

Abstract
Background:
Schizophrenia is a debilitating disorder that affects 1% of the US population. While the exogenous administration of cannabinoids such as tetrahydrocannabinol is reported to exacerbate psychosis in schizophrenia patients, augmenting the levels of endogenous cannabinoids has gained attention as a possible alternative therapy to schizophrenia due to clinical and preclinical observations. Thus, patients with schizophrenia demonstrate an inverse relationship between psychotic symptoms and levels of the endocannabinoid anandamide. In addition, increasing endocannabinoid levels (by blockade of enzymatic degradation) has been reported to attenuate social withdrawal in a preclinical model of schizophrenia. Here we examine the effects of increasing endogenous cannabinoids on dopamine neuron activity in the sub-chronic phencyclidine (PCP) model. Aberrant dopamine system function is thought to underlie the positive symptoms of schizophrenia.
Methods:
Using in vivo extracellular recordings in chloral hydrate–anesthetized rats, we now demonstrate an increase in dopamine neuron population activity in PCP-treated rats.
Results:
Interestingly, endocannabinoid upregulation, induced by URB-597, was able to normalize this aberrant dopamine neuron activity. Furthermore, we provide evidence that the ventral pallidum is the site where URB-597 acts to restore ventral tegmental area activity.
Conclusions:
Taken together, we provide preclinical evidence that augmenting endogenous cannabinoids may be an effective therapy for schizophrenia, acting in part to restore ventral pallidal activity.
Keywords: cannabinoids, hippocampus, phencyclidine, schizophrenia, ventral pallidum
Introduction
Schizophrenia is a debilitating disorder that affects up to 1% of the US population. The stereotypical psychosis associated with schizophrenia represents the positive symptoms of the disease, consisting of hallucinations and paranoid delusions. However, patients also display negative symptoms such as social withdrawal and blunted affect, and various cognitive impairments (American Psychiatric Association, 2000). Increases in dopamine system function have been demonstrated to contribute to the positive symptoms of schizophrenia (Laruelle et al., 1996; Epstein et al., 1999; Glenthoj et al., 2006). Furthermore, all antipsychotic drugs bind to dopamine D2 receptors and work by decreasing aberrant dopamine transmission (Meyer and Quenzer, 2005). Unfortunately, current antipsychotic drugs produce unpleasant side effects and are ineffective in a number of patients, which results in high rates of discontinuance (Lieberman et al., 2005). For this reason, alternative or adjunct therapies for schizophrenia are essential.
Modulation of the endocannabinoid system has gained attention as a possible alternative therapy for schizophrenia due to clinical (Leweke et al., 1999, 2012) and preclinical (Seillier et al., 2010, 2013) observations. At first, this may seem at odds with epidemiological data demonstrating that adolescent (Barnes et al., 2006; Chadwick et al., 2013) and regular (Arseneault et al., 2004; Moore et al., 2007; Hall and Degenhardt, 2008; Machielsen et al., 2010) cannabis use is a risk factor for schizophrenia in a sub-population of patients. Furthermore, it is known that marijuana can induce schizophrenia-like symptoms in the general population (Ames, 1958) and precipitate psychosis in patients (D’Souza et al., 2005, 2009). It is important to note, however, that a region-specific increase in endogenous cannabinoid transmission is not equivalent to the widespread CB1 receptor activation observed following cannabis administration. Indeed, while schizophrenia patients demonstrate increased levels of the endocannabinoid anandamide (AEA) in the cerebrospinal fluid (Leweke et al., 1999), those patients with the highest levels actually display lower positive and negative symptoms (Giuffrida et al., 2004). Thus, it seems increased AEA signaling may be a compensatory response to the disease and further increases in AEA may be beneficial as a therapy. Indeed, recent preclinical observations have demonstrated that AEA upregulation alleviates social withdrawal in a rat model of schizophrenia (Seillier et al., 2010, 2013). Furthermore, preliminary trials have demonstrated that the efficacy of increasing AEA in schizophrenia patients was comparable to conventional antipsychotics, but without the debilitating side effects (Leweke et al., 2012).
As mentioned above, increases in the dopamine system function are associated with the positive symptoms of schizophrenia and have been observed in both human patients (Laruelle et al., 1996; Epstein et al., 1999; Glenthoj et al., 2006) and a rodent model of the disease (Lodge and Grace, 2007). We have previously demonstrated that the augmented dopamine neuron activity in the methylazoxymethanol acetate (MAM) rodent model of schizophrenia (Chen et al., 2014) is directly attributable to a pathologically-enhanced drive from the ventral hippocampus (vHipp; Lodge and Grace, 2007; Perez et al., 2013; Perez and Lodge, 2013). This is in agreement with human imaging data in schizophrenia patients demonstrating hippocampal hyperactivity at rest, which is correlated with positive symptom severity (Malaspina et al., 2008; Schobel et al., 2009). However, whether this aberrant hippocampal regulation of dopamine neuron activity is observed across distinct animal models is not currently known. The sub-chronic phencyclidine (PCP) model of schizophrenia is well validated in reproducing positive, negative, and cognitive symptoms (for review see Mouri et al., 2007; Neill et al., 2010). We will utilize this model to examine the hippocampal regulation of the dopamine system function and to determine whether increasing endogenous AEA levels, by blockade of enzymatic degradation, will reverse aberrant dopamine neuron activity.
Method
Animals
All experiments were conducted in accordance with the guidelines outlined by the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of Texas Health Science Center.
Subchronic Drug Administration
Adult albino male Sprague-Dawley rats (300–400g) were used for all experiments. Rats were administered either PCP (5mg/kg, i.p.) or saline (1ml/kg, i.p.) twice a day (8 am and 8 pm) for 7 days and were examined following 7 days of withdrawal. This drug schedule has been previously demonstrated to induce deficits consistent with those observed in schizophrenia, without the confounds associated with an acute N-Methyl-d-aspartate (NMDA) receptor blockade (Mouri et al., 2007; Neill et al., 2010).
In Vivo Electrophysiology
Animals were anesthetized with 8% chloral hydrate (400mg/kg, i.p.) before being placed in a stereotaxic apparatus with a heating pad that maintained body temperature at 37oC. Subsequent boosters of chloral hydrate were given as needed to suppress the limb compression withdrawal reflex throughout the experiment.
For dopamine cell recordings, glass micro-electrodes (impedance 6–14 MΩ) were lowered into the ventral tegmental area (VTA) (coordinates from bregma: A/P -5.3mm, M/L +0.6mm, D/V – 6.5 to -9.0mm). Spontaneously-active cells are identified as putative dopamine neurons using previously-defined electrophysiological parameters recorded with open-filter settings (low pass: 30 Hz; high pass: 30kHz; Grace and Bunney, 1983). Indeed, results from recent single-cell labeling studies indicate that these criteria, if applied carefully, can be used to identify dopamine neurons based on their electrophysiological characteristics (Ungless and Grace, 2012). We examined the population activity (defined as the number of spontaneously-active dopamine cells per vertical electrode track), basal firing rate, and the proportion of action potentials occurring in bursts (Grace and Bunney, 1983). Between 4 and 9 tracks (separated by 0.2mm) were completed for each animal. For vHipp recordings, glass microelectrodes were lowered into the brain (coordinates from bregma: A/P -5.3mm, M/L +5.3mm, D/V -4.0 to -8.0mm) and putative pyramidal cells (neurons firing <2.0 Hz) were recorded for 3–5 minutes and firing rate compared between groups.
