

Abstract
Pseudomonas aeruginosa is an opportunistic pathogen that requires iron to cause infection, but it also must regulate the uptake of iron to avoid iron toxicity. The iron-responsive PrrF1 and PrrF2 small regulatory RNAs (sRNAs) are part of P. aeruginosa's iron regulatory network and affect the expression of at least 50 genes encoding iron-containing proteins. The genes encoding the PrrF1 and PrrF2 sRNAs are encoded in tandem in P. aeruginosa, allowing for the expression of a distinct, heme-responsive sRNA named PrrH that appears to regulate genes involved in heme metabolism. Using a combination of growth, mass spectrometry, and gene expression analysis, we showed that the ΔprrF1,2 mutant, which lacks expression of the PrrF and PrrH sRNAs, is defective for both iron and heme homeostasis. We also identified phuS, encoding a heme binding protein involved in heme acquisition, and vreR, encoding a previously identified regulator of P. aeruginosa virulence genes, as novel targets of prrF-mediated heme regulation. Finally, we showed that the prrF locus encoding the PrrF and PrrH sRNAs is required for P. aeruginosa virulence in a murine model of acute lung infection. Moreover, we showed that inoculation with a ΔprrF1,2 deletion mutant protects against future challenge with wild-type P. aeruginosa. Combined, these data demonstrate that the prrF-encoded sRNAs are critical regulators of P. aeruginosa virulence.
INTRODUCTION
Pseudomonas aeruginosa is a ubiquitous Gram-negative bacterium and versatile opportunistic pathogen. Iron is required for P. aeruginosa virulence (1–6) and is obtained through several mechanisms. In anaerobic environments, iron in its ferrous form is freely diffusible through the outer membrane (OM) and transported into the cytoplasm by the Feo inner membrane transport system (7, 8). However, the insolubility of ferric iron in aerobic environments limits accessibility to this nutrient. Moreover, the sequestration of iron by host proteins creates a substantial barrier to infection (9, 10). To overcome this barrier, P. aeruginosa synthesizes and secretes two siderophores, pyoverdine and pyochelin, which scavenge ferric iron (1–4). P. aeruginosa can also acquire heme, an abundant source of iron in the human host (11). Once internalized, heme is sequestered by the cytosolic PhuS heme chaperone (12). PhuS transfers heme to the iron-regulated HemO heme oxygenase, which degrades heme to biliverdin, releasing carbon monoxide and iron (13, 14). Several studies have shown that iron acquisition is essential for P. aeruginosa virulence (1–5) and biofilm formation (15–17), demonstrating the central role of this element in P. aeruginosa pathogenesis.
Despite its essentiality, iron and heme can be toxic due to their ability to catalyze the formation of reactive oxygen species. Thus, to maintain iron homeostasis, P. aeruginosa must be able to not only acquire iron but also regulate uptake of iron and heme from the environment, as well as iron use and storage. Expression of genes encoding iron and heme uptake systems in P. aeruginosa is therefore regulated by the Fur (ferric uptake regulator) protein (18–20). In iron-replete environments, Fur binds to iron and becomes an active transcriptional repressor, blocking expression of genes for iron acquisition (21). Fur also affects the expression of genes encoding virulence traits, including toxins and extracellular proteases (21–23). Fur regulation in P. aeruginosa mainly occurs through the regulation of cell surface signaling (CSS) systems, which are comprised of a TonB-dependent (OM) receptor, an extracytoplasmic function (ECF) sigma factor, and an anti-sigma factor that regulates the activity of the ECF sigma factor (24). As an example, Fur represses expression of pvdS, encoding an ECF sigma factor that directly activates expression of genes for pyoverdine biosynthesis and uptake, as well as multiple virulence factors (22, 25–27). Binding of ferripyoverdine to the FpvA OM receptor activates the PvdS sigma factor, which is normally sequestered at the inner membrane by its anti-sigma factor, FpvR (28, 29). This paradigm of Fur-mediated regulation via ECF sigma factors extends to the uptake systems for several additional iron sources used by P. aeruginosa (30, 31).
Fur also mediates positive regulation of gene expression via repression of the PrrF small regulatory RNAs (sRNAs) (32). The PrrF sRNAs negatively affect the expression of several genes, presumably by interaction with complementary regions of target mRNAs. Included in the PrrF regulon are genes encoding nonessential iron storage proteins and iron-containing metabolic enzymes (32, 33). Accordingly, PrrF regulation controls how P. aeruginosa uses and stores iron (iron utilization) by sparing this nutrient during growth in iron-limiting environments. The analogous iron-sparing system in Escherichia coli, which is mediated by the RyhB sRNA, was previously shown to promote growth under iron-depleted conditions by increasing intracellular pools of free iron (34). While most PrrF regulatory targets encode iron-containing proteins, it remains unknown if PrrF regulation similarly affects global iron usage or growth in low-iron environments. Regulation by the PrrF sRNAs was previously shown to allow for the production of the Pseudomonas quinolone signal (PQS), a secreted signaling molecule that activates expression of several virulence genes (35, 36). This effect is achieved through repression of the antABC mRNA, encoding iron-containing enzymes that metabolize anthranilate, which is the precursor for PQS (33). Due to their roles in iron homeostasis and PQS production, the PrrF sRNAs are predicted to contribute to pathogenesis of P. aeruginosa.
The PrrF sRNAs are encoded by two highly homologous genes, prrF1 and prrF2, which are located in tandem on the P. aeruginosa genome. This arrangement allows for the expression of a third, heme-responsive sRNA named PrrH (37). While the prrF1 and prrF2 genes are encoded by all pseudomonads, this tandem arrangement is unique to P. aeruginosa and potentially contributes to this species' capacity to cause disease. PrrH is hypothesized to regulate a specific subset of genes involved in heme metabolism (i.e., synthesis and degradation), heme uptake (acquisition of extracellular heme), and other cellular processes via its unique sequence derived from the prrF1-prrF2 intergenic region. By doing so, the PrrH sRNA is likely to contribute to overall heme homeostasis of the cell. Accordingly, we previously performed in silico analysis of the prrF1-prrF2 intergenic region combined with quantitative real-time PCR (qPCR) to identify nirL as a putative target of PrrH (37). The nirSMCFDLGHJEN gene cluster encodes dissimilatory nitrite reductase (NIR; cytochrome cd1) and includes genes for the biosynthesis of heme d1, a prosthetic group of NIR (38). Biosynthesis of heme d1 branches from the central heme biosynthetic pathway with NirF, NirJ, and NirE, catalyzing its production from uroporphyrinogen III. Thus, repression of NIR biosynthesis by PrrH may prioritize the function of the heme biosynthetic pathways under limiting heme concentrations, contributing to heme homeostasis (38).
In this study, we have characterized the effects of deleting the prrF locus (ΔprrF1,2) on multiple aspects of P. aeruginosa iron homeostasis and virulence. We show that the ΔprrF1,2 mutant is defective for iron and heme homeostasis. Additionally, we show that ΔprrF1,2 mutant biofilms are not enhanced by iron in the presence of aminoglycoside antibiotics, a known trait of P. aeruginosa biofilm formation (39, 40). Further, we demonstrate that iron and heme regulation of PrrF and PrrH is conserved in several P. aeruginosa clinical isolates. Moreover, we establish that the prrF locus is absolutely required for virulence in a murine model of acute lung infection. Combined, these results demonstrate the critical role of the PrrF and PrrH sRNAs in P. aeruginosa virulence.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Bacterial strains used in this work are listed in Table 1. Escherichia coli strains were routinely grown in L broth or on L agar plates, and P. aeruginosa strains were maintained in brain heart infusion (BHI) broth or on BHI agar plates. For growth curves, qPCR, mass spectrometry, and Northern blotting, strains were diluted from L broth overnights into M9 minimal medium purchased from Teknova containing 2% glucose and grown for 4 h, and then they were subcultured into fresh M9 for an additional 8 h at 37°C (41). For biofilm analysis, strains were grown in Chelex-treated, dialyzed Trypticase soy broth (DTSB) prepared as previously described (40). Ferric chloride (FeCl3) was added to a final concentration of 100 μM where indicated below. Heme stocks were freshly prepared prior to use in 0.1 N NaOH/20 mM Tris and adjusted to pH 7.0 with HCl, and stock concentration was determined by pyridine hemochrome assay as previously described (42), using the following extinction coefficients: ε = 170.0 mM−1 cm−1 at A418, ε = 17.5 mM−1 cm−1 at A525, and ε = 34.5 mM−1 cm−1 at A555. Heme was immediately diluted into M9 medium to a concentration of 5 μM where indicated below. Tetracycline was used at 60 μg/ml for growth of P. aeruginosa strains carrying the pVLT31 vector or its derivatives as indicated below.
TABLE 1.
Pseudomonas aeruginosa strains used in this study
| Strain | Description | Reference(s) |
|---|---|---|
| PAO1 | Wild-type strain used for mutational analysis in this and previous studies; originally isolated from a human wound in 1955 in Australia | 75 |
| ΔprrF1,2 mutant | Deletion mutant of entire prrF locus in PAO1 | 32 |
| PAO1/pVLT31 | Wild-type PAO1 strain carrying the pVLT31 broad-host-range vector (Tcr) | 33 |
| ΔprrF1,2/pVLT31 mutant | ΔprrF1,2 mutant carrying the pVLT31 broad-host-range vector (Tcr) | 33 |
| ΔprrF1,2/pVLT-prrF1,2 mutant | ΔprrF1,2 mutant carrying the prrF1,2 complementation construct (Tcr) | 32 |
| PA14 | Clinical isolate from 1995 at the Massachusetts General Hospital, Boston, MA; virulent in a variety of plant and animal models of infection | 56 |
| FRD1 | Mucoid clinical isolate in a sputum sample from a cystic fibrosis patient | 57 |
| WR5 | Clinical isolate from a burn patient at Walter Reed Army Hospital, Washington, DC, in 1976; natural toxA mutant isolate; virulent in exptl mouse models | 54, 55 |
Determination of cellular iron content.
