

Abstract
Background
In myocardial infarction (MI), repolarization alternans is a potent arrhythmia substrate that has been linked to Ca2+ cycling proteins, such as SERCA2a, located in the sarcoplasmic reticulum (SR). MI is also associated with oxidative stress and increased xanthine oxidase (XO) activity, an important source of reactive oxygen species (ROS) in the SR that may reduce SERCA2a function. We hypothesize that in chronic MI, XO mediated oxidation of SERCA2a is a mechanism of cardiac alternans.
Methods and Results
Male Lewis rats underwent ligation of the LAD (n=54) or sham procedure (n=24). At 4 weeks, optical mapping of intracellular Ca2+ and ROS were performed. ECG T-wave alternans (ECG ALT) and Ca2+ transient alternans (Ca2+ALT) were induced by rapid pacing (300–120ms) before and after the XO inhibitor allopurinol (ALLO, 50μmol/L). In MI, ECG ALT (2.32±0.41%) and Ca2+ ALT (22.3±4.5%) were significantly greater compared to sham (0.18±0.08%, p<0.001; 0.79±0.32%, p<0.01). Additionally, ROS was increased by 137% (p<0.01) and oxidation of SERCA2a by 30% (p<0.05) in MI compared to sham. Treatment with ALLO significantly decreased ECG ALT (−77±9%, p<0.05) and Ca2+ ALT (−56±7%, p<0.05) and, importantly, reduced ROS (−65%, p<0.01) and oxidation of SERCA2a (−38%, p<0.05). CaMKII inhibition and general antioxidant treatment had no effect on ECG ALT and Ca2+ ALT.
Conclusions
These results demonstrate, for the first time, that in MI increased ROS from XO is a significant cause of repolarization alternans. This suggests that targeting XO ROS production may be effective at preventing arrhythmia substrates in chronic MI.
Keywords: myocardial infarction, oxidative stress, arrhythmia (mechanisms), SERCA2a, allopurinol
Introduction
Sudden cardiac death following myocardial infarction (MI) is the most common cause of mortality from heart disease1. Furthermore, declining death rates from acute MI2 have increased the population of patients with chronic MI that are at risk for developing ventricular arrhythmias1. In patients with chronic MI, cardiac repolarization alternans is a potent arrhythmia substrate3–5. As we6–8 and others9, 10 have shown, abnormal intracellular Ca2+ cycling is a well-established mechanism of alternans. Specifically, we have recently shown that the sarcoplasmic reticulum (SR) Ca2+ ATPase (SERCA2a), an important regulator of SR calcium reuptake, can significantly modulate the magnitude of alternans8.
Reactive oxygen species (ROS) production is increased in many diseases, including ischemic heart disease11. Acute ROS exposure in isolated myocytes and whole hearts has been shown to significantly disrupt intracellular Ca2+ regulation12 and promote a broad range of arrhythmia substrates9, 13. Interestingly, however, heart disease patients do not see a reduction in cardiac events with general antioxidant therapy14–16 which raises two interesting possibilities: either increased oxidative stress is not a determinant of cardiac events, or targeting a specific source of ROS is more important. Support of the latter is evidenced by Sovari et al., who have recently shown that targeting mitochondrial ROS is effective at suppressing in vivo arrhythmias associated with connexin proteins in a non-ischemic heart failure model17.
Xanthine oxidase (XO) is a superoxide producing enzyme that is localized to the sarcoplasmic reticulum (SR)18, 19. We have recently shown that inhibition of XO activity can normalize the oxidative status of key SR calcium regulatory proteins20. Furthermore, in MI, XO activity is increased21–23. Based on this and the prominent role SERCA2a plays in Ca2+ cycling, we hypothesize that in chronic MI, XO mediated oxidation of SERCA2a is a mechanism of cardiac alternans.
Methods
Chronic Myocardial Infarction Model
This investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH publication no. 85–23, revised 1996) and was approved by the Institutional Animal Care and Use Committee of Case Western Reserve University. To create a model of chronic MI, we performed permanent ligation of the left anterior descending coronary artery (LAD) in male Lewis rats weighing 300–350 g (n = 54), as described previously24. Sham (n = 24) surgeries were performed as controls, and animals with no procedure (n=4) were used to assess nonspecific drug effects. After 4 weeks, animals were sacrificed for optical mapping and tissue sample analysis.
