

Abstract
Neuroblastoma (NB) is a pediatric tumor of neural crest origin with heterogeneous phenotypes. While low stage tumors carry a favorable prognosis, over 50% of high risk NB relapses after treatment with a fatal outcome. Thus, developing therapies targeting refractory NB remains an unsolved clinical problem. Brain-derived neurotrophic factor (BDNF) and its TrkB receptor are known to protect NB cells from chemotherapy-induced cell death, while neuropeptide Y (NPY), acting via its Y2 receptor (Y2R), is an autocrine proliferative and angiogenic factor crucial for maintaining NB tumor growth. Here, we show that in NB cells, BDNF stimulates the synthesis of NPY and induces expression of another one of its receptors, Y5R. In human NB tissues, the expression of NPY and Y5R positively correlated with the expression of BDNF and TrkB. Functionally, BDNF triggered Y5R internalization in NB cells, while Y5R antagonist inhibited BDNF-induced p44/42-MAPK activation and its pro-survival activity. These observations suggested TrkB-Y5R transactivation that resulted in cross-talk between their signaling pathways. Additionally, NPY and Y5R were up-regulated in a BDNF-independent manner in NB cells under pro-apoptotic conditions, such as serum deprivation and chemotherapy, as well as in cell lines and tissues derived from post-treatment NB tumors. Blocking Y5R in chemoresistant NB cells rich in this receptor sensitized them to chemotherapy-induced apoptosis and inhibited their growth in vivo by augmenting cell death. In summary, the NPY/Y5R axis is an inducible survival pathway activated in NB by BDNF or cellular stress. Upon such activation, Y5R augments the pro-survival effect of BDNF via its interactions with TrkB receptor and exerts an additional BDNF-independent anti-apoptotic effect, both of which contribute to NB chemoresistance. Therefore, the NPY/Y5R pathway may become a novel therapeutic target for patients with refractory NB, thus far an incurable form of this disease.
Keywords: neuroblastoma, neuropeptide Y, neuropeptide Y receptor Y5, chemoresistance
Introduction
Neuroblastoma (NB) is a pediatric malignancy with heterogeneous phenotypes.1 While low stage NB carries a favorable prognosis, 50–60% of high risk tumors relapse with a fatal outcome.2,3 At present, there is no adequate therapy for refractory NB and the mechanisms underlying its chemoresistance are poorly understood.
NB develops from precursors of sympathetic neurons and the level of neuronal differentiation is one of the major factors determining its clinical behavior, with undifferentiated tumors being the most aggressive.2,4,5 These tumors express neuronal markers and respond to factors regulating neuronal functions, such as brain derived neurotrophic factor (BDNF) and its receptor tyrosine kinase (RTK), TrkB.6,7 BDNF is known for its neuroprotective effects.8–10 In NB, BDNF is often endogenously expressed and acts as a pro-survival factor, enhancing resistance of these tumors to chemotherapy.11–14 These chemoresistance effects are mediated by the PI3K/Akt and p44/42 MAPK pathways.8–14 Consistently, a high level of TrkB in NB tumors is an adverse prognostic factor.7,15–18 Paradoxically, expression of TrkB in cell lines derived from advanced NBs is low or non-detectable.17 Therefore, NB cells stably transfected with TrkB are commonly used to study the role of BDNF in these tumors.11–14
Another neuronal protein, a sympathetic neurotransmitter neuropeptide Y (NPY), is highly expressed in NB and its elevated systemic levels have been associated with poor clinical outcome in high risk NB patients.19–21 NPY, acting via its G protein-coupled receptors (GPCRs), is known to stimulate cell proliferation and act as an angiogenic factor.22–27 Both NPY and its Y2 receptor (Y2R) are universally expressed in NBs.28,29 We have previously shown that this autocrine loop is essential for maintaining NB cell proliferation and tumor vascularization. Consequently, Y2R antagonist impairs tumor growth due to its anti-proliferative and anti-angiogenic effects.29 However, some NB cell lines additionally express another NPY receptor, Y5R.28 In contrast to Y2R, Y5R expression is variable and its role in NB remains unknown.
Similar to BDNF, NPY is also known for its neuroprotective activities mediated via p44/42 MAPK.30,31 Interestingly, BDNF stimulates NPY expression in neuronal cells.32–37 Thus, the goal of this study was to determine the molecular and functional interactions between BDNF and NPY systems in NB and their role in the biology of these tumors. We demonstrated that BDNF, acting via TrkB receptor, stimulates expression of NPY and its Y5R. This, in turn, facilitates cross-talk between TrkB and Y5R signaling pathways, which then enhances pro-survival activity of BDNF. In addition to this BDNF-induced activation, NPY and Y5R are also up-regulated in NB cells undergoing cellular stress, such as chemotherapy, and are highly expressed in cell lines and tissues derived from post-treatment patients. Such activation of the NPY/Y5R axis enhances NB cell survival, while blocking Y5R sensitizes them to chemotherapy. Taken together, we provide the first evidence for Y5R acting as an inducible pro-survival factor in NB, contributing to their chemoresistance.
Results
BDNF stimulates NPY system expression in NB
Given the low TrkB expression in NB cell lines, SH-SY5Y NB cells stably transfected with TrkB (SY5Y/TrkB) were used to determine interactions between NPY and BDNF systems.11,17 NPY release was elevated in SY5Y/TrkB cells, as compared to non-transfected SH-SY5Y cells (Fig. 1A), suggesting a TrkB-mediated stimulatory effect of endogenous BDNF on NPY expression. Indeed, in SY5Y/TrkB cells, BDNF increased NPY and Y5R mRNA, but not Y2R mRNA (Fig. 1B). The selective increase in Y5R expression induced by BDNF was confirmed on a protein level by Western blot (Fig. 1C). Trk inhibitor, K252a, blocked these stimulatory effects of BDNF on NPY and Y5R expression (Fig. 1B, C). No further increase in NPY release was detected in BDNF-treated SY5Y/TrkB cells, suggesting saturation of the system by endogenous neurotrophin.
Figure 1. BDNF up-regulates NPY and Y5R expression in NB cells.

A. NPY release from SH-SY5Y cells, non-transfected and transfected with TrkB cDNA (SY5Y/TrkB), measured in conditioned media by ELISA. B. mRNA levels of NPY and its receptors in SY5Y/TrkB transfectants treated with BDNF, with or without Trk antagonist, K252a, measured by real-time RT-PCR. C. Western blot analysis of NPY receptor expression in SY5Y/TrkB cells treated with BDNF, with or without Trk inhibitor. D. mRNA of NPY and Y5R in SK-N-BE(2) NB cells treated with BDNF in the presence or absence of Trk antagonist, measured by real-time RT-PCR. E. NPY released to culture media by control and BDNF-treated SK-N-BE(2) cells. F. Protein levels of Y5R in BDNF-treated SK-N-BE(2) cells detected by Western blot. G. A panel of NB cells derived from patients before (CHLA-15, SMS-KCN, SMS-KAN) and after chemotherapy (SK-N-BE(2), CHLA-20, SMS-KCNR, SMS-KANR) was treated with BDNF, and Y5R expression measured by western blot, as above. The Y5R band intensities were quantified by densitometry (three independent experiments per cell line) and averaged for each group. In all above experiments, cells were cultured in 1% FBS media for 24h and then treated with BDNF (0.1–1ng/ml) for 6–24h, with our without TrkB antagonist, K252a, at concentration 10−6M. PS – unspecific protein stain.
A similar stimulatory effect of BDNF on NPY system expression was observed in native NB cells. Despite a lower expression of TrkB receptor, non-transfected NB cells remained responsive to its ligand, as measured by BDNF-induced p44/42 MAPK phosphorylation (Supplementary Fig. 1). Consequently, BDNF increased NPY and Y5R mRNA in SK-N-BE(2) NB cells, and Trk antagonist blocked this effect (Fig. 1D). These increases in mRNA resulted in elevated NPY release and Y5R protein levels in BDNF-treated SK-N-BE(2) cells (Fig. 1E, F). To confirm this effect, we used a panel of NB cell lines derived from patients before (CHLA-15, SMS-KCN, SMS-KAN) and after chemotherapy (SK-N-BE(2), CHLA-20, SMS-KCNR, SMS-KANR).38 Interestingly, the BDNF-induced Y5R up-regulation was observed in post-treatment cell lines, while no such effect was detected in cells derived from patients at diagnosis (Fig. 1G).
