

Abstract
Genes identified as being mutated in Wilms’ tumour include TP53, a classic tumour suppressor gene (TSG); CTNNB1 (encoding β-catenin), a classic oncogene; WTX, which accumulating data indicate is a TSG; and WT1, which is inactivated in some Wilms’ tumours, similar to a TSG. However, WT1 does not always conform to the TSG label, and some data indicate that WT1 enhances cell survival and proliferation, like an oncogene. Is WT1 a chameleon, functioning as either a TSG or an oncogene, depending on cellular context? Are these labels even appropriate for describing and understanding the function of WT1?
Wilms’ tumour, a childhood embryonic tumour of the kidney, has long fascinated biomedical investigators, partly owing to its unique histology, which is suggestive of disorganized and incomplete kidney development, and partly owing to the early recognition that only a few rate-limiting alterations have key roles in tumorigenesis. Although this observation held the promise of a possible quick identification and functional understanding of these crucial gene alterations, this has not been the case. Mutations in Wilms tumour 1 (WT1), Wilms tumour gene on the X chromosome (WTX; also known as FAM123B and AMER1), β-catenin (CTNNB1) and TP53 have been identified in tumours1–4, although they occur, either singly or in combination, in only one-third of tumours5. CTNNB1 and TP53 are a well-established oncogene and a tumour suppressor gene (TSG), respectively, and accumulating data (reviewed below) suggest that WTX also functions as a TSG4–7. The observed tumour-specific ablation of WT1 led to its labelling as a TSG with the subsequent assumption that its normal cellular function is to inhibit cell proliferation. However, some observational and experimental data, and studies of haematopoiesis and acute myeloid leukaemia (AML), are suggestive of a role for WT1 as a survival and/or a growth-enhancing factor8–14. Furthermore, in some studies on certain cancers (for example, AML) WT1 expression has been associated with poor clinical outcome15–18, and neutralizing antibodies against WT1 have shown great promise in clinical trials19–21. Thus, the TSG label may mask a far more complex role for WT1 during development and tumorigenesis. In this Review, I present data that suggest an oncogenic role for WT1 during leukaemogenesis, I compare these data with data from studies on Wilms’ tumours and I discuss the effect of differentiation status and the presence of other gene mutations on the functional importance of WT1 mutation. I also present recent data on WTX that further support its tumour suppressor designation.
Wilms’ tumour genetics
Wilms’ tumour occurs at a frequency of ~1 in 10,000 live births. The mean age at diagnosis is 43–48 months, depending on gender, and 95% of cases are diagnosed by 10 years of age. Tumours occur unilaterally (in a single kidney) in 90–95% of cases, but can also occur bilaterally; bilateral cases present ~12 months earlier than unilateral cases22. Approximately 2% of cases have an affected relative, and Wilms’ tumour predisposition segregates in families as an autosomal dominant trait22–24. Familial Wilms’ tumour cases are generally associated with an increased frequency of bilateral disease and an earlier age of diagnosis, although variability exists between Wilms’ tumour families22–25. Similarly, patients with Wilms’ tumour with aniridia, genitourinary anomalies and mental retardation (known as WAGR syndrome) are more likely to develop bilateral disease and at an earlier age22. Statistical analysis of the age and frequency of bilateral tumours in both sporadic and familial patients with Wilms’ tumour suggested that, like retinoblastoma (another embryonic tumour occurring in childhood), the genetic aetiology of Wilms’ tumour fits a model in which two genetic alterations are rate-limiting (although not necessarily sufficient) for tumour development23. Patients with bilateral tumours, congenital anomalies and/or a family history were thought to inherit the first alteration and acquire the second event somatically. Patients with unilateral tumours and no family history were hypothesized to acquire both rate-limiting alterations somatically23.
Molecular analyses of patients and their tumours are consistent with this initial model. The presence of hetero zygous, cytogenetically detectable deletions encompassing chromosomal band 11p13 in most patients with WAGR immediately localized a Wilms’ tumour gene to this genomic region, and this was key to the identification of WT1, which encodes a transcription factor (described below). Tumours from patients with WAGR who are germline hemizygotes for WT1, that is, they have only one allele of WT1, invariably have somatic mutations in the remaining WT1 allele. Tumours from patients hetero zygous for an intragenic WT1 mutation are reduced to homozygosity for the germline mutation. Additionally, tumours from patients with no germline WT1 mutation are sometimes homozygous for somatic WT1 mutations or can carry compound heterozygous mutations2,26. In all these cases, the mutations are predicted to abolish WT1 function2,26. These observations strongly suggest that WT1 is a TSG, the homozygous inactivation of which is a crucial step in the development of Wilms’ tumour.
Wilms’ tumour is known to be genetically heterogeneous; WT1 mutations are present in only ~20% of tumours2,5. Other genes mutated in Wilms’ tumour include WTX (~20%), a gene identified through high-resolution comparative genome hybridization (CGH) analysis and which encodes a protein reported to negatively regulate the Wnt pathway; CTNNB1 (~15%), which encodes β-catenin and upregulates the Wnt pathway; and the well-known TSG, TP53 (5%)1,3–5,27 (TABLE 1). Loss of heterozygosity (LOH) studies and CGH studies have implicated other regions of the genome in the development of Wilms’ tumour, but the corresponding Wilms’ tumour gene has generally not yet been identified. Although WT1 germline mutations are observed in some patients with Wilms’ tumour, familial predisposition to Wilms’ tumour is rarely due to WT1 mutation, and predisposition in some families has been linked to loci on 19q and 17q28,29.
Table 1.
Commonly occurring gene mutations in Wilms’ tumour
| Gene | Type of mutation | Frequency | Germline or somatic? |
Tumour zygosity | Predicted effect | Refs |
|---|---|---|---|---|---|---|
| WT1 | Whole or partial gene deletion, insertion, nonsense and missense |
~20% | Both | Predominantly homozygous |
Inactivation of protein |
2,5 |
| WTX | Whole or partial gene deletion and nonsense |
~20% | Somatic only | Heterozygous and hemizygous |
Inactivation of protein |
4–7 |
| CTNNB1 | Inframe deletions and missense |
~15% | Somatic only | Heterozygous | Stabilization of protein |
3,90 |
| TP53 * | Missense | ~5% | Both | Heterozygous and homozygous |
Inactivation of protein |
1 |
Associated with anaplastic Wilms’ tumours.
Additional molecular alterations are observed in Wilms’ tumours, most notably LOH or loss of imprinting (LOI) on 11p15, which occurs in up to 70% of tumours30,31. This region is known to harbour genes for the somatic overgrowth syndrome, beckwith–Wiedemann syndrome (BWS), one feature of which is a predisposition to embryonal tumours, including Wilms’ tumour, embryonal rhabdomyosarcoma and hepatoblastoma32,33. Tumour range differs between BWS families, and those with cases of Wilms’ tumour carry germline mutations that alter the imprinting (methylation) of the insulin-like growth factor 2 (IGF2)–H19 gene cluster and result in biallelic expression of IGF2, which is normally expressed solely from the paternally derived allele34,35. IGF2 upregulation is also observed in Wilms’ tumours with 11p15 LOH or LOI30,31,36. These data strongly suggest that epigenetic upregulation of IGF2 can be an important, but not sufficient, alteration for tumorigenesis.
In summary, the known genetic alterations that occur in Wilms’ tumours are mutations of tumour-suppressor genes (WT1, WTX and TP53) and an oncogene (CTNNB1)1–4 (BOX 1). But are these labels appropriate? In some malignancies does WT1 function as an oncogene? Is WTX clearly a TSG? before exploring this issue, it is important to establish what is known about the function, expression, biological role and mutational consequence of these genes.
Box 1. CTNNB1 in nephrogenesis and Wilms’ tumour.
CTNNB1, which encodes β-catenin, has crucial roles during development and tumorigenesis, functioning both as a component of membrane adherens junctions and as an effector in the canonical Wnt signalling pathway99–101. In the developing kidney, CTNNB1 gene expression and signalling is detected in the ureteric bud, metanephric mesenchyme and condensed mesenchyme. Its expression is downregulated during differentiation and no expression is detected in adult kidney102. Consistent with this expression pattern, overexpression of a constitutively stable β-catenin mutant in tubule epithelium in transgenic mice results in renal cysts with associated increased cell proliferation in the tubular epithelium103. Global stabilization of β-catenin using pharmacological inactivation of glycogen synthase kinase 3 (GSK3) results in the induction of nephron development and also ectopic nephrogenesis in kidney organ culture104. Stabilization of β-catenin specifically in nephron progenitors in the condensed mesenchyme results in a similar phenotype of ectopic nephron induction in vivo in mice105. Interestingly, mature nephrons are not observed in these mice. Moreover, knock out of Ctnnb1 in the same nephron progenitor cells results in an almost complete loss of mature nephrons105. These data suggest a role for β-catenin in maintaining a balance between progenitor cell renewal and the initiation of nephron differentiation105. Although the role of β-catenin in normal development — and the effect of abnormal stabilization of β-catenin — is still not well understood, these data suggest that activation of β-catenin must be tightly regulated temporally and spatially for normal nephrogenesis. CTNNB1 mutations with known functional importance are observed in ~15% of Wilms’ tumours, and the majority are three nucleotide deletions or missense mutations that delete or mutate Ser45, one of the key targets for the phosphorylation and degradation of β-catenin3,90. CTNNB1 mutations are invariably somatic and are heterozygous (TABLE 1).
