

Conspectus
Redox-neutral methods for the functionalization of amine α-C–H bonds are inherently efficient because they avoid external oxidants and reductants and often do not generate unwanted byproducts. However, most of the current methods for amine α-C–H bond functionalization are oxidative in nature. While the most efficient variants utilize atmospheric oxygen as the terminal oxidant, many such transformations require the use of expensive or toxic oxidants, often coupled with the need for transition metal catalysts.
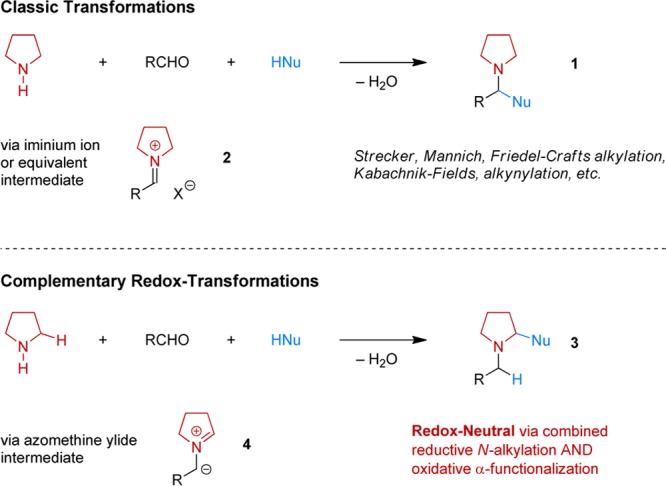
Redox-neutral amine α-functionalizations that involve intramolecular hydride transfer steps provide viable alternatives to certain oxidative reactions. These processes have been known for some time and are particularly well suited for tertiary amine substrates. A mechanistically distinct strategy for secondary amines has emerged only recently, despite sharing common features with a range of classic organic transformations. Among those are such widely used reactions as the Strecker, Mannich, Pictet–Spengler, and Kabachnik–Fields reactions, Friedel–Crafts alkylations, and iminium alkynylations. In these classic processes, condensation of a secondary amine with an aldehyde (or a ketone) typically leads to the formation of an intermediate iminium ion, which is subsequently attacked by a nucleophile. The corresponding redox-versions of these transformations utilize identical starting materials but incorporate an isomerization step that enables α-C–H bond functionalization. Intramolecular versions of these reactions include redox-neutral amine α-amination, α-oxygenation, and α-sulfenylation. In all cases, a reductive N-alkylation is effectively combined with an oxidative α-functionalization, generating water as the only byproduct.
Reactions are promoted by simple carboxylic acids and in some cases require no additives. Azomethine ylides, dipolar species whose usage is predominantly in [3 + 2] cycloadditions and other pericyclic processes, have been identified as common intermediates. Extension of this chemistry to amine α,β-difunctionalization has been shown to be possible by way of converting the intermediate azomethine ylides into transient enamines.
This Account details the evolution of this general strategy and the progress made to date. Further included is a discussion of related decarboxylative reactions and transformations that result in the redox-neutral aromatization of (partially) saturated cyclic amines. These processes also involve azomethine ylides, reactive intermediates that appear to be far more prevalent in condensation chemistry of amines and carbonyl compounds than previously considered. In contrast, as exemplified by some redox transformations that have been studied in greater detail, iminium ions are not necessarily involved in all amine/aldehyde condensation reactions.
1. Introduction
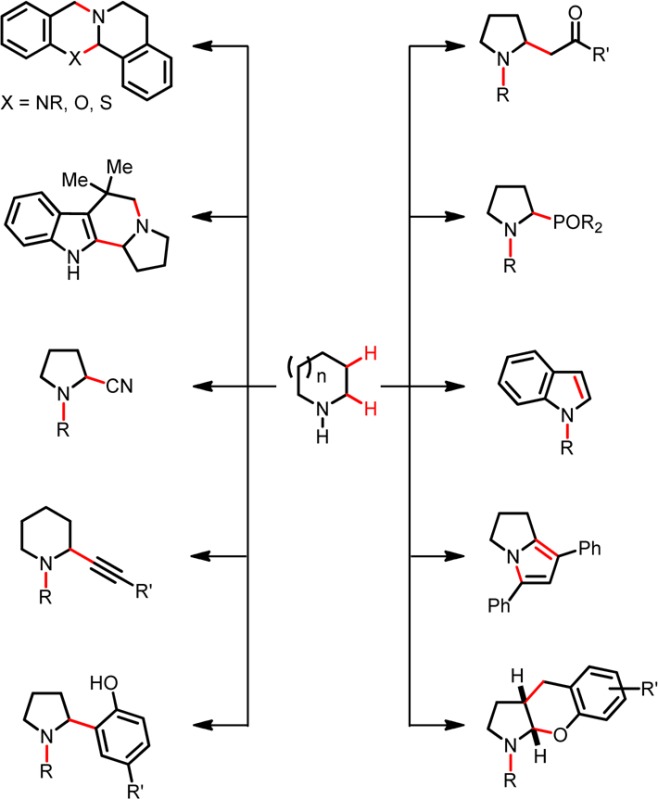
The broadly defined field of C–H functionalization continues to draw unwavering attention. A major focus is placed on the C–H functionalization of amines, mostly concerning moderately activated α-C–H bonds but recently also extending to less reactive β-C–H bonds.1−3 Given the privileged role of functionalized amines as bioactive materials, the tremendous level of interest in this substrate class is hardly surprising. Different strategies are available to accomplish the functionalization of amine α-C–H bonds, yet the field is dominated by oxidative approaches.3 Tertiary amines or protected amines (carbamates, amidines, N-aryl amines, etc.) are the main targets of these efforts. Redox-neutral4 methods that operate via hydride transfer mechanisms5,6 provide interesting alternatives, as do photoredox strategies,7 but these also typically rely on tertiary amines as starting materials.8 Our group has introduced a mechanistically distinct concept that provides a pathway for the C–H functionalization of secondary amines. This approach enables the formation of products that are regioisomers of those obtained in classic organic transformations such as the venerable Strecker, Mannich, and Friedel–Crafts reactions. A common feature of these widely utilized reactions is that they involve the condensation of a primary or secondary amine with an aldehyde (or ketone) and a (pro)nucleophile to afford products of type 1 (Scheme 1). In reactions with secondary amines (e.g., pyrrolidine), iminium ions such as 2 are typically invoked as intermediates. Identical starting materials can react with intervening internal redox isomerization to provide products of concomitant amine N-alkylation and α-C–H bond functionalization (e.g., 3). Just as in classic condensation reactions, water is produced as the sole byproduct. This method has proven to be highly generalizable across different types of reactions. Cyclic amines are the most attractive (and generally more reactive) substrates, leading to ring substitution and rapid access to compounds that may otherwise require lengthy synthetic procedures. Azomethine ylides (e.g., 4) have been identified as important intermediates, opening up a new chapter in the chemistry of these dipolar species whose transformations have largely been dominated by pericyclic processes.9−12 Here we provide the first account of this general strategy for amine C–H functionalization.