In human NB tumors, expression of BDNF and NPY systems positively correlate with each other
mRNA of NPY and BDNF systems were measured by real-time RT-PCR in 75 samples of NB tissues at diagnosis, a cohort enriched in undifferentiated and poorly differentiated tumors. These tumor subtypes typically carry a worse prognosis and are subsequently treated with chemotherapy. NPY and Y2R mRNA were present in all samples, while Y5R mRNA was detectable in 84% of cases. As summarized in Table 1, significant correlations were detected by the Pearson coefficient between BDNF and NPY systems, yet the magnitude and statistical significance of these correlations were dependent on NB differentiation. NPY mRNA had significant positive correlations with mRNA for both TrkB and BDNF, when all NB cases were analyzed together. Among NB subtypes, a statistically significant correlation between NPY and TrkB mRNAs was observed in undifferentiated tumors, while a correlation between NPY and BDNF mRNAs achieved statistical significance in differentiating NBs. Correlations between Y5R expression and TrkB and BDNF mRNA levels were statistically significant in poorly differentiated tumors. Similarly, a significant association was observed between Y5R and BDNF mRNA levels in differentiating tumors. This translated to a statistically significant correlation between these two factors in all NB cases. No statistically significant correlations between Y2R and BDNF system expression were observed.
Table 1.
Correlations of NPY and Y5R mRNAs with expression of BDNF and its TrkB receptor in NB tumors at various levels of differentiation.
| Correlation (r): | Undifferentiated (n=42) | Poorly differentiated (n=25) | Differentiating (n=8) | All NB cases (n=75) |
|---|---|---|---|---|
| NPY mRNA vs. TrkB mRNA | 0.371* | 0.141 | 0.608 | 0.347* |
| NPY mRNA vs. BDNF mRNA | 0.273 | 0.157 | 0.870** | 0.304* |
| Y5R mRNA vs. TrkB mRNA | −0.123 | 0.831*** | 0.057 | 0.088 |
| Y5R mRNA vs. BDNF mRNA | −0.073 | 0.700*** | 0.739* | 0.434*** |
p < 0.05,
p < 0.01,
p < 0.001
Correlations for NPY mRNA were performed on ΔCT, while for Y5R mRNA expression relative to β-actin was used to account for samples with non-detectable mRNA (expression = 0)
BDNF triggers internalization of Y5R
There is growing evidence for RTK-GPCR transactivation.39,40 To determine if coordinated expression of TrkB and Y5R results in such interactions, we transfected SY5Y/TrkB cells with Y5R cDNA fused to EGFP and used the internalization of Y5R-EGFP as a measure of its activation. Transfected cells were treated with BDNF, with or without Y5R and Trk antagonist, and Y5R-EGFP receptor internalization was monitored by time-lapse fluorescence microscopy. Within 2 min of BDNF stimulation, Y5R was recruited to the cell membrane and subsequently internalized, as observed 8 min after treatment (Fig. 2A). These changes were reflected by differences in the ratio of membrane/sub-membrane receptor fraction (Fig. 2B). No significant changes in Y5R distribution were observed upon pre-treatment with Y5R or Trk antagonist (Fig. 2A, B) or in non-treated cells (data not shown). BDNF-induced internalization of Y5R was confirmed by immunoprecipitation of membrane proteins. SY5Y/TrkB cells were treated with BDNF and subsequently the surface proteins were biotinylated, immunoprecipitated and immunoblotted with anti-Y5R antibody. Within 8 min, BDNF triggered the decrease in the cell surface fraction of Y5R, while no statistically significant effect was observed upon pre-treatment with Y5R or Trk antagonist (Fig. 2C).
Figure 2. BDNF stimulates internalization of Y5R in NB cells.

A. SY5Y/TrkB cells were transiently transfected with Y5R-EGFP cDNA and treated with BDNF (1ng/ml), in the presence or absence of Y5R or Trk antagonist (10−6M). Changes in Y5R-EGFP cellular distribution were detected by time-lapse fluorescence microscopy. Sequential images of representative cells are shown. B. Levels of membrane and sub-membrane Y5R content were quantified based on fluorescence intensities in the corresponding sub-cellular regions. Multiple regions with the initial membrane localization of Y5R (membrane/sub-membrane fluorescence ratio ≥ 1.3) from at least three independently treated cells per group were analyzed. C. SY5Y/TrkB cells were treated for 8 min with BDNF (1ng/ml) in the presence or absence of Y5R or Trk antagonists at concentrations 10−6M. Membrane proteins were then biotinylated and selectively immunoprecipitated. The fraction of Y5R present on the cell surface was detected by Western blot in immunoprecipitation eluates and normalized to Y5R levels in the original cell lysates. The results of three independent experiments were quantified by densitometry.
Blocking Y5R interferes with BDNF-induced p44/42 MAPK signaling
To determine if TrkB-Y5R interactions result in signaling cross-talk, we tested the effects of their receptor antagonists on the activation of downstream pathways. In SY5Y/TrkB cells, NPY activated the p44/42 MAPK, but not the Akt pathway (Fig. 3A). Y2R antagonist blocked NPY-driven p44/42 MAPK phosphorylation, while Y5R and Trk receptor antagonists had no effect. In contrast, BDNF-induced p44/42 MAPK activation was blocked by Trk inhibitor and significantly inhibited by Y5R antagonist, while Y2R antagonist had no effect (Fig. 3A). The BDNF-induced Akt activation was blocked by Trk inhibitor, but remained unaffected by NPY receptor antagonists. These results were confirmed using NPY receptor siRNAs. While both Y2R and Y5R siRNAs effectively inhibited their targets (Supplementary Fig. 2), only Y5R siRNA significantly decreased BDNF-induced p44/42 MAPK activation (Fig. 3B). Similarly, blocking Y5R reduced p44/42 MAPK phosphorylation triggered by BDNF in native SK-N-BE(2) cells (Fig. 3C), although no BDNF-induced Akt activation was observed (data not shown).
Figure 3. Blocking Y5R interferes with BDNF-induced p44/42 MAPK activation.

A. SY5Y/TrkB cells were cultured for 24h in serum-free media and then treated with BDNF (1ng/ml) or NPY (10−7M) for 5 min in the presence or absence of their cognate receptor inhibitors – Trk, Y2R or Y5R antagonists at concentrations 10−6M. Phosphorylated forms of p44/42 MAPK and Akt were detected by Western blot. The results of at least three independent experiments were quantified by densitometry. B. SY5Y/TrkB cells were transfected with negative control (NC), Y5R or Y2R siRNAs. 24h later, the cells were transferred for another 24h to serum free media and then treated with BDNF (1ng/ml) for 5 min. The results were quantified as above. C. SK-N-BE(2) cells were treated with NPY or BDNF with or without antagonists of their corresponding receptors, as described for panel A, and activated p44/42 MAPK was detected by Western blot.
Y5R contributes to pro-survival effects of BDNF
In SY5Y/TrkB cells cultured under low serum conditions, BDNF increased cell viability (Fig. 4A). This effect was decreased by Trk inhibitor, and further reduced by a combination of Trk and Y5R antagonists. Similarly, BDNF rescued NB cells from chemotherapy-induced cell death. For doxorubicin and vinblastine, the pro-survival effect of BDNF was significantly, yet partially reduced by TrkB and Y5R antagonists alone, while treatment with combined antagonists was required for its complete blockage (Fig. 4B, Supplementary Fig. 3). In contrast, Trk inhibitor completely blocked the pro-survival activity of BDNF upon treatment with etoposide and cisplatin, while Y5R antagonist alone had no effect, suggesting the prevalence of a Y5R-independent Akt pathway. Nevertheless, in all cases, the combination of Trk and Y5R antagonist was the most effective and decreased the cell viability below control levels, indicating a contribution of endogenous BDNF and NPY to NB survival. These changes in cell viability were accompanied by corresponding changes in p44/42 MAPK activation (Fig. 4B).
Figure 4. Blocking Y5R inhibits pro-survival effects of BDNF in NB cells.

A. SY5Y/TrkB cells were cultured for 24h in 1% FBS media and then treated with BDNF (1ng/ml), with or without 1h pre-incubation with Trk and Y5R antagonists (both at 10−6M). Cell viability was measured 24h later by MTS assay. B. SY5Y/TrkB cells were cultured and treated with BDNF and Trk or Y5R antagonists, as above. 2h after BDNF administration, cells were additionally treated with doxorubicin (1μg/ml) or etoposide (2.75 μg/ml). Cell extracts for detection of activated p44/42 MAPK by Western blot were collected 1h later, while cell viability was measured by MTS assay 24h later. C. SK-N-BE(2) cells were cultured as described for panel A, pre-incubated for 1h with Trk or Y5R antagonists at concentrations 10−6M, and then treated with BDNF (1ng/ml) followed by cisplatin (0.5μg/ml). 24h later, cell viability was measured by MTS assay and apoptosis was assessed by caspase 3/7 activity assay.
The pro-survival effect of BDNF was also observed in native SK-N-BE(2) cells. Upon cisplatin treatment, BDNF increased cell viability and decreased caspase 3/7 activity, suggesting inhibition of apoptosis as the mechanism of its actions (Fig. 4C). The pro-survival effect of BDNF in SK-N-BE(2) cells treated with cisplatin and other chemotherapeutics was completely blocked by Y5R antagonist (Fig. 4C, Supplementary Fig. 4).