Strains were grown overnight in LB, then diluted into M9 minimal medium purchased from Teknova containing 2% glucose and 60 μg/ml of tetracycline, and grown for 4 h to deplete intracellular iron stores. Strains were subcultured into fresh M9 medium for an additional 8 h at 37°C, with or without iron or heme supplementation, and harvested by centrifugation. Harvested bacteria from the secondary cultures were dissolved in 20% nitric acid and boiled overnight at 100°C. Inductively coupled plasma mass spectrometry (ICP-MS) was then used to determine the metal content of the dissolved bacteria (Agilent 7700 ICP-MS; Agilent Technologies). Raw ICP-MS data (μg/liter) were corrected for drift using values of internal controls (indium, scandium, and germanium) that were added to each sample during processing. Corrected values were then normalized to culture density as determined by the absorbance at 600 nm.
Real-time PCR.
Primers and probes used for quantitative real-time PCR (qPCR) are shown in the Table S1 in the supplemental material. RNA was extracted using the Qiagen RNeasy minikit according to manufacturer's directions. A total of 50 ng/μl of RNA was used to generate cDNA with the ImPromII cDNA synthesis kit (Promega) as previously described (37), and cDNA was analyzed using the StepOnePlus instrument (Applied Biosystems) and TaqMan reagents (LifeTechnologies). Standard curves were produced for each primer probe set by analyzing cDNA generated from serial dilutions of RNA and used to determine relative amounts of the RNAs. Relative RNA levels were then normalized to the levels of the oprF mRNA.
Biofilm growth and measurement.
The MBEC (minimal biofilm eradication concentration) assay physiology and genetics biofilm device (formerly known as the Calgary biofilm device; Innovotech) was used to assay biofilm formation and the MBEC of tobramycin against PAO1 and the ΔprrF1,2 mutant as previously described (40). Briefly, 96-well MBEC plates containing DTSB, supplemented with 100 μM FeCl3, were inoculated in duplicate with approximately 1 × 107 CFU ml−1 of bacteria. The plates were covered with the MBEC peg lids and incubated with gentle shaking (65 rpm) at 37°C for 24 h to allow biofilm formation on the pegs. Following incubation, the peg lid was gently washed in phosphate-buffered saline (PBS) to remove nonadherent bacteria and then transferred to a fresh 96-well “challenge plate” with DTSB, with or without 100 μM FeCl3, and various concentrations of tobramycin. Biofilms were incubated with tobramycin for 24 h at 37°C, at which point the pegs were stained with 0.1% crystal violet solution for 10 min and then rinsed in water. After drying, the pegs were destained in 200 μl of 30% acetic acid, and the amount of crystal violet dye released was determined by reading the wells for the optical density at 595 nm (OD595) in a Biotek plate reader.
Isotopic heme labeling studies.
Heme labeling studies were performed as previously described (41, 43). Briefly, heme was prepared from labeled [4-13C]δ-aminolevulinic acid (ALA) according to the method described by Rivera and Walker (44) and the yield calculated by the pyridine hemochrome assay (45). P. aeruginosa strains were grown in M9 medium for 4 h and then diluted into fresh M9 medium supplemented with 5 μM [13C]heme for an additional 8 h. Bilverdin (BVIX) isomers were purified from the culture supernatants and separated and analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) (Waters TQD triple quadrupole mass spectrometer with AQUITY H-class ultraperformance liquid chromatograph [UPLC]) as described previously (43). Fragmentation patterns of the parent ions at 583.21 (12C-BVIX) and 591.21 (13C-BVIX) were analyzed using multiple reaction monitoring (MRM). The fragmentation patterns of the respective BVIX isomers are shown in Scheme S1 in the supplemental material.
Northern blotting.
Northern analysis of the PrrF and PrrH sRNAs was performed as previously described (37), with some modifications. Briefly, 1.5 μg (PrrF1 and PrrF2 probes) or 4 μg (PrrH probe) of total RNA isolated on RNeasy minicolumns was run on a 6% polyacrylamide denaturing (7 M urea) gel and then transferred to a BrightStar membrane (Ambion) using a semidry transfer apparatus. Biotinylated probes complementary to the PrrF1, PrrF2, or PrrH intergenic region were purchased from Integrated DNA Technologies (IDT) and hybridized to the blot overnight at 42°C. The membrane was washed using the Ambion Northern Max low-stringency and high-stringency wash solutions according to the manufacturer's instructions. Detection of the biotinylated probes was carried out using streptavidin alkaline phosphatase (Life Technologies) and visualized by chemiluminescence using CDP-Star (Sigma-Aldrich) according to the manufacturer's instructions.
Mouse infections.
All mouse infections were performed in accordance with the Public Health Service policy on the humane care and use of laboratory animals and with a protocol approved by the University of Maryland Institutional Animal Care and Use Committee. C57BL/6 mice (female, 6 to 8 weeks old) were purchased from Jackson Laboratories. P. aeruginosa strains were grown overnight in L broth with 60 μg/ml of tetracycline and then diluted into fresh L broth with 60 μg/ml of tetracycline and grown for an additional 3 h at 37°C. Cells were harvested by centrifugation and resuspended in PBS, and appropriate dilutions were prepared according to the absorbance of the cultures at 600 nm. Mice were anesthetized by inhalation of isoflurane and were inoculated intranasally (i.n.) with 50 μl of inoculum (n = 5 mice for each inoculum of each bacterial strain). Inocula were also serially diluted and plated on L agar to enumerate the delivered dose. Mice were observed twice daily for survival and disease signs, and moribund mice were euthanized by carbon dioxide inhalation followed by cervical dislocation. Mice that survived infection with the ΔprrF1,2/pVLT31 mutant were challenged with a lethal dose (∼108 CFU) of wild-type PAO1 at 28 days postinfection. At 28 days after the final infection, all surviving mice were euthanized.
RESULTS
The ΔprrF1,2 mutant is defective for iron utilization.
It is established that the ΔprrF1,2 mutant displays increased expression of several genes encoding nonessential iron-containing proteins during iron-depleted growth (32, 33). However, it has not yet been determined how deregulated expression of these genes affects overall iron homeostasis of the ΔprrF1,2 mutant. As an initial assessment of the ability of these strains to maintain iron homeostasis, we performed growth curves for the wild-type PAO1 strain and isogenic ΔprrF1,2 mutant under iron-depleted conditions. The wild-type and ΔprrF1,2 mutant strains were grown for 4 h in M9 minimal medium to restrict intracellular iron stores. Cells were subsequently subcultured into M9 minimal medium with or without either 100 μM iron supplementation, and growth was monitored for 24 h. While the ΔprrF1,2 mutant grew similarly to the wild type under iron-replete conditions, growth of the ΔprrF1,2 mutant was significantly reduced compared to that of the wild type under iron-limiting conditions (Fig. 1A and B). Moreover, this growth defect was complemented by expression of the prrF genes in trans from the previously constructed pVLT-prrF1,2 vector (Fig. 1A and B) (32). Thus, deletion of the prrF locus results in a growth defect under iron-limiting conditions.
FIG 1.

The ΔprrF1,2 mutant is defective for growth in low-iron medium and with heme as a sole iron source. (A to C) The indicated strains were grown for 4 h in M9 minimal medium with 60 μg/ml of tetracycline to deplete intracellular iron stores and diluted into fresh M9 medium with or without FeCl3 (100 μM) or heme (5 μM) supplementation as indicated. Growth was monitored for 24 h in a Bioscreen C instrument. Significant differences in culture density of the wild type and ΔprrF1,2 mutant, as determined by two-tailed Student's t test, are indicated by a horizontal bar flanking the relevant time points. (D to F) The indicated strains were grown for 4 h in M9 minimal medium to deplete intracellular iron stores and then diluted into fresh M9 medium with or without FeCl3 (100 μM) or heme (5 μM) supplementation, grown for an additional 8 h in M9 minimal media as indicated, and subsequently analyzed for iron content by ICP-MS analysis. Error bars indicate the standard deviations of three independent experiments. The indicated P values were obtained by two-tailed Student's t test.
To confirm that the PrrF sRNAs were expressed and active under these growth conditions, we next analyzed expression of the PrrF sRNAs, as well as sodB, a known target of PrrF regulation (32), by qPCR. Our analysis showed that PrrF was expressed under these conditions and that transcription of the PrrF sRNAs was strongly repressed by supplementation of the medium with 100 μM iron (Fig. 2B). Moreover, expression of sodB was significantly derepressed by deletion of the prrF locus, and complementation of the ΔprrF1,2 mutant in trans restored sodB expression to wild-type levels (Fig. 3A). Surprisingly, iron exerted a very modest effect on expression of the PrrF sRNAs when expressed in trans (Fig. 2B). Sequencing of the prrF complementation construct revealed a single nucleotide change four residues upstream of the prrF1 Fur box (see Fig. S1 in the supplemental material), which we presume resulted in the loss of Fur-regulated PrrF expression. Nevertheless, this construct was capable of restoring growth (Fig. 1A) and sodB expression (Fig. 3A) to wild-type levels in the ΔprrF1,2 mutant grown in low-iron media and has been used to complement previously reported phenotypes of the ΔprrF1,2 mutant (32, 33). Thus, our data demonstrate that the PrrF sRNAs are expressed and functional under low-iron conditions and that deletion of the prrF locus results in deregulated expression of genes in the PrrF regulon.