To examine the vHipp regulation of the ventral pallidum (VP) a bipolar, concentric-stimulating electrode was lowered into the ventral hippocampus (A/P -5.3mm, M/L +5.3mm, D/V -7.5mm) and a glass recording electrode was lowered through the VP (A/P -0.0mm, M/L -2.3mm, D/V -7 to -9mm). Spontaneously-active pallidal neurons were examined for their response to single, isolated current pulses (1 mA, 0.25 msec) administered to the vHipp. Once a neuron receiving a polysynaptic input from the vHipp was identified, either vehicle or URB597 (0.3mg/kg i.v.) were administered via a lateral tail vein, and both spontaneous- and vHipp-evoked activity recorded. Only one neuron was examined per rat for these experiments.
Drug Administration
Systemic
When AEA is endogenously produced it is rapidly hydrolyzed by fatty acid amide hydrolase (FAAH; Piomelli, 2003). URB597 is a FAAH inhibitor, which will potentiate endogenous AEA signaling (Kathuria et al., 2003). Saline- or PCP-treated rats were administered either URB597 (0.3mg/kg i.p.) or vehicle (1ml/kg; 10% Tween-80, 10% Polyethylene Glycerol, 80% Saline) one hour before anesthesia. Dopamine cells were recorded in the VTA as described above. A second cohort of rats was treated, after 7 days of withdrawal from PCP/saline, chronically (for 7 days) with URB597 (0.3mg/kg/day i.p.) or a vehicle control.
Intracranial
For vHipp inactivation, PCP-treated rats were anesthetized and a guide cannula was lowered into the vHipp (A/P +5.6, M/L + 5.0, D/V -4.6mm from bregma). Tetrodotoxin (TTX; 7.5 µg in 0.75 µl) or vehicle (0.75 µl) was injected via a cannula protruding 2mm ventral of the guide. For intracranial URB-597 administration, the guide cannula was lowered into the VP (A/P -0.0, M/L -2.3, D/V -6.3mm) and either URB597 (0.1 µg in 0.5 µl) or vehicle (0.5 µl) was administered via a cannula protruding 2mm ventral of the guide. Thirty minutes after intracranial administration of URB-597, spontaneously-active dopamine cells were recorded in the VTA as described above.
Histology
At the cessation of the electrophysiology experiments, rats were killed by an overdose of anesthetic (chloral hydrate, additional 400mg/kg i.p.) and decapitated. Their brains were removed, fixed for at least 48 hours (8% w/v paraformaldehyde), and cryoprotected (25% w/v sucrose) until saturated. Brains were sectioned (50 µm coronal sections), mounted onto gelatin-chrom alum-coated slides and stained with Cresyl violet for histochemical verification of electrode and/or cannula sites. All histology was performed with reference to a stereotaxic atlas (Paxinos and Watson, 2009), and representative placements are depicted in Figure 1.
Figure 1.
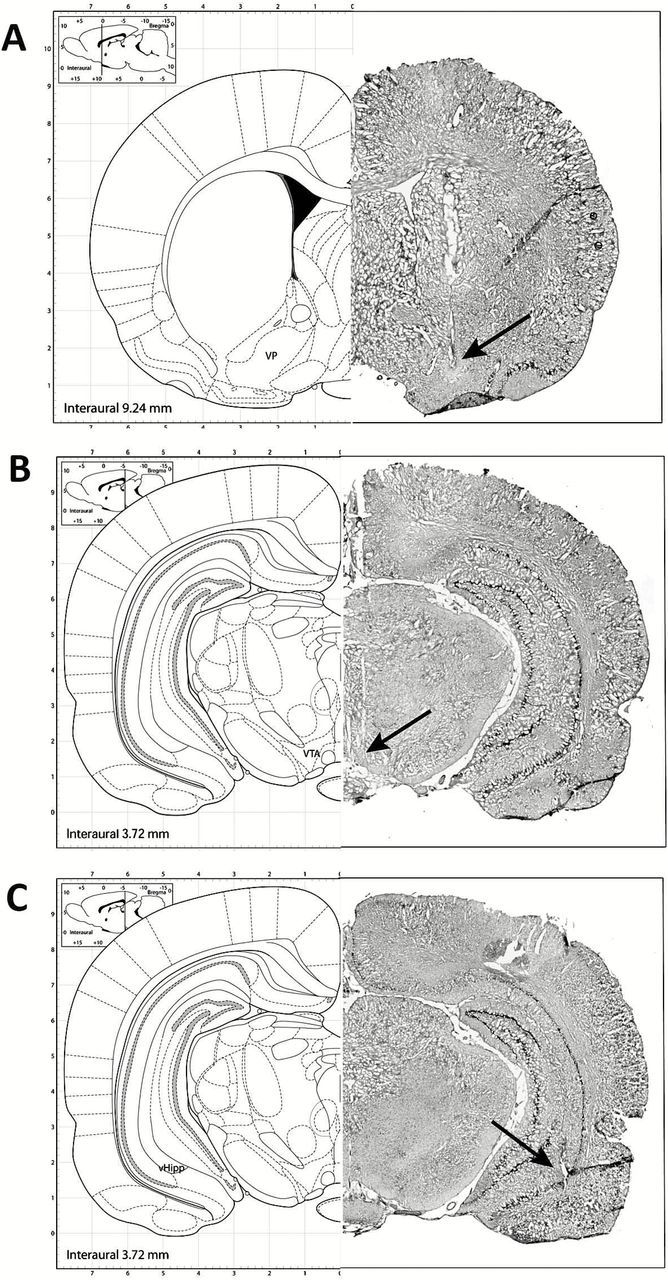
Representative photomicrographs demonstrating cannula placements in the ventral pallidum (A), an electrode track in the VTA (B), and a stimulating electrode located in the ventral hippocampus (C). The left side of each panel depicts the approximate location based on the atlas of Paxinos and Watson (Paxinos and Watson, 2009).
Analysis
Electrophysiological analysis of putative pyramidal neuron and dopamine neuron activity was performed with commercially-available computer software (LabChart version 7.1; ADInstruments) and plotted using Prism software (GraphPad Software). Data are represented as the mean ± SE, with n values representing the number of animals per experimental group or number of neurons per group where indicated. Statistics were calculated using SigmaPlot (Systat Software Inc.). Post hoc comparisons were performed using the Holm-Sidak method where appropriate.