Scheme 1. Classic Organic Reactions and Their Redox Variants.
2. Intramolecular Redox-Transformations
2.1. α-Amination
The first example of a redox-transformation leading to amine α-functionalization (via the general process outlined in Scheme 1) was discovered serendipitously by our group.13 In the course of conducting Friedländer condensations to access certain quinolines, we observed that ortho-aminobenzaldehydes undergo condensations with pyrrolidine to form cyclic aminals (Scheme 2). Detailed studies including a computational investigation conducted in collaboration with the Houk group provided insights into the mechanism of this transformation.14,15 Somewhat counterintuitively, simple iminium ions are apparently not involved as reactive intermediates. Addition of pyrrolidine to ortho-aminobenzaldehyde (5) initially affords N,O-acetal 7, which, rather than formally losing hydroxide to form an iminium ion, suffers direct elimination of water. The resulting azaquinone methide 8, which exhibits zwitterionic character (8′), undergoes 1,6-proton abstraction to form azomethine ylide 9. An ethanol-promoted proton transfer step (11) ensues, resulting in zwitterion 10, which then ring-closes to give aminal 6. The scope of this transformation is relatively broad. Aminobenzaldehydes with different substitution patterns readily undergo aminal-forming condensations with pyrrolidine under reflux conditions in ethanol. Heteroaromatic ortho-aminoaldehydes also participate in this transformation. Cyclic amines with benzylic α-C–H bonds such as 1,2,3,4-tetrahydroisoquinoline (THIQ) and tryptoline are among the most reactive substrates. In contrast, amines such as piperidine and morpholine require higher reaction temperatures. Shortly after our initial disclosure, the group of Dang and Bai reported their independent discovery of a similar aminal-forming process.16
Scheme 2. Intramolecular Redox Amination.

Upon further development of this reaction, we found that for most substrate combinations, brief exposure to high temperatures under microwave irradiation provides results superior to those obtained at lower temperatures and longer heating periods. Microwave conditions also facilitate reactions of secondary amine substrates that are unreactive under standard thermal conditions (e.g., reflux in n-butanol). Interestingly, at temperatures substantially higher than those required for the functionalization of its benzylic α-C–H bonds, THIQ undergoes a competing α-C–H functionalization at the 3-position (formation of 13). While the reasons for this reversal in regioselectivity are not fully understood at present, 13 was calculated to be thermodynamically more stable than its isomer 12.14 Using appropriate oxidation conditions, aminals can be directly converted to dihydroquinazoline and quinazolinone alkaloids.13,15,17,18
2.2. α-Oxygenation
The obvious extension of the chemistry outlined above using salicylaldehydes as substrates proved to be less straightforward than expected. In fact, two other research groups independently investigated this transformation and concluded that the reaction of secondary amines with various salicylaldehydes fails to afford the expected N,O-acetals, prompting them to develop oxidative approaches to these targets.19,20 Indeed, we also observed that conditions suitable for aminal synthesis are not applicable to the preparation of the corresponding N,O-acetals (Scheme 3). Following extensive experimentation, we identified a new set of reaction parameters that allow for the isolation of N,O-acetals. Under optimized conditions, salicylaldehyde undergoes condensation with THIQ to form product 14 in nearly quantitative yield. The key to a successful reaction is the use of a carboxylic acid (e.g., acetic acid) as a promoter. Reactions are greatly accelerated in the presence of molecular sieves.
Scheme 3. Intramolecular Redox Oxygenation.

When protic solvents are used or in the absence of a carboxylic acid promoter, only small amounts of 14 are observed, and 15 is isolated as the major product. This product of an apparent reductive amination likely results from the decomposition of N,O-acetal 14 via a (formal) retro-Diels–Alder reaction. In addition to 1,2-dihydroisoquinoline (DHIQ), which is observed as a byproduct, ortho-quinone methide 16 is formed as an intermediate and then intercepted by THIQ to afford 15. The overall mechanism of the desired N,O-acetal formation follows a path similar to that of the corresponding aminal process. The main difference is the nature of the rate limiting step, which is thought to be the formation of 18 from the corresponding azomethine ylide. Here lies the major role of acetic acid; it serves as a proton shuttle via 17, lowering the energy of the transition state by 8.6 kcal·mol–1 over the alternative process in which salicylaldehyde acts as the proton shuttle.21 The scope of the N,O-acetal formation with different THIQs and salicylaldehydes is relatively broad. Other amines also participate in this transformation. For reasons that remain unclear, the reaction of pyrrolidine with parent salicylaldehyde has thus far only provided the product of apparent reductive amination (not shown). Removal of water has a dramatic effect on the reaction rate. In the presence of a Dean–Stark apparatus, the reaction of THIQ and salicylaldehyde goes to completion in only 1 h (98% yield of 14). In stark contrast, without water removal, 14 is formed in only 52% yield after 24 h and the reaction remains incomplete.
An interesting precedent to the redox oxygenation was reported by Cohen et al.22 His group found that hydroxyketones such as 19 undergo reactions with proline to form N,O-acetals (e.g., 20). This decarboxylative transformation (vide infra) represents a rare and early example of a nonpericyclic reaction pathway of azomethine ylides, intermediates that are almost certainly involved in this process.