FIG 2.
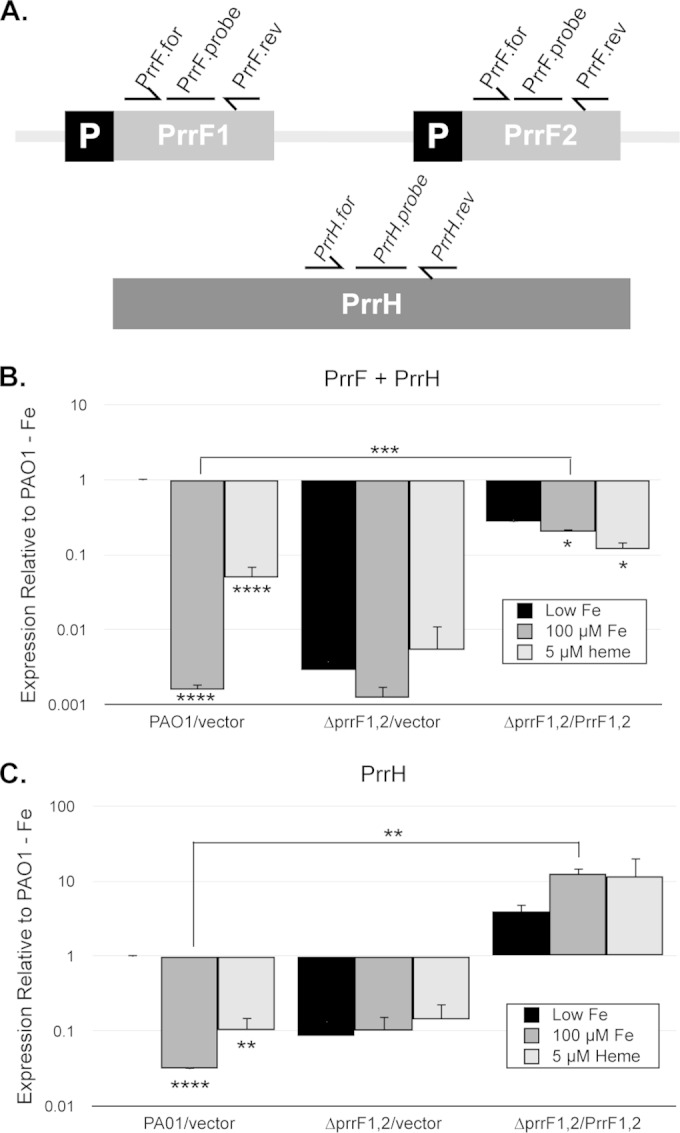
Iron and heme regulation of the PrrF and PrrH sRNAs in M9 media. (A) Map of the prrF locus, showing approximate locations of the PrrF.for and PrrF.rev primers and PrrF TaqMan probe, which detect the PrrF and PrrH sRNAs (B), and the PrrH.for and PrrH.rev primers and PrrH TaqMan Probe, which only detect PrrH (C). (B and C) The indicated strains were grown for 4 h in M9 medium with 60 μg/ml of tetracycline to deplete intracellular iron stores and then subcultured into fresh M9 medium without iron supplementation (black bars), with 100 μM FeCl3 supplementation (dark gray bars), or with 5 μM heme supplementation (light gray bars). Relative expression of PrrF and PrrH combined (B) and of PrrH (C), detected using the primers shown in panel A, was determined by standard curves as described in Materials and Methods, and expression of each sample was normalized to the PAO1/vector low-iron sample. Error bars indicate the standard deviations from three independent experiments. Asterisks indicate the following P values as determined by two-tailed Student's t test when comparing values upon heme or iron supplementation to that of the low-iron sample, or as otherwise indicated: *, P < 0.05; **, P < 0.001; ***, P < 0.0001; ****, P < 0.00001.
FIG 3.

The ΔprrF1,2 mutant shows altered expression of genes involved in heme homeostasis and virulence. (A to E) The indicated strains were grown for 4 h in M9 medium with 60 μg/ml of tetracycline to deplete intracellular iron stores and then subcultured into fresh M9 medium without iron supplementation. Relative expression of sodB (A), phuS (B), vreR (C), vreA (D), and vreI (E) was determined by standard curves as described in Materials and Methods, and expression of each sample was normalized to the PAO1/vector sample. Primers and probes used are shown in the supplemental material. Error bars indicate the standard deviations from three independent experiments. Complementarity between PrrF1 and the sodB mRNA as previously reported (32) and the PrrH unique region (“PrrH IG”) and the phuS mRNA are shown below the graphs in panels A and B, respectively. The translational start site of phuS is underlined. Asterisks indicate the following P value as determined by two-tailed Student's t test when comparing values upon heme or iron supplementation to that of the low-iron sample, or as otherwise indicated: ∧, P < 0.1; *, P < 0.05; **, P < 0.001; ***, P < 0.0001. (F) Map of the vreAIR operon, indicating the approximate location of complementarity with the PrrH unique region (PrrH IG).
Previous studies from Jacques and colleagues demonstrated that the iron-regulated RyhB sRNA of E. coli contributed to growth under iron-depleted conditions by controlling the expression of genes encoding iron-containing proteins, a function referred to as iron sparing (34). The ΔprrF1,2 mutant is similarly defective for expression of genes encoding iron-containing protein (32), yet to date the effects of gene expression changes of the ΔprrF1,2 mutant on growth and iron usage have not been examined. We therefore next sought to determine if the ΔprrF1,2 mutant exhibited changes in total iron content compared to the wild-type strain. Inductively coupled plasma mass spectrometry (ICP-MS) was used to measure intracellular iron levels of the wild type and ΔprrF1,2 mutant grown under iron-starved or iron-replete conditions. These data showed that iron content in the ΔprrF1,2 mutant under iron-depleted conditions was higher than that of the wild-type strain (Fig. 1D), indicating that the ΔprrF1,2 mutant induced expression of its iron uptake systems. In support of this idea, analysis of pyoverdine levels and iron scavenging activity in cell supernatants showed increased siderophore production by the ΔprrF1,2 mutant compared to that of the wild type (see Fig. S2 in the supplemental material). Combined, these data demonstrate that the growth defect of the ΔprrF1,2 mutant under iron-depleted conditions is not due to a defect in iron uptake. Instead, our data indicate that deregulated expression of nonessential iron-containing proteins resulted in the ΔprrF1,2 mutant sensing iron starvation, increased production of siderophores, and the growth defect observed in Fig. 1A.
The ΔprrF1,2 mutant biofilm is not enhanced by subinhibitory levels of tobramycin.
Iron uptake has been shown by several studies to be critical for P. aeruginosa biofilm formation (15, 16, 46), which is an important aspect of P. aeruginosa virulence and antibiotic resistance. Despite its defect in iron homeostasis as demonstrated above, the ΔprrF1,2 mutant was previously shown to form biofilms similar to those of the wild type (16). However, we wanted to determine if defective iron utilization of the ΔprrF1,2 mutant would affect biofilms formed under different growth conditions. For example, aminoglycoside antibiotics have been shown to increase biofilm formation when added to P. aeruginosa cultures at subinhibitory concentrations (39). Moreover, we recently showed that induction of biofilm formation by exposure to tobramycin, an aminoglycoside antibiotic that is often used to treat airway infections in cystic fibrosis patients, is dependent upon iron (40). We therefore determined if altered iron homeostasis in the ΔprrF1,2 mutant would affect the capacity of aminoglycosides to enhance biofilm formation. Biofilms of PAO1 and the ΔprrF1,2 mutant were formed under iron-replete growth conditions and then exposed to various concentrations of tobramycin in either high- or low-iron media. As expected, PAO1 biofilm mass was significantly higher when preformed biofilms were exposed to either 0.5 or 1 μg/ml of tobramycin under high- versus low-iron conditions (Fig. 4). In contrast, the ΔprrF1,2 mutant displayed significantly reduced biofilm mass compared to that of the wild type when the same concentrations of tobramycin were added to iron-replete medium (Fig. 4). Thus, our data suggest that the iron utilization defect of the ΔprrF1,2 mutant reduces the ability of this strain to enhance biofilm formation when exposed to tobramycin.
FIG 4.

The ΔprrF1,2 mutant is defective for antibiotic-induced biofilm formation. PAO1 and ΔprrF1,2 mutant biofilms were allowed to form on MBEC device pegs for 24 h in DTSB supplemented with 100 μM FeCl3. The biofilms were then washed in saline and challenged for 24 h with various concentrations of tobramycin in the presence or absence of 100 μM FeCl3 as indicated. Biofilm formation was then determined as described in Materials and Methods. Error bars show the standard deviations from three independent experiments. Asterisks indicate the following P value as determined by two-tailed Student's t test: **, P < 0.01.
The ΔprrF1,2 mutant is defective for heme homeostasis.
The tandem organization of the prrF1 and prrF2 genes in P. aeruginosa allows for the expression of a unique, heme-responsive sRNA named PrrH (37). The PrrH sRNA is predicted to contribute to heme homeostasis by regulating a distinct subset of genes involved in heme metabolism and uptake. Thus, it is possible that the ΔprrF1,2 mutant is defective for growth in the presence of heme as the sole iron source. To test this idea, we analyzed growth of the wild-type and ΔprrF1,2 mutant strains in the presence of 5 μM heme. These data showed that heme supplementation was not able to completely suppress the growth defect of the ΔprrF1,2 mutant in low iron (Fig. 1C). However, ICP-MS analysis demonstrated that the iron content of the ΔprrF1,2 mutant grown in the presence of heme is increased compared to that of the wild-type strain (Fig. 1F). These data suggest that the growth defect of the ΔprrF1,2 mutant under these conditions is not due to an inability to take up and degrade extracellular heme as an iron source but is instead due to other aspects of heme homeostasis.
