

Abstract
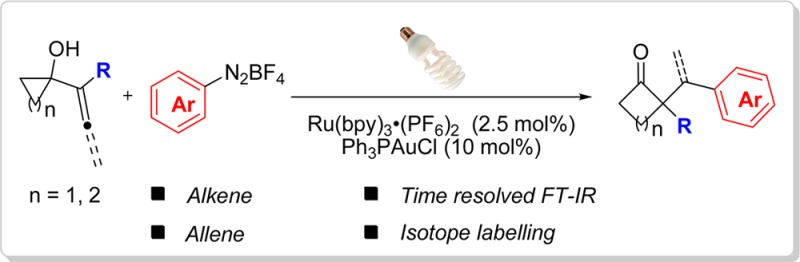
A combination of visible light photocatalysis and gold catalysis is applied to a ring expansion–oxidative arylation reaction. The reaction provides an entry into functionalized cyclic ketones from the coupling reaction of alkenyl and allenyl cycloalkanols with aryl diazonium salts. A mechanism involving generation of an electrophilic gold(III)–aryl intermediate is proposed on the basis of mechanistic studies, including time-resolved FT-IR spectroscopy.
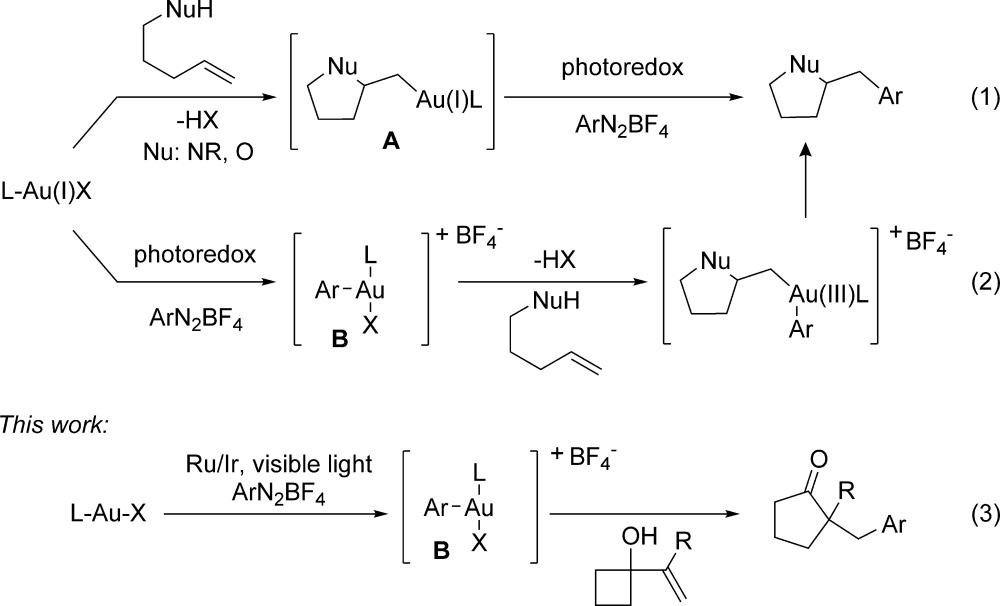
Homogeneous gold catalysis has received great attention in the past decade.1 The majority of reports rely on the π-acidity of either gold(I) or gold(III) complexes to activate multiple bonds toward nucleophilic attack, followed by protodeauration. On the other hand, gold complexes, in combination with a strong oxidant, have been shown to catalyze oxidative functionalization reactions of unactivated alkenes.2,3 In general, these reactions are proposed to proceed through alkylgold(III) intermediates that undergo coupling with nucleophiles such as arylsilanes and boronic acids.4 Recently, Glorius and co-workers proposed that aryl radicals, generated by photoredox reaction of aryldiazonium salts, could be employed to access analogous gold(III) species through oxidation of an alkylgold(I) intermediate (A, eq 1).5
 |
1 |
As an alternative, we envisioned that these reactions might proceed through a pathway in which photoredox catalysis6 occurs first, providing an entry into a cationic arylgold(III) intermediate (B) that subsequently reacts with the alkene to induce intramolecular heteroarylation (eq 2). Inspired by recent reports from the Gaunt laboratory,7 we hypothesized that gold(III) species B might be a potent electrophile, capable of engaging π-bonds and ultimately delivering a formal aryl electrophile (eq 3). Herein, we describe application of this hypothesis to the development of a gold-catalyzed arylative ring expansion and experiments distinguishing these mechanistic paths.