Drugs
URB597 was purchased from the Cayman Chemical Company. PCP, chloral hydrate, and Dulbecco’s phosphate-buffered saline were purchased from Sigma. URB597 was dissolved in a vehicle solution (10% Tween-80, 10% Polyethylene Glycerol, 80% Saline at 65oC), while PCP was dissolved in saline. All other chemicals and reagents were of either analytical or laboratory grade and purchased from various suppliers.
Results
VTA Dopamine Activity in PCP-Treated Rats
Consistent with our studies in the MAM model of schizophrenia, vehicle-administered PCP-treated rats displayed a significant increase in dopamine neuron population activity (n = 7–8; SAL-VEH: 1.05±0.13 c.f. PCP-VEH: 1.82±0.11; two-way ANOVA; PCP F(1,31) = 14.931; Holm-Sidak; t= 5.70; p < 0.05; Figure 2A) without any effects on the average firing rate (n = 7–8; SAL-VEH: 4.18±0.21 Hz c.f. PCP-VEH: 3.92±0.13 Hz; two-way ANOVA; PCP F(1,31) = 1.907; p > 0.05; Figure 2B) or burst firing (n = 7–8; SAL-VEH: 28.7±2.6 % c.f. PCP-VEH: 28.1±1.9 %; two-way ANOVA; PCP F(1,31) = 1.989; p > 0.05; Figure 2C). Given that we have previously demonstrated that increases in dopamine neuron population activity are related to aberrant vHipp activity in the MAM model (Lodge and Grace, 2007), we examined the effect of the vHipp blockade on dopamine neuron activity in the subchronic PCP model. Specifically, intra-vHipp TTX was able to completely reverse the aberrant dopamine neuron population activity in the PCP-treated rats (n = 10; PCP-VEH: 1.96±0.05 c.f. PCP-TTX: 1.23±0.03; student’s t-test; t = -11.967; p < 0.05; Figure 4).
Figure 2.
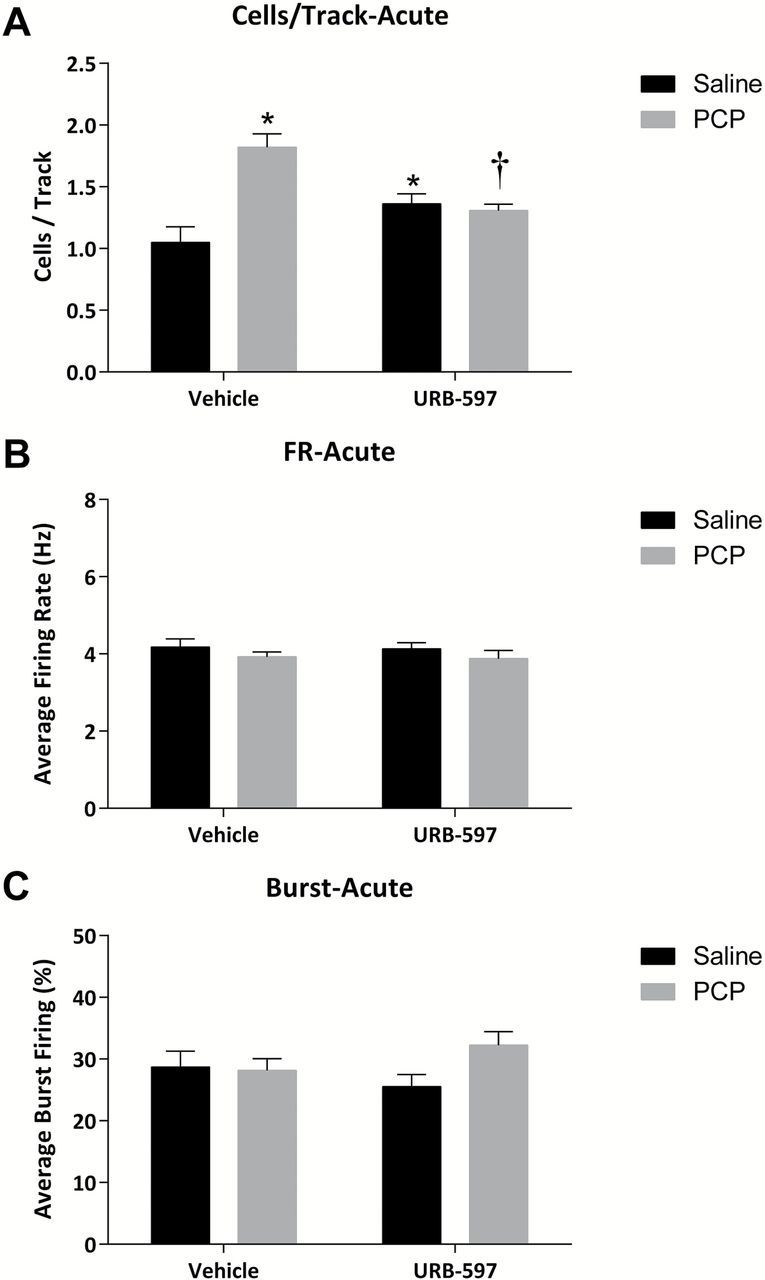
Acute URB-597 reverses aberrant dopamine neuron activity in the PCP-treated rat. (A) PCP-treated rats display increases in dopamine neuron population activity, which are reversed by systemic URB-597 administration. No changes in the average firing rate (B) or average percent burst firing (C) were observed. *significant difference from vehicle-treated saline rats; †significant difference from vehicle-treated PCP rats (two-way ANOVA).
Figure 4.