To more directly test the idea that heme homeostasis was altered in the ΔprrF1,2 mutant, we analyzed heme oxygenase activity of this strain using liquid chromatography-tandem mass spectrometry (LC-MS/MS) as previously described (43). This analysis is performed by extracting biliverdin IX isomers (BVIX), the breakdown products of heme by heme oxygenase, from the supernatants of P. aeruginosa cultures. BVIX isomers are then separated by LC, allowing spectral analysis of the BVIX isomers as a function of retention time. Furthermore, by providing cells with 13C-labeled heme as an extracellular iron source, MS/MS fragmentation can differentiate between BVIX resulting from degradation of extracellularly acquired (13C-BVIX) versus intracellularly synthesized (12C-BVIX) heme. Using this technique, it was previously shown that the iron-regulated HemO of P. aeruginosa degrades extracellular heme to BVIXδ and BVIXβ, while a second, non-iron-regulated heme oxygenase, BphO, degrades intracellular heme to BVIXα (see Scheme S1 in the supplemental material) (47). Similar results were obtained in the current study when we analyzed the PAO1 strain grown in 5 μM 13C-labeled heme: BphO predominantly degraded endogenous heme (blue trace) to BVIXα, while HemO degraded exogenous heme (red trace) to BVIXβ and BVIδ (Fig. 5A). The ΔprrF1,2 mutant also turned over extracellular heme via the catalytic action of HemO (Fig. 5B), indicating that this mutant is capable of using heme as an iron source, a conclusion that is consistent with our ICP-MS analysis in Fig. 1F. However, the ΔprrF1,2 mutant showed very little degradation of endogenous heme to BVIXα (Fig. 5B). qPCR analysis showed similar levels of bphO mRNA in the ΔprrF1,2 mutant and the wild type grown in 5 μM heme (see Fig. S3 in the supplemental material). Thus, decreased levels of BVIXα in the ΔprrF1,2 mutant were not due to PrrF- or PrrH-mediated repression of bphO.
FIG 5.

Heme metabolism is altered in the ΔprrF1,2 mutant. MS/MS fragmentation of BVIX isomers extracted from the culture supernatants of PAO1 (A) and the ΔprrF1,2 mutant (B) grown for 8 h in M9 minimal medium supplemented with 5 μM [13C]heme. LC-MS/MS was performed as described in Materials and Methods with multiple reaction monitoring.
Next, we determined if altered heme metabolism in the ΔprrF1,2 mutant was likely due to loss of the PrrH sRNA. For this analysis, qPCR primers and probes were designed within the prrF1-prrF2 intergenic region to specifically detect the PrrH sRNA transcript (Fig. 2A). Consistent with earlier studies (37), qPCR analysis demonstrated that PrrH was expressed under low-iron conditions (Fig. 2C). Our analysis also showed that expression of PrrH was significantly reduced (10-fold) by the addition of as little as 5 μM heme (Fig. 2C). However, similar to what was observed for PrrF in Fig. 2B, expression of PrrH from the prrF complementation construct was not repressed by either iron or heme (Fig. 2C). Thus, while PrrH expression is restored in the complemented strain grown in low iron, heme and iron regulation of PrrH is defective in this strain. Notably, since expression of PrrH is repressed by 5 μM heme in PAO1, it is unlikely that reduced BphO heme oxygenase activity in the ΔprrF1,2 mutant when grown in the presence of heme, as shown in Fig. 5B, is due to the loss of PrrH. However, the PrrF sRNAs are still expressed under this condition (Fig. 2B). Thus, it is possible that loss of the PrrF sRNAs is responsible for altered heme degradation by the ΔprrF1,2 mutant, potentially via their impact on overall iron homeostasis.
The ΔprrF1,2 mutant shows altered expression of genes for heme acquisition and virulence.
The ability to respond to extracellular heme via either the PrrF and/or PrrH sRNAs is likely to provide a competitive advantage in environments where heme is a potential iron source. We therefore sought to determine if the ΔprrF1,2 mutant showed altered expression of genes involved in heme uptake. Previous studies demonstrated that expression of phuS, which controls the flux of heme through the HemO heme oxygenase in PAO1, is regulated by heme (43). To determine if the sRNAs encoded by the prrF locus might be responsible for this regulation, we analyzed expression of phuS in PAO1 and the ΔprrF1,2 mutant. Our analysis shows that expression of phuS is significantly increased in the ΔprrF1,2 mutant compared to that in the wild type and is restored to wild-type levels in the complemented strain (Fig. 3B), indicating that one of the prrF-encoded sRNAs has a negative effect on phuS expression. While no complementarity was identified between the phuS mRNA and either of the PrrF sRNAs, analysis of the 5′ untranslated region (UTR) of phuS revealed a short, imperfect region of complementarity with the PrrH unique sequence (Fig. 3B). Thus, our data are consistent with a model in which heme modulates phuS mRNA levels via the PrrH sRNA.
Heme regulation via the PrrF and/or PrrH sRNAs may also provide a signaling pathway for the induction of virulence genes during infection. To explore this hypothesis, we used the updated TargetRNA2 algorithm (48) to search for potential PrrH-specific targets with links to virulence. This analysis identified an extensive region of complementarity between the PrrH unique sequence and the translational start site and coding sequence of the vreR mRNA (Fig. 3F). The vreR gene encodes an anti-sigma factor of the Vre extracytoplasmic function (ECF) cell surface signaling (CSS) system, which was previously shown to be required for P. aeruginosa virulence (49). To test the idea that vreR was regulated by either PrrF or PrrH, we assayed vreR expression by qPCR in PAO1 and the ΔprrF1,2 mutant. Similarly to the case with phuS, deletion of the prrF locus resulted in significantly increased levels of vreR (3-fold) (Fig. 3C). While vreR expression was not completely restored to wild-type levels in the complemented ΔprrF1,2 mutant, expression of vreR in this strain was not statistically different from that of the wild-type strain (Fig. 3C). Thus, our data indicate that vreR expression is negatively affected by expression of either the PrrF or PrrH sRNAs.
Previous studies showed that the VreA ECF receptor, the VreI sigma factor, and the VreR anti-sigma factor are encoded by a single operon (Fig. 3F) (50). The region of complementarity identified between the PrrH intergenic region and vreR is located downstream of the vreA and vreI genes. We therefore analyzed expression of vreA and vreI to determine if prrF1,2 deletion had a discoordinate effect on this operon. Our analysis showed that deletion of the prrF locus did not result in significantly increased expression of either vreA or vreI (Fig. 3D and E), indicating that prrF1,2 deletion specifically affected the expression of vreR. Combined, these data indicate that deletion of the prrF locus, potentially through the function of the PrrH sRNA, affects the expression of genes required for virulence in P. aeruginosa.
The ΔprrF1,2 mutant is attenuated for virulence in an acute mouse lung infection model.
Combined with previous studies, this report demonstrates that the prrF-encoded sRNAs contribute to iron and heme homeostasis of P. aeruginosa. Our studies also indicate that the RNAs encoded by the prrF locus contribute to virulence gene expression via PQS (33) and VreR (this study). We therefore assayed the ΔprrF1,2 mutant for its ability to cause lung infection in an intranasal (i.n.) murine infection model. C57BL/6 mice (n = 5) were inoculated with either the wild-type PAO1 vector control, the ΔprrF1,2 mutant vector control, or the complemented ΔprrF1,2 mutant. All mice inoculated with 108 CFU of either the wild type or the complemented mutant succumbed to lung infection within 4 days of inoculation (Fig. 6A and C). In contrast, all ΔprrF1,2 mutant-infected mice survived the entire 28-day course of the experiment (Fig. 6B). Thus, the prrF locus of P. aeruginosa is required for virulence in this model of acute mouse lung infection.
FIG 6.

The prrF locus is required for acute murine lung infection. (A to C) Eight-week old C57BL/6 mice were inoculated intranasally with 106, 107, or 108 CFU of wild-type PAO1 with vector (A), the ΔprrF1,2 mutant with vector (B), or the complemented ΔprrF1,2 mutant (C). Five mice received each inoculum of each of the indicated P. aeruginosa strains. Mice were monitored for survival as described in Materials and Methods. (D) C57BL/6 mice that survived infection with 108 CFU of the ΔprrF1,2 mutant (n = 5) were challenged with 108 CFU of the wild-type PAO1 strain (black squares) and monitored for survival as described in Materials and Methods. White squares indicate historical data obtained with naive C57BL/6 mice inoculated with 108 CFU of PAO1.
We next tested whether inoculation of mice with the ΔprrF1,2 mutant generated a protective immune response. Mice that received and survived the initial 108 CFU inoculum of the ΔprrF1,2 mutant (Fig. 6B) were subsequently inoculated i.n. with 108 CFU of wild-type PAO1. All mice primarily challenged with the ΔprrF1,2 mutant survived this subsequent lethal challenge (Fig. 6D, black squares), showing that intranasal inoculation of mice with the PAO1 ΔprrF1,2 mutant generated a protective immune response against the wild-type PAO1 strain. Therefore, the ΔprrF1,2 mutant presents a potential candidate for future vaccine development.
Iron and heme regulation of the prrF-encoded sRNAs is conserved in clinical isolates of P. aeruginosa.