2.3. α-Sulfenylation
Conditions similar to those developed for the synthesis of N,O-acetals are also applicable to the synthesis of the corresponding N,S-acetals from thiosalicylaldehydes and amines (Scheme 4).23 A catalytic amount of acetic acid provides higher yields than using one equivalent of this acid as a promoter. The scope with regard to the amine is unusually broad; even relatively unreactive substrates such as morpholine, thiomorpholine, piperazines, and dibenzylamine undergo this transformation.
Scheme 4. Intramolecular Redox Sulfenylation.

2.4. C–C Bond Formation
In addition to developing methods for the redox-neutral generation of C–X bonds, we investigated the possibility of performing redox cyclizations that involve C–C bond formation. Gratifyingly, we found that aldehydes linked to electron-rich aromatic rings readily undergo redox annulations with amines at elevated temperatures under microwave conditions (Scheme 5).24 In the absence of additives, indole-aldehydes engage amines such as THIQ, pyrrolidine, azepane, and proline esters to form the corresponding ring-fused products in moderate to good yields during brief reaction times. Interestingly, the aldehyde moiety can but does not need to be in conjugation with the nucleophile. These transformations can be considered as redox variants of classic Pictet–Spengler25 reactions. While some substrate combinations require relatively high reaction temperatures, it appears likely that addition of appropriate additives (e.g., carboxylic acids) will allow for these reactions to be performed at lower temperatures.
Scheme 5. Redox Annulation with Concurrent C–C Bond Formation.

3. Decarboxylative Three-Component Coupling Reactions
At the outset of our studies, it was not at all obvious whether it would be possible to conduct redox transformations in an intermolecular setting. While for the intramolecular variants described above, the classic reaction pathway is less favorable than the redox pathway (four-membered vs six-membered ring formation), any intermolecular redox transformation would necessarily have to compete with its classic counterpart. Because of the expected difficulties in realizing such transformations, we first sought to establish whether azomethine ylides can engage in intermolecular nonpericyclic reactions with appropriate (pro)nucleophiles. In this context, two interesting literature reports by the Li group drew our attention.26,27 These researchers reported metal-catalyzed, oxidative, decarboxylative reactions of N-benzyl proline that afford ring-substituted products 25, using alkynes, indoles, nitroalkanes, and naphthols as nucleophiles (Scheme 6). The intermediacy of (metal-bound) azomethine ylides was proposed.
Scheme 6. Decarboxylative Coupling Reactions.

We hypothesized that there might be a more direct pathway to products 25. Due to the pioneering contributions of Rizzi28 and Grigg,29 it is well-known that amino acids such as proline undergo decarboxylative condensations with aldehydes to furnish nonstabilized azomethine ylides 28/28′ via the intermediacy of 27. Exposure of 28/28′ to a moderately acidic substrate might result in protonation and formation of the regioisomeric ion pairs 29 or 30, which can collapse into products 25/26. Gratifyingly, we found that these three-component coupling reactions occur readily under simple reflux conditions in toluene.30 The key to obtaining high yields of the desired products is to keep the concentration of the aldehyde low, which is accomplished by adding the aldehyde slowly as a toluene solution via syringe pump. This prevents the well-known [3 + 2] cycloaddition of excess aldehyde with the intermediate azomethine ylide.31 Undesired regioisomers corresponding to 26 are occasionally observed as minor byproducts. In accord with the proposed reaction mechanism, N-methylindole, typically a better nucleophile than indole but lacking an acidic proton, fails to undergo the reaction under conditions suitable for indole itself. However, addition of benzoic acid as an additive enabled the corresponding coupling reaction. A likely role of benzoic acid is to protonate the azomethine ylide intermediate. With Cu(I)-bromide and TMEDA as the catalyst combination, the scope was extended to alkynes as nucleophiles.30 No oxidants are required in this process. A closely related approach to decarboxylative α-amino acid alkynylation was published by the Li group nearly simultaneously with our report.32 Subsequent to our initial disclosure, we developed a decarboxylative version of the Strecker reaction, a process that is most efficiently conducted under microwave irradiation.33 The above-mentioned redox annulations (Scheme 5) can also be conducted with α-amino acids under decarboxylative conditions.24
4. Intermolecular Redox Transformations
4.1. α-Cyanation
As another step toward the realization of a three-component redox transformation, we opted to explore the potential for isomerizing a classic three-component condensation product into its corresponding redox product. We reasoned that a compound derived from a nucleophile that also represents a good leaving group would be an ideal starting point. This line of reasoning brought our attention to α-aminonitriles, readily available products of Strecker reactions. Our investigations eventually resulted in the development of the first redox-Strecker reaction (Scheme 7).34 Initial attempts to isomerize 33 resulted in the striking observation that partial isomerization to 32 can be achieved in the absence of any additives. Simple heating of a toluene solution of 33 to 200 °C (microwave) for 20 min results in a 1:5 mixture of 32/33, with good overall α-aminonitrile recovery. Ultimately, it was found that the isomerization is vastly more efficient when conducted in the presence of catalytic amounts of benzoic acid. In this instance, an 18:1 ratio of 32/33 is obtained. This appears to correspond to the thermodynamic equilibrium ratio of the two compounds, given that the same 18:1 ratio is obtained when pure 32 is exposed to identical conditions. Redox-Strecker products can also be obtained directly from pyrrolidine and cyanohydrins. 2-Ethyl hexanoic acid (2-EHA) slightly outperforms benzoic acid in this two-component approach. A three-component approach with pyrrolidine, benzaldehyde and TMSCN is also feasible but leads to slightly inferior results (not shown).34 The two-component approach with pyrrolidine is applicable to a range of cyanohydrins derived from aromatic amines. Importantly, other amines such as piperidine and azepane also participate in redox-Strecker reactions, providing favorable product ratios. Interestingly, of the three possible α-aminonitriles that could potentially form with THIQ, the 3-substituted product (presumably the thermodynamically most stable α-aminonitrile) is obtained nearly exclusively. This represents a rare example of a C–H functionalization of THIQ that leads to substitution of the 3-position. Finally, 2-Me-pyrrolidine undergoes the redox-Strecker reaction with formation of two regioisomers; the standard Strecker product is not observed under these conditions.
