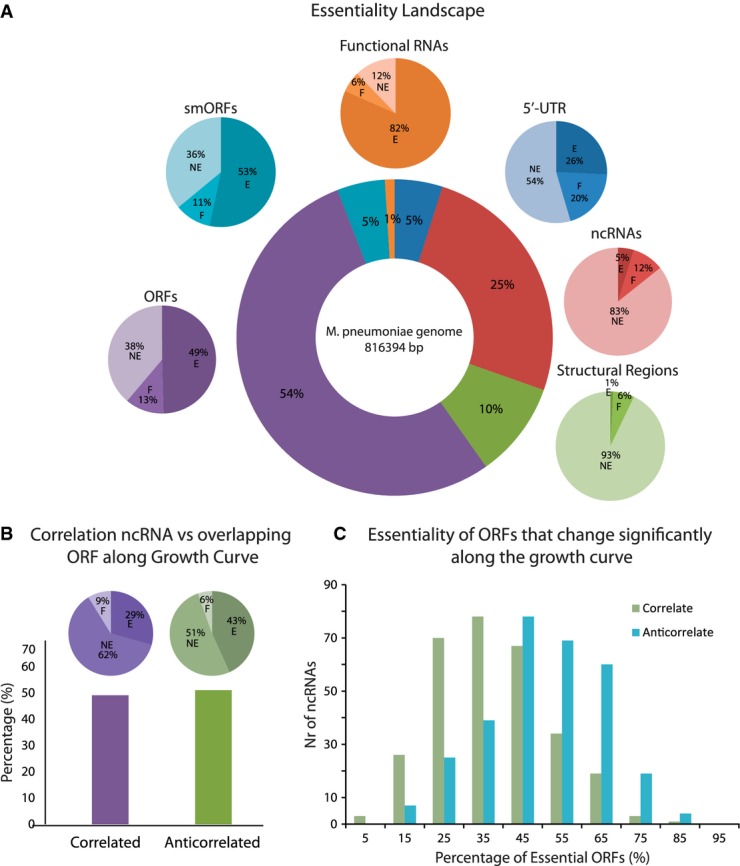
- Essentiality landscape of the M. pneumoniae genome. Circle in the center represents the percentages of the different genomic elements with respect to the chromosome length. The linked pie charts show the essentiality of each element (represented in the same color; E is essential, NE is non-essential, and F is fitness).
- The histogram represents the percentage of antisense ncRNAs that anti-correlate and correlate with their overlapping ORF along different time points of the growth curve (Supplementary Fig S7). The significant interactions (CLR score > 2.5) were determined for all pairs. Pearson correlations were used to determine whether the significant interaction is correlation or anti-correlation. The pie charts indicate the essentiality of the correlated and anti-correlated ncRNAs.
- The graph indicates the distributions of the percentages of essentiality of all ORFs whose expression is correlating and anti-correlating with a given ncRNA.
It is possible that some ncRNAs encode for smORFs similar to some long ncRNAs in eukaryotes (Cohen, 2014). In fact, smORFs have been found in bacteria associated with a diverse set of cellular functions (Hobbs et al, 2011; Samayoa et al, 2011). To investigate this, we translated all ncRNAs in the three reading frames and identified the putative ORFs by sequence searches and by combining mass spectroscopy (MS) with protein fractionation methodologies. Sequence conservation analysis with other bacterial species predicted eleven possible smORFs (Supplementary Fig S3, Supplementary Table S5, marked with α), of which four were identified by MS. Interesting examples are as follows: MPN391a, a cysteine-rich peptide predicted to be involved in peroxide resistance (Zimmerman & Herrmann, 2005), MPN347a, as part of an anti-toxin pair (Supplementary Fig S4) (Liu et al, 2008), and MPN155a that is homologous to a putative RNA-binding protein, YlxR, (Osipiuk et al, 2001) and is found in the same operon (Supplementary Fig S4). Interestingly, each fractionation methodology revealed new smORFs (Fig4A) extending the number from 32 annotated smORFs (25 detected proteins, mostly ribosomal, 56%) to a total of 67 smORFs (∽9% of the total ORFs). Additional fractionation experiments did not further increase the number of smORFs, suggesting that we are close to defining the complete M. pneumoniae small proteome (under the experimental limitation of identifiable peptides by MS for smORFs, Fig4A). As observed for the conventional ORFs, smORFs are often highly transcribed and essential (53%) (Supplementary Table S5, Fig3A).