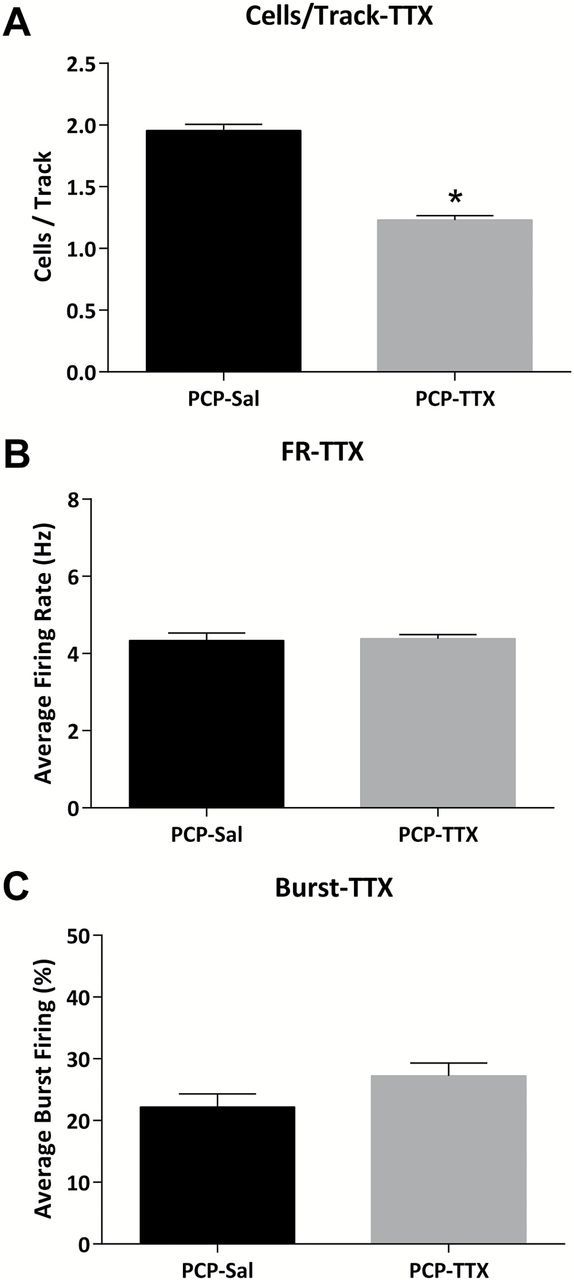
TTX inactivation of the vHipp reverses aberrant dopamine neuron activity in the PCP rat. Intra-vHipp TTX administration decreases dopamine neuron population activity (A) without affecting average firing rate (B) or burst firing (C) in PCP-treated rats. *Significant difference between groups (student’s t-test)
Aberrant VTA Dopamine Activity in PCP-Treated Rats is Normalized by URB597
The systemic administration of URB-597 (0.3mg/kg i.p.) one hour prior to anesthesia significantly reversed the aberrant dopamine neuron population activity in PCP-treated rats (n = 8–9; PCP-VEH: 1.82±0.11 c.f. PCP-URB: 1.31±0.05; two-way ANOVA; PCP F(1,31) = 14.931; PCP x URB F = 19.824; Holm-Sidak t= 4.04; p < 0.05; Figure 2A). Interestingly, a small but significant increase in dopamine neuron population activity was observed in control animals (n = 8–9; SAL-VEH: 1.05±0.13 c.f. SAL-URB: 1.36±0.08; Holm-Sidak t= 2.31; p < 0.05; Figure 2A). URB-597 administration did not alter the firing rate or pattern following acute administration (Figure 2B and C). Similar results were obtained with chronic (7 day) URB597 administration (Figure 3). Specifically, PCP-treated rats treated chronically with vehicle displayed a significant increase in spontaneously-active dopamine cells per track compared to saline-treated rats (n = 8–9; Sal: 1.17±0.07 c.f. PCP: 2.10±0.15; two-way ANOVA; PCP F(1,35) = 21.048; URB F(1,35) = 27.629; PCP x URB F(1,35) = 41.402; Holm-Sidak; t= 7.59; p < 0.05; Figure 3A). Chronic URB597 administration completely normalized this increase in dopamine neuron activity (n = 8–10; PCP-VEH: 2.10±0.15 c.f. PCP-URB: 1.12±0.05; Holm-Sidak; t= 8.24; p < 0.05; Figure 3A) without having an observable effect on population activity in control rats (n = 9; SAL-VEH: 1.17±0.07 c.f. SAL-URB: 1.27±0.06; Holm-Sidak; t= 0.84; p > 0.05; Figure 3A). It should be noted that chronic URB-597 produced small but significant effects on the average firing rate in both PCP- and saline-treated animals (Figure 3B).
Figure 3.
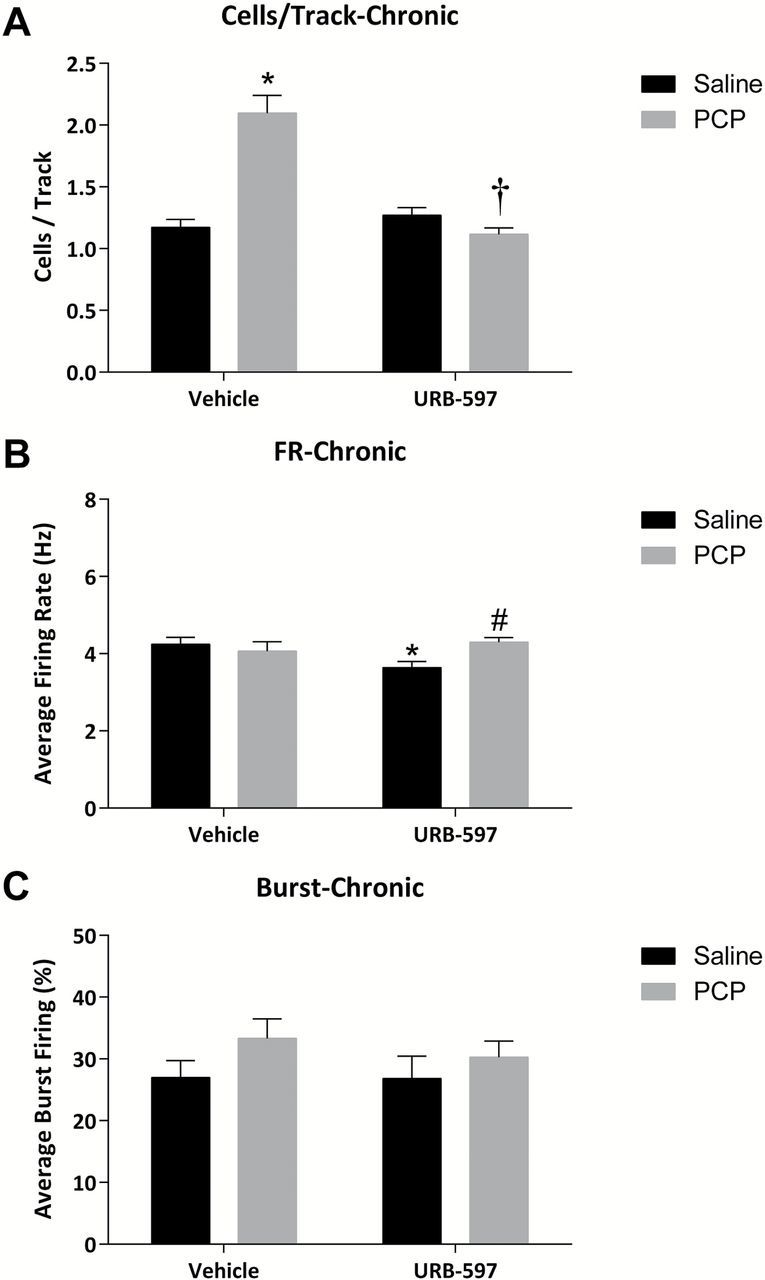
Chronic URB-597 reverses aberrant dopamine neuron activity in the PCP-treated rat. (A) PCP-treated rats display increases in dopamine neuron population activity, which are reversed by the chronic administration of URB-597. Small but significant alterations in the average firing rate were observed (B), with no effect on the average percent burst firing (C). *significant difference from vehicle-treated saline rats; †significant difference from vehicle-treated PCP rats; and #significant difference from URB-597–treated saline rats (two-way ANOVA).
The VP is the Site of Action for URB-597’s Effects on Dopamine Neuron Activity.