On the basis of this hypothesis, we reacted vinylcyclopropanol (1a) and phenyldiazonium 2a with 10% triphenylphosphinegold(I) and 5% Ru(bpy)3(PF6)2 under visible light.8 Under these conditions, 1a underwent ring expansion9 and coupling10 with 2a to afford cyclopentanone 3aa in 87% yield (Scheme 1, 2a). Only a trace amount of product was detected by NMR when the reaction was conducted in the absence of light, photocatalyst, or gold catalyst (Table S3). The reaction proceeded smoothly with diazonium salts having methyl substituted at each position of the aryl ring, giving good yields of desired products 2b–d. However, a more electron-rich substituent on the aryl ring gave low yield with the use of Ph3PAuCl as catalyst. A strong electron-withdrawing group on the ligand of the gold catalyst improved the yield of desired product (2e, method A vs C). The arylation of electron-deficient diazoniums also proceeded efficiently in the presence of Ru catalyst (2f–j). In the case of Ir(ppy)3-catalyzed arylation of 2d, a lower yield of desired product was obtained in 12 h, and a significant amount of diazonium salt was recovered (2d, method A vs B). However, improved results were obtained when the Ir photocatalyst was employed to couple electron-poor diazonium salts (2f and 2j, method A vs B).
Scheme 1. Reaction of 1a with Diazonium Compounds.
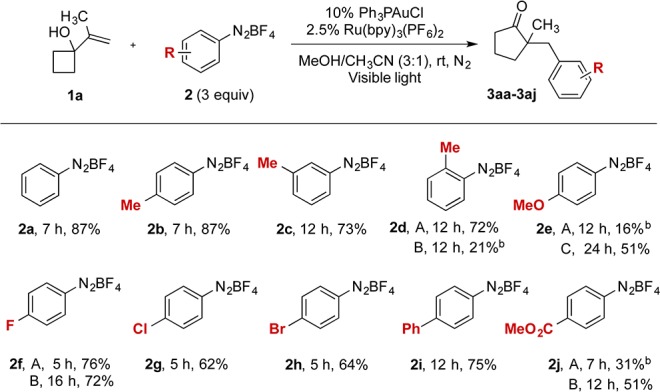
Unless noted otherwise, method A was used: visible light, 1a (0.2 mmol), ArN2BF4 (3 equiv), Ph3PAuCl (10 mol%), Ru(bpy)3(PF6)2 (2.5 mol%), degassed MeOH/CH3CN (3:1, 5 mL) under N2 at rt. Method B was used: Ph3PAuCl (10 mol%), Ir(ppy)3 (2.5 mol%). Method C was used: (4-CF3Ph)3PAuCl (10 mol%), Ru(bpy)3(PF6)2 (2.5 mol%). Isolated yields are given.
Yields were calculated on the basis of 1H NMR using internal standard.
The reaction proceeded efficiently with various alkyl-substituted alkenes (Table 1, entries 1–3). A substrate bearing a protected amine group was tolerated (entry 4). The Ru catalyst was inefficient in promoting the reaction of aryl-substituted substrates, and product 3faa was obtained in low yield with significant decomposition (entry 5); however, use of the Ir catalyst improved the reaction (entries 6–8).11 The reactivity was decreased when a sterically hindered aryl group was used (entry 9). Finally, good yields of cyclobutanones were obtained from the ring expansion/coupling reaction of vinylcyclopropanols (entries 10 and 11).
Table 1. Scope Studies of Vinylcyclobutanols and Vinylcyclopropanolsa.

| entry | n | R | Ar | time (h) | 3 (% yield) |
|---|---|---|---|---|---|
| 1 | 2 | n-hexyl (1b) | Ph | 6 | 3ba (90) |
| 2 | 2 | isopropyl (1c) | Ph | 6 | 3ca (81) |
| 3 | 2 | Bn (1d) | Ph | 6 | 3da (83) |
| 4 | 2 | PhthN(CH2)3 (1e) | Ph | 4 | 3ea (80) |
| 5 | 2 | Ph (1f) | Ph | 24 | 3fa (34) |
| 6b | 2 | Ph (1f) | 4-FPh | 24 | 3fb (71) |
| 7b | 2 | 4-ClPh (1g) | 4-FPh | 24 | 3ga (67) |
| 8b | 2 | 4-MePh (1h) | 4-FPh | 6 | 3ha (80) |
| 9b | 2 | 2-MePh (1i) | 4-FPh | 24 | 3ia (32) |
| 10 | 1 | Ph (1j) | Ph | 15 | 3ja (67) |
| 11 | 1 | Bn (1k) | Ph | 6 | 3ka (51) |
Unless noted otherwise, method A was used (see Scheme 1, footnote a). Isolated yields are given.