WT1
The WT1 gene consists of ten exons and encodes a zinc finger transcription factor containing an amino terminal proline glutamine-rich protein-interaction domain and four carboxy-terminal zinc finger domains37,38 (FIG. 1). Two different alternative splice sites result in four isoforms: proteins with and without exon 5, which encodes 17 amino acids, and proteins with and without three amino acids (lysine, threonine and serine (KTS)) between the third and fourth zinc finger domains encoded by exons 9 and 10, respectively. Mice homozygous for a Wt1 allele lacking exon 5 have no salient phenotype39. By contrast, a variety of data point to a profound functional importance of the alternative KTS splicing. For example, mice carrying either a KTS-positive (KTS+) or a KTS-negative (KTS−) Wt1 transgene display distinct phenotypes40, and X-ray crystallographic and structural studies along with DNA binding studies indicate that the KTS insertion destabilizes the interaction of WT1 with DNA41,42. A role for the KTS+ isoform in binding to RNA has also been reported43.
Figure 1. WT1 mutations observed in Wilms’ tumour and acute myeloid leukaemia (AML).

a ∣ WT1 gene and protein structure. Blue shading indicates alternatively spliced domains. Four isoforms result from exon 5 and KTS alternative splicing. b ∣ Germline mutations observed in patients with Wilms’ tumour-associated phenotypes. Similar mutations are observed somatically in tumours. c ∣ Somatic mutations observed in AML. Location of gene mutations are indicated by blue (insertion or deletions and frameshift mutations), purple (nonsense mutations), green (missense mutations) and orange (splice site mutations) ovals. Predicted mutant proteins are shown for deletion mutations. Asterisk indicates reduced KTS+/KTS− isoform ratio as a result of IVS9 splice site mutations. ZNF, zinc finger domain. Data from REFS 61,76–78.
In addition to splice variants, a non-AUG translation start site 5′ to the traditional AUG start site associated with the Kozak consensus sequence has been reported and results in an additional 68 amino acids at the N terminus. The functional importance of this variant is unknown; mice homozygous for an allele unable to encode this 68 amino acid extension display no phenotype44. An alternative exon 1 (exon 1a) containing an alternative AUG has also been reported, but the functional importance of this is unknown45. Editing of the WT1 transcript at an exon 6 codon has been proposed, based on sequence data from a rat testes cDNA library46. However, no evidence of such editing has been detected in Wilms’ tumours or in a genome-wide assessment of RNA editing47,48. WT1 has been reported to be post-translationally modified by phosphorylation, ubiquitylation and sumoylation49–51, but the effect of these modifications on protein function is still unclear.
Although WT1 has been reported to have a role in RNA processing43, the most well-documented function for WT1 is that of a transcription factor, and many genes have been identified by various approaches as being regulated, either positively or negatively, by WT1. Studies using Wt1 conditional knockout mice have identified Sox9, Snail and Cdh1 (encoding E-cadherin) as being deregulated in vivo following Wt1 ablation in embryonic testes or cardiovascular progenitor cells52,53, although evidence for a direct role for WT1 in transcriptional regulation is only available for Snail and Cdh1 (REF. 53).
WT1 in nephrogenesis
The histology of Wilms’ tumour suggests that tumorigenesis involves, at least in part, a perturbation of normal kidney development (BOX 2). Consistent with this, Wt1 is expressed early in nephrogenesis54,55. In the mouse, low levels of Wt1 expression are first detected at ~embryonic day (E) 9 in the intermediate mesoderm and later in the metanephric mesenchyme from which the nephrons and stroma of adult kidneys are derived. WT1 expression is widespread in the metanephric mesenchyme and is upregulated upon metanephric mesenchyme condensation. Subsequently, WT1 expression becomes increasingly restricted until, in the mature kidney, it is expressed in the highly differentiated glomerular podocytes.
Box 2. Kidney development and Wilms’ tumour histology.
The peculiar pathology of Wilms’ tumours first suggested a link between tumorigenesis and abnormal kidney development. The adult kidney arises during embryogenesis from intermediate mesoderm (reviewed in REF. 106). Its formation entails complex reciprocal interactions between the ureteric bud and the metanephric mesenchyme with the ureteric bud inducing mesenchyme condensation, which in turn induces ureteric bud growth and branching, to form the collecting duct system of the kidney. Condensed mesenchyme forms, in succession, distinctive structures (for example, renal vesicles, comma-shaped bodies and S-shaped bodies) that ultimately become the glomerulus and proximal and distal tubules of the nephron. During this morphogenesis, cells undergo a mesenchymal to epithelial transition and differentiate into the many specialized cells of the mature nephron. This process is asynchronous, with the ureteric bud continuously branching and invading new metanephric mesenchyme at the periphery of the developing organ. In humans, nephrogenesis is normally complete at birth; in mice it continues a few days post-natally.
Wilms’ tumours are thought to arise from metanephric mesenchyme and typically display a fascinating triphasic pathology in which histologically distinctive cell types derived from fetal mesenchyme (blastema, stroma and epithelial tubules) are observed (reviewed in REF. 10). These cells, however, are not normally differentiated and are aberrantly organized. In some tumours, heterologous elements, such as smooth muscle and cartilage, are observed. Thus, Wilms’ tumour is thought to be a malignancy of a relatively early mesenchymal progenitor cell that is able to differentiate, albeit aberrantly, and does not respond to normal growth control signals.
Observations of Wilms’ tumour and, as described below, early-onset renal failure in some patients with germline WT1 mutations first implicated WT1 as having important roles in kidney development and function. The manipulation of Wt1 in mouse embryos and the generation of Wt1 mutant mouse models have greatly elucidated these roles. In Wt1−/− mouse embryos metanephric blastema is present at E10.5, but quickly becomes apoptotic. By E12 no blastemal cells are detectable and embryos display complete renal agenesis56. Therefore, Wt1 is crucial, either directly or indirectly, for the survival of these early progenitor cells. However, somatic ablation of Wt1 at ~E13.5 results not in apoptosis but in a profound disruption of metanephric mesenchyme differentiation resulting in an almost complete absence of nephrons at birth57. A similar result was observed following downregulation of Wt1 using small interfering RNA (siRNA) in E11.5 kidney rudiment explant culture58. Thus, the cellular phenotype resulting from Wt1 knockdown at this later stage of nephrogenesis is compatible with its deduced role as a TSG in Wilms’ tumour.
WT1 alterations, genitourinary anomalies and Wilms’ tumour
Consistent with its expression and crucial role in the development of the kidney and gonad, in addition to predisposition to Wilms’ tumour, individuals who are heterozygous for WT1 germline mutations can also display genitourinary tract anomalies and glomerulosclerosis, leading to renal failure59. As depicted in FIG. 1, there are strong (but not inviolable) genotype–phenotype associations such that germline WT1 deletion or truncation mutations are associated with Wilms’ tumour and genitourinary anomalies (more frequently observed in males), and WT1 missense mutations at specific, highly conserved codons result in Denys–Drash syndrome60,61. Genitourinary anomalies and renal failure (albeit at a later age of onset) are also observed, along with gonadoblastoma, in Frasier syndrome62. Interestingly, patients with Frasier syndrome are typically heterozygous for IvS9 germline mutations that affect KTS splicing and reduce the KTS+ isoform/KTS− isoform ratio63. However, as patients with Frasier syndrome can present with WT1 mutations that are commonly seen in patients with Denys–Drash syndrome — and vice versa — this has led to the suggestion that Denys–Drash syndrome and Frasier syndrome represent two ends of a phenotypic range62. Mutations identified in these WT1-associated syndromes are also observed as homozygous somatic mutations in tumours from patients with Wilms’ tumour who do not have a germline mutation. Interestingly, given that WT1 ablation is considered to be a rate-limiting step in Wilms’ tumour development, those tumours that do not carry a WT1 mutation often express WT1 at substantially higher levels than cells from the adjacent normal kidneys resected with the tumours110, consistent with tumours arising from mesenchyme that robustly expresses WT1.
WT1 in haematopoiesis and leukaemogenesis
A role for WT1 during haematopoiesis was first suggested by the observation of WT1 expression in thymus and spleen55. Although no WT1 expression is observed in long-term haematopoietic stem cells from bone marrow14,15,64–66, it is expressed in a small (~1%) subfraction of CD34+ multi-potent progenitor cells, both in the uncommitted, quiescent CD38− fraction, where its expression levels are comparable to those seen in K562 leukaemic cells66, and in the committed, proliferating CD38+ fractions14,15,64–66. WT1 expression is upregulated during early myeloid differentiation, and during this stage ~4% of common myeloid progenitors (CMPs), ~2% of granulocyte-monocyte progenitors (GMPs) and ~17% of megakaryocyteerythroid progenitors (MEPs) express WT1. WT1 expression is downregulated at later stages of differentiation, and only a small proportion (<1%) of fully differentiated cells are positive for WT1 (REF. 14).
Fetal liver cells from Wt1−/− mouse embryos can reconstitute the haematopoietic system in lethally irradiated recipients67. However, in Wt1−/−;Wt1+/+ adult chimaeras, Wt1−/− blood cells are not detectable. These data in aggregate suggest that, although Wt1 is not essential for haematopoietic progenitor cell viability, its loss results in impaired haematopoiesis67. Ectopic expression of Wt1 in long-term mouse haematopoietic stem cells (HSCs) leads to a reduction in the numbers of HSCs and in HSC progeny, including CMPs, GMPs and MEPs, which normally express WT1 (REF. 14). Exogenous expression of Wt1 in non-committed progenitors results in quiescence, not proliferation, and exogenous Wt1 expression in committed progenitors induces proliferation9–11,13. This changing role for WT1 during haematopoietic differentiation is similar to that observed during nephrogenesis. WT1 in early undifferentiated cells is important for the viability or the quiescence of uncommitted cells (intermediate mesoderm and CD34+CD38− cells), but in committed cells (metanephric mesenchyme and CD34+CD38+ cells), WT1 is important for differentiation.