Given that the increase in dopamine neuron population activity is attributable to hyperactivity within the vHipp, we examined the effects of URB-597 on putative pyramidal neuron activity throughout the vHipp. Interestingly, initial studies demonstrated that neither saline- nor PCP-treated rats displayed alterations in vHipp activity following URB597, suggesting that the site of action of URB-597 was downstream of the vHipp.
It should be noted that the vHipp regulation of dopamine neuron activity occurs via a polysynaptic pathway involving the nucleus accumbens and ventral pallidum. Thus, activating the vHipp drives the nucleus accumbens, which, in turn, inhibits the ventral pallidum. Given that the pallidum provides a tonic inhibitory drive of the VTA, vHipp activation leads to disinhibition of dopamine neuron activity. Indeed, consistent with this circuit (outlined in Figure 5), vHipp stimulation produced a long-latency inhibition of spontaneous VP activity (Figure 5). Therefore, the effect of URB597 (0.3mg/kg) on vHipp evoked ventral pallidal activity was compared to vehicle 30 mins following i.v. administration (selected for its rapid kinetics compared to intraperitoneal administration). Interestingly, the vHipp-induced inhibition of VP activity was attenuated by URB-597 (analysis of treatment across time in two-way ANOVA; URB F(1,269) = 9.660, p < 0.05; Figure 6A; analysis of normalized data via Mann-Whitney Rank Sum test, U Statistic = 22.000, p < 0.05) implicating the VP as a key site for URB-597. To confirm this, we administered URB-597 directly into the VP and examined the effect on dopamine neuron activity. Specifically, PCP-treated rats receiving intra-VP infusions of the vehicle displayed a significant increase in dopamine neuron population activity (n = 5–7; SAL-VEH: 0.97±0.13 c.f. PCP-VEH: 1.65±0.14; two-way ANOVA; PCP x URB F(1,27) = 6.46; Holm-Sidak; t= 3.06; p < 0.05; Figure 7A), consistent with the observations detailed above. This was reversed by the intra-VP administration of URB-597, such that there was no longer any difference between vehicle- and PCP-treated rats (n = 5–7; SAL-URB: 1.36±0.14 c.f. PCP-URB: 1.29±0.14; Holm-Sidak; t= 0.37; p < 0.05; Figure 7A).
Figure 5.
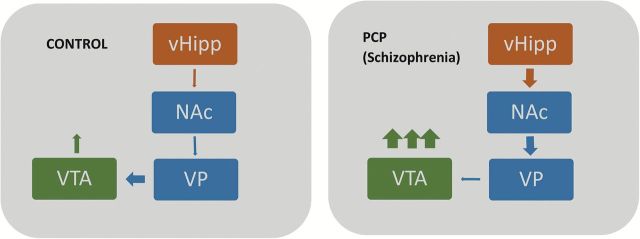
Schematic demonstrating the aberrant vHipp regulation of dopamine neuron activity in the PCP model of schizophrenia. Aberrant vHipp activity drives the nucleus accumbens (NAc) that, in turn, inhibits spontaneous activity within the ventral pallidum (VP). This results in the disinhibition of VTA dopamine neurons and an increase in the number of spontaneously-active neurons.
Figure 6.
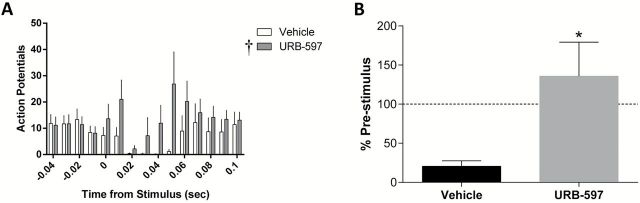
URB-597 inhibits the vHipp regulation of the VP. Stimulation of the vHipp induces a long-latency inhibition of spontaneous activity in VP neurons (A). This results in a decreased number of action potentials when compared to an equivalent pre-stimulus time (B). URB-597 attenuates this vHipp regulation of the VP. †main effect of treatment (two-way ANOVA); *significant difference between groups (Mann Whitney Rank Sum test).
Figure 7.
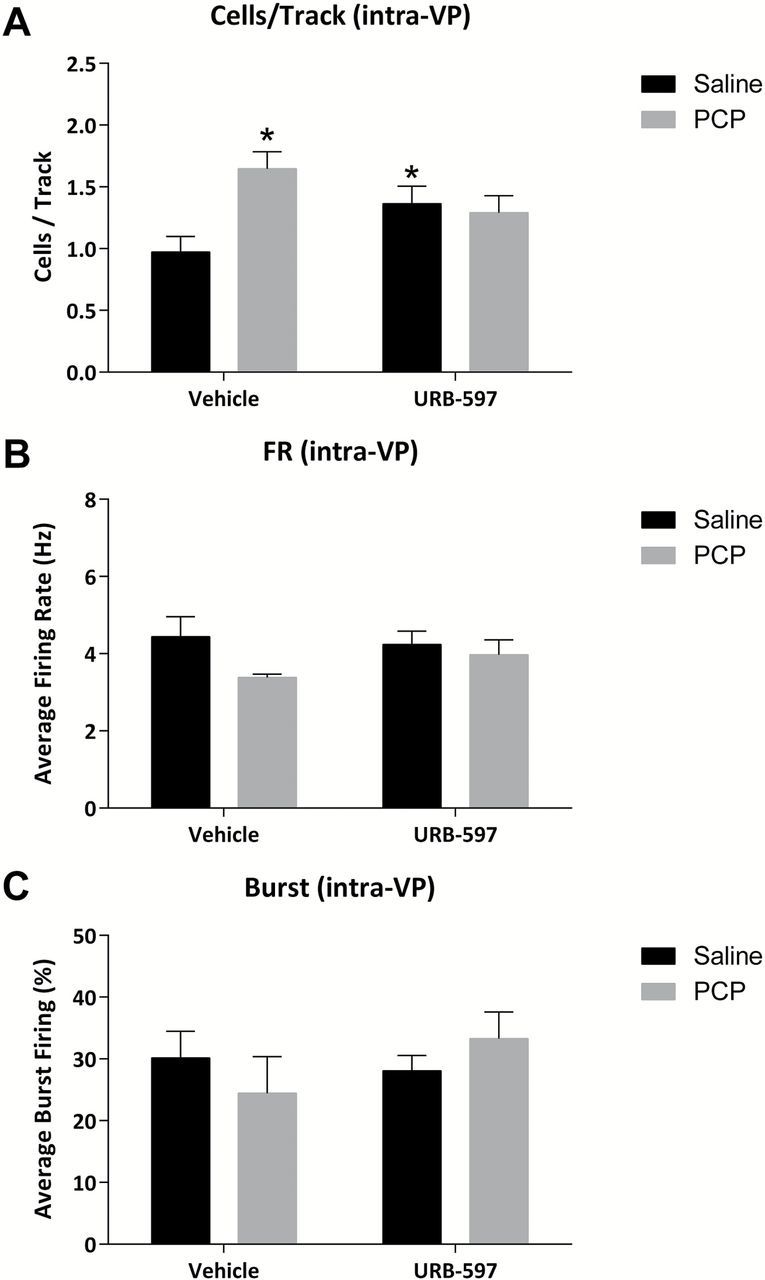
Intra-VP URB-597 reverses aberrant dopamine neuron activity in the PCP-treated rat. (A) The increase in dopamine neuron population activity observed in PCP-treated rats is not present following the direct administration of URB-597 into the VP (A). Consistent with the acute systemic administration of URB-597 detailed above, no effects were observed in either the average firing rate (B) or average percent burst firing (C). *significant difference from vehicle-treated saline rats (two-way ANOVA).