Scheme 7. Redox-Strecker Reaction.

4.2. α-Phosphonation
We subsequently developed a redox variant of the Kabachnik–Fields reaction (Scheme 8).35 This transformation is best conducted as a three-component process. The optimized conditions differ slightly from those developed for the redox-Strecker reaction in that 2-EHA acid is replaced with benzoic acid. In addition, the reaction is conducted at an increased concentration (0.5 vs 0.1 M). As seen for the corresponding α-aminonitriles, Kabachnik–Fields products 35 readily isomerize to the apparently thermodynamically preferred redox-Kabachnik–Fields products 34. The scope of the three-component approach to the ring-substituted products is relatively broad with regard to the aromatic aldehyde; heteroaromatic aldehydes are also viable reaction partners. The scope of the amine is more limited. Even with 2,6-dichlorobenzaldehyde (vide infra), product ratios are not favorable for piperidine and azepane. Prior to the publication of our report, the Wang group disclosed a decarboxylative version of the Kabachnik–Fields reaction.36
Scheme 8. Redox-Kabachnik–Fields Reaction.

4.3. α-Alkynylation
We then turned our attention to classic organic transformations in which the nucleophile is not a particularly good leaving group. The A3 reaction (coupling of an amine, an aldehyde, and an alkyne) was identified as a suitably challenging example of such a process.37 In considering what factors might allow for the development of a redox-A3 reaction, we reasoned that the presumably required transformation of iminium ions such as 2 to azomethine ylides 4 might be accelerated when 2 is derived from a relatively electron-poor aldehyde.38 In addition, the classic pathway (copper acetylide addition to 2) should be slowed for iminium ions generated from a bulky aldehyde. 2,6-Dichlorobenzaldehyde was identified as a promising candidate that might fulfill these requirements. Indeed, under optimized conditions with Cu(II) 2-ethylhexanoate (Cu(2-EH)2) as the catalyst, redox-A3 product 36 is obtained nearly exclusively over the standard A3 product 37 (Scheme 9). Cu(2-EH)2 is believed to play a dual role in the overall reaction. First, it serves as a source of Cu(I), which is required in the formation of the copper acetylide nucleophile, also releasing 2-EHA in the process. The latter appears to be crucial in the isomerization of 2 to 4. Product isomerization appears to play virtually no role in the overall selectivity of the reaction. Exposure of 37 to the reaction conditions only leads to minor amounts of 36, with most of 37 being recovered. The use of 2,6-dichlorobenzaldehyde in the initial development of the redox-A3 reaction turned out to have been a good choice. Electronically similar 2,4-dichlorobenzaldehyde, under otherwise identical conditions, only shows a 2.6:1 preference for the redox-A3 product. The ratio drops to 1:1 with 3,4-dichlorobenzaldehyde. Nevertheless, it is notable that even with simple benzaldehyde, an appreciable amount of redox-A3 product is obtained. Overall, the sterics of the aldehyde seems to outweigh electronic factors. A case in point is mesitaldehyde, which provides an 11:1 ratio in favor of the redox-A3 product. Redox-A3 reactions with pyrrolidine and 2,6-dichlorobenzaldehyde give favorable results with a range of alkynes. Piperidine and azepane also participate in redox-A3 reactions but generally provide lower selectivities. Morpholine is a poor substrate, although selectivities for this heterocycle greatly depend on subtle sterics on the alkyne. Subsequently to our seminal report on the redox-A3 reaction, the Yu group published a related report on the redox-alkynylation of THIQ.39 With CuI as the catalyst, a broad range of aldehydes and alkynes undergo the redox-A3 reaction with THIQ under relatively mild conditions (50 °C in toluene). Nearly at the same time, Ma and co-workers reported the first catalytic enantioselective redox-A3 reaction with THIQ.40
Scheme 9. Redox-A3 Reaction.

4.4. α-Arylation
Initial attempts to extend the concept to redox arylations using β-naphthol as the model nucleophile met with limited success; a mixture of various products was typically isolated.41 Good yields of redox-arylation products were eventually obtained with 2,6-dichlorobenzaldehyde (Scheme 10). Key to a selective and high-yielding reaction is the slow addition of the aldehyde to a mixture of pyrrolidine and β-naphthol. Interestingly, with naphthols and phenols as nucleophiles, slightly improved yields are obtained in the absence of a carboxylic acid catalyst. This suggests that these substrates are sufficiently acidic to catalyze the redox isomerization. The scope of this transformation is relatively broad with regard to the nucleophile. In addition to β-naphthol, simple phenols, indoles, and pyrroles participate in redox arylations with pyrrolidine and 2,6-dichlorobenzaldehyde. Due to their reduced acidities, reactions with pyrrole or indole require the addition of 2-EHA as an additive. Piperidine is also a viable reaction partner, although lower yields are typically obtained. Nearly simultaneously to the publication of our report on the redox arylation, the group of Jana published a very similar strategy, employing fluorenone as a coupling partner in place of an aldehyde.42
Scheme 10. Redox-Arylation of Amines.

4.5. α-Alkylation
The Mannich reaction is a classic transformation that enables the synthesis of β-aminoketones from amines, ketones, and aldehydes (generally nonenolizable).43 Following extensive experimentation, we succeeded in developing a redox version of the Mannich reaction with amines such as pyrrolidine and THIQ (Scheme 11).44 The optimized conditions for pyrrolidine call for a slow addition of a mixture of the aldehyde and the ketone to an excess of the amine in refluxing toluene. While this reaction is surprisingly tolerant of deviations from the optimized conditions, the presence of a carboxylic acid catalyst is a strict requirement. However, the nature or the amount of acid is less important. For pyrrolidine, aldehydes other than 2,6-dichlorobenzaldehyde generally lead to low yields of redox-Mannich products. In case of THIQ, a broad range of aromatic and heteroaromatic aldehydes participate in redox-Mannich reactions with ketones such as acetophenone or acetone. Milder reaction conditions (50 °C in toluene) are sufficient, and slow addition is not required. An interesting aspect of these transformations is that regular Mannich products are never isolated as byproducts, although they are believed to form in a competing process. These undesired products readily eliminate amine under the reaction conditions to form the corresponding chalcones, products that then undergo other side reactions with the amine.44 In contrast, redox-Mannich products are quite stable, since elimination of amine would lead to an open-chain product that can readily revert to the corresponding redox-Mannich compound via ring closure (not shown). Nitroalkanes are also competent nucleophiles in this reaction.