Figure 4.

- Venn diagram showing the number of smORF proteins identified by MS in different experiments. SDS gels: smORFs identified in SDS gels (n = 4); solution: smORFs identified from total protein extracts (n = 4); SEC_MS: smORFs identified in size-exclusion chromatography (n = 2); DNAC_MS: smORFs identified in DNA–cellulose columns (n = 3). The graph represents the increase in the number of detected new smORFs (in orange) and known smORFs (in blue) by using different MS approaches. The names of MS approaches are as follows: Tris-Gly (n = 2) and Bis-Tis (n = 2) indicate smORFs identified by different SDS gels; solution: smORFs identified from total protein extracts (n = 4); SEC_MS1: smORFs identified in the first experiment of size-exclusion chromatography (n = 1) and SEC_MS2: smORFs identified in the second experiment of size-exclusion chromatography (n = 1); DNAC_RNA, DNAC_C and DNAC_NaCl: smORFs identified in DNA–cellulose columns, eluted by using RNA, chromatin or NaCl, respectively (n = 3); and sucrose indicates the number of smORFs identified by sucrose cushion.
- The graph represents the size-exclusion chromatography elution profiles of different proteins: MPN516 (rpoB, subunit of the RNA polymerase complex), MPN606 (enolase, 50 kDa) and MPN476 (cytidylate kinase, 24 kDa). The main elution peak for the identified smORFs is indicated by dots. MPN155a and MPN060a were found after overexpression as a TAP fusion.
- The histogram represents the percentages of proteins in each category that have been identified to interact with DNA in affinity chromatography after receiving operating characteristic curve (ROC) analysis (Supplementary Materials and Methods). “All proteins” represent the percentage of all proteins excluding known DNA- and RNA-binding proteins (Supplementary Materials and Methods). “DNA binding” proteins show the percentage of known poly-nucleotide directly binding proteins (Supplementary Materials and Methods). Black bars indicate the standard deviation after considering replicates from three different experiments.
In order to get insight into the reasons behind the high essentiality of the smORFs, we first investigated whether they are part of large protein complexes as previously suggested for some smORFs (Gassel et al, 1999). By size-exclusion chromatography coupled to MS (SEC-MS), we found that the vast majority (31 out of 34; 11 new) of the detectable smORFs eluted in fractions of significantly higher molecular weight than expected from the size of the individual proteins. This indicates that smORFs are frequently associated within larger protein complexes (Fig4B, Supplementary Table S6) and probably this is the case for the majority of the smORF. For example, overexpressing two smORFs, MPN060a and MPN155a, not detected in the original SEC-MS experiments, we find them eluting in high molecular weight fractions (Fig4B, Supplementary Fig S3B). Second, we used DNA–cellulose (DNAC) affinity chromatography coupled to MS to analyze DNA- or RNA-binding properties (Mai et al, 1998). We found that out of 35 smORFs identified in this experiment (14 previously unknown, Fig4A), 42% of new smORFs (including the putative RNA-binding protein MPN155a, YlxR) bind to DNA/RNA, compared to 15% of the conventional ORFs (excluding well-known DNA and RNA directly binding proteins) in M. pneumoniae (Fig4C, Supplementary Materials and Methods).
Understanding the minimal set of essential genetic elements is important for several applications, ranging from synthetic biology approaches to drug targets identification in pathogenic bacteria (Gallagher et al, 2007). Based on our analysis, we conclude that essentiality should be considered at a protein domain resolution and that smORFs as well as regulatory elements (5′-UTRs and ncRNAs) are frequently essential genomic elements, considerably increasing the repertoire of building blocks that need to be considered for a minimal genome. Furthermore, we revealed a previously unknown layer of essentiality composed of smORFs that are likely to play important roles in protein complex functionality and DNA transcriptional regulation. Thus, it is crucial to more carefully consider smORFs in genome annotations as they can comprise 9% of the genome ORFs.