Optical Mapping
Rats were anesthetized (0.3cc Ketamine/0.1 cc Xylazine, intraperitoneal), and their hearts were rapidly removed via medial thoracotomy and Langendorff perfused with oxygenated (95% O2-5% CO2) Tyrode’s solution containing (mmol/L): 137 NaCl, 5.4 KCl, 3.0 CaCl2, 1 MgSO4, 5.0 dextrose, and 10 HEPES (pH 7.45, 34°C). Perfusion pressure was maintained between 50 and 70 mmHg by regulating coronary flow using a pulsatile flow system. To measure intracellular calcium, hearts were stained with the calcium-sensitive indicator Indo-1 AM (Invitrogen) at a final concentration of 10 μmol/L for 30 min at room temperature to minimize dye compartmentalization, followed by a 15-min washout period. The same procedure was used in separate animals to load the ROS sensitive dye, 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (DCF, Invitrogen), at a final concentration of 20 μmol/L. In all experiments, 5 μmol/L of Blebbistatin (Enzo) was used to ensure that motion artifact did not influence our results.
Perfused hearts were placed in a Lexan chamber, and the mapping field was positioned over anterior-superior aspect of the heart. The ECG was monitored by using three Ag-AgCl disk electrodes fixed to the chamber in positions corresponding to ECG limb leads I, II, and III. ECG signals were filtered (0.3–300 Hz), amplified (1,000x), and displayed on an oscilloscope. A fine-gauge (0.003″ diameter), bipolar electrode was placed on the left ventricular anterior wall to stimulate the heart at twice diastolic threshold current. Physiological stability of the preparation was assured by monitoring the ECG, coronary pressure, coronary flow, and perfusion temperature continuously throughout each experiment.
To measure Ca2+ transients, Indo-1 was excited by light from a 365 nM, 500 mW LED (Nichea). Fluorescent light from the preparation was collected with a tandem lens configuration, and a 445 nm long-pass filter (Chroma Technology) that transmitted light to a 16 × 16 element photodiode array (Hamamatsu). For calcium measurements, an optical magnification of x1.24 was used, resulting in a total mapping field of 14.2 mm × 14.2 mm, with 0.9-mm spatial resolution and 0.81-mm2 pixel size.
To measure ROS separately in the whole hearts, DCF was excited by light from a cyan Luxeon Rebel LED (Philips) filtered through a 485±20 nm optical bandpass filter. Fluorescent light from the preparation was filtered with a 535±30 nm optical bandpass filter (which also blocks NADH auto fluorescence) and recorded using a MiCam02-HR CCD camera (SciMedia) at the same time in every experiment immediately after DCF loading. For ROS measurements, an optical magnification of x0.41 was used, resulting in a total mapping field of 11.7 mm × 15.6 mm.
Experimental Protocol
Decremental rapid pacing (300 – 120 ms) was used to elicit calcium transient alternans under baseline conditions in sham and MI hearts. To inhibit XO, allopurinol (ALLO, 50 μmol/L, Sigma-Aldrich), was perfused for 15 min. To test for CaMKII activity, KN-93 (0.5 μmol/L) or its inactive analogue KN-92 (0.5 μmol/L), was perfused for 15 min. The nonspecific antioxidant N-acetylcysteine (NAC, 10 μmol/L) was perfused for 15 min to scavenge ROS. After each treatment (performed in separate animals), the alternans induction protocol was repeated. Finally, arrhythmia incidence was assessed by quantifying the occurrence of extra beats or VT/VF following termination of rapid pacing (120 ms) with one-to-one capture.
ROS Measurement in Frozen Sections
Fresh heart tissue was frozen in Tissue-Tek O.C.T., sectioned into 20 μm slices along the short axis, washed with PBS, and stained with 10 μmol/L DCF for 30 min at 37° C 25. DCF fluorescence was then imaged using an epifluorescence microscope (Olympus).