Method D was used: 4-FPhN2BF4 (4 equiv), Ir(ppy)3 (2.5 mol%), and (4-CF3Ph)3PAuCl (10 mol%).
To probe the mechanism of this dual catalysis, time-resolved FT-IR spectroscopy in the attenuated total reflection (ATR) mode was employed to detect intermediates under reaction conditions. Catalysis in methanol/acetonitrile (3/1) solution containing reactants 1a and 2b, catalyst (CF3Ph)3PAuCl, and sensitizer Ru(bpy)3(PF6)2 held in a liquid cell was initiated with a 458 nm laser pulse (150 mW, Ar ion laser) of 12 s duration. Arrival of the laser pulse was synchronized with the acquisition of rapid-scan FT-IR spectra over a period of 96 s. Reaction of the phenyl radical generated by photoactivation of the sensitizer was monitored by characteristic infrared absorptions of the reactants and final cyclopentanone product, which were identified by recording spectra of authentic samples (compound 2b, 1583 cm–1, νAr(C–C); compound 1a, 1467 cm–1, ν(C=C) and 1250 cm–1, ν(C–O); additional spectral data are presented in Table S5 and Figure S7). The kinetic behavior for the formation of the final product 3ab (phenyl C–C mode at 1515 cm–1) displays an induction period relative to the kinetics for consumption of the diazonium compound and suggests the formation of an intermediate involved in the rate-limiting step. As shown in Figure 1, the absorption band at 1488 cm–1 was identified to be associated with this intermediate, increasing upon onset of the light pulse and decreasing concurrently with the growth of the final product. The kinetic relevancy of this intermediate was directly demonstrated by the matching rates of its consumption and the rise of the product band at 1515 cm–1. The absorption band at 1730 cm–1, which is the ν(C=O) mode of the expanded ring, is also associated with this intermediate. By simultaneous least-squares fitting of the temporal behavior of the 1250 cm–1 reactant, the 1488 cm–1 intermediate, and the 1515 cm–1 final product bands according to the model
kinetic parameters were obtained, on the basis of which the temporal behavior of the 1730 cm–1 C=O stretch absorption was simulated. As shown in Figure S8, excellent agreement of the simulated curve with the experimental absorbance at 1730 cm–1 was obtained. The result confirms that both the intermediate and the final product absorb at 1730 cm–1. Notably, these bands were not observed when the experiment was carried out in the absence of gold (Figure S7), strongly suggesting that the gold catalyst is required for ring expansion to occur. These observations are consistent with a kinetically relevant intermediate that requires decomposition of the diazonium compound for its formation and contains an expanded five-membered-ring ketone (1730 cm–1), but has yet to undergo reductive elimination to form the C–C bond (methyl-substituted analogue of 11, see Scheme 2).
Figure 1.
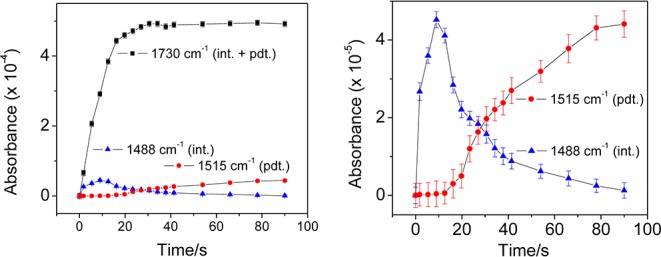
Temporal behavior of intermediate (1488 cm–1) and final product (1515 cm–1) of arylation–ring expansion of vinylcyclobutanol in liquid solution monitored by rapid-scan ATR FT-IR spectroscopy. The panel on the right shows the temporal behavior on an expanded absorbance scale for clarity. The band at 1730 cm–1 is the C=O mode of the expanded ring.
Scheme 2. Proposed Mechanism for Photoredox-Promoted, Gold-Catalyzed Ring Expansion–Arylation Reaction.