Materials and Methods
The mini-transposon mutant libraries of M. pneumoniae were obtained after transforming with pMT85 and pMTnTetM438 vectors and doing serial passages (Supplementary Materials and Methods) (Pich et al, 2006). Genomic DNAs were collected using the Illustrabacteria genomic kit (GE) and sequenced with the HITS approach (Fig1A and B) using standard Illumina paired-end sequencing. Raw reads were filtered by inverted repeats (IR) and then mapped to the M. pneumoniae reference genome (NC_000912, NCBI) using BLASTs (Supplementary Table S7).
Two gold standard sets were manually assembled; one contained 37 protein-coding genes that are known to be essential, and the other contained 29 NE ORFs (Supplementary Table S1). The two datasets were evaluated using our mini-transposon library, and then, a scoring system was developed that consisted of two parameters, PE, the probability for a genomic region of being essential, and PNE, the probability of being non-essential rounded to two decimals (Supplementary Table S2). This analysis revealed three distinct groups of genes with 99% confidence (Supplementary Table S2): those that are essential (E; PE > 0 and PNE = 0), those that are non-essential (NE; PE = 0 and PNE > 0) and a third group with an intermediate essentiality score that we define as fitness (F; PE > 0; PNE > 0 or PE = 0; PNE = 0). The fitness category includes those genes that essentiality could depend on condition and transposon insertions and despite having an impact on growth, they do not affect cell viability.
To study whether the protein products of smORFs could be involved in protein complexes, ten smORFs were selected and cloned into vector pMT85-clpB-TAPtag SfiI/NotI (Kuhner et al, 2009). After transforming M. pneumoniae, the protein complexes were studied by molecular weight exclusion chromatography coupled to Western blot. Fractions from molecular weight exclusion chromatography were trypsin-digested and then subjected to MS (Supplementary Materials and Methods). DNA/RNA-binding proteins were identified by DNA–cellulose (DNAC) affinity chromatography coupled to MS (Supplementary Materials and Methods).
Data availability
The raw data of transposon libraries and RNAseq have been submitted to the ArrayExpress database (http://www.ebi.ac.uk/arrayexpress) and assigned the identifier E-MTAB-3075 and E-MTAB-3076, respectively. Additionally, genome re-annotation and MS data used for identification of smORFs have been submitted to ProteomeXchange via the PRIDE database (http://www.ebi.ac.uk/pride) and assigned the identifier PXD001611.
Acknowledgments
We thank Dr. Christina Kiel for her comments and the Genomics, Proteomics and Protein Technologies Core Facilities at CRG. Also we thank to Dr. Marc Güell and Dr. Hinnerk Eilers for fruitful discussions. Besray Unal was co-funded by Marie Curie Actions. This work was supported by the European Research Council (ERC), the Fundación Marcelino Botin, the Spanish Ministerio de Economía y Competitividad BIO2007-61762 and the ISCIII (PI10/01702).
Author contributions
LS and PB conceived the study; MLS, JDB and WHC assembled and analyzed the data and wrote the manuscript; PB, LS, JS and ACG revised the manuscript; FJO, TF, MLS and VvN performed experiments of protein complexes; JAHW generated the database of ORFs. VLLR and EBU helped with the analyses of the transcriptome data; EY and SM did DNA-binding experiments; MLS and AV developed HITS technique. RJN obtained the DNA samples of transposon libraries at the different passages. AS participated in isolation of M. pneumoniae mutants from the library, all authors have read and approved the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary File S1
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Table S7
Supplementary Table S8
Supplementary Table S9
Supplementary Table S10
Supplementary Table S11
Supplementary Information
Review Process File
References