WT1 expression and mutation in leukaemias
Data regarding WT1 mutation and expression in leukaemia and the subsequent inferences about its role in leukaemogenesis have been contradictory. The observation of WT1 expression in immature myeloid leukaemias and during chronic myelogenous leukaemia (CML) blast crisis suggested a tumour-promoting or oncogenic role for WT1 in leukaemogenesis8. However, the early identification of a somatic WT1 mutation in the leukaemic cells of a patient with WAGR who later developed AML suggested that, as in Wilms’ tumour, WT1 functioned as a TSG in leukaemia68.
Subsequent data from patients with leukaemia continue to be seemingly contradictory. WT1 is expressed in most acute leukaemias and is a useful marker for the detection of residual disease15,69,70. Additionally, but controversially, the level of its expression has been reported to be of prognostic importance with higher levels associated, variably, with decreased attainment of remission, poor disease-free survival and/or poor overall survival15–18. Other studies have found that there is no prognostic relevance to the presence or absence of WT1 expression or when WT1 expression is coded as low versus high71,72. In one study of paediatric AML, higher levels of WT1 correlated with better overall survival73. These results and conclusions are confounded by technical differences between studies in quantifying WT1 expression and in the patient populations examined, with differences in patient age and histological and cytogenetic subtypes of leukaemia being notable confounding factors. Furthermore, data indicating that WT1 protein stability is modulated through interactions with HSP90 in myeloid leukaemia xenografts74 further complicates the interpretation of WT1 gene expression data.
Additionally, and importantly, WT1 is mutated in a considerable proportion of both adult and childhood AML75–77. The vast majority of alterations are insertions or deletions occurring in exon 7 or exon 9 and, to a lesser extent, missense mutations in exon 9 like those observed in patients with Denys–Drash syndrome (FIG. 1c). WT1 heterozygous, homozygous and compound heterozygous mutations have been reported at different frequencies in AML. Interestingly, WT1 mutation is mainly observed in cytogenetically normal AML or in those with mutations in the kinase FLT3 (REFS 75–77). In a study of childhood AML, WT1 mutation (either heterozygous or homozygous) was significantly and independently associated with poor prognosis, and most patients with a relapse carried the same WT1 mutation as that observed at diagnosis78. In ~10% of cases, relapsed patients had WT1 mutations even though at initial diagnosis leukaemic cells had none77.
Initial data suggested that transgenic overexpression of Wt1 was required for leukaemogenesis in mice, and mice transplanted with bone marrow cells expressing the AML1–ETO fusion gene did not develop leukaemia, whereas those with bone marrow cells expressing both AML1–ETO and WT1 did develop leukaemia13. Subsequent work using two different mouse models of leukaemia (mice expressing the oncogenic fusion proteins TEL–PDGFBR and AML1–ETO or BCR–ABL1 alone) found that Wt1 expression was not required for disease development. Although an increased frequency of Wt1+ cells, especially immature leukaemic cells, was observed in both these models, Wt1− leukaemic cells were also present. Additionally, Wt1− and Wt1+ subfractions were equally leukaemogenic when transplanted into recipient animals. These contrasting data may be due to the different mutational contexts in which the leukaemic potential of Wt1 expression was tested.
Interestingly, one of the oncogenes used in these studies, TEL–PDGFBR, is reported to activate the Ras–ERK pathway79, and contrasting roles for WT1 in RAS-mediated cell proliferation have been observed, with WT1 expression either inhibiting HRAS-dependent transformation or being required for KRAS-mediated cell proliferation80,81. WT1 has also been reported to negatively regulate inhibitors of the Ras–ERK pathway82,83.
WTX
WTX expression has been observed in 22 adult and six fetal tissues assessed, although the level of expression varies4,84. The gene contains two exons and is transcribed as a ~7.5 kb mRNA, which encodes a 1,135 amino acid protein (FIG. 2). Use of alternative splice donor and splice acceptor sites, both located within exon 2, results in the production of an isoform lacking 277 amino acids in the N-terminal portion of the protein27. The full-length WTX protein resides at the plasma membrane and cytoplasm and possesses three adenomatous polyposis coli (APC) binding domains. These facilitate its interaction with APC at the plasma membrane84,85. The short isoform localizes primarily to the nucleus85,86. Nuclear WTX forms a complex with β-catenin and the destruction complex (AXIN1, β-transducin repeat-containing protein 2 and APC) and promotes ubiquitylation and degradation of β-catenin27. Therefore, it is the short WTX isoform that negatively regulates the Wnt-signalling pathway. A C-terminal region of both WTX isoforms has been reported to interact with the WT1 protein in vitro, and, interestingly, enhancement of WT1 function as a transcriptional activator is observed, but only in the case of the short isoform86.
Figure 2. WTX mutations in Wilms’ tumour and osteopathia striata congenita with cranial sclerosis (OSCS).
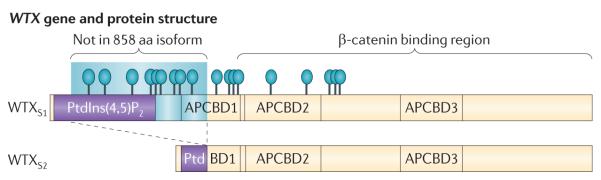
The full-length WTX protein possesses two phosphatidylinositol(4,5)-bisphosphate (PtdIns(4,5)P2) binding domains that mediate the localization of the protein to the plasma membrane, whereas the smaller isoform lacks these domains and localizes primarily to the nucleus84–86. Both WTX isoforms contain a β-catenin binding domain. Additionally, three adenomatous polyposis coli (APC) binding domains are in the full-length protein84 ; one of these is truncated in the shorter WTX isoform85,86. As denoted, mutations of known functional importance include whole-gene deletions and mutations (blue ovals) resulting in truncated protein products (lines). Blue shading denotes a region of the protein present only in the larger isoform owing to alternative intra-exonic splicing. aa, amino acid.
WTX alterations in Wilms’ tumour
The WTX gene is altered in 7–29% of Wilms’ tumours, with most of these tumours (about two-thirds) carrying deletions of the entire WTX gene4–6,87,88. The remaining one-third of WTX-mutated Wilms’ tumours carry truncating mutations (nonsense mutations, and insertions and deletions that cause frameshifts, resulting in termination codons) or missense mutations. The functional importance of the missense alterations is unclear, as the missense alteration can be present in normal tissue from the same patient, whereas the deletion and truncation mutations are always specific to the tumour. The truncation mutations result in loss of most or all of the β-catenin-binding region (FIG. 2). Truncated WTX results in an increase in nuclear β-catenin in vitro27, and these data suggest that the WTX mutations observed in Wilms’ tumour have a similar in vivo role and stabilize β-catenin. A correlation between WTX mutations and increased expression of Wnt pathway target genes is observed in Wilms’ tumours displaying a predominantly blastemal histology, but not in epithelial-predominant tumours87. These data suggest that, as noted above with respect to different phenotypic effects of WT1 mutations at different stages of kidney development, differentiation status may also have an important effect on the functional importance of WTX mutations.
WTX germline mutations are observed in both the familial and sporadic forms of the rare sclerosing bone dysplasia, osteopathia striata congenita with cranial sclerosis (OSCS) (BOX 3). Interestingly, individuals with OSCS display no increased risk for Wilms’ tumour or any cancer85, and this observation led to the questioning of the labelling of WTX as a TSG. However, nephrogenic rests, potential Wilms’ tumour precursor lesions, have recently been reported in the kidney of one male germline WTX mutation carrier7, suggesting that such mutations can predispose to Wilms’ tumour. The lack of overt Wilms’ tumours in patients with OSCS might be due to the generally infrequent progression of the precursors to tumours and the rarity of WTX-mutated OSCS cases. Another possibility is that WTX mutation, in the absence of other genetic alterations, is not tumorigenic. This is similar to what is observed with WT1. Although WT1-ablated nephrogenic rests have been reported in some patients89, in mice Wt1 ablation alone does not result in tumours57.
Box 3. WTX mutations in osteopathia striata congenita with cranial sclerosis (OSCS).
Mutations in WTX have been observed in both the familial and sporadic forms of osteopathia striata congenita with cranial sclerosis (OSCS)85. Similar skeletal phenotypes are observed when the Wnt signalling pathway is upregulated via other gene alterations85; that WTX mutations result in a similar phenotype is consistent with both the experimental and observational data above. Whole-gene deletions constitute the major class of WTX mutations in Wilms’ tumours4–6,87,88, whereas truncating mutations are more common in OSCS. However, the range of WTX truncating mutations that is observed in tumours is identical to that observed in OSCS (FIG. 2). Some of these mutant alleles affect only the full-length protein; the normal WTX isoform 2 protein would be generated in carriers of these alleles. However, the presence of the normal smaller isoform is not sufficient to rescue the lethal effect of the WTX mutation in affected males with OSCS85, suggesting that the function of the smaller WTX isoform 2 protein may be distinct from that of the larger isoform, consistent with its localization to the nucleus rather than to the plasma membrane27. The smaller WTX isoform does retain the β-catenin binding region and so may have a role in binding to β-catenin in the nucleus and regulating its activity85.