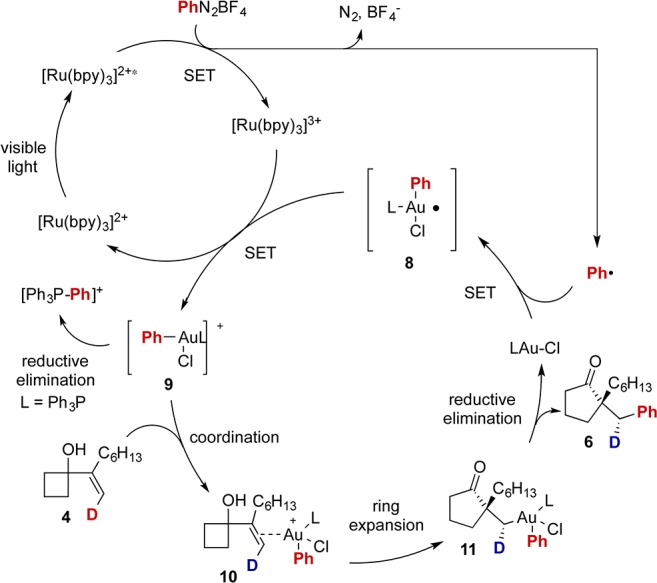
The stereochemical course of the gold-catalyzed ring expansion–arylation reaction was probed with deuterium-labeled alkenes (eq 4). In the presence of gold catalysts, the ring expansion reaction occurred with high levels of stereospecificity.12 Moreover, the observed stereochemistry of the corresponding products is consistent with the previously observed anti-migration13 and carbon–carbon bond formation with stereochemical retention from alkylgold(III)–phenyl species.4a
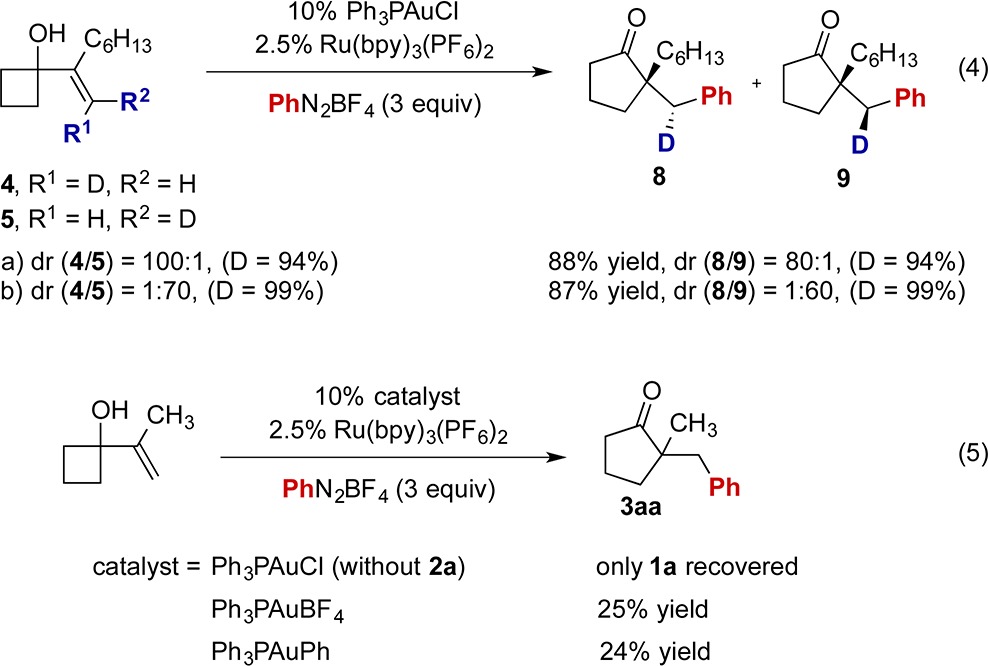
The current ring expansion–arylation reaction appears to begin with gold(III) or gold(I) coordination with the alkene. However, Ph3PAuCl on its own does not promote additions to alkenes, and employing Ph3PAuBF4 as catalyst produced inferior results (eq 4 and Figure S6). The reaction of Ph3PAuCl with phenyl radical could generate Ph3PAuPh,14 which would catalyze the title reaction. However, employing the independently prepared phenylgold(I) compound as a catalyst produced 3aa in inferior yield (eq 5).