Monobromobimane Assay
SERCA2a oxidation (SERCA2a-SOx) was measured using the free-thiol specific fluorescent tag, monobromobimane (mBB, Enzo), similar to that published previously9, 26. Briefly, fresh heart tissue was frozen using liquid-N2, powdered, and suspended in HEN buffer (mmoL/L): Hepes 50, 1 EDTA 1, and neocuproine 0.1 (pH 7.7). Membrane bound proteins were isolated by centrifugation at 16,000 × g for 25 min (4° C). The supernatant was then split and incubated for 30 min with the reducer N-Ethylmaleimide (NEM, 4 mmol/L), oxidizer Dithiothreitol (DTT, 5 mmol/L), or untreated. All samples were then treated with mBB (5 mmol/L) for 1 hour. Samples were run on a 7.5% SDS-PAGE gel and mBB fluorescence was imaged using a Gel Doc™ XR+ (Bio-Rad). Subsequently, Brilliant Blue G (Sigma-Aldrich) staining was used to normalize for SERCA2a loading within each sample. Finally, untreated fluorescence measurements were normalized to the maximum (DTT) and minimum (NEM) fluorescence values.
ICa,L Measurements
Myocytes were isolated enzymatically from rat hearts using the enzymatic dispersion technique described previously27. ICa,L was recorded by ruptured-patch whole cell voltage clamp at 35° C. Microelectrodes were fabricated from TW150F borosilicate glass capillaries and filled with a solution of (mmol/L): CsMES 130, TEA Cl 20, MgCl2 1, HEPES 10, EGTA 10, TRIS GTP 0.3, Phosphocreatine 14, Mg ATP 4, and Creatine phosphokinase 2 (pH 7.2). Isolated myocytes were placed in a solution of (mmol/L): NaCl 137, CsCl 5.4, MgCl21.8, CaCl22, glucose 10, and HEPES 10 (pH 7.3). ICa,L was elicited from a holding potential of −40 mV with depolarizing voltage pulses from −30 mV to 60 mV for 300 ms. Ionic current density (pA/pF) was calculated from the ratio of current amplitude to cell capacitance. Command and data acquisition were operated with an Axopatch 200B patch clamp amplifier controlled by a PC using a Digidata 1200 acquisition board driven by pCLAMP 7.0 (Axon Instruments).
Data Analysis
ECG T-wave alternans (ECG ALT) was measured by signal averaging consecutive beat pairs over a 5 sec window. The beat averages were time aligned by the pacing artifact, and the peak-to-peak amplitude was normalized to 100%. Then, the average percent difference between beat pairs from the end of the QRS to the end of the T wave was calculated28. Calcium transient alternans (Ca2+ ALT) were measured as the difference in amplitude between two consecutive beats, expressed as a percentage of the largest beat6 and averaged across all 256 channels within the mapping field. For regional analysis, any site within 2 mm of the visible scar was considered border zone (BZ) and all other sites were considered remote (REM). Calcium transient duration was calculated from the time of release to 85% of diastolic values. For the optical mapping measurements of ROS, DCF fluorescence (F) at each site was normalized to the mean background fluorescence (FO) after staining from an area of viable tissue representing 5% of the mapping field. For each heart, DCF fluorescence was quantified by averaging over all recording sites. In a couple of experiments, signal quality near the scar was not suitable for analysis or 1-to-1 pacing capture was lost. Such experiments were not included in the analysis. For unpaired comparisons with small sample size, Wilcoxon rank-sum test was used. Otherwise, student’s paired and unpaired t-tests were performed where appropriate. In situations with unequal data variance, we used the Welch t-test with unpooled variance and Satterthwaite equation. Fisher’s exact test was used for statistical analysis of categorical data. Comparisons were considered significant for p-values < 0.05. Values reported are mean±SE.