Mutation associations
In the absence of any associations — positive or negative — between gene mutations, the frequency of the co-occurrence of two mutations in Wilms’ tumours will be the product of their independent frequencies. Interestingly, some combinations of mutations occur more frequently than is predicted by chance.
A strong positive association was initially observed between the presence of WT1 and CTNNB1 mutations90 and has been confirmed in subsequent studies4,5,87,91–96. Compiled data from these studies (a few of which enriched their samples for WT1-mutant tumours) show that of 154 tumours with CTNNB1 mutations, 121 (79%) also have mutations in WT1, a significant co-occurrence considering the frequency of WT1 mutations (~20%) in Wilms’ tumours overall. Interestingly, assessment of tumours and nephrogenic rests from two patients with Wilms’ tumour revealed that, although tumours carried both WT1 and CTNNB1 mutations, the autologous nephrogenic rests carried only WT1 mutations; CTNNB1 mutations occurred at a later step in tumour development97 (BOX 4).
Box 4. Other genetic alterations modulate WT1-mutant phenotype.
Wilms’ tumours with WT1 mutations often display aberrant biallelic expression of the imprinted gene insulin-like growth factor 2 (IGF2) and/or mutation of CTNNB1 (REFS 90,108). Do these epigenetic and genetic changes alter the phenotypic effect of WT1 mutations? The observation of CTNNB1 mutations occurring in tumours, but not in WT1-mutant precursor lesions (nephrogenic rests), from the same patient97 suggests a model whereby developmentally arrested mesenchyme becomes fully malignant as a result of CTNNB1 activation. Although it is tempting to speculate that IGF2 upregulation or β-catenin stabilization confers a proliferative capacity to WT1-mutant mesenchyme, the differentiation of which has been disrupted, the observation of variability in mitotic activity both in prelesions and tumours109 suggests that the role of these alterations with respect to WT1-associated tumorigenesis is far more complex and may alter differentiation programmes that result in the unique, heterogeneous type of histology typical of Wilms’ tumours. What is implicit in this model is that in WT1-mutant Wilms’ tumours, WT1 mutations alone are not sufficient for tumorigenesis; that the rate-limiting two hits proposed in the original genetic model for Wilms’ tumour are not the mutation of one WT1 allele and then the mutation and loss of the remaining allele, but the functional loss of WT1 and the alteration of a second locus. This is strongly supported by the recent generation of a mouse model for Wilms’ tumour in which somatic, mosaic ablation of Wt1 results in endogenous tumours in the context of constitutional upregulation of Igf2, but not in the absence of Igf2 upregulation57.
WTX mutations in Wilms’ tumours were initially reported to be negatively correlated with mutations in WT1 (REF. 4), but subsequent studies showed WTX mutations to be approximately equally frequent in tumours with and without mutations in WT1 (REFS 5,87). Thus, although WTX enhancement of WT1 function has been reported86, existing mutation data do not consistently support a model whereby WTX and WT1 mutations are functionally redundant.
By contrast, inactivating mutations in WTX (deletions and truncations or frameshift mutations) are reported to be negatively correlated with activating mutations in exon 3 of CTNNB1 in Wilms’ tumours, according to two studies5,87. Compiled data from four studies4,5,87,96 indicate that of the 60 tumours with CTNNB1 exon 3 mutations studied, only two (3%) also carried WTX mutations, far fewer than the ~20% expected on the basis of the frequency of WTX in Wilms’ tumours overall. These observations suggest a functional redundancy in which ablation of WTX, which normally destabilizes β-catenin, has a similar effect to directly stabilizing β-catenin through mutation of the exon 3-encoded phosphorylation sites. However, the lack of an association between WTX and WT1 mutations when such a strong association exists between CTNNB1 and WT1 mutations suggests that, although WTX and CTNNB1 mutations may share some functional effect, mutation of CTNNB1 has an additional consequence that relates to its co-occurrence with WT1 ablation that WTX mutation does not. Plausible explanations for this possible phenotypic difference include differing roles of β-catenin and WTX in Wnt signalling in adherens junctions and/or in fetal kidney development.
WTX: a TSG?
Current data support the TSG label for WTX. Deletion and resulting loss of expression is observed in tumours, consistent with a TSG function. Although the absence of Wilms’ tumour in patients with OSCS initially raised the question of whether WTX germline mutation acted as the classic first hit for tumorigenesis, as would be expected for a TSG, the recent case report7 of Wilms’ tumour precursor lesions in an OSCS male fetus suggests that WTX mutation does predispose to Wilms’ tumour, as the TSG name implies.
WT1: a TSG, an oncogene, both or neither?
Data from human malignancies seem to suggest a contradictory role for WT1: that of a TSG in Wilms’ tumour and that of an oncogene in leukaemia. Can these roles be reconciled? Could potential tissue-specific differences in post-translational modification of WT1 alter its regulation of downstream targets? As a transcription factor, does WT1 positively regulate growth-enhancing genes at one stage of development and then positively regulate growth-suppressing genes at a later stage of differentiation? Or does WT1 regulate a similar set of target genes, but the consequence of their expression differs depending on the differentiation status of the cell? Or is the role of WT1 more related to regulating differentiation, and loss of its function at different stages of differentiation results in varying responses by cells to a disruption in that process? Similarly, is the phenotypic consequence of WT1 mutation affected by the presence of other gene alterations? Are there parallels in the role of WT1 in kidney and haematopoietic development and malignancy that can potentially help in understanding the seemingly contradictory data regarding its tumour suppressor role in Wilms’ tumour and its oncogenic role in leukaemia? Are there alternative interpretations of the data?
A simple depiction of WT1 expression during nephrogenesis and haematopoiesis and the effect of its loss or upregulation, based on observational and experimental data from in vivo or ex vivo studies, is shown in FIG. 3. Although the two developmental processes are not strictly analogous, there are many similarities. In both tissue types WT1 is expressed in lineage-committed precursor cells, upregulated during differentiation and then downregulated in all but a subset of the resulting fully differentiated cells. In both, loss of WT1 has been observed to result in a block in, or severe retardation of, further differentiation. In both, WT1 mutation is observed in malignancies, and in these malignancies WT1 expression is often increased relative to fully differentiated cell types. This observation has not led to the moniker of oncogene for WT1 in Wilms’ tumour for various reasons: because of patient data that implicated WT1 as a TSG even before it was cloned, the subsequent molecular data that confirmed this tumour suppressor role and the knowledge that WT1 was expressed in the developing kidney. Additionally, the realization that Wilms’ tumour is genetically heterogeneous provided a reasonable explanation for the observation of WT1 overexpression in some tumours compared with mature kidney tissue: the genetic aetiology of these tumours was due to the alteration of other Wilms’ tumour genes, and tumours displayed robust WT1 expression owing to their origin from a cell type in which WT1 was robustly expressed.
Figure 3. WT1 expression during kidney and myeloid differentiation, and phenotypic consequences of altered expression or gene ablation.

a ∣ WT1 loss of function in the developing kidney results in cell death when ablated in early metanephric mesenchyme56 or results in a disruption of differentiation if ablated in committed condensed mesenchyme57,58. The observation of WT1 mutations in Wilms’ tumours implies that its loss — in conjunction with other alterations — ultimately results in proliferation (tumours). b ∣ During haematopoiesis, loss of WT1 retards cell proliferation and/or differentiation67. Exogenous expression of WT1 in the CD34+CD38+ committed precursor population results in quiescence, whereas such expression in the more differentiated common myeloid progenitors results in proliferation9–13). Ectopic expression of WT1 in long-term haematopoietic stem cells results in reduced haematopoiesis14. As with Wilms’ tumour, the observation of WT1 mutations in some leukaemias links its loss at some stage of haematopoiesis with a proliferative phenotype.
Can the same reasoning be applied to the role of WT1 in leukaemia? As in Wilms’ tumours, WT1-inactivating mutations are observed in a subset of AMLs, notably those that are cytogenetically normal and those that carry FLT3 mutations75–77. Interestingly, although in most WT1-mutant Wilms’ tumours the mutations are homozygous, in leukaemias WT1 mutations are often heterozygous, implying a functional importance for reduced, but not absent, WT1 function. Alternatively, factors that reduce WT1 protein stability in leukaemias74 might result in a net loss of WT1 function, despite the presence of one wild-type WT1 allele.
Similar to the higher level of WT1 expression in Wilms’ tumours compared with normal kidney, AMLs express higher levels of WT1 than normal bone marrow. However, WT1 is expressed and is upregulated and downregulated in specific subsets of cells during haematopoiesis. These data are consistent with and support previous suggestions that the WT1 overexpression observed in some human leukaemias may be a reflection of cancer ontogeny11,65,66,98. As in Wilms’ tumour, malignant cells have arisen from undifferentiated cells that express WT1 at higher levels than the normal, differentiated cells with which leukaemic cells are being compared. Together, the observations of WT1 mutation and expression in AML suggest that, as in Wilms’ tumour, loss of WT1 function, not WT1 overexpression, is important, although not sufficient, for the development of a subset of AMLs.