Discussion
Here we demonstrate that PCP-treated rats display an aberrant increase in dopamine neuron population activity, consistent with that observed in other preclinical models of schizophrenia (Lodge and Grace, 2007; Perez et al., 2013; Perez and Lodge, 2013; Chen et al., 2014). Moreover, this was attributable to an increase in the vHipp activity and was completely normalized by a vHipp blockade, as TTX inactivation of the hippocampus proved sufficient to restore normal spontaneous dopamine activity in the VTA. This is particularly relevant as we have now demonstrated a similar neurophysiological phenotype in multiple, diverse rodent models of schizophrenia. Indeed, the MAM model is produced by the administration of a mitotoxin during gestation (GD17), whereas the PCP model requires sub-chronic NMDA receptor–antagonist treatment for a week in adulthood. The finding that two diverse models both lead to an aberrant vHipp drive of the dopamine system function adds support to the hypothesis that this may be a key site of pathology in schizophrenia (Lodge and Grace, 2007; Perez et al., 2013; Perez and Lodge, 2013). Indeed, imaging studies in schizophrenia patients demonstrate elevated hippocampal activity at rest using MRI (Malaspina et al., 2008), increased regional cerebral blood volume using fMRI (Schobel et al., 2009), and increased cerebral blood flow with PET (Friston et al., 1992) or SPECT (Malaspina et al., 2004). Moreover, increases in hippocampal activity are correlated with symptom severity (Malaspina et al., 2008; Schobel et al., 2009).
Data from clinical and preclinical studies suggest that increasing AEA may provide a therapeutic intervention for schizophrenia (Seillier et al., 2010, 2013; Leweke et al., 2012). Indeed, we now demonstrate that URB-597 reverses aberrant dopamine neuron population activity in the PCP model. Given that AEA is produced on demand in an activity-dependent fashion (Piomelli, 2003) we administered URB-597 one hour preceding anesthesia (for acute experiments) or for an entire week preceding the experiment (as pharmacotherapies typically require chronic administration). In both cases, URB-597 restored aberrant dopamine system function in PCP-treated rats. It should be noted that acute URB597 produced a small but significant increase in dopamine neuron population activity in saline-treated rats. This is consistent with previous reports demonstrating increases in dopamine neuron activity (French, 1997; French et al., 1997) and dopamine release (Tanda et al., 1997; Cheer et al., 2004) following cannabinoid administration. After confirming that URB597 was able to reverse aberrant dopamine neuron activity in PCP-treated rats, we examined the neuronal brain regions underlying this effect. As detailed above, hyperactivity within the vHipp underlies the augmented dopamine neuron activity (Lodge and Grace, 2007, 2011; Perez et al., 2013; Perez and Lodge, 2013). For this reason we examined whether the beneficial effects of AEA upregulation on dopamine neuron function were due to an effect on hippocampal activity. We have recently demonstrated that treatment with URB597 or subchronic PCP did not significantly alter the firing rates of vHipp neurons (unpublished observation). This is in agreement with previous literature demonstrating a lack of effect of URB597 on hippocampal firing under physiological conditions (Coomber et al., 2008).
Previous studies have detailed a circuit whereby the vHipp drives the dopamine system via the nucleus accumbens and VP (see Figure 5; Lodge and Grace, 2007, 2011). Using this characterized circuit as a guide, we identified several areas with the potential to be modified by AEA upregulation and to affect VTA activity. CB1 receptors exist in the hippocampus (Egertova et al., 2003), nucleus accumbens (Yuan et al., 2013), and ventral pallidum (Harkany et al., 2003); however, the nucleus accumbens displays relatively little FAAH expression when compared to the VP (Egertova et al., 2003; Harkany et al., 2003). For this reason, we examined whether the VP was involved in URB597’s ability to decrease augmented dopamine system activity. We tested this hypothesis by stimulating the ventral hippocampus and recording from GABAergic cells in the VP. vHipp stimulation transiently inhibited activity in the ventral pallidum with a latency of 15–25 msec. This delay represents the time for the signal to propagate through the nucleus accumbens to the ventral pallidum (Yang and Mogenson, 1985). vHipp regulation of the VP was attenuated by the administration of URB597, suggesting that the VP may be the site of action for the effects of this compound. This is consistent with previous studies demonstrating that the pallidal response to striatal stimulation is blocked by cannabinoid drugs (Miller and Walker, 1996). To verify that the VP was indeed the site whereby systemic URB-597 produced its beneficial effect on dopamine system function, we examined the effect of intracranial administration of URB-597, directly into the VP, on the activity of VTA dopamine neurons. Consistent with the data detailed above, PCP-treated rats receiving vehicle infusions into the VP demonstrated significantly more spontaneously active dopamine cells in the VTA than saline-treated controls. However, this was attenuated by the intra-VP administration of URB597, demonstrating that this is likely the brain region whereby systemic URB-597 acts to reverse aberrant dopamine neuron activity. In addition, it should be noted that midbrain dopamine activity, in turn, has a strong influence on VP firing activity (Turner et al., 2002), emphasizing an important feedback relationship between these two structures.
When looking at social interaction preclinically, the beneficial effects of AEA upregulation are due to CB1r stimulation (Seillier et al., 2013). Indeed, many patients with schizophrenia already self-medicate with cannabis (Barnes et al., 2006; Madigan et al., 2013), which contains the CB1r agonist delta-9-tetrahydrocannabinol (THC). Surprisingly, there is strong evidence that THC exacerbates psychotic symptoms in patients with schizophrenia (D’Souza et al., 2005, 2009; Mason et al., 2009; Machielsen et al., 2010). Based on this information, human trials explored CB1r antagonism as a potential therapy for schizophrenia with negative results (Meltzer et al., 2004). The reasoning behind increased cannabis consumption in patients with schizophrenia may have more to do with the levels of cannabidiol rather than THC in each individual strain (Morgan and Curran, 2008). Cannabidiol treatment significantly increases serum AEA levels in patients with schizophrenia, and reduces positive and negative symptoms as effectively as amisulpride, but without the side effects seen with conventional antipsychotics (Leweke et al., 2012).