Results
Cardiac alternans is significantly increased in MI compared to sham operated animals. Shown in figure 1A are representative ECG recordings from isolated sham and MI hearts measured during steady state pacing at a cycle length (CL) of 120 ms. In the sham heart, two consecutive beats (a and b) are superimposed and demonstrate the absence of ECG ALT (beats a and b are identical). In contrast, ECG ALT is evident in the MI heart. Over all animals tested, ECG ALT measured at the same CL is significantly higher in MI (2.32±0.41%, n=20) compared to shams (0.18±0.08%, n=8, p < 0.001). Similarly, Ca2+ transient alternans (Ca2+ ALT) is larger in MI hearts compared to shams. This can be seen in the Ca2+ recordings shown in Panel B that demonstrate the absence of beat-to-beat Ca2+ ALT in a sham heart, and large Ca2+ ALT in a heart with MI paced at the same CL (120 ms). Similar to ECG ALT, Ca2+ ALT is significantly higher in MI (22.3±4.5%, n=7) compared to shams (0.79±0.32%, n=8, p < 0.01). Shortening fraction is significantly less in MI 25±4% compared to shams 55±2% (p<0.01). These data demonstrate that Ca2+ ALT and global repolarization alternans (ECG ALT) are higher in MI compared to shams.
Figure 1.

A: Representative ECG of two consecutive paced beats (120 ms CL) normalized peak-to-peak and superimposed (a and b) to demonstrate ECG T-wave alternans (ECG ALT) in an MI heart but not in a sham. Summary data (right) show that over all experiments MI hearts (n=20) had significantly higher ECG ALT compared to sham hearts (n=8). B: Representative intracellular Ca2+ transients from a sham and MI heart at a pacing cycle length of 120 ms and normalized peak-to-peak demonstrating beat-to-beat Ca2+ transient amplitude alternans (Ca2+ ALT) in MI but not shams. Summary data show that over all experiments, MI hearts (n=7) had significantly higher Ca2+ ALT as compared to sham hearts (n=8).
Previous studies have associated a slower decay of the Ca2+ transient with greater cardiac alternans8. In the present study we also found that the duration of the Ca2+ transient, which depends on the decay phase of the Ca2+ transient, is longer in MI compared to shams. Figure 2A shows representative examples of Ca2+ transients recorded from a sham and MI heart at the same pacing CL (300 ms). When measured at 85% of Ca2+ transient amplitude, the durations are on average significantly longer in MI (n=6) compared to sham (n=5, p<0.05). The decay of the Ca transient is also slower in MI (119±7 ms) compared to sham (101±6 ms). Interestingly, we found no significant change in the expression of SERCA2a (Panel B) in animals with MI (1.2±0.03 au) compared to shams (1.1±0.12 au, p = 0.31). These results suggest that the increase in cardiac alternans observed in MI is due to slower Ca2+ cycling, which may be explained by decreased SERCA2a function rather than reduced expression.
Figure 2.

A: Representative intracellular Ca2+ transients recorded at a slow pacing cycle length (300 ms) and normalized peak-to-peak demonstrate a longer duration (measured at 85% of diastole) in MI compared to sham hearts. Summary data shows that Ca2+ transient durations are significantly prolonged in MI hearts (n=6) compared to shams (n=5). B: Western blots against SERCA2a and actin (loading control) for sham and MI hearts. Summary data shows that across all hearts (n=3 for each) SERCA2a protein expression was unchanged in MI.
When Ca2+ ALT is compared across the heart surface, the largest levels are observed near the scar. Shown in Figure 3 are representative examples of the spatial distribution of Ca2+ ALT in a sham and MI heart paced at the same CL (120ms). In the sham heart, no Ca2+ ALT is observed across the mapping field. In contrast, in the MI heart Ca2+ ALT is maximum near the scar (dashed outline). In the center of the scar no Ca2+ ALT is observed, however this is because Ca2+ signals are absent due to poor dye perfusion or the presence of non-viable tissue.
Figure 3.