Do the data and the above discussion imply that WT1 is a TSG in the sense that its role is to negatively regulate cell growth? or could the role of WT1 in malignancy relate more to its effect on differentiation? As presented in FIG. 3, WT1 mutation or induced expression in vivo or ex vivo variably result in apoptosis, quiescence, enhanced differentiation and a block in differentiation, depending on the differentiation status of the affected cell. In both nephrogenesis and haematopoiesis, loss of expression of WT1 in committed precursors results in disrupted or reduced differentiation and has an important role in tumorigenesis in some Wilms’ tumours and leukaemias. In more differentiated haematopoietic cells, exogenous WT1 expression results in enhanced proliferation. These combined data indicate that developmental stage has a profound effect on the phenotypic consequence of WT1 ablation or overexpression and suggest a model in which alteration of WT1 directly affects the differentiation status of the cell and this, in turn, results indirectly in a change in cell viability and/or proliferative status. Additional to the effect of differentiation status — and its unique gene expression profile — on the phenotypic result of WT1 mutations is the potential effect of other gene mutations. As discussed (BOX 4) in Wilms’ tumours, CTNNB1 mutation and/or IGF2 upregulation are sometimes observed in WT1-mutant tumours, and data from both humans and mice indicate that these alterations have a direct effect on the phenotypic effect of WT1 mutations. Conversely, WT1 mutation may also have a strong effect on the phenotypic consequence of cells carrying mutations in other genes (as recently described for Kras-mutated cells81) and it is likely that this effect is variable, depending on what other gene is mutated.
Therefore, the labels of TSG or oncogene may not be appropriate for describing the function of WT1. Doing so may misleadingly put WT1 in the same category as, for example, receptor tyrosine kinases that have a direct role in stimulating cell proliferation, or p53 that has a direct role in regulating the cell cycle and apoptosis. The role for WT1 for which the strongest data exist to date is that of a gene that regulates key differentiation genes. The oncogenic or tumour suppressive effect of WT1 alteration is likely to be a result of how a cell at a particular stage of development responds to perturbations in the expression of those genes. Perhaps the TSG and oncogene labels should be retired for WT1. They are a convenient shorthand notation, but, rather than describing the cellular function of WT1, they more accurately describe the effect of WT1 alteration in the varying contexts of cell type, differentiation status, presence of other gene alterations and microenvironment.
Conclusion
Genes mutated in Wilms’ tumours include TSGs (WTX and TP53), an oncogene (CTNNB1) and a chameleon gene, WT1. Observational data from Wilms’ tumours and AML along with an improving knowledge of the temporal and spatial expression of WT1 during nephrogenesis and haematopoiesis indicate that loss of WT1 function has an important role in a subset of these malignancies. However, in vivo and ex vivo experimental data indicate that loss of WT1 has differing phenotypic consequences — apoptosis, quiescence or proliferation — depending on the differentiation status of the cell. The presence of other gene mutations and the microenvironment of the mutant cells are likely to be additional confounding factors that change the effect of WT1 alteration. Teasing out the direct downstream effect of WT1 loss versus the subsequent cellular response to that initial effect, all in the context of varying stages of differentiation and the presence of other gene mutations and differing microenvironments, will be challenging. However, accomplishing this will be a major step in understanding the role of WT1 during development and how its loss, in addition to other molecular alterations, results in malignancy.
At a glance.
WT1 and WTX seem to function as tumour suppressor genes (TSGs) in Wilms’ tumours, but questions have arisen about these labels.
The lack of an increased frequency of Wilms’ tumours or other malignancies in patients with osteopathia striata congenita with cranial sclerosis (OSCS) with WTX germline mutations initially challenged its designation as a TSG, but a recent observation of Wilms’ tumour precursor lesions in a patient with OSCS supports this label.
In Wilms’ tumours, WT1 conforms to a TSG label: patients heterozygous for WT1 germline mutations are predisposed to Wilms’ tumour and WT1 is inactivated in tumours. These data link loss of WT1 function with enhanced cell viability and/or proliferation.
By contrast, ablation of WT1 at the initial stages of kidney development results in apoptosis and renal agenesis, indicating that it has a crucial role in maintaining cell viability.
In some leukaemias, the increased expression of WT1 compared with normal bone marrow cells, along with some reports of WT1 expression being a marker of poor prognosis, suggest that WT1 functions as an oncogene. By contrast, observations of WT1 inactivating mutations in leukaemias suggest it functions as a TSG.
WT1 has important roles in regulating normal differentiation in various organs and cell types. During both nephrogenesis and haematopoiesis, loss of WT1 or overexpression of WT1 is associated with differing phenotypic consequences, depending on the differentiation status of the cell.
The oncogenic or tumour suppressive effect of WT1 alteration is likely to be a result of how a cell at a particular stage of development responds to perturbations in normal differentiation. In short, either label may be misleading and/or inadequate when used to describe the function of WT1.
Acknowledgements
The author would like to thank C. Ruteshouser for her assistance. The author’s work is supported by US NIH grants CA34936 and DK069599, NCI CCSG grant CA16672 and CPRIT grants RP100329 and RP110234.
Glossary
- Autosomal dominant trait
A trait that is the result of being heterozygous for an allele for a gene that is present on any non-sex (not the X or Y) chromosome.
- Aniridia
Lack of development of the iris of the eye.
- Rhabdomyosarcoma
A tumour of skeletal muscle.
- Intermediate mesoderm
Region of the embryonic mesoderm from which the kidneys and gonads arise.
- Metanephric blastema
A subpopulation of the intermediate mesoderm that induces the outgrowth of the ureteric bud and from which the nephrons and stroma of the mature kidney arise.
- Renal agenesis
Lack of development of the kidney.
- Kidney rudiment explant culture
Embryonic kidneys as early as the stage at which the ureteric bud begins to invade the metanephric mesenchyme can be grown in culture chambers on filters. The reciprocal inductive interactions between the ureteric bud and the metanephric mesenchyme occur in culture, resulting in a three-dimensional structure of nephrons and collecting ducts like a normal kidney but without capillary invasion.
- Ureteric bud
A developmental structure that buds off the mesonephric (Wolffian) duct, invades the metanephric mesenchyme and develops into the collecting duct system of the kidney.
- Glomerulosclerosis
A general term to describe scarring of the glomerulus, the primary filtration structure in the kidney predominantly composed of capillaries and podocytes.
- Denys–Drash syndrome
A phenotypic triad of Wilms’ tumour, congenital genitourinary anomalies and early-onset renal failure.
- Gonadoblastoma
A tumour arising in the developing ovary or testes.
- TEL–PDGFBR and AML1–ETO
Two different fusion proteins resulting from chromosomal translocations and observed in some myeloid leukaemias. In TEL–PDGFBR the N-terminal region of TEL is fused with the domain of transmembrane and cytoplasmic domains of the receptor kinase protein, PDGFBR. In AML1–ETO the DNA binding domain of AML1 is fused with the ETO co-repressor protein.
- Comma-shaped and S-shaped bodies
Two morphological stages observed as induced metanephric mesenchyme differentiates and epithelializes to form mature nephrons.
- Osteopathia striata congenita with cranial sclerosis
(OSCS). X-linked dominant condition in which increased bone density and aberrant development of the skull is commonly observed. Fetal or perinatal lethality is often observed in males.
Footnotes
Competing interests statement The author declares no competing financial interests.
References
- 1.Bardeesy N, et al. Anaplastic Wilms’ tumour, a subtype displaying poor prognosis, harbours p53 gene mutations. Nature Genet. 1994;7:91–97. doi: 10.1038/ng0594-91. [DOI] [PubMed] [Google Scholar]
- 2.Huff V. Wilms tumor genetics. Am. J. Med. Genet. 1998;79:260–267. doi: 10.1002/(sici)1096-8628(19981002)79:4<260::aid-ajmg6>3.0.co;2-q. Although an older paper, this still provides a good basic summary of Wilms’ tumour genetics along with primary data and an overall description of the type of WT1 mutations observed in patients with Wilms’ tumour and their tumours that is still valid.
- 3.Koesters R, et al. Mutational activation of the β-catenin proto-oncogene is a common event in the development of Wilms’ tumors. Cancer Res. 1999;59:3880–3882. [PubMed] [Google Scholar]
- 4.Rivera MN, et al. An X chromosome gene, WTX, is commonly inactivated in Wilms tumor. Science. 2007;315:642–645. doi: 10.1126/science.1137509. This is the original report identifying WTX as a Wilms’ tumour gene. It presents a nice story of going from observing a gene copy number change over a very short genomic region to identifying a new cancer-related gene.
- 5.Ruteshouser EC, Robinson SM, Huff V. Wilms tumor genetics: mutations in WT1, WTX, and CTNNB1 account for only about one-third of tumors. Genes Chromosomes Cancer. 2008;47:461–470. doi: 10.1002/gcc.20553. This paper presents data from more than 100 Wilms’ tumours regarding the type, frequency and co-occurrence of WT1, WTX and CTNNB1 mutations.