- Christen B, Abeliuk E, Collier JM, Kalogeraki VS, Passarelli B, Coller JA, Fero MJ, McAdams HH, Shapiro L. The essential genome of a bacterium. Mol Syst Biol. 2011;7:528. doi: 10.1038/msb.2011.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SM. Everything old is new again: (linc)RNAs make proteins! EMBO J. 2014;33:937–938. doi: 10.1002/embj.201488303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French CT, Lao P, Loraine AE, Matthews BT, Yu H, Dybvig K. Large-scale transposon mutagenesis of Mycoplasma pulmonis. Mol Microbiol. 2008;69:67–76. doi: 10.1111/j.1365-2958.2008.06262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher LA, Ramage E, Jacobs MA, Kaul R, Brittnacher M, Manoil C. A comprehensive transposon mutant library of Francisella novicida, a bioweapon surrogate. Proc Natl Acad Sci USA. 2007;104:1009–1014. doi: 10.1073/pnas.0606713104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gassel M, Mollenkamp T, Puppe W, Altendorf K. The KdpF subunit is part of the K(+)-translocating Kdp complex of Escherichia coli and is responsible for stabilization of the complex in vitro. J Biol Chem. 1999;274:37901–37907. doi: 10.1074/jbc.274.53.37901. [DOI] [PubMed] [Google Scholar]
- Gil R, Silva FJ, Pereto J, Moya A. Determination of the core of a minimal bacterial gene set. Microbiol Mol Biol Rev. 2004;68:518–537. doi: 10.1128/MMBR.68.3.518-537.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass JI, Assad-Garcia N, Alperovich N, Yooseph S, Lewis MR, Maruf M, Hutchison CA, III, Smith HO, Venter JC. Essential genes of a minimal bacterium. Proc Natl Acad Sci USA. 2006;103:425–430. doi: 10.1073/pnas.0510013103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosjean H, Breton M, Sirand-Pugnet P, Tardy F, Thiaucourt F, Citti C, Barre A, Yoshizawa S, Fourmy D, de Crecy-Lagard V, Blanchard A. Predicting the minimal translation apparatus: lessons from the reductive evolution of mollicutes. PLoS Genet. 2014;10:e1004363. doi: 10.1371/journal.pgen.1004363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güell M, van Noort V, Yus E, Chen WH, Leigh-Bell J, Michalodimitrakis K, Yamada T, Arumugam M, Doerks T, Kühner S, Rode M, Suyama M, Schmidt S, Gavin AC, Bork P, Serrano L. Transcriptome complexity in a genome-reduced bacterium. Science. 2009;326:1268–1271. doi: 10.1126/science.1176951. [DOI] [PubMed] [Google Scholar]
- Hobbs EC, Fontaine F, Yin X, Storz G. An expanding universe of small proteins. Curr Opin Microbiol. 2011;14:167–173. doi: 10.1016/j.mib.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser BK, Clifton MC, Shen BW, Stoddard BL. The structure of a bacterial DUF199/WhiA protein: domestication of an invasive endonuclease. Structure. 2009;17:1368–1376. doi: 10.1016/j.str.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhner S, van Noort V, Betts MJ, Leo-Macias A, Batisse C, Rode M, Yamada T, Maier T, Bader S, Beltran-Alvarez P, Castaño-Diez D, Chen WH, Devos D, Güell M, Norambuena T, Racke I, Rybin V, Schmidt A, Yus E, Aebersold R, et al. Proteome organization in a genome-reduced bacterium. Science. 2009;326:1235–1240. doi: 10.1126/science.1176343. [DOI] [PubMed] [Google Scholar]
- Liu M, Zhang Y, Inouye M, Woychik NA. Bacterial addiction module toxin Doc inhibits translation elongation through its association with the 30S ribosomal subunit. Proc Natl Acad Sci USA. 2008;105:5885–5890. doi: 10.1073/pnas.0711949105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lluch-Senar M, Vallmitjana M, Querol E, Pinol J. A new promoterless reporter vector reveals antisense transcription in Mycoplasma genitalium. Microbiology. 2007;153:2743–2752. doi: 10.1099/mic.0.2006/007559-0. [DOI] [PubMed] [Google Scholar]