NPY system is activated in pro-apoptotic conditions
Having determined TrkB-Y5R interactions, we sought to identify other factors activating Y5R in NB. To this end, SY5Y/TrkB cells were transfected with Y5R siRNA and cultured under various conditions. Upon 24h culture in low serum media, Y5R siRNA had no effect on cell survival (Fig. 5A). However, in cells subjected to more severe cellular stress, such as chemotherapy or serum deprivation for 48h, Y5R knock-down significantly decreased cell viability. In line with these observations, Y5R protein was not detectable in SY5Y/TrkB cells cultured in 10% FBS media, while its expression was induced under serum-free conditions (Fig. 5B), implicating NPY/Y5R axis as an inducible pro-survival pathway.
Figure 5. NPY system is up-regulated in NB cells under pro-apoptotic conditions.
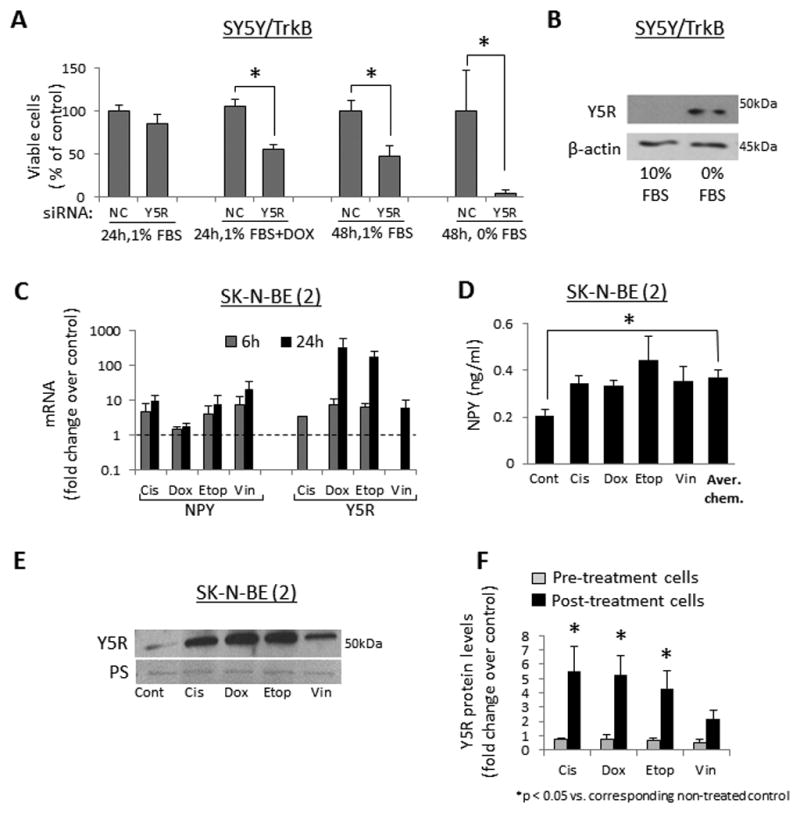
A. SY5Y/TrkB cells were transfected with negative control (NC) or Y5R siRNAs and cultured in 1% or 0% FBS culture media for 24h–48h, with or without doxorubicin (1μg/ml). Cell viability was measured by MTS assay. B. SY5Y/TrkB cells were cultured for 24h in 10% or 0% FBS media and protein levels of Y5R were detected by Western blot. C. SK-N-BE(2) cells were cultured for 24h in 1% FBS media and then treated with four different chemotherapeutics – cisplatin (Cis, 0.5μg/ml), doxorubicin (Dox, 0.5μg/ml), etoposide (Etop, 2.5μg/ml) or vinblastine (Vin, 1μg/ml) for 6 or 24h. mRNA levels of NPY and Y5R were measured by real-time RT-PCR. D. SK-N-BE(2) cells were treated with chemotherapy as above and NPY released to the cell culture media was measured by ELISA. E. Protein levels of Y5R were assessed by Western blot in chemotherapy-treated SK-N-BE(2) cells. F. A panel of NB cells derived from patients before (CHLA-15, SMS-KCN, SMS-KAN) and after chemotherapy (SK-N-BE(2), CHLA-20, SMS-KCNR, SMS-KANR) was treated for 6–12h with chemotherapeutic agents, as above, and Y5R detected by Western blot. Expression levels from at least three independent experiments for each cell line were quantified by densitometry and the results for all pre- and post-treatment cells were averaged. PS – unspecific protein staining.
Similarly, activation of the NPY/Y5R axis under cellular stress was observed in non-transfected NB cells expressing low levels of TrkB. In SK-N-BE(2) cells, chemotherapeutics increased mRNA of NPY and Y5R (Fig. 5C). Consequently, treatment with all compounds increased NPY release, which resulted in a significantly elevated NPY concentration in conditioned media from chemotherapy-treated SK-N-BE(2) cells (Fig. 5D). The increase in Y5R expression was confirmed by Western Blot (Fig. 5E). Trk inhibitor did not block this effect, suggesting its BDNF-independent regulation (data not shown). As demonstrated for sensitivity to BDNF, the chemotherapy-induced increase in Y5R expression was detectable in a panel of cell lines derived from previously treated NB patients, but not in pre-treatment cells (Fig. 5F).38
NPY exerts pro-survival effects in chemotherapy-treated NB cells
Changes in the NPY system suggested its Y5R-mediated pro-survival activity. Indeed, in SK-N-BE(2) cells treated with various chemotherapeutics, NPY increased cell viability (Fig. 6A). Y5R antagonist blocked this effect and decreased NB cell survival below baseline, indicating a pro-survival effect of the endogenous peptide. The NPY-induced increase in cell viability was associated with reduced caspase 3/7 activity in chemotherapy-treated NB cells, suggesting the inhibition of apoptosis as the main mechanism of its action (Fig. 6B).
Figure 6. NPY promotes survival of chemotherapy-treated NB cells.
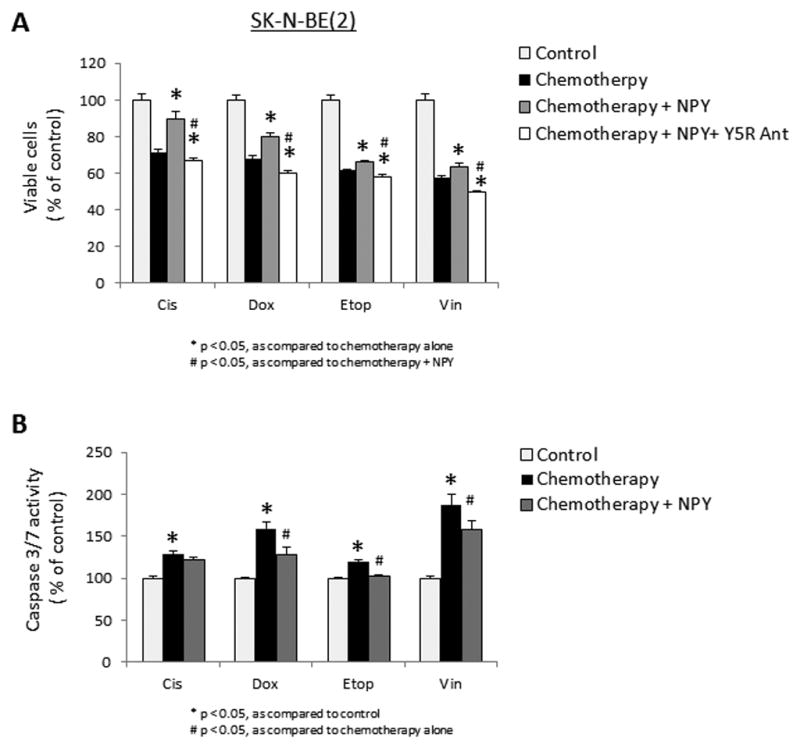
A. SK-N-BE(2) cells were cultured in 1% FBS media and treated with cisplatin (0.5μg/ml), doxorubicin (0.5μg/ml), etoposide (2.5μg/ml) or vinblastine (1μg/ml) for 24h with or without 10−7M NPY and Y5R antagonist (10−6M). Cell survival was measured by MTS assay. B. SK-N-BE(2) cells were treated with chemotherapy with or without NPY, as above, and apoptosis was assessed by caspase 3/7 activity assay.
NPY system is up-regulated in post-treatment NB cells
To confirm the clinical relevance of NPY system activation observed upon short-term treatment, pairs of NB cell lines derived from patients before and after chemotherapy were compared in terms of NPY system expression.38 On an mRNA level, the treatment-induced increase in NPY and Y5R expression was observed in CHLA-15/CHLA-20 and SMS-KCN/SMS-KCNR cell lines, while in the SMS-KAN/SMS-KANR pair, only Y5R mRNA was elevated (Fig. 7A). In line with RT-PCR results, NPY cellular content was elevated in post-treatment CHLA-20 and SMS-KCNR, but not SMS-KANR cells (Fig. 7B), while Y5R protein levels were increased in all post-treatment cells (Fig. 7C).