- 6.Perotti D, et al. Functional inactivation of the WTX gene is not a frequent event in Wilms’ tumors. Oncogene. 2008;27:4625–4632. doi: 10.1038/onc.2008.93. [DOI] [PubMed] [Google Scholar]
- 7.Fukuzawa R, et al. WTX mutations can occur both early and late in the pathogenesis of Wilms tumour. J. Med. Genet. 2010;47:791–794. doi: 10.1136/jmg.2010.080663. [DOI] [PubMed] [Google Scholar]
- 8.Miwa H, Beran M, Saunders GF. Expression of the Wilms’ tumor gene (WT1) in human leukemias. Leukemia. 1992;6:405–409. [PubMed] [Google Scholar]
- 9.Yamagami T, et al. Growth inhibition of human leukemic cells by WT1 (Wilms tumor gene) antisense oligodeoxynucleotides: implications for the involvement of WT1 in leukemogenesis. Blood. 1996;87:2878–2884. [PubMed] [Google Scholar]
- 10.Tsuboi A, et al. Constitutive expression of the Wilms’ tumor gene WT1 inhibits the differentiation of myeloid progenitor cells but promotes their proliferation in response to granulocyte-colony stimulating factor (G-CSF) Leuk. Res. 1999;23:499–505. doi: 10.1016/s0145-2126(99)00037-5. [DOI] [PubMed] [Google Scholar]
- 11.Ellisen LW, Carlesso N, Cheng T, Scadden DT, Haber DA. The Wilms tumor suppressor WT1 directs stage-specific quiescence and differentiation of human hematopoietic progenitor cells. EMBO J. 2001;20:1897–1909. doi: 10.1093/emboj/20.8.1897. This early study assessed the effect of upregulation of WT1 in normal haematopoietic progenitor cells in addition to that in leukaemia cell lines. The discussion provides a thorough and balanced overview of the data on WT1 in leukaemia and haematopoiesis up to the time of the study.
- 12.Svedberg H, Richter J, Gullberg U. Forced expression of the Wilms tumor 1 (WT1) gene inhibits proliferation of human hematopoietic CD34+ progenitor cells. Leukemia. 2001;15:1914–1922. doi: 10.1038/sj.leu.2402303. This is a similar study to reference 11 on the effect of upregulation of WT1 in normal haematopoietic progenitor cells. A model on the variable phenotypic effects of WT1 expression during haematopoiesis is presented in the discussion.
- 13.Nishida S, et al. AML1-ETO rapidly induces acute myeloblastic leukemia in cooperation with the Wilms tumor gene, WT1. Blood. 2006;107:3303–3312. doi: 10.1182/blood-2005-04-1656. [DOI] [PubMed] [Google Scholar]
- 14.Hosen N, et al. The Wilms’ tumor gene WT1-GFP knock-in mouse reveals the dynamic regulation of WT1 expression in normal and leukemic hematopoiesis. Leukemia. 2007;21:1783–1791. doi: 10.1038/sj.leu.2404752. This is an excellent paper describing an extensive analysis of the temporal and spatial expression of Wt1 during mouse haematopoiesis and the role of Wt1 in leukaemogenesis using a Wt1 promoter-driven GFP reporter gene.
- 15.Inoue K, et al. WT1 as a new prognostic factor and a new marker for the detection of minimal residual disease in acute leukemia. Blood. 1994;84:3071–3079. [PubMed] [Google Scholar]
- 16.Bergmann L, et al. High levels of Wilms’ tumor gene (wt1) mRNA in acute myeloid leukemias are associated with a worse long-term outcome. Blood. 1997;90:1217–1225. [PubMed] [Google Scholar]
- 17.Trka J, et al. Real-time quantitative PCR detection of WT1 gene expression in children with AML: prognostic significance, correlation with disease status and residual disease detection by flow cytometry. Leukemia. 2002;16:1381–1389. doi: 10.1038/sj.leu.2402512. [DOI] [PubMed] [Google Scholar]
- 18.Garg M, Moore H, Tobal K, Liu Yin JA. Prognostic significance of quantitative analysis of WT1 gene transcripts by competitive reverse transcription polymerase chain reaction in acute leukaemia. Br. J. Haematol. 2003;123:49–59. doi: 10.1046/j.1365-2141.2003.04552.x. [DOI] [PubMed] [Google Scholar]
- 19.Oka Y, et al. Induction of WT1 (Wilms’ tumor gene)-specific cytotoxic T lymphocytes by WT1 peptide vaccine and the resultant cancer regression. Proc. Natl Acad. Sci. USA. 2004;101:13885–13890. doi: 10.1073/pnas.0405884101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keilholz U, et al. A clinical and immunologic phase 2 trial of Wilms tumor gene product 1 (WT1) peptide vaccination in patients with AML and MDS. Blood. 2009;113:6541–6548. doi: 10.1182/blood-2009-02-202598. [DOI] [PubMed] [Google Scholar]
- 21.Maslak PG, et al. Vaccination with synthetic analog peptides derived from WT1 oncoprotein induces T cell responses in patients with complete remission from acute myeloid leukemia. Blood. 2010;116:171–179. doi: 10.1182/blood-2009-10-250993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Breslow N, Beckwith JB, Ciol M, Sharples K. Age distribution of Wilms’ tumor: report from the National Wilms’ Tumor Study. Cancer Res. 1988;48:1653–1657. [PubMed] [Google Scholar]
- 23.Knudson AG, Strong LC. Mutation and cancer: a model for Wilms’ tumor of the kidney. J. Natl Cancer Inst. 1972;48:313–324. [PubMed] [Google Scholar]
- 24.Breslow NE, Beckwith JB, Perlman EJ, Reeve AE. Age distributions, birth weights, nephrogenic rests, and heterogeneity in the pathogenesis of Wilms tumor. Pediatr. Blood Cancer. 2006;47:260–267. doi: 10.1002/pbc.20891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huff V, et al. Evidence for genetic heterogeneity in familial Wilms’ tumor. Cancer Res. 1997;57:1859–1862. [PubMed] [Google Scholar]
- 26.Huff V, Villalba F, Strong LC, Saunders GF. Alteration of the WT1 gene in patients with Wilms’ tumor and genitourinary anomalies. Am. J. Hum. Genet. 1991;49:44. [Google Scholar]
- 27.Major MB, et al. Wilms tumor suppressor WTX negatively regulates WNT/β-catenin signaling. Science. 2007;316:1043–1046. doi: 10.1126/science/1141515. [DOI] [PubMed] [Google Scholar]
- 28.Rahman N, et al. Evidence for a familial Wilms’ tumour gene (FWT1) on chromosome 17q12-q21. Nature Genet. 1996;13:461–463. doi: 10.1038/ng0896-461. [DOI] [PubMed] [Google Scholar]
- 29.McDonald JM, et al. Linkage of familial Wilms’ tumor predisposition to chromosome 19 and a two-locus model for the etiology of familial tumors. Cancer Res. 1998;58:1387–1390. [PubMed] [Google Scholar]
- 30.Ogawa O, et al. Relaxation of insulin-like growth factor II gene imprinting implicated in Wilms’ tumour. Nature. 1993;362:749–751. doi: 10.1038/362749a0. [DOI] [PubMed] [Google Scholar]
- 31.Steenman MJ, et al. Loss of imprinting of IGF2 is linked to reduced expression and abnormal methylation of H19 in Wilms’ tumour. Nature Genet. 1994;7:433–439. doi: 10.1038/ng0794-433. [DOI] [PubMed] [Google Scholar]
- 32.Wiedemann H. Tumours and hemihypertrophy associated with Wiedemann-Beckwith syndrome. Eur. J. Pediatr. 1983;141:129. [Google Scholar]
- 33.Weksberg R, Shuman C, Smith AC. Beckwith-Wiedemann syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2005;137C:12–23. doi: 10.1002/ajmg.c.30058. [DOI] [PubMed] [Google Scholar]
- 34.Sparago A, et al. Microdeletions in the human H19 DMR result in loss of IGF2 imprinting and Beckwith-Wiedemann syndrome. Nature Genet. 2004;36:958–960. doi: 10.1038/ng1410. [DOI] [PubMed] [Google Scholar]
- 35.Prawitt D, et al. Microdeletion of target sites for insulator protein CTCF in a chromosome 11p15 imprinting center in Beckwith-Wiedemann syndrome and Wilms’ tumor. Proc. Natl Acad. Sci. USA. 2005;102:4085–4090. doi: 10.1073/pnas.0500037102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rainier S, et al. Relaxation of imprinted genes in human cancer. Nature. 1993;362:747–749. doi: 10.1038/362747a0. [DOI] [PubMed] [Google Scholar]
- 37.Call KM, et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell. 1990;60:509–520. doi: 10.1016/0092-8674(90)90601-a. [DOI] [PubMed] [Google Scholar]
- 38.Gessler M, et al. Homozygous deletion in Wilms tumours of a zinc-finger gene identified by chromosome jumping. Nature. 1990;343:774–778. doi: 10.1038/343774a0. [DOI] [PubMed] [Google Scholar]
- 39.Natoli TA, et al. A mammal-specific exon of WT1 is not required for development or fertility. Mol. Cell. Biol. 2002;22:4433–4438. doi: 10.1128/MCB.22.12.4433-4438.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hammes A, et al. Two splice variants of the Wilms’ tumor 1 gene have distinct functions during sex determination and nephron formation. Cell. 2001;106:319–329. doi: 10.1016/s0092-8674(01)00453-6. [DOI] [PubMed] [Google Scholar]
- 41.Laity JH, Dyson HJ, Wright PE. Molecular basis for modulation of biological function by alternate splicing of the Wilms’ tumor suppressor protein. Proc. Natl Acad. Sci. USA. 2000;97:11932–11935. doi: 10.1073/pnas.97.22.11932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stoll R, et al. Structure of the Wilms tumor suppressor protein zinc finger domain bound to DNA. J. Mol. Biol. 2007;372:1227–1245. doi: 10.1016/j.jmb.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 43.Larsson SH, et al. Subnuclear localization of WT1 in splicing or transcription factor domains is regulated by alternative splicing. Cell. 1995;81:391–401. doi: 10.1016/0092-8674(95)90392-5. [DOI] [PubMed] [Google Scholar]
- 44.Miles CG, et al. Mice lacking the 68-amino-acid, mammal-specific N-terminal extension of WT1 develop normally and are fertile. Mol. Cell. Biol. 2003;23:2608–2613. doi: 10.1128/MCB.23.7.2608-2613.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dallosso AR, et al. Genomic imprinting at the WT1 gene involves a novel coding transcript (AWT1) that shows deregulation in Wilms’ tumours. Hum. Mol. Genet. 2004;13:405–415. doi: 10.1093/hmg/ddh038. [DOI] [PubMed] [Google Scholar]
- 46.Sharma PM, Bowman M, Yu BF, Sukumar S. A rodent model for Wilms tumors: embryonal kidney neoplasms induced by N-nitroso-N’-methylurea. Proc. Natl Acad. Sci. USA. 1994;91:9931–9935. doi: 10.1073/pnas.91.21.9931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gunning KB, Cohn SL, Tomlinson GE, Strong LC, Huff V. Analysis of possible WT1 RNA processing in primary Wilms tumors. Oncogene. 1996;13:1179–1185. [PubMed] [Google Scholar]
- 48.Li JB, et al. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science. 2009;324:1210–1213. doi: 10.1126/science.1170995. [DOI] [PubMed] [Google Scholar]
- 49.Ye Y, Raychaudhuri B, Gurney A, Campbell CE, Williams BR. Regulation of WT1 by phosphorylation: inhibition of DNA binding, alteration of transcriptional activity and cellular translocation. EMBO J. 1996;15:5606–5615. [PMC free article] [PubMed] [Google Scholar]
- 50.Smolen GA, Vassileva MT, Wells J, Matunis MJ, Haber DA. SUMO-1 modification of the Wilms’ tumor suppressor WT1. Cancer Res. 2004;64:7846–7851. doi: 10.1158/0008-5472.CAN-04-1502. [DOI] [PubMed] [Google Scholar]
- 51.Makki MS, Heinzel T, Englert C. TSA downregulates Wilms tumor gene 1 (Wt1) expression at multiple levels. Nucleic Acids Res. 2008;36:4067–4078. doi: 10.1093/nar/gkn356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gao F, et al. The Wilms tumor gene, Wt1, is required for Sox9 expression and maintenance of tubular architecture in the developing testis. Proc. Natl Acad. Sci. USA. 2006;103:11987–11992. doi: 10.1073/pnas.0600994103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martinez-Estrada OM, et al. Wt1 is required for cardiovascular progenitor cell formation through transcriptional control of Snail and E-cadherin. Nature Genet. 2010;42:89–93. doi: 10.1038/ng.494. This is an excellent paper describing the role of WT1 in epithelial to mesenchymal transition in cardiovascular progenitors cells through its regulation of two genes crucial for this process.