Scheme 11. Redox-Mannich Reaction.

5. Redox-Neutral α,α-Difunctionalization
Based on precedent by Soeder and Cartaya-Marin,45 we developed a distinct type of redox transformation that leads to amine α,α-difunctionalization (Scheme 12).46 Specifically, pyrrolidine undergoes a p-TSA facilitated condensation with 1,3-diketone 40 to form the fused pyrrole 41 in moderate yield. Relatively high reaction temperatures are required, and N-benzoyl pyrrolidine is formed as a byproduct via a competing retro-Claisen process (not shown).46 A potential mechanism involves enaminone 42 (also isolated as a byproduct). This compound could undergo a 1,6-proton transfer to form conjugated azomethine ylide 43, followed by 6π-electrocyclization to afford intermediate 44. Finally, elimination of water yields pyrrole 40. The scope of this transformation is relatively limited with regard to the amine. Pyrrolidine and THIQ represent the most susceptible substrates. The 1,3-diketone has to bear at least one aryl group; the yields are moderate to good.
Scheme 12. Redox α,α-Difunctionalization.

6. Redox-Neutral Aromatization
A number of interesting literature reports describe the formation of aromatic heterocycles from (partially) saturated cyclic amines and aldehydes (Scheme 13). While these transformations, at least on first sight, might appear to be oxidative in nature, they are in fact redox-neutral. These reports have served as a source of inspiration in the development of our redox transformations, which may alternatively be considered as examples of “interrupted aromatization.” An early example of amine aromatization is the acetic acid promoted formation of 3,5-dibenzylpyridine from piperidine and benzaldehyde, reported by Poirier in 1961.47 Remarkably, a closely related process with N-benzoyl piperidine as the starting material was already discovered by Rügheimer in 1891.48 It was later shown that the first step in the Rügheimer procedure involves amide hydrolysis and formation of piperidine. Another report by Burrows describes the synthesis of 4-benzylisoquinoline from THIQ and benzaldehyde under very similar conditions.49 This process was later improved and studied in detail by Dannhardt who provided conclusive evidence for the intermediacy of endocyclic enamines, in line with the observed β-functionalization.50 More recently, the Oda group reported the synthesis of 1,3-dibenzylpyrrole from pyrrolidine and benzaldehyde.51 Tunge and co-workers disclosed the benzoic acid catalyzed formation of N-alkyl pyrroles from 3-pyrroline and aldehydes or ketones.52 Earlier, Cook et al. had observed the formation of N-cyclohexyl pyrrole from cyclohexanone and 3-pyrroline under thermal conditions.53 It should be pointed out that these transformations are distinctly different from the pyrrole formation shown in Scheme 12 where the amine that serves as the starting material does not undergo aromatization.
Scheme 13. Redox-Neutral Aromatization of (Partially) Saturated Amines, Implication of Azomethine Ylide Intermediates.

We developed an analogous procedure for the preparation of N-alkyl indoles from indoline.54 A closely related independent report was published earlier by Pan et al.55 The main impetus for our study was the goal to obtain further insights into the mechanism of these redox aromatizations. Mechanistic considerations led us to propose azomethine ylides as key intermediates in all of these processes. To obtain supporting evidence, we performed the condensation of 3-pyrroline with aldehyde 45, bearing a pendent dipolarophile. Indeed, under conditions that lead to pyrrole formation with simple aldehydes, [3 + 2] product 46 is obtained. Similarly, the reaction of indoline with 45 results in the formation of 47.54 However, in this case aromatization is a competitive process and 48 is also obtained. This strategy for trapping intermediate azomethine ylides was adopted from Grigg et al., who had previously shown that THIQ, upon reaction with 49, undergoes azomethine ylide formation with subsequent dipolar cycloaddition to give product 50.56 Regarding the mechanism of azomethine ylide formation, it is known since the pioneering work of Huisgen that the deprotonation of iminium ions represents a viable pathway.57 Given the mildly basic nature of carboxylate anions, we proposed that they might deprotonate intermediate iminium ions. An interesting alternative was proposed by the Yu group, who conducted a computational study of Tunge’s N-alkyl pyrrole formation.58 According to their analysis, iminium ions are not involved in the reaction sequence, and azomethine ylide 52 is formed directly from N,O-acetal 51 via elimination of benzoic acid.
7. Redox-Neutral α,β-Difunctionalization
A particularly promising avenue for further development is the utilization of in situ generated endocyclic enamines (e.g., 60, Scheme 14) in α,β-difunctionalization or β-functionalization reactions. As pointed out in our discussion on redox aromatizations of piperidine and pyrrolidine (vide supra), enamines are clearly formed as intermediates in these processes. With appropriate reaction partners present, further transformation of an enamine intermediate might be diverted away from aromatization to a range of other processes. In the course of our work on the redox arylation (Scheme 10),41 we came across an opportunity to develop a direct redox-neutral amine α,β-difunctionalization reaction (Scheme 14).59 Specifically, upon attempting the isomerization of 54 into its corresponding redox-arylation product (not shown), we noticed the formation of unexpected product 55, a compound that is obtained as a single diastereomer. A subsequent detailed study revealed that polycyclic product 55 can be prepared in high yield by exposing 54 to a slight excess of benzaldehyde. As seen in redox arylations with β-naphthol, carboxylic acid additives are not required. A plausible mechanism for the reaction was established through two complementary experiments. While a combination of 54 with p-chlorobenzaldehyde gives rise to product 56, the p-chlorobenzaldehyde-derived 57, upon reacting with benzaldehyde, affords the regioisomeric product 58. Both, 56 and 58 are obtained in very similar yields. A likely course of events leading to the formation of 58 initiates with the fragmentation of 57 to ortho-quinone methide 59 with concurrent release of pyrrolidine. The latter engages benzaldehyde and through a series of steps generates enamine 60, which undergoes a (potentially stepwise) endo-selective hetero-Diels–Alder reaction with 59 (via 61) to afford product 58. It is quite possible that the success of this transformation is dependent on a low concentration of pyrrolidine being present at all times. This is ensured by its slow release from the starting material. The scope of this transformation is remarkably broad with regard to the amine. Challenging substrates containing piperidine, morpholine, thiomorpholine, and piperazine moieties readily participate in this unprecedented α,β-difunctionalization. Notably, this reaction can also be performed as a three-component reaction, using a one-pot, two-stage approach (Scheme 14).