Figure 7. NPY system is up-regulated in cells and tissues from chemotherapy-treated tumors.

A. mRNA levels of NPY and Y5R were measured by real-time RT-PCR and compared within pairs of cells derived from the same patients before and after therapy. B. NPY levels in the above panel of cells were measured by ELISA. C. Protein levels of Y5R in pre- and post-treatment cells were detected by Western blot and quantified by densitometry. PS – unspecific protein staining. D. Y5R immunostaining of human NB tissues derived from primary tumors at diagnosis (representative images) and after chemotherapy (3 independent cases).
To validate findings in the cell lines, we compared Y5R immunostaining in primary NB tumors derived from non-treated patients and those after chemotherapy. In non-treated tumors, Y5R expression was observed in single NB cells (an average 30% of positive tumor cells in undifferentiated and poorly differentiated NBs) (Fig. 7D). In contrast, in chemotherapy-treated NB tumors, 100% of surviving NB cells was Y5R positive.
Blocking Y5R sensitizes NB cells to chemotherapy
Chemotherapy-induced changes in NPY system expression suggested a role for Y5R in NB chemoresistance. To test the extent and specificity of this effect, we used CHLA-15 and CHLA-20 cells, a pair of pre- and post-treatment cell lines that are rich in Y5R, but also express Y2R.29 Cells were treated with increasing doses of chemotherapy in the presence of Y2R or Y5R antagonist. In both cell lines, blocking Y5R decreased cell viability, while Y2R inhibitor had no effect (Fig. 8A). These changes were associated with corresponding increases in caspase 3/7 activity upon Y5R, but not Y2R antagonist treatment (Fig. 8B).
Figure 8. Blocking Y5R augments NB cell death.

A. Dose response curves of CHLA-15 and CHLA-20 NB cells (pre- and post-treatment, respectively) to doxorubicin and vinblastine in the presence or absence of Y2R and Y5R antagonists (10−6M). B. Survival and caspase 3/7 activity in CHLA-15 and CHLA-20 cells treated with doxorubicin (0.5 and 1 μg/ml, respectively) in the presence of Y2R and Y5R antagonists (10−6M). C. Chemoresistant CHLA-20 cells were injected subcutaneously into nude mice and treated with Y5R antagonist (10−6M) or placebo. Tumor volumes were measured daily and the growth rate compared between the experimental groups. D. The level of cell death was quantified in the placebo and Y5R antagonist-treated CHLA-20 xenograft tissues by area of positive TUNEL staining. Representative pictures are shown for each experimental group.
Y5R antagonist inhibits growth of chemoresistant NB cells in vivo
To validate the pro-survival effect of Y5R in vivo, subcutaneous xenografts obtained from chemoresistant CHLA-20 cells38 were treated with Y5R antagonist. Blocking Y5R resulted in significant inhibition of CHLA-20 xenograft growth (Fig. 8C), which was associated with increased tumor cell death (Fig. 8D). No significant changes were observed in NB cell proliferation or tumor vascularization measured by Ki67 and von Willebrand factor (vWF) immunostaining, respectively (data not shown).
Discussion
BDNF and NPY are important factors in NB biology. BDNF, acting via its TrkB receptor, is mainly a pro-survival factor for NB cells, protecting them from chemotherapy-induced cell death, but also promoting angiogenesis and invasiveness.6,7,41 Consequently, high expression of TrkB in tumor tissue correlates with poor prognosis in NB patients.7,15,17 Similarly, elevated systemic levels of NPY are associated with a poor outcome of NB.19–21 Actions of endogenous NPY mediated by its Y2R are essential to maintain NB cell proliferation and tumor vascularization.19–21,28,29 However, functions of Y5R, which is also detectable in some NB cell lines, were not well defined.28 Moreover, despite similar functions of NPY and BDNF, as well as a known stimulatory effect of BDNF on NPY synthesis in neurons, interactions of these two systems in NB remained unknown.32–37 Here, we have shown that BDNF up-regulates the NPY system in NB, which in turn enhances its pro-survival functions via interactions between TrkB and Y5R receptors.
In NB cells, BDNF increased synthesis of NPY and induced expression of its Y5R, while no consistent up-regulation of Y2R was observed. These results are in agreement with the presence of binding sites for two transcription factors known to mediate neurotrophins’ effects, AP-1 and CREB, in the promoters of NPY and Y5R, but not Y2R.42,43 Importantly CREB is activated by p44/42 MAPK, the pathway previously shown to mediate BDNF-induced increase in NPY expression.34,43 The enhanced responsiveness of post-treatment NB cells to BDNF stimulation could be associated with chemotherapy-induced over-expression of TrkB. We observed a trend toward increased TrkB expression in these cells, albeit not statistically significant (data not shown).
The specificity of BDNF’s stimulatory effect on the NPY system was confirmed by positive correlations between BDNF/TrkB and NPY/Y5R mRNAs, but not Y2R mRNA, observed in human NB tissues. The strength of these correlations depended on tumor differentiation grade, which may be associated with the variability in TrkB and BDNF expression, as well as in the presence of stromal cells known to release BDNF and regulate NB growth.44–46 Interestingly, the strongest associations of NPY and Y5R levels with BDNF system expression were observed in different NB subsets, suggesting that other factors differentially expressed in these tumors modify BDNF actions. Importantly, NPY is highly expressed in all NB subtypes, while non-treated NBs express Y5R at much lower levels.28,29 Thus, Y5R expression rather than NPY level is the limiting factor for NPY/Y5R axis activation. Altogether, our data implicate NPY and Y5R, but not Y2R, as direct BDNF targets.
Expression of Y5R stimulated by BDNF resulted in TrkB-Y5R transactivation and signaling cross-talk, as demonstrated by Y5R internalization upon BDNF treatment and the decrease in BDNF-induced p44/42 MAPK activation while Y5R was blocked. Such RTK-GPCR transactivation has been shown for other receptors and often involves phosphorylation of GPCRs by RTKs or their signaling mediators, physical interactions between the receptors, or simultaneous activation of p44/42 MAPK by both receptors.39,40,47–50 Further studies are required to determine which of these mechanisms mediate TrkB-Y5R interactions.
TrkB-Y5R transactivation enhanced BDNF-induced p44/42 MAPK phosphorylation and augmented its anti-apoptotic effect in chemotherapy-treated NB cells. However, these interactions did not affect the BDNF-activated PI3K/Akt pathway, suggesting its sole dependence on TrkB. Such a selective activation of signaling pathways is common for RTK-GPCR transactivation and may result from allosteric changes that occur due to receptor interactions.49,51–54 Our results are in contrast to previous reports implicating the PI3K/Akt axis as the main pathway mediating pro-survival effects of BDNF in chemotherapy-treated NB cells.11,12,14 However, the role of p44/42 MAPK in BDNF-induced survival has been demonstrated in paclitaxel-treated NB cells.13 Similarly, in normal neurons, neuroprotective actions of BDNF and NPY are largely p44/42 MAPK-mediated.8–10,30 Our results indicate that BDNF-induced p44/42 MAPK is dependent on TrkB-Y5R transactivation. Such RTK-GPCR interactions are regulated by RTK ligand and receptor availability.40,47,55 In cells expressing high levels of RTKs or exposed to high concentrations of their ligand, no GPCR involvement is observed and the signaling events solely depend on RTKs. In contrast, the RTK-GPCR cross-talk is activated in low concentrations of RTK and/or its agonist when the signal from RTK is not sufficient to trigger the biological response. In line with this, previous studies demonstrating PI3K/Akt pathway as solely responsible for BDNF’s pro-survival effect relied on TrkB transfectants treated with high doses of neurotrophin (100ng/ml), while markedly lower concentrations (0.1–1ng/ml) were used in our study.11,12,14 Moreover, we observed no Akt pathway activation in non-transfected NB cells which express low levels of TrkB, yet respond to BDNF by p44/42 MAPK phosphorylation. Consequently, the pro-survival effect of BDNF in these cells was completely blocked by Y5R antagonist. Thus, in agreement with reports on other RTK-GPCR systems, our results indicate that TrkB-Y5R interactions serve as a rescue system activating p44/42 MAPK and enhancing pro-survival effects of BDNF when the activation of the BDNF/TrkB axis alone is not sufficient to protect the cells. Therefore, in NB tumors with low TrkB expression and/or low BDNF content, the p44/42 MAPK pathway may contribute to their chemoresistance and become a therapeutic target along with Akt.
The TrkB-Y5R transactivation is in agreement with the ability of Y5R to interact with other receptors. Y5R forms heterodimerms with Y1R, and enhances Y1R-mediated proliferation of vascular smooth muscle cells in low NPY concentrations.56,57 It also augments functions of the α1-adrenergic receptor in cardiomyocytes.24,58 Here, we have shown that Y5R contributed to BDNF-induced, but not NPY-induced, p44/42 MAPK phosphorylation. This selectivity in Y5R signaling may depend on cellular context, such as the prevalence of Y2R expression, which binds the majority of NPY and/or high expression of TrkB, which recruits Y5R to its signaling complex.