- 54.Pritchard-Jones K, et al. The candidate Wilms’ tumour gene is involved in genitourinary development. Nature. 1990;346:194–197. doi: 10.1038/346194a0. [DOI] [PubMed] [Google Scholar]
- 55.Buckler AJ, Pelletier J, Haber DA, Glaser T, Housman DE. Isolation, characterization, and expression of the murine Wilms’ tumor gene (WT1) during kidney development. Mol. Cell. Biol. 1991;11:1707–1712. doi: 10.1128/mcb.11.3.1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kreidberg JA, et al. WT-1 is required for early kidney development. Cell. 1993;74:679–691. doi: 10.1016/0092-8674(93)90515-r. This paper describes the phenotype effect of germline ablation of Wt1 in the mouse and demonstrates that Wt1 is crucial for the normal development of many organ systems, including the kidney and gonads (as would be predicted by the phenotypes exhibited by patients carrying germline WT1 mutations).
- 57.Hu Q, et al. Wt1 ablation and Igf2-upregulation in mice result in Wilms tumors with elevated ERK1/2 phosphorylation. J. Clin. Invest. 2010 Dec 1; doi: 10.1172/JCI43772. (doi: 10.1172/JCI43772). This paper describes the first mouse model for Wilms’ tumour, which demonstrates that mosaic somatic Wt1 ablation in conjunction with Igf2 biallelic expression is sufficient for Wilms’ tumours. Additionally, complete somatic ablation of Wt1 results in a complete block in kidney development.
- 58.Davies JA, et al. Development of an siRNA-based method for repressing specific genes in renal organ culture and its use to show that the Wt1 tumour suppressor is required for nephron differentiation. Hum. Mol. Genet. 2004;13:235–246. doi: 10.1093/hmg/ddh015. [DOI] [PubMed] [Google Scholar]
- 59.Pelletier J, et al. Germline mutations in the Wilms’ tumor suppressor gene are associated with abnormal urogenital development in Denys-Drash syndrome. Cell. 1991;67:437–447. doi: 10.1016/0092-8674(91)90194-4. [DOI] [PubMed] [Google Scholar]
- 60.Huff V. Genotype/phenotype correlations in Wilms’ tumor. Med. Pediatr. Oncol. 1996;27:408–414. doi: 10.1002/(SICI)1096-911X(199611)27:5<408::AID-MPO4>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 61.Royer-Pokora B, et al. Twenty-four new cases of WT1 germline mutations and review of the literature: genotype/phenotype correlations for Wilms tumor development. Am. J. Med. Genet. A. 2004;127:249–257. doi: 10.1002/ajmg.a.30015. [DOI] [PubMed] [Google Scholar]
- 62.Koziell A, et al. Frasier syndrome, part of the Denys Drash continuum or simply a WT1 gene associated disorder of intersex and nephropathy? Clin. Endocrinol. 2000;52:519–524. doi: 10.1046/j.1365-2265.2000.00980.x. [DOI] [PubMed] [Google Scholar]
- 63.Barbaux S, et al. Donor splice-site mutations in WT1 are responsible for Frasier syndrome. Nature Genet. 1997;17:467–470. doi: 10.1038/ng1297-467. [DOI] [PubMed] [Google Scholar]
- 64.Baird PN, Simmons PJ. Expression of the Wilms’ tumor gene (WT1) in normal hemopoiesis. Exp. Hematol. 1997;25:312–320. [PubMed] [Google Scholar]
- 65.Maurer U, Weidmann E, Karakas T, Hoelzer D, Bergmann L. Wilms tumor gene (wt1) mRNA is equally expressed in blast cells from acute myeloid leukemia and normal CD34+ progenitors. Blood. 1997;90:4230–4232. [PubMed] [Google Scholar]
- 66.Hosen N, et al. Very low frequencies of human normal CD34+ haematopoietic progenitor cells express the Wilms’ tumour gene WT1 at levels similar to those in leukaemia cells. Br. J. Haematol. 2002;116:409–420. doi: 10.1046/j.1365-2141.2002.03261.x. [DOI] [PubMed] [Google Scholar]
- 67.Alberta JA, et al. Role of the WT1 tumor suppressor in murine hematopoiesis. Blood. 2003;101:2570–2574. doi: 10.1182/blood-2002-06-1656. This was the first study to examine the role of Wt1 in haematopoiesis in vivo. Creating chimaeras of Wt1-wild-type and Wt1-null cells, this work established that Wt1 is not required for normal haematopoiesis, but is important for robust haematopoiesis.
- 68.Pritchard-Jones K, Renshaw J, King-Underwood L. The Wilms tumour (WT1) gene is mutated in a secondary leukaemia in a WAGR patient. Hum. Mol. Genet. 1994;3:1633–1637. doi: 10.1093/hmg/3.9.1633. [DOI] [PubMed] [Google Scholar]
- 69.Menssen HD, et al. Presence of Wilms’ tumor gene (wt1) transcripts and the WT1 nuclear protein in the majority of human acute leukemias. Leukemia. 1995;9:1060–1067. [PubMed] [Google Scholar]
- 70.Inoue K, et al. Long-term follow-up of minimal residual disease in leukemia patients by monitoring WT1 (Wilms tumor gene) expression levels. Blood. 1996;88:2267–2278. [PubMed] [Google Scholar]
- 71.Gaiger A, et al. Detection of the WT1 transcript by RT-PCR in complete remission has no prognostic relevance in de novo acute myeloid leukemia. Leukemia. 1998;12:1886–1894. doi: 10.1038/sj.leu.2401213. [DOI] [PubMed] [Google Scholar]
- 72.Yanada M, et al. Multiplex real-time RT-PCR for prospective evaluation of WT1 and fusion gene transcripts in newly diagnosed de novo acute myeloid leukemia. Leuk. Lymphoma. 2004;45:1803–1808. doi: 10.1080/10428190410001693551. [DOI] [PubMed] [Google Scholar]
- 73.Rodrigues PC, et al. Prognostic significance of WT1 gene expression in pediatric acute myeloid leukemia. Pediatr. Blood Cancer. 2007;49:133–138. doi: 10.1002/pbc.20953. [DOI] [PubMed] [Google Scholar]
- 74.Bansal H, et al. Heat shock protein 90 regulates the expression of Wilms tumor 1 protein in myeloid leukemias. Blood. 2010;116:4591–4599. doi: 10.1182/blood-2009-10-247239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Summers K, et al. Wilms’ tumour 1 mutations are associated with FLT3-ITD and failure of standard induction chemotherapy in patients with normal karyotype AML. Leukemia. 2007;21:550–551. doi: 10.1038/sj.leu.2404514. author reply 552. [DOI] [PubMed] [Google Scholar]
- 76.Paschka P, et al. Wilms’ tumor 1 gene mutations independently predict poor outcome in adults with cytogenetically normal acute myeloid leukemia: a cancer and leukemia group B study. J. Clin. Oncol. 2008;26:4595–4602. doi: 10.1200/JCO.2007.15.2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hollink IH, et al. Clinical relevance of Wilms tumor 1 gene mutations in childhood acute myeloid leukemia. Blood. 2009;113:5951–5960. doi: 10.1182/blood-2008-09-177949. This study of a large panel of paediatric AMLs not only confirmed earlier reports of WT1 mutations being an independent indicator of poor prognosis, but it also provided novel data regarding the association of WT1 mutations with other genetic alterations and an assessment of WT1 mutation status in paired samples taken at diagnosis and relapse.