Numerous studies have demonstrated the plasticity of the P. aeruginosa genome, resulting in highly variable expression of virulence factors in different strains (51–53). We therefore determined the ability of several P. aeruginosa laboratory strains and clinical isolates to express and regulate the PrrF and PrrH sRNAs in response to iron and heme. Included in our analysis were PA14 and WR5, which are both wound isolates (54–56), and FRD1, a mucoid strain isolated from the lung of a cystic fibrosis patient (57). Northern analysis demonstrated that the PrrF1, PrrF2, and PrrH sRNAs are expressed by each of these strains when grown in M9 minimal media, although PrrH expression was barely apparent in strain WR5 (Fig. 7A). We also assayed iron- and heme-regulated expression of the PrrF and PrrH sRNAs in each of these isolates by qPCR. Supplementation of the PAO1, PA14, and FRD1 strains with either 100 μM FeCl3 or 5 μM heme resulted in significant repression of PrrH expression (Fig. 7B and C). In contrast, heme supplementation of WR5 cultures had no effect on PrrH expression, and iron repression of PrrH in strain WR5 was less substantial than that of other strains analyzed in this study (Fig. 7B and C). Consistent with our Northern blot analysis, PrrH also appeared to be expressed at lower levels in the WR5 strain grown in low iron than in PAO1 and other isolates that we tested, although this difference was not statistically significant (Fig. 7C). Overall, our results indicate that expression of the PrrF and PrrH sRNAs is conserved across P. aeruginosa, although the mechanisms that guide iron and heme regulation of these sRNAs are somewhat variable.
FIG 7.
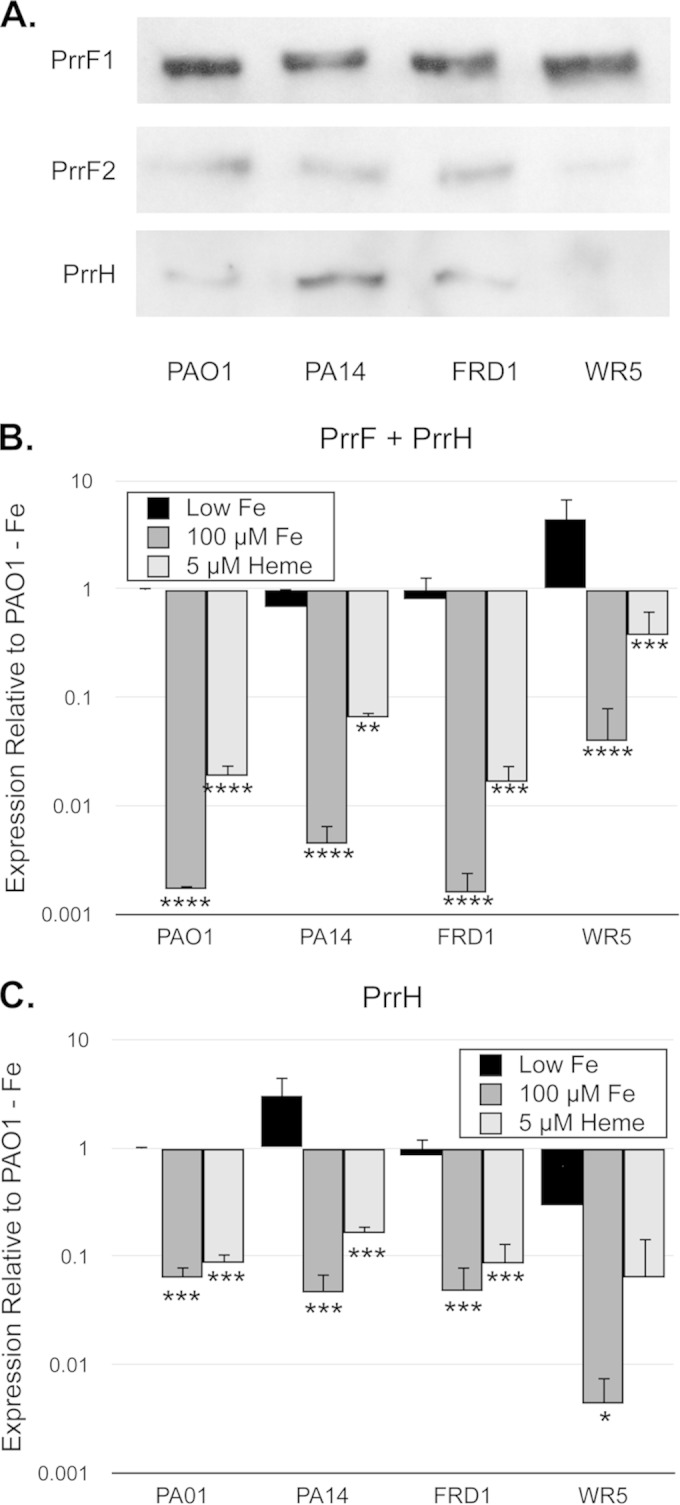
The PrrF and PrrH sRNAs are expressed by multiple clinical isolates of P. aeruginosa. (A) The indicated strains were grown for 4 h in M9 medium to deplete intracellular iron stores and then subcultured into fresh M9 medium without iron supplementation and grown for an additional 8 h. RNA was analyzed by Northern blotting as described in Materials and Methods to detect the PrrF1, PrrF2, and PrrH sRNAs. (B and C) The indicated strains were grown for 4 h in M9 medium to deplete intracellular iron stores and then subcultured into fresh M9 medium without iron supplementation (black bars) or with supplementation of 100 μM FeCl3 (dark gray bars) or 5 μM heme (light gray bars). Relative expression of PrrF and PrrH combined (B) and PrrH (C), detected using the primers shown in Fig. 2A, was determined by standard curves as described in Materials and Methods, and expression of each sample was normalized to the PAO1 low-iron sample. Error bars indicate the standard deviations from three independent experiments. Asterisks indicate the following P values as determined by two-tailed Student's t test when comparing values upon heme or iron supplementation to that of the low-iron sample: *, P < 0.05; **, P < 0.001; ***, P < 0.0001; ****, P < 0.00001.
DISCUSSION
Several studies have demonstrated the essentiality of iron for pathogenesis of P. aeruginosa (1–5). While iron acquisition plays a critical role in maintaining iron homeostasis of P. aeruginosa, appropriate utilization of this element is also essential for bacterial survival, as evidenced by the broad conservation of iron regulatory factors. Iron-responsive regulators in bacteria have largely been shown to contribute to iron homeostasis by one of two mechanisms: repressing the expression of iron uptake systems under iron-replete conditions and reducing the production of iron-containing proteins in iron-limiting environments. The latter of these functions is mediated in many bacterial species by iron-responsive sRNAs (32, 34, 58–65), and in some pathogenic species, these iron-regulated sRNAs affect the expression of virulence traits (33, 63, 64, 66, 67). Here, we show that deletion of the locus encoding the iron-responsive PrrF sRNAs of P. aeruginosa causes defects of iron and heme homeostasis, alters biofilm formation, affects virulence gene expression, and, most strikingly, attenuates virulence. Combined with a previous study demonstrating the role of the PrrF sRNAs in production of the PQS quorum sensing molecule (33), these results demonstrate the central role of the prrF locus in the physiology and pathogenesis of P. aeruginosa.
A recent report demonstrated that a mutant for the iron-regulated RyhB sRNA of uropathogenic E. coli (UPEC) was defective for bladder colonization in a murine model of urinary tract infection (68). Notably, the ryhB mutant of E. coli was also shown in this study to be defective for siderophore production, a known contributor to UPEC virulence (69). As previously shown with nonpathogenic E. coli (70), the UPEC ΔryhB mutant displayed decreased expression of shiA, encoding a permease for shikimate, a required substrate of enterobactin biosynthesis. Moreover, this study also showed that genes encoding biosynthetic enzymes for the hydroxamate siderophore aerobactin were downregulated in the ΔryhB mutant (68). Thus, the RyhB sRNA of UPEC contributes to siderophore production by multiple mechanisms, likely promoting colonization and survival during urinary tract infection. These findings are in stark contrast to those for the P. aeruginosa ΔprrF1,2 mutant, which is not defective for siderophore production (see Fig. S2 in the supplemental material). Moreover, we show that the ΔprrF1,2 mutant is capable of utilizing heme as an iron source (Fig. 5B). Thus, our data indicate that defects in iron uptake, via either siderophore or heme acquisition pathways, are not the source of virulence attenuation in the P. aeruginosa ΔprrF1,2 mutant.
Iron is a known requirement for P. aeruginosa biofilm formation (16, 40, 46). Results of a previous study indicated that either active uptake of chelated iron (through siderophores or ferric citrate) or intracellular iron levels were responsible for this dependency (16). This same study established that the ΔprrF1,2 mutant formed biofilms similar to those of the wild-type strain. Thus, our finding that the ΔprrF1,2 mutant shows reduced biofilm formation compared to that of the wild type in the presence of iron and subinhibitory concentrations of tobramycin is intriguing. While subinhibitory concentrations of tobramycin and other aminoglycosides are known to induce biofilm formation (39), the underlying mechanism for this phenomenon remains unclear. We previously showed that iron supplementation is required for this phenomenon (40). Our data here suggest that appropriate iron utilization, mediated by the PrrF sRNAs, may also play a role in tobramycin-induced biofilm formation. The PrrF sRNAs are also known to mediate production of the PQS quorum sensing molecule, which itself contributes to multicellular behavior (33). Thus, the inability of the ΔprrF1,2 mutant biofilm to respond to iron may be the result of pleiotropic effects on iron utilization, PQS biosynthesis, and potentially other unknown factors. More studies into the formation and structure of the PAO1 and ΔprrF1,2 mutant biofilms in the presence of subinhibitory concentrations of aminoglycosides will be required to fully understand the basis for this phenotype.