- Lluch-Senar M, Luong K, Llorens-Rico V, Delgado J, Fang G, Spittle K, Clark TA, Schadt E, Turner SW, Korlach J, Serrano L. Comprehensive methylome characterization of Mycoplasma genitalium and Mycoplasma pneumoniae at single-base resolution. PLoS Genet. 2013;9:e1003191. doi: 10.1371/journal.pgen.1003191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai VQ, Chen X, Hong R, Huang L. Small abundant DNA binding proteins from the thermoacidophilic archaeon Sulfolobus shibatae constrain negative DNA supercoils. J Bacteriol. 1998;180:2560–2563. doi: 10.1128/jb.180.9.2560-2563.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier T, Schmidt A, Guell M, Kuhner S, Gavin AC, Aebersold R, Serrano L. Quantification of mRNA and protein and integration with protein turnover in a bacterium. Mol Syst Biol. 2011;7:511. doi: 10.1038/msb.2011.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Noort V, Seebacher J, Bader S, Mohammed S, Vonkova I, Betts MJ, Kuhner S, Kumar R, Maier T, O'Flaherty M, Rybin V, Schmeisky A, Yus E, Stülke J, Serrano L, Russell RB, Heck AJ, Bork P, Gavin AC. Cross-talk between phosphorylation and lysine acetylation in a genome-reduced bacterium. Mol Syst Biol. 2012;8:571. doi: 10.1038/msb.2012.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osipiuk J, Gornicki P, Maj L, Dementieva I, Laskowski R, Joachimiak A. Streptococcus pneumonia YlxR at 1.35 A shows a putative new fold. Acta Crystallogr D Biol Crystallogr. 2001;57:1747–1751. doi: 10.1107/s0907444901014019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pich OQ, Burgos R, Planell R, Querol E, Pinol J. Comparative analysis of antibiotic resistance gene markers in Mycoplasma genitalium: application to studies of the minimal gene complement. Microbiology. 2006;152:519–527. doi: 10.1099/mic.0.28287-0. [DOI] [PubMed] [Google Scholar]
- Samayoa J, Yildiz FH, Karplus K. Identification of prokaryotic small proteins using a comparative genomic approach. Bioinformatics. 2011;27:1765–1771. doi: 10.1093/bioinformatics/btr275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidl SR, Gronau K, Pietack N, Hecker M, Becher D, Stulke J. The phosphoproteome of the minimal bacterium Mycoplasma pneumoniae: analysis of the complete known Ser/Thr kinome suggests the existence of novel kinases. Mol Cell Proteomics. 2010;9:1228–1242. doi: 10.1074/mcp.M900267-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PC, Karpowich N, Millen L, Moody JE, Rosen J, Thomas PJ, Hunt JF. ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol Cell. 2002;10:139–149. doi: 10.1016/s1097-2765(02)00576-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wodke JA, Alibes A, Cozzuto L, Hermoso A, Yus E, Lluch-Senar M, Serrano L, Roma G. MyMpn: a database for the systems biology model organism Mycoplasma pneumoniae. Nucleic Acids Res. 2014 doi: 10.1093/nar/gku1105. doi: 10.1093/nar/gku1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SM, Gawronski JD, Lapointe D, Akerley BJ. High-throughput insertion tracking by deep sequencing for the analysis of bacterial pathogens. Methods Mol Biol. 2011;733:209–222. doi: 10.1007/978-1-61779-089-8_15. [DOI] [PubMed] [Google Scholar]
- Yus E, Maier T, Michalodimitrakis K, van Noort V, Yamada T, Chen WH, Wodke JA, Guell M, Martinez S, Bourgeois R, Raineri E, Letunic I, Kalinina OV, Rode M, Herrmann R, Gutiérrez-Gallego R, Russell RB, Gavin AC, Bork P, et al. Impact of genome reduction on bacterial metabolism and its regulation. Science. 2009;326:1263–1268. doi: 10.1126/science.1177263. [DOI] [PubMed] [Google Scholar]
- Yus E, Guell M, Vivancos AP, Chen WH, Lluch-Senar M, Delgado J, Claude Gavin A, Bork P, Serrano L. Transcription start site associated RNAs in bacteria. Mol Syst Biol. 2012;8:585. doi: 10.1038/msb.2012.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman CU, Herrmann R. Synthesis of a small, cysteine-rich, 29 amino acids long peptide in Mycoplasma pneumoniae. FEMS Microbiol Lett. 2005;253:315–321. doi: 10.1016/j.femsle.2005.09.054. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary File S1
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Table S7
Supplementary Table S8
Supplementary Table S9
Supplementary Table S10
Supplementary Table S11
Supplementary Information
Review Process File
Data Availability Statement
The raw data of transposon libraries and RNAseq have been submitted to the ArrayExpress database (http://www.ebi.ac.uk/arrayexpress) and assigned the identifier E-MTAB-3075 and E-MTAB-3076, respectively. Additionally, genome re-annotation and MS data used for identification of smORFs have been submitted to ProteomeXchange via the PRIDE database (http://www.ebi.ac.uk/pride) and assigned the identifier PXD001611.