 |
4 |
On the basis of these observations and the experimental results described above, a possible mechanism is proposed in Scheme 2. The reaction of PhN2BF4 with photoexcited Ru(bpy)32+* generates Ru(bpy)33+ 10 and phenyl radical, and that reacts with the gold(I) catalyst to initially generate gold(II)–aryl 8.15 This gold(II) intermediate is further oxidized by RuIII, giving gold(III)–aryl 9(16) and regenerating the photocatalyst.17 The proposed gold(III) intermediate 9 accounts for the formation of ligand and diazonium-derived phosphonium salts ([Ph3P-Ph]+ in Scheme 2) after consumption of alkene substrate or in the reaction without alkene (Figure S1).18 The anti-ring expansion of complex 10 furnishes alkylgold(III) intermediate 11, which undergoes a reductive elimination to give the desired product 6 and regenerate the gold(I) catalyst.
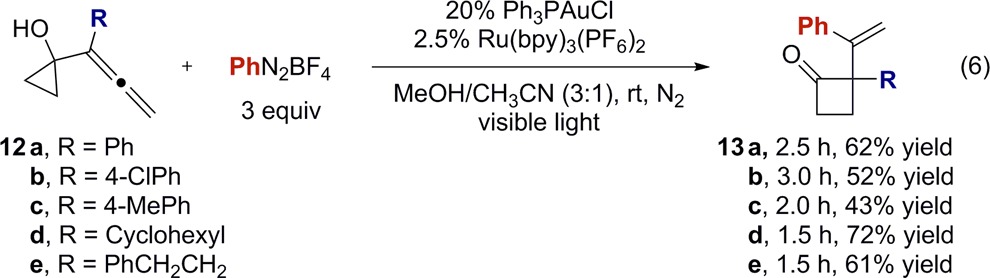
Previous gold-catalyzed oxidative functionalization reactions have precluded the use of allenes as substrates because of the propensity of vinylgold intermediates to undergo facile protodeauration. In contrast, the mechanism outlined in Scheme 2 implies that the gold(III)–aryl is formed directly from the ring expansion event, and therefore reductive elimination might become competitive with protodeauration. In order to further probe this hypothesis and expand the scope of the dual catalysis, the standard reaction conditions were applied to allenes 12a–e (eq 6). These allene substrates were more reactive than alkenes and generally gave moderate yields of the ring-expanded products in shorter time.
 |
6 |
In conclusion, we have reported a ring expansion–oxidative arylation reactions of small sized ring-substituted alkenes and allenes. The reaction was promoted by dual visible light photoredox catalysis and gold catalysis at ambient conditions. The mechanism of the reaction was studied by time-resolved FT-IR spectroscopy and is consistent with formal oxidative addition of gold(I) to aryldiazonium salts to form gold(III)–Ar species preceding the reaction with the alkene. These oxidized intermediates react as potent electrophiles toward π-bonds and ultimately provide an entry into formal electrophilic aryl species.
Acknowledgments
We acknowledge support from the Director, Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Geological and Biosciences, of the U.S. DOE under contract DE-AC02-05CH11231. F.D.T. gratefully acknowledge NIHGMS (RO1 GM073932) for financial support.