A notable characteristic of P. aeruginosa is the heterogeneity of genome sequences from different clinical and environmental isolates. In many cases, this genetic variation results in altered expression of virulence factors or a loss altogether of genes encoding certain virulence determinants (51–53). To date, all sequenced isolates of P. aeruginosa possess the tandem arrangement of the prrF genes (71), indicating that the region encoding both the PrrF and PrrH sRNAs is part of the core genome of this species. In this study, we present additional evidence that iron and heme regulation of the PrrF and PrrH sRNAs is conserved across several clinical isolates of P. aeruginosa. Moreover, we recently showed that the PrrF and PrrH sRNAs are maintained and expressed by a variety of isolates of P. aeruginosa during CF lung infection (41). Combined, these data support the hypothesis that the unique arrangement of the P. aeruginosa prrF genes is a critical asset during infection of the human host.
One of the most fascinating aspects of the PrrF sRNAs in P. aeruginosa is the tandem arrangement of the prrF1 and prrF2 genes, allowing for expression of the heme-responsive PrrH sRNA (37). Heme is an abundant source of iron in the human host, and strains that are capable of using and responding to this nutrient are likely to exhibit a competitive advantage during infection. It is therefore predicted that heme regulated expression of PrrH and PrrF contributes to heme homeostasis and, potentially, the pathogenic capacity of this organism. In support of this hypothesis, the present study demonstrates that the ΔprrF1,2 mutant is altered for heme metabolism; specifically, our data indicate that the BphO heme oxygenase, which degrades endogenously synthesized heme, is expressed yet relatively inactive in the ΔprrF1,2 mutant (Fig. 5B; see also Fig. S3 in the supplemental material). However, the mechanism underlying this phenotype is not yet known. One possibility is that the iron starvation phenotype of the ΔprrF1,2 mutant, resulting from increased production of iron-containing proteins under iron-depleted conditions, precludes endogenous heme biosynthesis. Alternatively, altered heme metabolism in the ΔprrF1,2 mutant may be due to changes in the production of distinct factors required for endogenous heme biosynthesis and/or degradation. Future studies into how the ΔprrF1,2 mutant responds to changes in heme availability on a genomic scale will be key to distinguishing between these possibilities.
We previously showed that the prrF locus allows for heme-regulated expression of the nirL gene, potentially due to a region of complementarity identified between the unique region of the PrrH sRNA and the 5′ end of the nirL mRNA (37). In silico analysis in the current study identified additional mRNAs that shared complementarity with the PrrH unique sequence: vreR, encoding a regulator of virulence gene expression (49), and phuS, encoding a heme-responsive regulator of heme acquisition and iron homeostasis (43). Our data further showed that the ΔprrF1,2 mutant exhibits increased levels of the vreR and phuS mRNAs, suggesting that the PrrH sRNA is capable of interacting with and modulating the expression of these genes. Notably, deletion of the prrF locus had no significant effect on the expression levels of vreA and vreI, which are cotranscribed with vreR (50). Combined with the finding that the unique region of PrrH shares complementarity with a region of the vreAIR operon immediately upstream of vreR, these data support the model that PrrH is responsible for prrF-mediated regulation of vreR. However, more studies are clearly required to determine the mechanism by which the sRNAs encoded by the prrF locus affect expression of each of these genes, as well as whether this regulation occurs by a direct or indirect mechanism.
One complicating factor of our studies was the finding that iron and heme regulation of the PrrF and PrrH sRNAs expressed from the pVLT-prrF1,2 complementation construct was diminished (Fig. 2B and C). Sequencing analysis of the complementation construct identified a single nucleotide substitution immediately upstream of the prrF1 Fur binding site, within the −35 region of the prrF1 promoter (see Fig. S1 in the supplemental material), which is presumably the cause of deregulated PrrF and PrrH expression. While this construct complements many of the previously described phenotypes of the ΔprrF1,2 mutant (32, 33), as well as those described in this report, it is clear that more refined genetic studies will be necessary for teasing apart the individual regulatory roles of the PrrF and PrrH sRNAs during pathogenesis. Unfortunately, subsequent attempts at cloning the prrF locus have resulted in similar promoter mutations, and clones lacking the native prrF promoters carry mutations within the prrF genes. We are therefore pursuing alternative strategies to assess the individual functions of the PrrF and PrrH sRNAs.
In spite of these issues, the current prrF expression construct was able to complement the virulence defect of the ΔprrF1,2 mutant, demonstrating the requirement for at least one of the prrF-encoded sRNAs in virulence. Even more striking, our studies show that intranasal inoculation of mice with the ΔprrF1,2 mutant generated a protective immune response, presenting this strain as a potential candidate for future vaccine development in vulnerable populations. To date, our studies indicate that virulence attenuation of the ΔprrF1,2 mutant may be due to any one or a combination of the following phenotypes: reduced PQS production, loss of iron homeostasis, loss of heme homeostasis via deregulated expression of nirL and/or phuS, and/or deregulated expression of the Vre regulon. Because of the potential multifactorial role of the PrrF and PrrH sRNAs in regulating virulence, developing resistance to a PrrF and/or PrrH inhibitor would likely require mutations in both the PrrF and PrrH sRNAs and several of the target mRNAs they regulate. Thus, this strategy may raise the bar for infecting strains to develop resistance, potentially reducing future burdens of antimicrobial-resistant P. aeruginosa. Delivery of antisense oligonucleotides (AS-ODNs) via anionic liposomes has been successful in several bacterial species (72–74) and may provide a novel means for targeting sRNAs, such as PrrF and PrrH, to block virulence. Future work determining how each of the ΔprrF1,2 mutant phenotypes contributes to P. aeruginosa virulence attenuation will be critical for the development of AS-ODN molecules that effectively block pathogenesis.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Michael Schurr for sharing the FRD1 mucoid cystic fibrosis isolate and to Mike Vasil for the WR5 acute wound isolate. We also thank Lauren Hittle for helpful discussions about heme regulation and metabolism in P. aeruginosa.
This work was supported by NIH-NIAID grant AI089776 and start-up funding from the University of Maryland School of Pharmacy (to A.G.O.-S.) and NIH grant AI102883 (to A.W.).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02707-14.
REFERENCES
- 1.Meyer JM, Neely A, Stintzi A, Georges C, Holder IA. 1996. Pyoverdine is essential for virulence of Pseudomonas aeruginosa. Infect Immun 64:518–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Takase H, Nitanai H, Hoshino K, Otani T. 2000. Impact of siderophore production on Pseudomonas aeruginosa infections in immunosuppressed mice. Infect Immun 68:1834–1839. doi: 10.1128/IAI.68.4.1834-1839.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nadal Jimenez P, Koch G, Papaioannou E, Wahjudi M, Krzeslak J, Coenye T, Cool RH, Quax WJ. 2010. Role of PvdQ in Pseudomonas aeruginosa virulence under iron-limiting conditions. Microbiology 156:49–59. doi: 10.1099/mic.0.030973-0. [DOI] [PubMed] [Google Scholar]
- 4.Xiong YQ, Vasil ML, Johnson Z, Ochsner UA, Bayer AS. 2000. The oxygen- and iron-dependent sigma factor pvdS of Pseudomonas aeruginosa is an important virulence factor in experimental infective endocarditis. J Infect Dis 181:1020–1026. doi: 10.1086/315338. [DOI] [PubMed] [Google Scholar]
- 5.Takase H, Nitanai H, Hoshino K, Otani T. 2000. Requirement of the Pseudomonas aeruginosa tonB gene for high-affinity iron acquisition and infection. Infect Immun 68:4498–4504. doi: 10.1128/IAI.68.8.4498-4504.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cox CD. 1982. Effect of pyochelin on the virulence of Pseudomonas aeruginosa. Infect Immun 36:17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y, Wilks JC, Danhorn T, Ramos I, Croal L, Newman DK. 2011. Phenazine-1-carboxylic acid promotes bacterial biofilm development via ferrous iron acquisition. J Bacteriol 193:3606–3617. doi: 10.1128/JB.00396-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marshall B, Stintzi A, Gilmour C, Meyer JM, Poole K. 2009. Citrate-mediated iron uptake in Pseudomonas aeruginosa: involvement of the citrate-inducible FecA receptor and the FeoB ferrous iron transporter. Microbiology 155:305–315. doi: 10.1099/mic.0.023531-0. [DOI] [PubMed] [Google Scholar]