Aside from BDNF-induced up-regulation of the NPY/Y5R axis, expression of NPY and Y5R was increased under cellular stress, such as serum deprivation or chemotherapy. As seen with the response to BDNF, more pronounced up-regulation of the NPY system was observed in cells derived from chemotherapy-treated NBs. This effect was not blocked by Trk inhibitor, suggesting BDNF-independent pro-survival activity of the NPY/Y5R pathway. Indeed, in chemotherapy treated NB cells, NPY increased cell viability in a Y5R-dependent manner. Thus, while after short-term stimulation, such as that used to detect p44/42 MAPK activation, Y5R predominantly interacts with TrkB, under prolonged cellular stress it mediates also anti-apoptotic effects of NPY, Importantly, Y5R antagonist decreased the viability of chemotherapy-treated NB cells in the absence of exogenous survival factors, confirming the role of endogenous NPY and BDNF. The saturation of NPY receptors with the autocrine peptide may also explain the modest effect of exogenous NPY in these cells.
The expression of two types of NPY receptors in NB - Y2R and Y5R - raises a question regarding the specificity of their functions. Previously, we have demonstrated that Y2R is constitutively expressed in NB cell lines and tumors, as well as in endothelium within tumor vasculature, and mediates proliferative and angiogenic actions of endogenous NPY.28,29 Here, we demonstrated that Y5R, but not Y2R, is an inducible, pro-survival NPY receptor. Altogether, our results indicate that actions of Y2R and Y5R in NB depend on the cellular context. During exponential growth, proliferative and pro-angiogenic actions of Y2R predominate (Fig. 9A).28,29 However, upon BDNF stimulation and under cellular stress, Y5R expression is induced and its TrkB-dependent and -independent pro-survival functions are activated, while Y2R does not play a major role (Fig. 9B, C). Ultimately, the effects of Y2R and Y5R antagonist on NB growth depend on the balance between tumor cell proliferation and apoptosis. Interestingly, despite their different biological functions, actions of both receptors are mediated by p44/42 MAPK. This could result from Y5R interactions with other receptors, which modify its function, but also from differences in Y2R and Y5R signaling, since Y2R regulates intracellular Ca++ in addition to the decrease in cAMP stimulated by both receptors (Fig. 9).59,60
Figure 9. Proposed mechanisms of BDNF and NPY interactions and functions in NB cells.
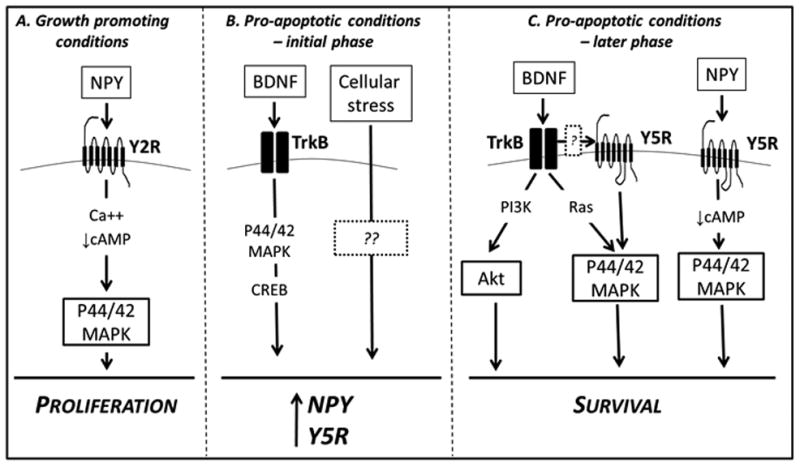
A. Under growth promoting conditions, NPY and Y2R are constitutively expressed in NB cells. This NPY/Y2R autocrine loop stimulates NB cell proliferation via p44/42 MAPK activation. This effect is most likely triggered by signaling events known to mediate Y2R actions – changes in intracellular Ca++ and decrease in cAMP levels. B. BDNF, a known survival factor for NB cells, stimulates the synthesis of NPY and induces expression of its Y5R. This effect is mediated by p44/42 MAPK and CREB. Expression of NPY and its Y5R is further up-regulated under cellular stress, such as serum deprivation or chemotherapy, in a BDNF-independent manner. C. Induction of Y5R expression in pro-apoptotic conditions activates its pro-survival actions. Upon BDNF stimulation, its receptor, TrkB, transactivates Y5R, which augments BDNF-induced p44/42 MAPK activation and its anti-apoptotic effect. TrkB-Y5R interactions do not affect BDNF-induced Akt activation. Y5R, acting via p44/42 MAPK, also mediates the pro-survival effect of NPY. This effect could be initiated by a decrease in cAMP levels, a signaling event triggered upon NPY/Y5R axis activation. Solid boxes – molecules tested in the current study; dashed boxes – unknown factors; no outline – molecules previously implicated in BDNF or NPY signaling in NB.
The clinical relevance of Y5R activation was confirmed by its expression in all surviving NB cells within chemotherapy-treated tumors. This phenomenon may result from up-regulation of Y5R in all tumor cells or selection of the Y5R-positive NB clones. In vivo, blocking Y5R increased apoptosis and inhibited growth of xenografts derived from Y5R-rich chemoresistant CHLA-20 cells.38 This apoptotic effect was in agreement with increased caspase 3/7 activity observed in NB cells treated with Y5R antagonist in vitro. This is in contrast to anti-proliferative and anti-angiogenic effects of Y2R blockade, which also inhibit NB xenograft growth.29 Thus, our in vivo data corroborate the proliferative and pro-survival actions of Y2R and Y5R in NB, respectively. Although in the current study the xenografts were not treated with chemotherapy, cell death occurs in tumors naturally (e.g. due to hypoxia). As shown in Ewing sarcoma, these conditions also up-regulate Y5R.61 Further studies are required to determine if Y5R antagonists, alone or in combination with Y2R antagonist or Trk inhibitors, have a synergistic effect with chemotherapy in vivo. Importantly, Y5R antagonist has already been approved for clinical trials for other disorders.62,63
Overall, we present the first evidence for TrkB-Y5R interactions and their contribution to the anti-apoptotic effect of BDNF. We also demonstrate that Y5R, acting in a BDNF-dependent or -independent manner, is a pro-survival factor for NB cells, up-regulated in pro-apoptotic conditions. Consequently, Y5R blockage increases NB cell death and sensitizes them to chemotherapy. These findings, along with our previous results implicating Y2R as a crucial proliferative and pro-angiogenic factor in NB, underscore the importance of the NPY system in NB biology and validate the peptide and its receptors as therapeutic targets. Importantly, while the NPY/Y2R axis is active in all NB tumors, implicating Y2R as a universal therapeutic target, Y5R may be the most relevant to chemoresistant, refractory NBs that lack an adequate treatment.
Methods
Materials
Recombinant human BDNF was purchased from R&D Systems (Minneapolis, MN); NPY from Bachem (San Carlos, CA); Y5R antagonist, CGP71683, and Y2R antagonist, BIIE0246, from Tocris (Ellisville, MO); Trk inhibitor, K252a and chemotherapeutics from Sigma (St. Louis, MO).
Human NB samples
mRNA and paraffin-embedded sections from 75 NB cases at diagnosis were obtained from Children’s Oncology Group, while 4 paraffin-embedded samples from NB tumors upon induction chemotherapy were collected by Dr. Izycka-Swieszewska in compliance with institutional regulations. Use of these samples was approved by Georgetown University Institutional Review Board.
Cell culture
Human NB cells – SK-N-BE(2), SH-SY5Y were obtained from ATCC (Manassas, VA), SH-SY5Y cells stably transfected with TrkB cDNA (SY5Y/TrkB) from Dr. Brodeur (University of Pennsylvania, Philadelphia, PA)11 and pairs of NB cell lines derived from patients at diagnosis and during induction chemotherapy – SMS-KCN and SMS-KCNR, SMS-KAN and SMS-KANR, CHLA-15 and CHLA-20 - from Dr. Reynolds (Children’s Hospital of Los Angeles, CA).38 Cells were cultured according to the suppliers’ recommendation.
Real time RT-PCR and NPY ELISA were performed as previously described.28,64
Western blot for NPY receptors was performed on membrane proteins isolated as previously described,65 using goat polyclonal anti-Npy2r (My Biosource, Camarillo, CA) and anti-Y5R (Everest Biotech, Ramona, CA) antibodies, while unspecific protein staining (Pierce Reversible Protein Stain Kit for Nitrocellulose Membranes, Thermo, Rockford, IL) served as a loading control. Signaling molecules were detected in whole cell extracts using the following antibodies: mouse monoclonal anti-phospho p44/42 E10, rabbit polyclonal anti-phospho Akt Thr308 and Ser473 (Cell Signaling Technology, Boston, MA) and mouse monoclonal anti-β-actin (Sigma, St. Louis, MO). Densitometry was conducted using ImageJ software.