An obvious contradiction arises since CB1r stimulation is beneficial following AEA upregulation but detrimental when activated by THC administration. As mentioned previously, AEA is produced “on demand” in an activity-dependent fashion (Piomelli, 2003). As such, drugs such as URB597 or cannabidiol will potentiate endogenous CB1 signaling that will be localized to regions already producing AEA. On the other hand, THC is a lipophilic CB1r agonist, so its administration will likely activate CB1 receptors across the entire neuraxis. We posit that this difference in specificity may underlie the differences between THC and URB-597; indeed, we have electrophysiological data in freely-behaving animals demonstrating different effects of URB-597 and THC (unpublished observations).
In conclusion, we demonstrate that AEA upregulation by URB-597 shows potential therapeutic efficacy by reducing hyperactive dopamine system function in a rodent model of schizophrenia. This builds onto experiments that show AEA upregulation normalizes social withdrawal in the same model (Seillier et al., 2010, 2013). Furthermore, we now demonstrate that the VP is the likely site of action for the beneficial effects of URB-597 on dopamine neuron activity. Taken together, we posit that FAAH inhibitors may represent an alternative therapy for patients with schizophrenia and that the ventral pallidum may prove to be an attractive target for reducing psychotic episodes in patients with schizophrenia and improving their quality of life.
Statement of Interest
Dr Lodge discloses receiving consulting fees from Dey Pharmaceuticals, whereas Drs Chen and Aguilar have nothing to disclose.
Acknowledgments
The authors thank Dr Angie Boley for assistance with the histology and Drs Alex Seillier and Andrea Giuffrida for their helpful discussions. This work was supported by the NIH (R01: MH091130 and R25: GM095480).
References
- American Psychiatric Association (2000). Diagnostic and statistical manual of mental disorders, fourth edition, text revision (DSM-IV-TR). Washington, DC: Author. [Google Scholar]
- Ames F. (1958). A clinical and metabolic study of acute intoxication with Cannabis sativa and its role in the model psychoses. Br J Psychiatry 104:972–999. [DOI] [PubMed] [Google Scholar]
- Arseneault L, Cannon M, Witton J, Murray RM. (2004). Causal association between cannabis and psychosis: examination of the evidence. Brit J Psychiatry 184:110–117. [DOI] [PubMed] [Google Scholar]
- Barnes TR, Mutsatsa SH, Hutton SB, Watt HC, Joyce EM. (2006). Comorbid substance use and age at onset of schizophrenia. Br J Psychiatry 188:237–242. [DOI] [PubMed] [Google Scholar]
- Chadwick B, Miller ML, Hurd YL. (2013). Cannabis use during adolescent development: susceptibility to psychiatric illness. Front Psychiatry 4:129. 10.3389/fpsyt.2013.00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheer JF, Wassum KM, Heien ML, Phillips PE, Wightman RM. (2004). Cannabinoids enhance subsecond dopamine release in the nucleus accumbens of awake rats. J Neurosci 24:4393–4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Perez SM, Lodge DJ. (2014). An augmented dopamine system function is present prior to puberty in the methylazoxymethanol acetate rodent model of schizophrenia. Develop Neurobiol 74:907–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coomber B, O’Donoghue MF, Mason R. (2008). Inhibition of endocannabinoid metabolism attenuates enhanced hippocampal neuronal activity induced by kainic acid. Synapse 62:746–755. [DOI] [PubMed] [Google Scholar]
- D’Souza DC, Abi-Saab WM, Madonick S, Forselius-Bielen K, Doersch A, Braley G, Gueorguieva R, Cooper TB, Krystal JH. (2005). Delta-9-tetrahydrocannabinol effects in schizophrenia: implications for cognition, psychosis, and addiction. Biol Psychiatry 57:594–608. [DOI] [PubMed] [Google Scholar]
- D’Souza DC, Sewell RA, Ranganathan M. (2009). Cannabis and psychosis/schizophrenia: human studies. Eur Arch Psychiatry Clin Neurosci 259:413–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egertova M, Cravatt BF, Elphick MR. (2003). Comparative analysis of fatty acid amide hydrolase and cb(1) cannabinoid receptor expression in the mouse brain: evidence of a widespread role for fatty acid amide hydrolase in regulation of endocannabinoid signaling. Neuroscience 119:481–496. [DOI] [PubMed] [Google Scholar]
- Epstein J, Stern E, Silbersweig D. (1999). Mesolimbic activity associated with psychosis in schizophrenia. Symptom-specific PET studies. Ann NY Acad Sci 877:562–574. [DOI] [PubMed] [Google Scholar]
- French ED. (1997). delta9-Tetrahydrocannabinol excites rat VTA dopamine neurons through activation of cannabinoid CB1 but not opioid receptors. Neurosci Lett 226:159–162. [DOI] [PubMed] [Google Scholar]
- French ED, Dillon K, Wu X. (1997). Cannabinoids excite dopamine neurons in the ventral tegmentum and substantia nigra. Neuroreport 8:649–652. [DOI] [PubMed] [Google Scholar]
- Friston KJ, Liddle PF, Frith CD, Hirsch SR, Frackowiak RS. (1992). The left medial temporal region and schizophrenia. A PET study. Brain 115(Pt 2):367–382. [DOI] [PubMed] [Google Scholar]
- Giuffrida A, Leweke FM, Gerth CW, Schreiber D, Koethe D, Faulhaber J, Klosterkotter J, Piomelli D. (2004). Cerebrospinal anandamide levels are elevated in acute schizophrenia and are inversely correlated with psychotic symptoms. Neuropsychopharmacology 29:2108–2114. [DOI] [PubMed] [Google Scholar]
- Glenthoj BY, Mackeprang T, Svarer C, Rasmussen H, Pinborg LH, Friberg L, Baare W, Hemmingsen R, Videbaek C. (2006). Frontal dopamine D(2/3) receptor binding in drug-naive first-episode schizophrenic patients correlates with positive psychotic symptoms and gender. Biol Psychiatry 60:621–629. [DOI] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. (1983). Intracellular and extracellular electrophysiology of nigral dopaminergic neurons--1. Identification and characterization. Neuroscience 10:301–315. [DOI] [PubMed] [Google Scholar]
- Hall W, Degenhardt L. (2008). Cannabis use and the risk of developing a psychotic disorder. World Psychiatry 7:68–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkany T, Hartig W, Berghuis P, Dobszay MB, Zilberter Y, Edwards RH, Mackie K, Ernfors P. (2003). Complementary distribution of type 1 cannabinoid receptors and vesicular glutamate transporter 3 in basal forebrain suggests input-specific retrograde signalling by cholinergic neurons. Eur J Neurosci 18:1979–1992. [DOI] [PubMed] [Google Scholar]
- Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. (2003). Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med 9:76–81. [DOI] [PubMed] [Google Scholar]