Scheme 14. Redox α,β-Difunctionalization.

8. Summary and Outlook
We have outlined a new strategy for the redox-neutral α-C–H functionalization of simple secondary amines. These reactions combine a reductive amination with an oxidative α-functionalization and can be conducted intra- or intermolecularly. Azomethine ylides have been identified as important intermediates, whereas simple iminium ions are not necessarily involved. These transformations provide regioisomeric products of important classic reactions and offer a number of additional attractive features. Among those are the ease with which these redox reactions can be conducted and the fact that only benign and inexpensive catalysts (if any) are required. For azomethine ylides bearing α-protons (C–H bonds β to nitrogen in the original amine), these species can be converted to enamines, enabling amine β-functionalization and α,β-difunctionalization.
The general strategies described in this Account have already been shown to be applicable to a wide range of reactions. Nevertheless, it is reasonable to expect many further developments, the emergence of additional enantioselective variants, and applications to complex target synthesis.
Acknowledgments
I am deeply indebted to my co-workers, whose names appear in the appropriate references, for their crucial experimental and intellectual contributions to this program. Financial support from the NIH–NIGMS (Grant R01GM101389-01) is gratefully acknowledged.
Biography
Daniel Seidel studied chemistry at the Friedrich-Schiller-Universität Jena, Germany, and at the University of Texas at Austin (Diplom 1998). He performed his graduate studies in the lab of Prof. Jonathan L. Sessler, obtaining his Ph.D. in 2002. From 2002 to 2005, he was an Ernst Schering Postdoctoral Fellow in the group of Prof. David A. Evans at Harvard University. He started his independent career at Rutgers University in 2005 and was promoted to Full Professor in 2014. Research in his group is focused on new concepts for asymmetric catalysis and synthetic methodology.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Campos K. R. Direct sp(3) C-H Bond Activation Adjacent to Nitrogen in Heterocycles. Chem. Soc. Rev. 2007, 36, 1069–1084. [DOI] [PubMed] [Google Scholar]
- Mitchell E. A.; Peschiulli A.; Lefevre N.; Meerpoel L.; Maes B. U. W. Direct alpha-Functionalization of Saturated Cyclic Amines. Chem.—Eur. J. 2012, 18, 10092–10142. [DOI] [PubMed] [Google Scholar]
- Girard S. A.; Knauber T.; Li C.-J. The Cross-Dehydrogenative Coupling of C sp 3-H Bonds: A Versatile Strategy for C-C Bond Formations. Angew. Chem., Int. Ed. 2014, 53, 74–100. [DOI] [PubMed] [Google Scholar]
- Burns N. Z.; Baran P. S.; Hoffmann R. W. Redox Economy in Organic Synthesis. Angew. Chem., Int. Ed. 2009, 48, 2854–2867. [DOI] [PubMed] [Google Scholar]
- Peng B.; Maulide N. The Redox-Neutral Approach to C-H Functionalization. Chem.—Eur. J. 2013, 19, 13274–13287. [DOI] [PubMed] [Google Scholar]
- Haibach M. C.; Seidel D. C-H Bond Functionalization through Intramolecular Hydride Transfer. Angew. Chem., Int. Ed. 2014, 53, 5010–5036. [DOI] [PubMed] [Google Scholar]
- Prier C. K.; Rankic D. A.; MacMillan D. W. C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a review on the redox-neutral α-C–H bond functionalization of alcohols, see:Ketcham J. M.; Shin I.; Montgomery T. P.; Krische M. J. Catalytic Enantioselective C-H Functionalization of Alcohols by Redox-Triggered Carbonyl Addition: Borrowing Hydrogen, Returning Carbon. Angew. Chem., Int. Ed. 2014, 53, 9142–9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padwa A.; Pearson W. H.. Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products; Wiley: Chichester, U.K., 2002; Vol. 59. [Google Scholar]
- Coldham I.; Hufton R. Intramolecular Dipolar Cycloaddition Reactions of Azomethine Ylides. Chem. Rev. 2005, 105, 2765–2809. [DOI] [PubMed] [Google Scholar]
- Pinho e Melo T. M. V. D. Conjugated Azomethine Ylides. Eur. J. Org. Chem. 2006, 2873–2888. [Google Scholar]
- Nyerges M.; Toth J.; Groundwater P. W. 1,7-Electrocyclizations of Azomethine Ylides: Scope and Synthetic Aspects. Synlett 2008, 1269–1278. [Google Scholar]
- Zhang C.; De C. K.; Mal R.; Seidel D. α-Amination of Nitrogen Heterocycles: Ring-Fused Aminals. J. Am. Chem. Soc. 2008, 130, 416–417. [DOI] [PubMed] [Google Scholar]