Y5R-EGFP transfections and live cell imaging
Human Y5R cDNA was cloned into pEGFP-N1 vector (Clontech Laboratories, Mountain View, CA) and transfected into SY5Y/TrkB cells using Lipofectamine 2000 (Life Techonologies, Grand Island, NY). Transfected cells were imaged 24h later with a Zeiss 510 LSM/META confocal microscope during 30 min incubation with BDNF (1ng/ml), with or without 15 min pre-incubation with Y5R or Trk antagonist (10−6M). Fluorescence intensities on the cell membrane and in the sub-membrane areas were measured using ImageJ software. For the analysis, multiple cellular regions with initial membrane Y5R-EGFP localization (membrane/sub-membrane fluorescence ratio ≥ 1.3) were selected.
Cell surface protein immunoprecipitation
SY5Y/TrkB cells were pre-incubated for 24h in serum-free media and treated for 8 min with BDNF (1ng/ml), with or without 15 min pre-treatment with TrkB or Y5R antagonists (10−6M). Then, cell surface proteins were biotynylated and immunoprecipitated using Cell Surface Protein Isolation Kit (Thermo Scientific, Rockford, IL). Y5R in cell lysates and immunoprecipitation eluates was detected by Western blot.
Cell viability and apoptosis assays
Cells plated in 96-well plates were cultured in 1% FBS media for 24h and treated as desired. After 24h, the number of viable cells was measured using MTS-based CellTiter 96®AQueous One Solution Cell Proliferation Assay, while caspase activity was measured using Apo-ONE Homogenous Caspase 3/7 reagent (Promega, Madison, WI).
siRNA transfection
SY5Y/TrkB cells were transfected with 30nM Y2R or negative control siRNA (Life Technologies, Grand Island, NY), as previously described29 or with Y5R siGENOME SMART pool siRNA (Thermo Scientific, Rockford, IL) using Trans IT-TKO (Mirus, Madison, WI). 48h later, knockdown efficiency was tested by Western blot, while cell viability and signaling pathway activation were assessed as above.
NB xenografts
107 of CHLA-20 cells suspended in 0.1ml of Matrigel (BD Biosciences, San Jose, CA) were injected subcutaneously into the dorsal flank of 4–6 weeks old female nude mice. When tumors reached a volume of 100mm3, daily treatment with local injections of Y5R antagonist (10−6M, 0.1ml) was initiated. Mice were euthanized once tumors reached a volume of approximately 1cm3. This study was approved by an Institutional Animal Care and Use Committee of the Georgetown University.
Tissue analyses
Immunohistochemistry was performed using the following rabbit polyclonal antibodies: anti-Y5R (Novus Biologicals, LLC, Littleton, CO), anti-Ki67 (Abcam, Cambridge, MA) and anti-vWF (DAKO, Carpinteria, CA). Cell death was detected by TUNEL (Roche, Indianapolis, IN). All tissues were assessed by a pathologist. Staining area was quantified using ImageJ software.
Statistical analysis
Statistical analyses were performed using SigmaStat®, GraphPad and SPSS software. Between-group comparisons were assessed using one-way or two-way repeated measures ANOVA with post-hoc t-test, independent-samples t-tests or paired-samples t-tests, as appropriate. Significant associations were assessed at an alpha level of 0.05. All experiments were repeated at least three times. Data is presented as mean ± standard errors. Bivariate associations are reported using the Pearson product-moment correlation coefficient.
Supplementary Material
A. Western blot analysis of TrkB expression in SY5Y/TrkB and SK-N-BE(2) cells. B. Time course of p44/42 MAPK activation in SK-N-BE(2) cells cultured for 24h in serum free media and subsequently treated with BDNF (1ng/ml).
SY5Y/TrkB cells were transfected with Y2R or Y5R siRNAs and 48h later the corresponding receptors were detected by Western blot. The efficiency of knockdown was measured by densitometry. PS – unspecific protein staining.
SY5Y/TrkB cells were cultured for 24h in 1% FBS media and then treated with BDNF (1ng/ml), with or without 1h pre-incubation with Trk and Y5R antagonists (both at 10−6M). 2h later, cells were treated with vinblastine (0.55μg/ml) or cisplatin (2.75μg/ml). After 24h, cell viability was assessed by MTS assay.
SK-N-BE(2) cells were cultured for 24h in 1% FBS media and then pre-incubated with Y5R antagonists (10−6M) for 1h followed by 2h incubation with BDNF (1ng/ml). Subsequently, the cells were treated with various chemotherapeutic agents – doxorubicin (0.5μg/ml), etoposide (2.5μg/ml) or vinblastine (1μg/ml). 24h later, cell viability was measured by MTS assay.
Acknowledgments
Human NB samples at diagnosis were provided by the Children’s Oncology Group. The authors thank Dr. Garret Brodeur for providing SY5Y/TrkB transfectants. This work was supported by National Institutes of Health grants: UL1TR000101 (previously UL1RR031975) from the National Center for Advancing Translational Sciences (NCATS), through the Clinical and Translational Science Awards Program (CTSA), 1R01CA123211 and 1R03CA178809 to J. Kitlinska, as well as funding from Children’s Cancer Foundation (Baltimore, MD) to J. Kitlinska, and Georgetown Undergraduate Research Opportunities Program to M. Horton and D. Christian. Experiments were performed with use of the Lombardi Comprehensive Cancer Center Shared Resources (Histopathology & Tissue, Biostatistics & Bioinformatics, Microscopy & Imaging, Animal Model and Tissue Culture) partially supported by NIH/NCI grant P30-CA051008. The authors thank Drs. Aykut Üren, Ruijun Han and Kristi Graves for their critical reading of the manuscript.
Footnotes
Conflict of interest:
The authors declare no conflict of interest.
References
- 1.Maris JM. The biologic basis for neuroblastoma heterogeneity and risk stratification. Curr Opin Pediatr. 2005;17:7–13. doi: 10.1097/01.mop.0000150631.60571.89. [DOI] [PubMed] [Google Scholar]
- 2.Maris JM. Recent advances in neuroblastoma. N Engl J Med. 2010;362:2202–11. doi: 10.1056/NEJMra0804577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park JR, Bagatell R, London WB, Maris JM, Cohn SL, Mattay KM, et al. Children’s Oncology Group’s 2013 blueprint for research: neuroblastoma. Pediatric blood & cancer. 2013;60:985–93. doi: 10.1002/pbc.24433. [DOI] [PubMed] [Google Scholar]
- 4.Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer. 2003;3:203–16. doi: 10.1038/nrc1014. [DOI] [PubMed] [Google Scholar]
- 5.Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007;369:2106–20. doi: 10.1016/S0140-6736(07)60983-0. [DOI] [PubMed] [Google Scholar]
- 6.Thiele CJ, Li Z, McKee AE. On Trk--the TrkB signal transduction pathway is an increasingly important target in cancer biology. Clin Cancer Res. 2009;15:5962–7. doi: 10.1158/1078-0432.CCR-08-0651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brodeur GM, Minturn JE, Ho R, Simpson AM, Iyer R, Varela CR, et al. Trk receptor expression and inhibition in neuroblastomas. Clin Cancer Res. 2009;15:3244–50. doi: 10.1158/1078-0432.CCR-08-1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Han BH, Holtzman DM. BDNF protects the neonatal brain from hypoxic-ischemic injury in vivo via the ERK pathway. J Neurosci. 2000;20:5775–81. doi: 10.1523/JNEUROSCI.20-15-05775.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klocker N, Kermer P, Weishaupt JH, Labes M, Ankerhold R, Bahr M. Brain-derived neurotrophic factor-mediated neuroprotection of adult rat retinal ganglion cells in vivo does not exclusively depend on phosphatidyl-inositol-3′-kinase/protein kinase B signaling. J Neurosci. 2000;20:6962–7. doi: 10.1523/JNEUROSCI.20-18-06962.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Skaper SD, Floreani M, Negro A, Facci L, Giusti P. Neurotrophins rescue cerebellar granule neurons from oxidative stress-mediated apoptotic death: selective involvement of phosphatidylinositol 3-kinase and the mitogen-activated protein kinase pathway. J Neurochem. 1998;70:1859–68. doi: 10.1046/j.1471-4159.1998.70051859.x. [DOI] [PubMed] [Google Scholar]