Supporting Information Available
Experimental procedures and 1H NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- For selected recent reviews of homogeneous Au catalysis, see:; a Obradors C.; Echavarren A. M. Chem. Commun. 2014, 50, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Garayalde D.; Nevado C. ACS Catal. 2012, 2, 1462. [Google Scholar]; c Pérez-Temprano M. H.; Casares J. A.; Espinet P. Chem.—Eur. J. 2012, 18, 1864. [DOI] [PubMed] [Google Scholar]; d Liu L.-P.; Hammond G. B. Chem. Soc. Rev. 2012, 41, 3129. [DOI] [PubMed] [Google Scholar]; e Rudolph M.; Hashmi A. S. K. Chem. Soc. Rev. 2012, 41, 2448. [DOI] [PubMed] [Google Scholar]; f Shapiro N. D.; Toste F. D. Synlett 2010, 675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For reviews on gold-catalyzed coupling reactions, see:; a Wegner H. A.; Auzias M. Angew. Chem., Int. Ed. 2011, 50, 8236. [DOI] [PubMed] [Google Scholar]; b Hopkinson M. N.; Gee A. D.; Gouverneur V. Chem.—Eur. J. 2011, 17, 8248. [DOI] [PubMed] [Google Scholar]; c Garcia P.; Malacria M.; Aubert C.; Gandon V.; Fensterbank L. ChemCatChem. 2010, 2, 493. [Google Scholar]
- a Ball L. T.; Lloyd-Jones G. C.; Russell C. A. Chem.—Eur. J. 2012, 18, 2931. [DOI] [PubMed] [Google Scholar]; b Melhado A. D.; Brenzovich W. E. Jr.; Lackner A. D.; Toste F. D. J. Am. Chem. Soc. 2010, 132, 8885. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Brenzovich W. E. Jr.; Brazeau J. F.; Toste F. D. Org. Lett. 2010, 12, 4728. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Brenzovich W. E. Jr.; Benitez D.; Lackner A. D.; Shunatona H. P.; Tkatchouk E.; Goddard W. A. III; Toste F. D. Angew. Chem., Int. Ed. 2010, 49, 5519. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Zhang G.; Cui L.; Wang Y.; Zhang L. J. Am. Chem. Soc. 2010, 132, 1474. [DOI] [PubMed] [Google Scholar]; f Ball L. T.; Green M.; Lloyd-Jones G. C.; Russell C. A. Org. Lett. 2010, 12, 4724. [DOI] [PubMed] [Google Scholar]
- a Tkatchouk E.; Mankad N. P.; Benitez D.; Goddard W. A.; Toste F. D. J. Am. Chem. Soc. 2011, 133, 14293. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mankad N.; Toste F. D. J. Am. Chem. Soc. 2010, 132, 12859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahoo B.; Hopkinson M. N.; Glorius F. J. Am. Chem. Soc. 2013, 135, 5505. [DOI] [PubMed] [Google Scholar]
- For combination of visible light photocatalysis with palladium catalysis, see:; a Kalyani D.; McMurtrey K. B.; Neufeldt S. R.; Sanford M. S. J. Am. Chem. Soc. 2011, 133, 18566. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Neufeldt S. R.; Sanford M. S. Adv. Synth. Catal 2012, 354, 3517. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Osawa M.; Nagai H.; Akita M. Dalton Trans. 2007, 827. [DOI] [PubMed] [Google Scholar]; With copper catalysis, see:; d Ye Y.; Sanford M. S. J. Am. Chem. Soc. 2012, 134, 9034. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Ye Y.; Künzi S. A.; Sanford M. S. Org. Lett. 2012, 14, 4979. [DOI] [PMC free article] [PubMed] [Google Scholar]; For reviews and perspectives on visible light photoredox catalysis, see:; f Prier C. K.; Rankic D. A.; MacMillan D. W. C. Chem. Rev. 2013, 113, 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Narayanam J. M. R.; Stephenson C. R. J. Chem. Soc. Rev. 2011, 40, 102. [DOI] [PubMed] [Google Scholar]; h Teply F. Collect. Czech. Chem. Commun. 2011, 76, 859. [Google Scholar]; i Yoon T. P.; Ischay M. A.; Du J. Nat. Chem. 2010, 2, 527. [DOI] [PubMed] [Google Scholar]
- a Walkinshaw A. J.; Xu W.; Suero M. G.; Gaunt M. J. J. Am. Chem. Soc. 2013, 135, 12532. [DOI] [PubMed] [Google Scholar]; b Suero M. G.; Bayle E. D.; Collins B. S. L.; Gaunt M. J. J. Am. Chem. Soc. 2013, 135, 5332. [DOI] [PubMed] [Google Scholar]; c Collins B. S. L.; Suero M. G.; Gaunt M. J. Angew. Chem., Int. Ed. 2013, 52, 5799. [DOI] [PubMed] [Google Scholar]; d Phipps R. J.; McMurray L.; Ritter S.; Duong H. A.; Gaunt M. J. J. Am. Chem. Soc. 2012, 134, 10773. [DOI] [PubMed] [Google Scholar]
- For details and additional conditions examined, see Supporting Information.