- Dieckmann A.; Richers M. T.; Platonova A. Y.; Zhang C.; Seidel D.; Houk K. N. Metal-Free α-Amination of Secondary Amines: Computational and Experimental Evidence for Azaquinone Methide and Azomethine Ylide Intermediates. J. Org. Chem. 2013, 78, 4132–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richers M. T.; Deb I.; Platonova A. Y.; Zhang C.; Seidel D. Facile Access to Ring-Fused Aminals via Direct alpha-Amination of Secondary Amines with o-Aminobenzaldehydes: Synthesis of Vasicine, Deoxyvasicine, Deoxyvasicinone, Mackinazolinone, and Ruteacarpine. Synthesis 2013, 45, 1730–1748. [PMC free article] [PubMed] [Google Scholar]
- Zheng L.; Yang F.; Dang Q.; Bai X. A Cascade Reaction with Iminium Ion Isomerization as the Key Step Leading to Tetrahydropyrimido[4,5-d]pyrimidines. Org. Lett. 2008, 10, 889–892. [DOI] [PubMed] [Google Scholar]
- Zhang C.; De C. K.; Seidel D. o-Aminobenzaldehyde, Redox-Neutral Aminal Formation and Synthesis of Deoxyvasicinone. Org. Synth. 2012, 89, 274–282. [Google Scholar]
- Richers M. T.; Zhao C. F.; Seidel D. Selective Copper(II) Acetate and Potassium Iodide Catalyzed Oxidation of Aminals to Dihydroquinazoline and Quinazolinone Alkaloids. Beilstein J. Org. Chem. 2013, 9, 1194–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deb M. L.; Dey S. S.; Bento I.; Barros M. T.; Maycock C. D. Copper-Catalyzed Regioselective Intramolecular Oxidative α-Functionalization of Tertiary Amines: An Efficient Synthesis of Dihydro-1,3-Oxazines. Angew. Chem., Int. Ed. 2013, 52, 9791–9795. [DOI] [PubMed] [Google Scholar]
- Mahato S.; Haldar S.; Jana C. K. Diastereoselective alpha-C-H Functionalization of Aliphatic N-Heterocycles: An Efficient Route to Ring Fused Oxazines. Chem. Commun. 2014, 50, 332–334. [DOI] [PubMed] [Google Scholar]
- Richers M. T.; Breugst M.; Platonova A. Y.; Ullrich A.; Dieckmann A.; Houk K. N.; Seidel D. Redox-Neutral α-Oxygenation of Amines: Reaction Development and Elucidation of the Mechanism. J. Am. Chem. Soc. 2014, 136, 6123–6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen N.; Blount J. F.; Lopresti R. J.; Trullinger D. P. A Novel Sterically Mediated Transformation of Proline. J. Org. Chem. 1979, 44, 4005–4007. [Google Scholar]
- Jarvis C. L.; Richers M. T.; Breugst M.; Houk K. N.; Seidel D. Redox-Neutral α-Sulfenylation of Secondary Amines: Ring-Fused N,S-Acetals. Org. Lett. 2014, 16, 3556–3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.; Das D.; Seidel D. Azomethine Ylide Annulations: Facile Access to Polycyclic Ring Systems. Chem. Sci. 2011, 2, 233–236. [Google Scholar]
- Stockigt J.; Antonchick A. P.; Wu F. R.; Waldmann H. The Pictet-Spengler Reaction in Nature and in Organic Chemistry. Angew. Chem., Int. Ed. 2011, 50, 8538–8564. [DOI] [PubMed] [Google Scholar]
- Bi H.-P.; Zhao L.; Liang Y.-M.; Li C.-J. The Copper-Catalyzed Decarboxylative Coupling of the sp3-Hybridized Carbon Atoms of alpha -Amino Acids. Angew. Chem., Int. Ed. 2009, 48, 792–795. [DOI] [PubMed] [Google Scholar]
- Bi H.-P.; Chen W.-W.; Liang Y.-M.; Li C.-J. A Novel Iron-Catalyzed Decarboxylative Csp3–Csp2 Coupling of Proline Derivatives and Naphthol. Org. Lett. 2009, 11, 3246–3249. [DOI] [PubMed] [Google Scholar]
- Rizzi G. P. Evidence for an Azomethine Ylide Intermediate in the Carbonyl- Assisted Decarboxylation of Sarcosine. Novel Synthesis of dl-Phenylephrine Hydrochloride. J. Org. Chem. 1970, 35, 2069–2072. [Google Scholar]
- Grigg R.; Idle J.; McMeekin P.; Vipond D. The Decarboxylative Route to Azomethine Ylides. Mechanism of 1,3-Dipole Formation. J. Chem. Soc., Chem. Commun. 1987, 49–51. [Google Scholar]
- Zhang C.; Seidel D. Nontraditional Reactions of Azomethine Ylides: Decarboxylative Three-Component Couplings of α-Amino Acids. J. Am. Chem. Soc. 2010, 132, 1798–1799. [DOI] [PubMed] [Google Scholar]
- Orsini F.; Pelizzoni F.; Forte M.; Destro R.; Gariboldi P. 1,3-Dipolar Cycloadditions of Azomethine Ylides with Aromatic Aldehydes. Syntheses of 1-Oxapyrrolizidines and 1,3-Oxazolidines. Tetrahedron 1988, 44, 519–541. [Google Scholar]
- Bi H.-P.; Teng Q.; Guan M.; Chen W.-W.; Liang Y.-M.; Yao X.; Li C.-J. Aldehyde- and Ketone-Induced Tandem Decarboxylation-Coupling (Csp3–Csp) of Natural α-Amino Acids and Alkynes. J. Org. Chem. 2010, 75, 783–788. [DOI] [PubMed] [Google Scholar]
- Das D.; Richers M. T.; Ma L.; Seidel D. The Decarboxylative Strecker Reaction. Org. Lett. 2011, 13, 6584–6587. [DOI] [PubMed] [Google Scholar]
- Ma L.; Chen W.; Seidel D. Redox-Neutral α-Cyanation of Amines. J. Am. Chem. Soc. 2012, 134, 15305–15308. [DOI] [PubMed] [Google Scholar]
- Das D.; Seidel D. Redox-Neutral α-C–H Bond Functionalization of Secondary Amines with Concurrent C–P Bond Formation/N-Alkylation. Org. Lett. 2013, 15, 4358–4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D.; Zhao D.; Mao L.; Wang L.; Wang R. Copper/DIPEA-Catalyzed, Aldehyde-Induced Tandem Decarboxylation-Coupling of Natural α-Amino Acids and Phosphites or Secondary Phosphine Oxides. J. Org. Chem. 2011, 76, 6426–6431. [DOI] [PubMed] [Google Scholar]