- Laruelle M, Abi-Dargham A, van Dyck CH, Gil R, D’Souza CD, Erdos J, McCance E, Rosenblatt W, Fingado C, Zoghbi SS, Baldwin RM, Seibyl JP, Krystal JH, Charney DS, Innis RB. (1996). Single photon emission computerized tomography imaging of amphetamine-induced dopamine release in drug-free schizophrenic subjects. Proc Nat Acad Sci USA 93:9235–9240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leweke FM, Giuffrida A, Wurster U, Emrich HM, Piomelli D. (1999). Elevated endogenous cannabinoids in schizophrenia. Neuroreport 10:1665–1669. [DOI] [PubMed] [Google Scholar]
- Leweke FM, Piomelli D, Pahlisch F, Muhl D, Gerth CW, Hoyer C, Klosterkotter J, Hellmich M, Koethe D. (2012). Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia. Transl Psychiatry 2:e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, Keefe RS, Davis SM, Davis CE, Lebowitz BD, Severe J, Hsiao JK. ; Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators (2005) . Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. New Eng J Med 353:1209–1223. [DOI] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. (2007). Aberrant hippocampal activity underlies the dopamine dysregulation in an animal model of schizophrenia. J Neurosci 27:11424–11430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. (2011). Hippocampal dysregulation of dopamine system function and the pathophysiology of schizophrenia. Trends Pharmacol Sci 32:507–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machielsen M, van der Sluis S, de Haan L. (2010). Cannabis use in patients with a first psychotic episode and subjects at ultra high risk of psychosis: impact on psychotic- and pre-psychotic symptoms. Aus New Zeal J Psychiatry 44:721–728. [DOI] [PubMed] [Google Scholar]
- Madigan K, Brennan D, Lawlor E, Turner N, Kinsella A, O’Connor JJ, Russell V, Waddington JL, O’Callaghan E. (2013). A multi-center, randomized controlled trial of a group psychological intervention for psychosis with comorbid cannabis dependence over the early course of illness. Schizophr Res 143:138–142. [DOI] [PubMed] [Google Scholar]
- Malaspina D, Harkavy-Friedman J, Corcoran C, Mujica-Parodi L, Printz D, Gorman JM, Van Heertum R. (2004). Resting neural activity distinguishes subgroups of schizophrenia patients. Biol Psychiatry 56:931–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaspina D, Schobel SS, Lewandowski LM, Corcoran CM, Brown T, Small SA. (2008). Hippocampal dysfunction in CA1 is associated with schizophrenia. Schizophr Res 102:1–279.18514488 [Google Scholar]
- Mason O, Morgan CJ, Dhiman SK, Patel A, Parti N, Patel A, Curran HV. (2009). Acute cannabis use causes increased psychotomimetic experiences in individuals prone to psychosis. Psychol Med 39:951–956. [DOI] [PubMed] [Google Scholar]
- Meltzer HY, Arvanitis L, Bauer D, Rein W, Meta-Trial Study G. (2004). Placebo-controlled evaluation of four novel compounds for the treatment of schizophrenia and schizoaffective disorder. Am J Psych 161:975–984. [DOI] [PubMed] [Google Scholar]
- Meyer JS, Quenzer LF. (2005). Psychopharmacology: drugs, the brain, and behavior. Sunderland, Massachusetts: Sinauer Associates, Inc. [Google Scholar]
- Miller AS, Walker JM. (1996). Electrophysiological effects of a cannabinoid on neural activity in the globus pallidus. Eur J Pharmacol 304:29–35. [DOI] [PubMed] [Google Scholar]
- Moore TH, Zammit S, Lingford-Hughes A, Barnes TR, Jones PB, Burke M, Lewis G. (2007). Cannabis use and risk of psychotic or affective mental health outcomes: a systematic review. Lancet 370:319–328. [DOI] [PubMed] [Google Scholar]
- Morgan CJ, Curran HV. (2008). Effects of cannabidiol on schizophrenia-like symptoms in people who use cannabis. Br J Psychiatry 192:306–307. [DOI] [PubMed] [Google Scholar]
- Mouri A, Noda Y, Enomoto T, Nabeshima T. (2007). Phencyclidine animal models of schizophrenia: approaches from abnormality of glutamatergic neurotransmission and neurodevelopment. Neurochem Int 51:173–184. [DOI] [PubMed] [Google Scholar]
- Neill JC, Barnes S, Cook S, Grayson B, Idris NF, McLean SL, Snigdha S, Rajagopal L, Harte MK. (2010). Animal models of cognitive dysfunction and negative symptoms of schizophrenia: focus on NMDA receptor antagonism. Pharmacol Ther 128:419–432. [DOI] [PubMed] [Google Scholar]
- Paxinos GW, Watson C. (2009). The rat brain in stereotaxic coordinates, Compact 6th edition. Sydney, Australia: Academic Press. [Google Scholar]
- Perez SM, Lodge DJ. (2013). Hippocampal interneuron transplants reverse aberrant dopamine system function and behavior in a rodent model of schizophrenia. Mol Psychiatry 18:1193–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez SM, Shah A, Asher A, Lodge DJ. (2013). Hippocampal deep brain stimulation reverses physiological and behavioural deficits in a rodent model of schizophrenia. Int J Neuropsychop 16:1331–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piomelli D. (2003). The molecular logic of endocannabinoid signalling. Nat Rev Neurosci 4:873–884. [DOI] [PubMed] [Google Scholar]
- Schobel SA, Lewandowski NM, Corcoran CM, Moore H, Brown T, Malaspina D, Small SA. (2009). Differential targeting of the CA1 subfield of the hippocampal formation by schizophrenia and related psychotic disorders. Arch Gen Psychiatry 66:938–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seillier A, Advani T, Cassano T, Hensler JG, Giuffrida A. (2010). Inhibition of fatty-acid amide hydrolase and CB1 receptor antagonism differentially affect behavioural responses in normal and PCP-treated rats. Int J Neuropsychop 13:373–386. [DOI] [PubMed] [Google Scholar]
- Seillier A, Martinez AA, Giuffrida A. (2013). Phencyclidine-induced social withdrawal results from deficient stimulation of cannabinoid CB(1) receptors: implications for schizophrenia. Neuropsychopharmacology 38:1816–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanda G, Pontieri FE, Di Chiara G. (1997). Cannabinoid and heroin activation of mesolimbic dopamine transmission by a common mu1 opioid receptor mechanism. Science 276:2048–2050. [DOI] [PubMed] [Google Scholar]
- Turner MS, Mignon L, Napier TC. (2002). Alterations in responses of ventral pallidal neurons to excitatory amino acids after long-term dopamine depletion. J Pharm Exp Ther 301:371–381. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Grace AA. (2012). Are you or aren’t you? Challenges associated with physiologically identifying dopamine neurons. Trends Neurosci 35:422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CR, Mogenson GJ. (1985). An electrophysiological study of the neural projections from the hippocampus to the ventral pallidum and the subpallidal areas by way of the nucleus accumbens. Neuroscience 15:1015–1024. [DOI] [PubMed] [Google Scholar]
- Yuan WX, Heng LJ, Ma J, Wang XQ, Qu LJ, Duan L, Kang JJ, Chen LW, Gao GD. (2013). Increased expression of cannabinoid receptor 1 in the nucleus accumbens core in a rat model with morphine withdrawal. Brain Res 1531:102–112. [DOI] [PubMed] [Google Scholar]