- a Mack D. J.; Njardarson J. T. ACS Catal. 2013, 3, 272. [Google Scholar]; b Song Z.-L.; Fan C.-A.; Tu Y.-Q. Chem. Rev. 2011, 111, 7523. [DOI] [PubMed] [Google Scholar]; c Wang B.-M.; Tu Y.-Q. Acc. Chem. Res. 2011, 44, 1207. [DOI] [PubMed] [Google Scholar]; d Li C.-W.; Pati K.; Lin G.-Y.; Sohel S. M. A.; Hung H.-H.; Liu R.-S. Angew. Chem., Int. Ed. 2010, 49, 9891. [DOI] [PubMed] [Google Scholar]
- For reviews on diazonium applied in cross-coupling, see:; a Bonin H.; Fouquet E.; Felpin F. X. Adv. Synth. Catal. 2011, 353, 3063. [Google Scholar]; b Roglans A.; Pla-Quintana A.; Moreno-Manas M. Chem. Rev. 2006, 106, 4622. [DOI] [PubMed] [Google Scholar]; For reviews on radicals generated from diazonium, see:; c Heinrich M. R. Chem.—Eur. J. 2009, 15, 821. [Google Scholar]; d Galli C. Chem. Rev. 1988, 88, 765. [Google Scholar]; For selected examples on the application of diazonium in visible light catalysis, see:; e Hari D. P.; Schroll P.; König B. J. Am. Chem. Soc. 2012, 134, 2958. [DOI] [PubMed] [Google Scholar]; f Schroll P.; Hari D. P.; König B. ChemistryOpen 2012, 1, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Hari D. P.; Hering T.; König B. Org. Lett. 2012, 14, 5334. [DOI] [PubMed] [Google Scholar]; h Xiao T.; Dong X.; Tang Y.; Zhou L. Adv. Synth. Catal. 2012, 354, 3195. [Google Scholar]; i Cano-Yelo H.; Deronzier A. J. Chem. Soc., Perkin Trans. 2 1984, 1093. [Google Scholar]
- The reaction of 1h gave 61% yield of 3ha under the conditions of method D when Ru(bpy)3(PF6)2 was used instead of Ir(ppy)3.
- The reaction shown in Table 1, entry 6, has a slow background reaction in the absence of gold catalyst, which allows for probing the stereochemical course of the phenyl radical-mediated ring expansion. In the absence of gold catalyst, the formation of deuterio-3fb was low-yielding and not stereospecific (21% yield, 1:1 dr; see Supporting Information). For radical arylation of styrenes, see ref (10f) and; Fumagalli G.; Boyd S.; Greaney M. F. Org. Lett. 2013, 15, 4398. [DOI] [PubMed] [Google Scholar]
- a Kleinbeck F.; Toste F. D. J. Am. Chem. Soc. 2009, 131, 9178. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Markham J. P.; Staben S. T.; Toste F. D. J. Am. Chem. Soc. 2005, 127, 9708. [DOI] [PubMed] [Google Scholar]
- For radical trapping by gold chlorides, see:; Aprile C.; Boronat M.; Ferrer B.; Corma A.; Garcia H. J. Am. Chem. Soc. 2006, 128, 8388. [DOI] [PubMed] [Google Scholar]
- For reviews on Au(II) compounds see:; a Mirzadeh N.; Bennett M. A.; Bhargava S. K. Coord. Chem. Rev. 2013, 257, 2250. [Google Scholar]; b Laguna A.; Laguna M. Coord. Chem. Rev. 1999, 193–195, 837. [Google Scholar]; For radical oxidation of Au(I) to Au(III) via Au(II) intermediate, see:; c Johnson A.; Puddephatt R. J. Chem. Soc., Dalton Trans. 1976, 1360. [Google Scholar]; d Johnson A.; Puddephatt R. J. J. Chem. Soc., Dalton Trans. 1975, 115. [Google Scholar]
- The observation of a significant increase in the amount of 4,4′-difluorobiphenyl formation when Ph3PAuCl was added to 4-FPhN2BF4 under photocatalysis conditions in the absence of alkene (Figure S5) is consistent with the generation of a gold(III)–phenyl intermediate.
- An alternative mechanism to that shown in Scheme 2, in which 8 reacts with phenyldiazomethane to give 9 and a phenyl radical, without regeneration of the photocatalyst, could not be discounted.
- The phosphonium salt has been reported to be generated by reductive elimination from high-valent compound Ar-Mn+2(PPh3). For related works, see:; a Tappe F. M. J.; Trepohl V. T.; Oestreich M. Synthesis 2010, 3037. [Google Scholar]; b Marcoux D.; Charette A. B. Adv. Synth. Catal. 2008, 350, 2967. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.