- 9.Otto BR, Verweij-van Vught AM, MacLaren DM. 1992. Transferrins and heme-compounds as iron sources for pathogenic bacteria. Crit Rev Microbiol 18:217–233. doi: 10.3109/10408419209114559. [DOI] [PubMed] [Google Scholar]
- 10.Nairz M, Schroll A, Sonnweber T, Weiss G. 2010. The struggle for iron—a metal at the host-pathogen interface. Cell Microbiol 12:1691–1702. doi: 10.1111/j.1462-5822.2010.01529.x. [DOI] [PubMed] [Google Scholar]
- 11.Ochsner UA, Johnson Z, Vasil ML. 2000. Genetics and regulation of two distinct haem-uptake systems, phu and has, in Pseudomonas aeruginosa. Microbiology 146(Part 1):185–198. [DOI] [PubMed] [Google Scholar]
- 12.Kaur AP, Lansky IB, Wilks A. 2009. The role of the cytoplasmic heme-binding protein (PhuS) of Pseudomonas aeruginosa in intracellular heme trafficking and iron homeostasis. J Biol Chem 284:56–66. doi: 10.1074/jbc.M806068200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lansky IB, Lukat-Rodgers GS, Block D, Rodgers KR, Ratliff M, Wilks A. 2006. The cytoplasmic heme-binding protein (PhuS) from the heme uptake system of Pseudomonas aeruginosa is an intracellular heme-trafficking protein to the delta-regioselective heme oxygenase. J Biol Chem 281:13652–13662. doi: 10.1074/jbc.M600824200. [DOI] [PubMed] [Google Scholar]
- 14.Ratliff M, Zhu W, Deshmukh R, Wilks A, Stojiljkovic I. 2001. Homologues of neisserial heme oxygenase in gram-negative bacteria: degradation of heme by the product of the pigA gene of Pseudomonas aeruginosa. J Bacteriol 183:6394–6403. doi: 10.1128/JB.183.21.6394-6403.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patriquin GM, Banin E, Gilmour C, Tuchman R, Greenberg EP, Poole K. 2008. Influence of quorum sensing and iron on twitching motility and biofilm formation in Pseudomonas aeruginosa. J Bacteriol 190:662–671. doi: 10.1128/JB.01473-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Banin E, Vasil ML, Greenberg EP. 2005. Iron and Pseudomonas aeruginosa biofilm formation. Proc Natl Acad Sci U S A 102:11076–11081. doi: 10.1073/pnas.0504266102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh PK, Parsek MR, Greenberg EP, Welsh MJ. 2002. A component of innate immunity prevents bacterial biofilm development. Nature 417:552–555. doi: 10.1038/417552a. [DOI] [PubMed] [Google Scholar]
- 18.Hassett DJ, Sokol PA, Howell ML, Ma JF, Schweizer HT, Ochsner U, Vasil ML. 1996. Ferric uptake regulator (Fur) mutants of Pseudomonas aeruginosa demonstrate defective siderophore-mediated iron uptake, altered aerobic growth, and decreased superoxide dismutase and catalase activities. J Bacteriol 178:3996–4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ochsner UA, Vasil AI, Johnson Z, Vasil ML. 1999. Pseudomonas aeruginosa fur overlaps with a gene encoding a novel outer membrane lipoprotein, OmlA. J Bacteriol 181:1099–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong SM, Mekalanos JJ. 2000. Genetic footprinting with mariner-based transposition in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 97:10191–10196. doi: 10.1073/pnas.97.18.10191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ochsner UA, Vasil AI, Vasil ML. 1995. Role of the ferric uptake regulator of Pseudomonas aeruginosa in the regulation of siderophores and exotoxin A expression: purification and activity on iron-regulated promoters. J Bacteriol 177:7194–7201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilderman PJ, Vasil AI, Johnson Z, Wilson MJ, Cunliffe HE, Lamont IL, Vasil ML. 2001. Characterization of an endoprotease (PrpL) encoded by a PvdS-regulated gene in Pseudomonas aeruginosa. Infect Immun 69:5385–5394. doi: 10.1128/IAI.69.9.5385-5394.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ochsner UA, Vasil ML. 1996. Gene repression by the ferric uptake regulator in Pseudomonas aeruginosa: cycle selection of iron-regulated genes. Proc Natl Acad Sci U S A 93:4409–4414. doi: 10.1073/pnas.93.9.4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Llamas MA, Imperi F, Visca P, Lamont IL. 2014. Cell-surface signaling in Pseudomonas: stress responses, iron transport, and pathogenicity. FEMS Microbiol Rev 38:569–597. doi: 10.1111/1574-6976.12078. [DOI] [PubMed] [Google Scholar]
- 25.Wilson MJ, Lamont IL. 2000. Characterization of an ECF Sigma Factor Protein from Pseudomonas aeruginosa. Biochem Biophys Res Commun 273:578–583. doi: 10.1006/bbrc.2000.2996. [DOI] [PubMed] [Google Scholar]
- 26.Ochsner UA, Johnson Z, Lamont IL, Cunliffe HE, Vasil ML. 1996. Exotoxin A production in Pseudomonas aeruginosa requires the iron-regulated pvdS gene encoding an alternative sigma factor. Mol Microbiol 21:1019–1028. doi: 10.1046/j.1365-2958.1996.481425.x. [DOI] [PubMed] [Google Scholar]
- 27.Cunliffe HE, Merriman TR, Lamont IL. 1995. Cloning and characterization of pvdS, a gene required for pyoverdine synthesis in Pseudomonas aeruginosa: PvdS is probably an alternative sigma factor. J Bacteriol 177:2744–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beare PA, For RJ, Martin LW, Lamont IL. 2003. Siderophore-mediated cell signaling in Pseudomonas aeruginosa: divergent pathways regulate virulence factor production and siderophore receptor synthesis. Mol Microbiol 47:195–207. [DOI] [PubMed] [Google Scholar]
- 29.Lamont IL, Beare PA, Ochsner U, Vasil AI, Vasil ML. 2002. Siderophore-mediated signaling regulates virulence factor production in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 99:7072–7077. doi: 10.1073/pnas.092016999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Llamas MA, Mooij MJ, Sparrius M, Vandenbroucke-Grauls CM, Ratledge C, Bitter W. 2008. Characterization of five novel Pseudomonas aeruginosa cell-surface signalling systems. Mol Microbiol 67:458–472. doi: 10.1111/j.1365-2958.2007.06061.x. [DOI] [PubMed] [Google Scholar]
- 31.Llamas MA, Sparrius M, Kloet R, Jimenez CR, Vandenbroucke-Grauls C, Bitter W. 2006. The heterologous siderophores ferrioxamine B and ferrichrome activate signaling pathways in Pseudomonas aeruginosa. J Bacteriol 188:1882–1891. doi: 10.1128/JB.188.5.1882-1891.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilderman PJ, Sowa NA, FitzGerald DJ, FitzGerald PC, Gottesman S, Ochsner UA, Vasil ML. 2004. Identification of tandem duplicate regulatory small RNAs in Pseudomonas aeruginosa involved in iron homeostasis. Proc Natl Acad Sci U S A 101:9792–9797. doi: 10.1073/pnas.0403423101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oglesby AG, Farrow JM III, Lee JH, Tomaras AP, Greenberg EP, Pesci EC, Vasil ML. 2008. The influence of iron on Pseudomonas aeruginosa physiology: a regulatory link between iron and quorum sensing. J Biol Chem 283:15558–15567. doi: 10.1074/jbc.M707840200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jacques JF, Jang S, Prevost K, Desnoyers G, Desmarais M, Imlay J, Masse E. 2006. RyhB small RNA modulates the free intracellular iron pool and is essential for normal growth during iron limitation in Escherichia coli. Mol Microbiol 62:1181–1190. doi: 10.1111/j.1365-2958.2006.05439.x. [DOI] [PubMed] [Google Scholar]
- 35.Déziel E, Gopalan S, Tampakaki AP, Lepine F, Padfield KE, Saucier M, Xiao G, Rahme LG. 2005. The contribution of MvfR to Pseudomonas aeruginosa pathogenesis and quorum sensing circuitry regulation: multiple quorum sensing-regulated genes are modulated without affecting lasRI, rhlRI or the production of N-acyl-L-homoserine lactones. Mol Microbiol 55:998–1014. doi: 10.1111/j.1365-2958.2004.04448.x. [DOI] [PubMed] [Google Scholar]
- 36.Calfee MW, Coleman JP, Pesci EC. 2001. Interference with Pseudomonas quinolone signal synthesis inhibits virulence factor expression by Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 98:11633–11637. doi: 10.1073/pnas.201328498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oglesby-Sherrouse AG, Vasil ML. 2010. Characterization of a heme-regulated non-coding RNA encoded by the prrF locus of Pseudomonas aeruginosa. PLoS One 5:e9930. doi: 10.1371/journal.pone.0009930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kawasaki S, Arai H, Kodama T, Igarashi Y. 1997. Gene cluster for dissimilatory nitrite reductase (nir) from Pseudomonas aeruginosa: sequencing and identification of a locus for heme d1 biosynthesis. J Bacteriol 179:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoffman LR, D'Argenio DA, MacCoss MJ, Zhang Z, Jones RA, Miller SI. 2005. Aminoglycoside antibiotics induce bacterial biofilm formation. Nature 436:1171–1175. doi: 10.1038/nature03912. [DOI] [PubMed] [Google Scholar]
- 40.Oglesby-Sherrouse AG, Djapgne L, Nguyen AT, Vasil AI, Vasil ML. 2014. The complex interplay of iron, biofilm formation, and mucoidy affecting antimicrobial resistance of Pseudomonas aeruginosa. Pathog Dis 70:307–320. doi: 10.1111/2049-632X.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nguyen AT, O'Neill MJ, Watts AM, Robson CL, Lamont IL, Wilks A, Oglesby-Sherrouse AG. 2014. Adaptation of iron homeostasis pathways by a Pseudomonas aeruginosa pyoverdine mutant in the cystic fibrosis lung. J Bacteriol 196:2265–2276. doi: 10.1128/JB.01491-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sinclair PR, Gorman N, Jacobs JM. 2001. Measurement of heme concentration. Curr Protoc Toxicol Chapter 8:Unit 8.3. doi: 10.1002/0471140856.tx0803s00. [DOI] [PubMed] [Google Scholar]