- Peshkov V. A.; Pereshivko O. P.; Van der Eycken E. V. A Walk around the A(3)-Coupling. Chem. Soc. Rev. 2012, 41, 3790–3807. [DOI] [PubMed] [Google Scholar]
- Das D.; Sun A. X.; Seidel D. Redox-Neutral Copper (II) Carboxylate Catalyzed α-Alkynylation of Amines. Angew. Chem., Int. Ed. 2013, 52, 3765–3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q.-H.; Meng W.; Jiang G.-J.; Yu Z.-X. CuI-Catalyzed C1-Alkynylation of Tetrahydroisoquinolines (THIQs) by A3 Reaction with Tunable Iminium Ions. Org. Lett. 2013, 15, 5928–5931. [DOI] [PubMed] [Google Scholar]
- Lin W.; Cao T.; Fan W.; Han Y.; Kuang J.; Luo H.; Miao B.; Tang X.; Yu Q.; Yuan W.; Zhang J.; Zhu C.; Ma S. Enantioselective Double Manipulation of Tetrahydroisoquinolines with Terminal Alkynes and Aldehydes under Copper(I) Catalysis. Angew. Chem., Int. Ed. 2014, 53, 277–281. [DOI] [PubMed] [Google Scholar]
- Chen W.; Wilde R. G.; Seidel D. Redox-Neutral α-Arylation of Amines. Org. Lett. 2014, 16, 730–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldar S.; Mahato S.; Jana C. K. Metal- and Oxidant-Free Direct sp3 C-H Arylation of Pyrrolidine. Asian J. Org. Chem. 2014, 3, 44–47. [Google Scholar]
- Arend M.; Westermann B.; Risch N. Modern Variants of the Mannich Reaction. Angew. Chem., Int. Ed. 1998, 37, 1044–1070. [DOI] [PubMed] [Google Scholar]
- Chen W.; Seidel D. The Redox-Mannich Reaction. Org. Lett. 2014, 16, 3158–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soeder R. W.; Bowers K.; Pegram L. D.; Cartaya-Marin C. P. A One Pot Synthesis of 5,7-Diphenyl-2,3-dihydro-1H-pyrrolizine. Synth. Commun. 1992, 22, 2737–2740. [Google Scholar]
- Deb I.; Seidel D. Retro-Claisen Condensation versus Pyrrole Formation in Reactions of Amines and 1,3-Diketones. Tetrahedron Lett. 2010, 51, 2945–2947. [Google Scholar]
- Poirier R. H.; Morin R. D.; McKim A. M.; Bearse A. E. Abnormal Condensation of Piperidinium Acetate with Aromatic Aldehydes. J. Org. Chem. 1961, 26, 4275–4278. [Google Scholar]
- Rügheimer L. Einführung zweiwerthiger Radicale in das Piperidin. Ber. Dtsch. Chem. Ges. 1891, 24, 2186–2187. [Google Scholar]
- Burrows W. D.; Burrows E. P. β-Condensation Reactions of Cyclic Amines with Benzaldehyde: Evidence for the Enamine Pathway. J. Org. Chem. 1963, 28, 1180–1182. [Google Scholar]
- Dannhardt G.; Roelcke J. Investigations Concerning the Benzylation of Tetrahydroisoquinolines with Benzaldehyde and Substituted Benzaldehydes (Ruegheimer-Burrows-Reaction). Arch. Pharm. (Weinheim, Ger.) 1992, 325, 671–677. [Google Scholar]
- Oda M.; Fukuchi Y.; Ito S.; Thanh N. C.; Kuroda S. A Facile Non-Oxidative Method for Synthesizing 1,3-Disubstituted Pyrroles from Pyrrolidine and Aldehydes. Tetrahedron Lett. 2007, 48, 9159–9162. [Google Scholar]
- Pahadi N. K.; Paley M.; Jana R.; Waetzig S. R.; Tunge J. A. Formation of N-Alkylpyrroles via Intermolecular Redox Amination. J. Am. Chem. Soc. 2009, 131, 16626–16627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook A. G.; Switek K. A.; Cutler K. A.; Witt A. N. Synthesis and Isomerization of 3-Pyrroline Enamines. Lett. Org. Chem. 2004, 1, 1–5. [Google Scholar]
- Deb I.; Das D.; Seidel D. Redox Isomerization via Azomethine Ylide Intermediates: N-Alkyl Indoles from Indolines and Aldehydes. Org. Lett. 2011, 13, 812–815. [DOI] [PubMed] [Google Scholar]
- Mao H.; Xu R.; Wan J.; Jiang Z.; Sun C.; Pan Y. Tunable Hydride Transfer in the Redox Amination of Indoline with Aldehyde: An Attractive Intramolecular Hydrogen-Bond Effect. Chem.—Eur. J. 2010, 16, 13352–13355. [DOI] [PubMed] [Google Scholar]
- Ardill H.; Fontaine X. L. R.; Grigg R.; Henderson D.; Montgomery J.; Sridharan V.; Surendrakumar S. X:Y-ZH Compounds As Potential 1,3-Dipoles. Part 29. The Iminium Ion Route to Azomethine Ylides. Reaction of Cyclic Secondary Amines with Mono- And Bifunctional Aldehydes. Tetrahedron 1990, 46, 6449–6466. [Google Scholar]
- Huisgen R.; Grashey R.; Steingruber E. Azomethinylides and Their 1,3-Dipolar Cycloadditions. Tetrahedron Lett. 1963, 1441–1445. [Google Scholar]
- Xue X. S.; Yu A.; Cai Y.; Cheng J. P. A Computational Reinvestigation of the Formation of N-Alkylpyrroles via Intermolecular Redox Amination. Org. Lett. 2011, 13, 6054–6057. [DOI] [PubMed] [Google Scholar]
- Chen W.; Kang Y.; Wilde R. G.; Seidel D. Redox-Neutral α,β-Difunctionalization of Cyclic Amines. Angew. Chem., Int. Ed. 2014, 53, 5179–5182. [DOI] [PMC free article] [PubMed] [Google Scholar]