- 11.Ho R, Eggert A, Hishiki T, Minturn JE, Ikegaki N, Foster P, et al. Resistance to chemotherapy mediated by TrkB in neuroblastomas. Cancer research. 2002;62:6462–6. [PubMed] [Google Scholar]
- 12.Jaboin J, Kim CJ, Kaplan DR, Thiele CJ. Brain-derived neurotrophic factor activation of TrkB protects neuroblastoma cells from chemotherapy-induced apoptosis via phosphatidylinositol 3′-kinase pathway. Cancer research. 2002;62:6756–63. [PubMed] [Google Scholar]
- 13.Li Z, Zhang J, Liu Z, Woo CW, Thiele CJ. Downregulation of Bim by brain-derived neurotrophic factor activation of TrkB protects neuroblastoma cells from paclitaxel but not etoposide or cisplatin-induced cell death. Cell death and differentiation. 2007;14:318–26. doi: 10.1038/sj.cdd.4401983. [DOI] [PubMed] [Google Scholar]
- 14.Jaboin J, Hong A, Kim CJ, Thiele CJ. Cisplatin-induced cytotoxicity is blocked by brain-derived neurotrophic factor activation of TrkB signal transduction path in neuroblastoma. Cancer Lett. 2003;193:109–14. doi: 10.1016/s0304-3835(02)00723-1. [DOI] [PubMed] [Google Scholar]
- 15.Brodeur GM, Nakagawara A, Yamashiro DJ, Ikegaki N, Liu XG, Azar CG, et al. Expression of TrkA, TrkB and TrkC in human neuroblastomas. J Neurooncol. 1997;31:49–55. doi: 10.1023/a:1005729329526. [DOI] [PubMed] [Google Scholar]
- 16.Nakagawara A. Trk receptor tyrosine kinases: a bridge between cancer and neural development. Cancer Lett. 2001;169:107–14. doi: 10.1016/s0304-3835(01)00530-4. [DOI] [PubMed] [Google Scholar]
- 17.Nakagawara A, Azar CG, Scavarda NJ, Brodeur GM. Expression and function of TRK-B and BDNF in human neuroblastomas. Mol Cell Biol. 1994;14:759–67. doi: 10.1128/mcb.14.1.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakagawara A, Brodeur GM. Role of neurotrophins and their receptors in human neuroblastomas: a primary culture study. Eur J Cancer. 1997;33:2050–3. doi: 10.1016/s0959-8049(97)00280-3. [DOI] [PubMed] [Google Scholar]
- 19.Cohen PS, Cooper MJ, Helman LJ, Thiele CJ, Seeger RC, Israel MA. Neuropeptide Y expression in the developing adrenal gland and in childhood neuroblastoma tumors. Cancer research. 1990;50:6055–61. [PubMed] [Google Scholar]
- 20.Dotsch J, Christiansen H, Hanze J, Lampert F, Rascher W. Plasma neuropeptide Y of children with neuroblastoma in relation to stage, age and prognosis, and tissue neuropeptide Y. Regul Pept. 1998;75–76:185–90. doi: 10.1016/s0167-0115(98)00067-6. [DOI] [PubMed] [Google Scholar]
- 21.Kogner P, Bjork O, Theodorsson E. Plasma neuropeptide Y in healthy children: influence of age, anaesthesia and the establishment of an age-adjusted reference interval. Acta Paediatr. 1994;83:423–7. doi: 10.1111/j.1651-2227.1994.tb18134.x. [DOI] [PubMed] [Google Scholar]
- 22.Hansel DE, Eipper BA, Ronnett GV. Neuropeptide Y functions as a neuroproliferative factor. Nature. 2001;410:940–4. doi: 10.1038/35073601. [DOI] [PubMed] [Google Scholar]
- 23.Lee EW, Michalkiewicz M, Kitlinska J, Kalezic I, Switalska H, Yoo P, et al. Neuropeptide Y induces ischemic angiogenesis and restores function of ischemic skeletal muscles. J Clin Invest. 2003;111:1853–62. doi: 10.1172/JCI16929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Movafagh S, Hobson JP, Spiegel S, Kleinman HK, Zukowska Z. Neuropeptide Y induces migration, proliferation, and tube formation of endothelial cells bimodally via Y1, Y2, and Y5 receptors. Faseb J. 2006;20:1924–6. doi: 10.1096/fj.05-4770fje. [DOI] [PubMed] [Google Scholar]
- 25.Pons J, Kitlinska J, Ji H, Lee EW, Zukowska Z. Mitogenic actions of neuropeptide Y in vascular smooth muscle cells: synergetic interactions with the beta-adrenergic system. Can J Physiol Pharmacol. 2003;81:177–85. doi: 10.1139/y02-166. [DOI] [PubMed] [Google Scholar]
- 26.Sheriff S, Ali M, Yahya A, Haider KH, Balasubramaniam A, Amlal H. Neuropeptide Y Y5 receptor promotes cell growth through extracellular signal-regulated kinase signaling and cyclic AMP inhibition in a human breast cancer cell line. Mol Cancer Res. 2010;8:604–14. doi: 10.1158/1541-7786.MCR-09-0301. [DOI] [PubMed] [Google Scholar]
- 27.Tilan JU, Everhart LM, Abe K, Kuo-Bonde L, Chalothorn D, Kitlinska J, et al. Platelet neuropeptide Y is critical for ischemic revascularization in mice. FASEB J. 2013 doi: 10.1096/fj.12-213546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kitlinska J, Abe K, Kuo L, Pons J, Yu M, Li L, et al. Differential effects of neuropeptide Y on the growth and vascularization of neural crest-derived tumors. Cancer research. 2005;65:1719–28. doi: 10.1158/0008-5472.CAN-04-2192. [DOI] [PubMed] [Google Scholar]
- 29.Lu C, Everhart L, Tilan J, Kuo L, Sun CC, Munivenkatappa RB, et al. Neuropeptide Y and its Y2 receptor: potential targets in neuroblastoma therapy. Oncogene. 2010 doi: 10.1038/onc.2010.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Decressac M, Pain S, Chabeauti PY, Frangeul L, Thiriet N, Herzog H, et al. Neuroprotection by neuropeptide Y in cell and animal models of Parkinson’s disease. Neurobiol Aging. 2012;33:2125–37. doi: 10.1016/j.neurobiolaging.2011.06.018. [DOI] [PubMed] [Google Scholar]
- 31.Malva JO, Xapelli S, Baptista S, Valero J, Agasse F, Ferreira R, et al. Multifaces of neuropeptide Y in the brain--neuroprotection, neurogenesis and neuroinflammation. Neuropeptides. 2012;46:299–308. doi: 10.1016/j.npep.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 32.Williams AG, Hargreaves AC, Gunn-Moore FJ, Tavare JM. Stimulation of neuropeptide Y gene expression by brain-derived neurotrophic factor requires both the phospholipase Cgamma and Shc binding sites on its receptor, TrkB. Biochem J. 1998;333 (Pt 3):505–9. doi: 10.1042/bj3330505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wirth MJ, Patz S, Wahle P. Transcellular induction of neuropeptide Y expression by NT4 and BDNF. Proc Natl Acad Sci U S A. 2005;102:3064–9. doi: 10.1073/pnas.0404712102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barnea A, Roberts J. Induction of functional and morphological expression of neuropeptide Y (NPY) in cortical cultures by brain-derived neurotrophic factor (BDNF): evidence for a requirement for extracellular-regulated kinase (ERK)-dependent and ERK-independent mechanisms. Brain Res. 2001;919:57–69. doi: 10.1016/s0006-8993(01)02999-7. [DOI] [PubMed] [Google Scholar]
- 35.Reibel S, Vivien-Roels B, Le BT, Larmet Y, Carnahan J, Marescaux C, et al. Overexpression of neuropeptide Y induced by brain-derived neurotrophic factor in the rat hippocampus is long lasting. The European journal of neuroscience. 2000;12:595–605. doi: 10.1046/j.1460-9568.2000.00941.x. [DOI] [PubMed] [Google Scholar]
- 36.Yoshimura R, Ito K, Endo Y. Differentiation/maturation of neuropeptide Y neurons in the corpus callosum is promoted by brain-derived neurotrophic factor in mouse brain slice cultures. Neurosci Lett. 2009;450:262–5. doi: 10.1016/j.neulet.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 37.Xapelli S, Bernardino L, Ferreira R, Grade S, Silva AP, Salgado JR, et al. Interaction between neuropeptide Y (NPY) and brain-derived neurotrophic factor in NPY-mediated neuroprotection against excitotoxicity: a role for microglia. The European journal of neuroscience. 2008;27:2089–102. doi: 10.1111/j.1460-9568.2008.06172.x. [DOI] [PubMed] [Google Scholar]
- 38.Keshelava N, Seeger RC, Groshen S, Reynolds CP. Drug resistance patterns of human neuroblastoma cell lines derived from patients at different phases of therapy. Cancer research. 1998;58:5396–405. [PubMed] [Google Scholar]
- 39.Delcourt N, Bockaert J, Marin P. GPCR-jacking: from a new route in RTK signalling to a new concept in GPCR activation. Trends Pharmacol Sci. 2007;28:602–7. doi: 10.1016/j.tips.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 40.Pyne NJ, Waters C, Moughal NA, Sambi BS, Pyne S. Receptor tyrosine kinase-GPCR signal complexes. Biochemical Society transactions. 2003;31:1220–5. doi: 10.1042/bst0311220. [DOI] [PubMed] [Google Scholar]