- 43.O'Neill MJ, Wilks A. 2013. The P. aeruginosa heme binding protein PhuS is a heme oxygenase titratable regulator of heme uptake. ACS Chem Biol 8:1794–1802. doi: 10.1021/cb400165b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rivera M, Walker FA. 1995. Biosynthetic preparation of isotopically labeled heme. Anal Biochem 230:295–302. doi: 10.1006/abio.1995.1477. [DOI] [PubMed] [Google Scholar]
- 45.Teale FW. 1959. Cleavage of the haem-protein link by acid methylethylketone. Biochim Biophys Acta 35:543. doi: 10.1016/0006-3002(59)90407-X. [DOI] [PubMed] [Google Scholar]
- 46.Kaneko Y, Thoendel M, Olakanmi O, Britigan BE, Singh PK. 2007. The transition metal gallium disrupts Pseudomonas aeruginosa iron metabolism and has antimicrobial and antibiofilm activity. J Clin Invest 117:877–888. doi: 10.1172/JCI30783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barker KD, Barkovits K, Wilks A. 2012. Metabolic flux of extracellular heme uptake in Pseudomonas aeruginosa is driven by the iron-regulated heme oxygenase (HemO). J Biol Chem 287:18342–18350. doi: 10.1074/jbc.M112.359265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kery MB, Feldman M, Livny J, Tjaden B. 2014. TargetRNA2: identifying targets of small regulatory RNAs in bacteria. Nucleic Acids Res 42:W124–W129. doi: 10.1093/nar/gku317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Llamas MA, van der Sar A, Chu BC, Sparrius M, Vogel HJ, Bitter W. 2009. A novel extracytoplasmic function (ECF) sigma factor regulates virulence in Pseudomonas aeruginosa. PLoS Pathog 5:e1000572. doi: 10.1371/journal.ppat.1000572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Faure LM, Llamas MA, Bastiaansen KC, de Bentzmann S, Bigot S. 2013. Phosphate starvation relayed by PhoB activates the expression of the Pseudomonas aeruginosa sigmavreI ECF factor and its target genes. Microbiology 159:1315–1327. doi: 10.1099/mic.0.067645-0. [DOI] [PubMed] [Google Scholar]
- 51.Shen K, Sayeed S, Antalis P, Gladitz J, Ahmed A, Dice B, Janto B, Dopico R, Keefe R, Hayes J, Johnson S, Yu S, Ehrlich N, Jocz J, Kropp L, Wong R, Wadowsky RM, Slifkin M, Preston RA, Erdos G, Post JC, Ehrlich GD, Hu FZ. 2006. Extensive genomic plasticity in Pseudomonas aeruginosa revealed by identification and distribution studies of novel genes among clinical isolates. Infect Immun 74:5272–5283. doi: 10.1128/IAI.00546-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ernst RK, D'Argenio DA, Ichikawa JK, Bangera MG, Selgrade S, Burns JL, Hiatt P, McCoy K, Brittnacher M, Kas A, Spencer DH, Olson MV, Ramsey BW, Lory S, Miller SI. 2003. Genome mosaicism is conserved but not unique in Pseudomonas aeruginosa isolates from the airways of young children with cystic fibrosis. Environ Microbiol 5:1341–1349. doi: 10.1111/j.1462-2920.2003.00518.x. [DOI] [PubMed] [Google Scholar]
- 53.Spencer DH, Kas A, Smith EE, Raymond CK, Sims EH, Hastings M, Burns JL, Kaul R, Olson MV. 2003. Whole-genome sequence variation among multiple isolates of Pseudomonas aeruginosa. J Bacteriol 185:1316–1325. doi: 10.1128/JB.185.4.1316-1325.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bjorn MJ, Vasil ML, Sadoff JC, Iglewski BH. 1977. Incidence of exotoxin production by Pseudomonas species. Infect Immun 16:362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pavlovskis OR, Pollack M, Callahan LT III, Iglewski BH. 1977. Passive protection by antitoxin in experimental Pseudomonas aeruginosa burn infections. Infect Immun 18:596–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rahme LG, Stevens EJ, Wolfort SF, Shao J, Tompkins RG, Ausubel FM. 1995. Common virulence factors for bacterial pathogenicity in plants and animals. Science 268:1899–1902. doi: 10.1126/science.7604262. [DOI] [PubMed] [Google Scholar]
- 57.Ohman DE, Chakrabarty AM. 1981. Genetic mapping of chromosomal determinants for the production of the exopolysaccharide alginate in a Pseudomonas aeruginosa cystic fibrosis isolate. Infect Immun 33:142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mellin JR, Goswami S, Grogan S, Tjaden B, Genco CA. 2007. A novel fur- and iron-regulated small RNA, NrrF, is required for indirect fur-mediated regulation of the sdhA and sdhC genes in Neisseria meningitidis. J Bacteriol 189:3686–3694. doi: 10.1128/JB.01890-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jung YS, Kwon YM. 2008. Small RNA ArrF regulates the expression of sodB and feSII genes in Azotobacter vinelandii. Curr Microbiol 57:593–597. doi: 10.1007/s00284-008-9248-z. [DOI] [PubMed] [Google Scholar]
- 60.Deng Z, Meng X, Su S, Liu Z, Ji X, Zhang Y, Zhao X, Wang X, Yang R, Han Y. 2012. Two sRNA RyhB homologs from Yersinia pestis biovar microtus expressed in vivo have differential Hfq-dependent stability. Res Microbiol 163:413–418. doi: 10.1016/j.resmic.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 61.Kim JN, Kwon YM. 2013. Genetic and phenotypic characterization of the RyhB regulon in Salmonella Typhimurium. Microbiol Res 168:41–49. doi: 10.1016/j.micres.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 62.Leclerc JM, Dozois CM, Daigle F. 2013. Role of the Salmonella enterica serovar Typhi Fur regulator and small RNAs RfrA and RfrB in iron homeostasis and interaction with host cells. Microbiology 159:591–602. doi: 10.1099/mic.0.064329-0. [DOI] [PubMed] [Google Scholar]
- 63.Murphy ER, Payne SM. 2007. RyhB, an iron-responsive small RNA molecule, regulates Shigella dysenteriae virulence. Infect Immun 75:3470–3477. doi: 10.1128/IAI.00112-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mey AR, Craig SA, Payne SM. 2005. Characterization of Vibrio cholerae RyhB: the RyhB regulon and role of ryhB in biofilm formation. Infect Immun 73:5706–5719. doi: 10.1128/IAI.73.9.5706-5719.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Massé E, Vanderpool CK, Gottesman S. 2005. Effect of RyhB small RNA on global iron use in Escherichia coli. J Bacteriol 187:6962–6971. doi: 10.1128/JB.187.20.6962-6971.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Oglesby AG, Murphy ER, Iyer VR, Payne SM. 2005. Fur regulates acid resistance in Shigella flexneri via RyhB and ydeP. Mol Microbiol 58:1354–1367. doi: 10.1111/j.1365-2958.2005.04920.x. [DOI] [PubMed] [Google Scholar]
- 67.Kim JN, Kwon YM. 2013. Identification of target transcripts regulated by small RNA RyhB homologs in Salmonella: RyhB-2 regulates motility phenotype. Microbiol Res 168:621–629. doi: 10.1016/j.micres.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 68.Porcheron G, Habib R, Houle S, Caza M, Lepine F, Daigle F, Masse E, Dozois CM. 2014. The small RNA RyhB contributes to siderophore production and virulence of uropathogenic Escherichia coli. InfectI Immun 82:5056–5068. doi: 10.1128/IAI.02287-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Garcia EC, Brumbaugh AR, Mobley HL. 2011. Redundancy and specificity of Escherichia coli iron acquisition systems during urinary tract infection. Infect Immun 79:1225–1235. doi: 10.1128/IAI.01222-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Prévost K, Salvail H, Desnoyers G, Jacques JF, Phaneuf E, Masse E. 2007. The small RNA RyhB activates the translation of shiA mRNA encoding a permease of shikimate, a compound involved in siderophore synthesis. Mol Microbiol 64:1260–1273. doi: 10.1111/j.1365-2958.2007.05733.x. [DOI] [PubMed] [Google Scholar]
- 71.Winsor GL, Lam DK, Fleming L, Lo R, Whiteside MD, Yu NY, Hancock RE, Brinkman FS. 2011. Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res 39:D596–D600. doi: 10.1093/nar/gkq869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Meng J, Wang H, Hou Z, Chen T, Fu J, Ma X, He G, Xue X, Jia M, Luo X. 2009. Novel anion liposome-encapsulated antisense oligonucleotide restores susceptibility of methicillin-resistant Staphylococcus aureus and rescues mice from lethal sepsis by targeting mecA. Antimicrob Agents Chemother 53:2871–2878. doi: 10.1128/AAC.01542-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Harth G, Zamecnik PC, Tang JY, Tabatadze D, Horwitz MA. 2000. Treatment of Mycobacterium tuberculosis with antisense oligonucleotides to glutamine synthetase mRNA inhibits glutamine synthetase activity, formation of the poly-L-glutamate/glutamine cell wall structure, and bacterial replication. Proc Natl Acad Sci U S A 97:418–423. doi: 10.1073/pnas.97.1.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fillion P, Desjardins A, Sayasith K, Lagace J. 2001. Encapsulation of DNA in negatively charged liposomes and inhibition of bacterial gene expression with fluid liposome-encapsulated antisense oligonucleotides. Biochim Biophys Acta 1515:44–54. doi: 10.1016/S0005-2736(01)00392-3. [DOI] [PubMed] [Google Scholar]
- 75.Martin DW, Holloway BW, Deretic V. 1993. Characterization of a locus determining the mucoid status of Pseudomonas aeruginosa: AlgU shows sequence similarities with a Bacillus sigma factor. J Bacteriol 175:1153–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.