- 78.Virappane P, et al. Mutation of the Wilms’ tumor 1 gene is a poor prognostic factor associated with chemotherapy resistance in normal karyotype acute myeloid leukemia: the United Kingdom Medical Research Council Adult Leukaemia Working Party. J. Clin. Oncol. 2008;26:5429–5435. doi: 10.1200/JCO.2008.16.0333. [DOI] [PubMed] [Google Scholar]
- 79.Dobbin E, et al. Tel/PDGFRβeta induces stem cell differentiation via the Ras/ERK and STAT5 signaling pathways. Exp. Hematol. 2009;37:111–121. doi: 10.1016/j.exphem.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 80.Luo XN, et al. The tumor suppressor gene WT1 inhibits ras-mediated transformation. Oncogene. 1995;11:743–750. [PubMed] [Google Scholar]
- 81.Vicent S, et al. Wilms tumor 1 (WT1) regulates KRAS-driven oncogenesis and senescence in mouse and human models. J. Clin. Invest. 2010;120:3940–3952. doi: 10.1172/JCI44165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gross I, et al. The receptor tyrosine kinase regulator Sprouty1 is a target of the tumor suppressor WT1 and important for kidney development. J. Biol. Chem. 2003;278:41420–41430. doi: 10.1074/jbc.M306425200. [DOI] [PubMed] [Google Scholar]
- 83.Morrison DJ, Kim MK, Berkofsky-Fessler W, Licht JD. WT1 induction of mitogen-activated protein kinase phosphatase 3 represents a novel mechanism of growth suppression. Mol. Cancer Res. 2008;6:1225–1231. doi: 10.1158/1541-7786.MCR-08-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Grohmann A, Tanneberger K, Alzner A, Schneikert J, Behrens J. AMER1 regulates the distribution of the tumor suppressor APC between microtubules and the plasma membrane. J. Cell Sci. 2007;120:3738–3747. doi: 10.1242/jcs.011320. [DOI] [PubMed] [Google Scholar]
- 85.Jenkins ZA, et al. Germline mutations in WTX cause a sclerosing skeletal dysplasia but do not predispose to tumorigenesis. Nature Genet. 2009;41:95–100. doi: 10.1038/ng.270. This paper was the first to describe the phenotypic consequence of germline WTX mutations.
- 86.Rivera MN, et al. The tumor suppressor WTX shuttles to the nucleus and modulates WT1 activity. Proc. Natl Acad. Sci. USA. 2009;106:8338–8343. doi: 10.1073/pnas.0811349106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fukuzawa R, Anaka MR, Weeks RJ, Morison IM, Reeve AE. Canonical WNT signalling determines lineage specificity in Wilms tumour. Oncogene. 2009;28:1063–1075. doi: 10.1038/onc.2008.455. [DOI] [PubMed] [Google Scholar]
- 88.Wegert J, et al. WTX inactivation is a frequent, but late event in Wilms tumors without apparent clinical impact. Genes Chromosomes Cancer. 2009;48:1102–1111. doi: 10.1002/gcc.20712. [DOI] [PubMed] [Google Scholar]
- 89.Park S, et al. Inactivation of WT1 in nephrogenic rests, genetic precursors to Wilms’ tumour. Nature Genet. 1993;5:363–367. doi: 10.1038/ng1293-363. [DOI] [PubMed] [Google Scholar]
- 90.Maiti S, Alam R, Amos CI, Huff V. Frequent association of β-catenin and WT1 mutations in Wilms tumors. Cancer Res. 2000;60:6288–6292. [PubMed] [Google Scholar]
- 91.Fukuzawa R, et al. Myogenesis in Wilms’ tumors is associated with mutations of the WT1 gene and activation of Bcl-2 and the Wnt signaling pathway. Pediatr. Dev. Pathol. 2004;7:125–137. doi: 10.1007/s10024-003-3023-8. [DOI] [PubMed] [Google Scholar]
- 92.Satoh Y, et al. Genetic and epigenetic alterations on the short arm of chromosome 11 are involved in a majority of sporadic Wilms’ tumours. Br. J. Cancer. 2006;95:541–547. doi: 10.1038/sj.bjc.6603302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fukuzawa R, et al. Wilms tumour histology is determined by distinct types of precursor lesions and not epigenetic changes. J. Pathol. 2008;215:377–387. doi: 10.1002/path.2366. [DOI] [PubMed] [Google Scholar]
- 94.Haruta M, et al. Duplication of paternal IGF2 or loss of maternal IGF2 imprinting occurs in half of Wilms tumors with various structural WT1 abnormalities. Genes Chromosomes Cancer. 2008;47:712–727. doi: 10.1002/gcc.20572. [DOI] [PubMed] [Google Scholar]
- 95.Royer-Pokora B, et al. Clinical relevance of mutations in the Wilms tumor suppressor 1 gene WT1 and the cadherin-associated protein β1 gene CTNNB1 for patients with Wilms tumors: results of long-term surveillance of 71 patients from International Society of Pediatric Oncology Study 9/Society for Pediatric Oncology. Cancer. 2008;113:1080–1089. doi: 10.1002/cncr.23672. [DOI] [PubMed] [Google Scholar]
- 96.Corbin M, et al. WNT/β-catenin pathway activation in Wilms tumors: a unifying mechanism with multiple entries? Genes Chromosomes Cancer. 2009;48:816–827. doi: 10.1002/gcc.20686. [DOI] [PubMed] [Google Scholar]
- 97.Fukuzawa R, Heathcott RW, More HE, Reeve AE. Sequential WT1 and CTNNB1 mutations and alterations of β-catenin localization in intralobar nephrogenic rests and associated Wilms tumours: two case studies. J. Clin. Pathol. 2007;60:1013–1016. doi: 10.1136/jcp.2006.043083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Inoue K, et al. Aberrant overexpression of the Wilms tumor gene (WT1) in human leukemia. Blood. 1997;89:1405–1412. [PubMed] [Google Scholar]
- 99.Daugherty RL, Gottardi CJ. Phospho-regulation of β-catenin adhesion and signaling functions. Physiology. 2007;22:303–309. doi: 10.1152/physiol.00020.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.MacDonald BT, Tamai K, He X. Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev. Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Heuberger J, Birchmeier W. Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb. Perspect. Biol. 2010;2:a002915. doi: 10.1101/cshperspect.a002915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Iglesias DM, et al. Canonical WNT signaling during kidney development. Am. J. Physiol. Renal Physiol. 2007;293:F494–F500. doi: 10.1152/ajprenal.00416.2006. [DOI] [PubMed] [Google Scholar]
- 103.Saadi-Kheddouci S, et al. Early development of polycystic kidney disease in transgenic mice expressing an activated mutant of the β-catenin gene. Oncogene. 2001;20:5972–5981. doi: 10.1038/sj.onc.1204825. [DOI] [PubMed] [Google Scholar]
- 104.Kuure S, Popsueva A, Jakobson M, Sainio K, Sariola H. Glycogen synthase kinase-3 inactivation and stabilization of β-catenin induce nephron differentiation in isolated mouse and rat kidney mesenchymes. J. Am. Soc. Nephrol. 2007;18:1130–1139. doi: 10.1681/ASN.2006111206. [DOI] [PubMed] [Google Scholar]
- 105.Park JS, Valerius MT, McMahon AP. Wnt/β-catenin signaling regulates nephron induction during mouse kidney development. Development. 2007;134:2533–2539. doi: 10.1242/dev.006155. [DOI] [PubMed] [Google Scholar]
- 106.Vainio S, Lin Y. Coordinating early kidney development: lessons from gene targeting. Nature Rev. Genet. 2002;3:533–543. doi: 10.1038/nrg842. [DOI] [PubMed] [Google Scholar]
- 107.Beckwith JB. In: Renal Pathology with Clinical and Functional Correlations. Tisher CC, Brenner BM, editors. J. B. Lippincott Company; Philadelphia: 1994. This presents an excellent description of the histology of Wilms’ tumours, along with a model regarding the ontogenic relationship between histologically different Wilms’ tumours and other kidney tumours.
- 108.Kaneda A, et al. Enhanced sensitivity to IGF-II signaling links loss of imprinting of IGF2 to increased cell proliferation and tumor risk. Proc. Natl Acad. Sci. USA. 2007;104:20926–20931. doi: 10.1073/pnas.0710359105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bove KE, Lewis C, Debrosse BK. Proliferation and maturation indices in nephrogenic rests and Wilms tumor; the emergence of heterogeneity from dormant nodular renal blastema. Pediatr. Pathol. Lab. Med. 1995;15:223–244. doi: 10.3109/15513819509026959. [DOI] [PubMed] [Google Scholar]
- 110.Miwa H, et al. RNA expression of the WT1 gene in Wilms’ tumors in relation to histology. J. Natl Cancer Inst. 1992;84:181–187. doi: 10.1093/jnci/84.3.181. [DOI] [PubMed] [Google Scholar]