- 41.Schramm A, Schulte JH, Astrahantseff K, Apostolov O, Limpt V, Sieverts H, et al. Biological effects of TrkA and TrkB receptor signaling in neuroblastoma. Cancer Lett. 2005;228:143–53. doi: 10.1016/j.canlet.2005.02.051. [DOI] [PubMed] [Google Scholar]
- 42.Takei N, Sasaoka K, Higuchi H, Endo Y, Hatanaka H. BDNF increases the expression of neuropeptide Y mRNA and promotes differentiation/maturation of neuropeptide Y-positive cultured cortical neurons from embryonic and postnatal rats. Brain Res Mol Brain Res. 1996;37:283–9. doi: 10.1016/0169-328x(95)00299-8. [DOI] [PubMed] [Google Scholar]
- 43.Pandey SC. Anxiety and alcohol abuse disorders: a common role for CREB and its target, the neuropeptide Y gene. Trends Pharmacol Sci. 2003;24:456–60. doi: 10.1016/S0165-6147(03)00226-8. [DOI] [PubMed] [Google Scholar]
- 44.Dudas J, Bitsche M, Schartinger V, Falkeis C, Sprinzl GM, Riechelmann H. Fibroblasts produce brain-derived neurotrophic factor and induce mesenchymal transition of oral tumor cells. Oral Oncol. 2011;47:98–103. doi: 10.1016/j.oraloncology.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilhelm JC, Xu M, Cucoranu D, Chmielewski S, Holmes T, Lau KS, et al. Cooperative roles of BDNF expression in neurons and Schwann cells are modulated by exercise to facilitate nerve regeneration. J Neurosci. 2012;32:5002–9. doi: 10.1523/JNEUROSCI.1411-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zeine R, Salwen HR, Peddinti R, Tian Y, Guerrero L, Yang Q, et al. Presence of cancer-associated fibroblasts inversely correlates with Schwannian stroma in neuroblastoma tumors. Mod Pathol. 2009;22:950–8. doi: 10.1038/modpathol.2009.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flajolet M, Wang Z, Futter M, Shen W, Nuangchamnong N, Bendor J, et al. FGF acts as a co-transmitter through adenosine A(2A) receptor to regulate synaptic plasticity. Nature neuroscience. 2008;11:1402–9. doi: 10.1038/nn.2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garcia-Sainz JA, Romero-Avila MT, del Medina LC. Dissecting how receptor tyrosine kinases modulate G protein-coupled receptor function. European journal of pharmacology. 2010;648:1–5. doi: 10.1016/j.ejphar.2010.08.049. [DOI] [PubMed] [Google Scholar]
- 49.Borroto-Escuela DO, Tarakanov AO, Guidolin D, Ciruela F, Agnati LF, Fuxe K. Moonlighting characteristics of G protein-coupled receptors: focus on receptor heteromers and relevance for neurodegeneration. IUBMB life. 63:463–72. doi: 10.1002/iub.473. [DOI] [PubMed] [Google Scholar]
- 50.Waters CM, Connell MC, Pyne S, Pyne NJ. c-Src is involved in regulating signal transmission from PDGFbeta receptor-GPCR(s) complexes in mammalian cells. Cellular signalling. 2005;17:263–77. doi: 10.1016/j.cellsig.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 51.Rajagopal R, Chen ZY, Lee FS, Chao MV. Transactivation of Trk neurotrophin receptors by G-protein-coupled receptor ligands occurs on intracellular membranes. J Neurosci. 2004;24:6650–8. doi: 10.1523/JNEUROSCI.0010-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci. 2003;4:299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- 53.Lou X, Yano H, Lee F, Chao MV, Farquhar MG. GIPC and GAIP form a complex with TrkA: a putative link between G protein and receptor tyrosine kinase pathways. Molecular biology of the cell. 2001;12:615–27. doi: 10.1091/mbc.12.3.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee FS, Rajagopal R, Chao MV. Distinctive features of Trk neurotrophin receptor transactivation by G protein-coupled receptors. Cytokine & growth factor reviews. 2002;13:11–7. doi: 10.1016/s1359-6101(01)00024-7. [DOI] [PubMed] [Google Scholar]
- 55.Rakhit S, Pyne S, Pyne NJ. Nerve growth factor stimulation of p42/p44 mitogen-activated protein kinase in PC12 cells: role of G(i/o), G protein-coupled receptor kinase 2, beta-arrestin I, and endocytic processing. Molecular pharmacology. 2001;60:63–70. doi: 10.1124/mol.60.1.63. [DOI] [PubMed] [Google Scholar]
- 56.Gehlert DR, Schober DA, Morin M, Berglund MM. Co-expression of neuropeptide Y Y1 and Y5 receptors results in heterodimerization and altered functional properties. Biochem Pharmacol. 2007;74:1652–64. doi: 10.1016/j.bcp.2007.08.017. [DOI] [PubMed] [Google Scholar]
- 57.Pons J, Kitlinska J, Jacques D, Perreault C, Nader M, Everhart L, et al. Interactions of multiple signaling pathways in neuropeptide Y-mediated bimodal vascular smooth muscle cell growth. Can J Physiol Pharmacol. 2008;86:438–48. doi: 10.1139/y08-054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pellieux C, Sauthier T, Domenighetti A, Marsh DJ, Palmiter RD, Brunner HR, et al. Neuropeptide Y (NPY) potentiates phenylephrine-induced mitogen-activated protein kinase activation in primary cardiomyocytes via NPY Y5 receptors. Proc Natl Acad Sci U S A. 2000;97:1595–600. doi: 10.1073/pnas.030533197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pedrazzini T, Pralong F, Grouzmann E. Neuropeptide Y: the universal soldier. Cell Mol Life Sci. 2003;60:350–77. doi: 10.1007/s000180300029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sheriff S, Dayal R, Kasckow J, Regmi A, Chance W, Fischer J, et al. NPY upregulates genes containing cyclic AMP response element in human neuroblastoma cell lines bearing Y1 and Y2 receptors: involvement of CREB. Regul Pept. 1998;75–76:309–18. doi: 10.1016/s0167-0115(98)00083-4. [DOI] [PubMed] [Google Scholar]
- 61.Tilan JU, Lu C, Galli S, Izycka-Swieszewska E, Earnest JP, Shabbir A, et al. Hypoxia shifts activity of neuropeptide Y in Ewing sarcoma from growth-inhibitory to growth-promoting effects. Oncotarget. 2013;4:2487–501. doi: 10.18632/oncotarget.1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Erondu N, Gantz I, Musser B, Suryawanshi S, Mallick M, Addy C, et al. Neuropeptide Y5 receptor antagonism does not induce clinically meaningful weight loss in overweight and obese adults. Cell metabolism. 2006;4:275–82. doi: 10.1016/j.cmet.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 63.Erondu N, Wadden T, Gantz I, Musser B, Nguyen AM, Bays H, et al. Effect of NPY5R antagonist MK-0557 on weight regain after very-low-calorie diet-induced weight loss. Obesity (Silver Spring, Md. 2007;15:895–905. doi: 10.1038/oby.2007.620. [DOI] [PubMed] [Google Scholar]
- 64.Lu C, Tilan JU, Everhart L, Czarnecka M, Soldin SJ, Mendu DR, et al. Dipeptidyl Peptidases as Survival Factors in Ewing Sarcoma Family of Tumors: IMPLICATIONS FOR TUMOR BIOLOGY AND THERAPY. The Journal of biological chemistry. 2011;286:27494–505. doi: 10.1074/jbc.M111.224089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pfeiffer M, Koch T, Schroder H, Klutzny M, Kirscht S, Kreienkamp HJ, et al. Homo- and heterodimerization of somatostatin receptor subtypes. Inactivation of sst(3) receptor function by heterodimerization with sst(2A) The Journal of biological chemistry. 2001;276:14027–36. doi: 10.1074/jbc.M006084200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A. Western blot analysis of TrkB expression in SY5Y/TrkB and SK-N-BE(2) cells. B. Time course of p44/42 MAPK activation in SK-N-BE(2) cells cultured for 24h in serum free media and subsequently treated with BDNF (1ng/ml).
SY5Y/TrkB cells were transfected with Y2R or Y5R siRNAs and 48h later the corresponding receptors were detected by Western blot. The efficiency of knockdown was measured by densitometry. PS – unspecific protein staining.
SY5Y/TrkB cells were cultured for 24h in 1% FBS media and then treated with BDNF (1ng/ml), with or without 1h pre-incubation with Trk and Y5R antagonists (both at 10−6M). 2h later, cells were treated with vinblastine (0.55μg/ml) or cisplatin (2.75μg/ml). After 24h, cell viability was assessed by MTS assay.
SK-N-BE(2) cells were cultured for 24h in 1% FBS media and then pre-incubated with Y5R antagonists (10−6M) for 1h followed by 2h incubation with BDNF (1ng/ml). Subsequently, the cells were treated with various chemotherapeutic agents – doxorubicin (0.5μg/ml), etoposide (2.5μg/ml) or vinblastine (1μg/ml). 24h later, cell viability was measured by MTS assay.