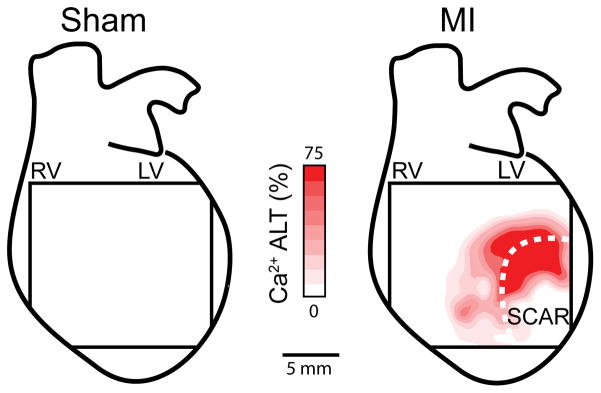
Representative contour maps of epicardial Ca2+ ALT magnitude induced by rapid pacing (120 ms) in a sham and MI heart, overlaid on a schematic of the whole heart. The size of the mapping field corresponds to 14.2 × 14.2 mm. Ca2+ ALT is absent in the sham, however, large Ca2+ ALT are present in the region of the scar (dashed line) of the MI heart.
The high level of Ca2+ ALT and prolonged Ca2+ transient duration observed in MI hearts may be related to increased ROS associated with MI. Images of DCF fluorescence measured in an isolated MI heart using optical mapping techniques reveal increased ROS, especially near the scar (Figure 4). In contrast, ROS is much lower from the same region in a sham heart. When ROS levels are normalized for each animal, significantly higher levels (136.9±39%, p < 0.01) are observed in MI hearts (n=8) compared to shams (n=7). In separate experiments, several hearts were prepared for in situ ROS detection in tissue slices using DCF (Panel B). This method, which is independent of dye perfusion (unlike measurements in the whole heart), also showed increased levels of ROS associated with MI. Shown at the bottom (Panel C) is Ca2+ ALT plotted against ROS for SHAM hearts and for MI hearts that were divided into remote (REM) and border zone (BZ) regions. Overall, Ca2+ ALT is positively correlated with ROS levels.
Figure 4.

A: Contour maps of ROS (green), determined by measuring DCF fluorescence from the epicardial surface of a sham, MI, and MI+ALLO heart, overlaid on a schematic of the whole heart. B: Shown are consecutive transverse cross-sections (20 μm) from an MI heart. The first slice was stained with H&E (left) and the second was superfused with DCF (right). Black arrows point to the epicardial scar. C: Correlation between ROS and Ca2+ ALT (measured in separate animals) for all sham hearts along with the BZ and REM regions of all MI hearts.
It is possible that increased ROS associated with MI is responsible for the increased Ca2+ ALT we observed. In MI hearts treated with ALLO, a specific inhibitor of XO, ROS (1.3±0.1 F/Fo, n=8) was significantly lower compared to MI (3.7±2.8 F/Fo, n=8, p < 0.005), as demonstrated in a representative example (Figure 4A, ALLO+MI). Cardiac alternans was also measured before and after the administration of ALLO. Shown in Figure 5A are ECG ALT and Ca2+ ALT in MI before (MI) and then after ALLO administration (MI+ALLO). The representative traces show that ALLO essentially eliminated beat-to-beat ECG ALT and Ca2+ ALT. On average, ALLO significantly decreased ECG ALT (−77±9%, p < 0.05) and Ca2+ ALT (−56±7%, p <0.05) in paired comparisons (n=5). Ca2+ ALT contour maps from an MI heart before and after ALLO demonstrate a significant reduction of Ca2+ ALT (Panel B). Panel C demonstrates the effectiveness of ALLO (open circles) compared to MI alone (filled circles) on Ca2+ ALT and ROS for BZ and REM regions. Time controls in MI animals showed no change in alternans over a similar time period (0.1±4%, p = ns). Finally, arrhythmia incidence was reduced in MI+ALLO (11%) compared to MI (44%), but this did not reach statistical significance.
Figure 5.

Acute treatment with allopurinol (ALLO) decreases alternans in chronic MI. A: Shown (left) are examples of two consecutively paced beats (a and b, 120ms) normalized peak-to-peak that have been superimposed to demonstrate ECG ALT from the same heart, before (MI) and after ALLO (MI+ALLO). Shown (right) is a representative example of Ca2+ ALT from the same location normalized peak-to-peak before and after ALLO. Summary data show that across all experiments average ECG ALT (n=5) and Ca2+ ALT (n=5) in MI was significantly reduced following ALLO administration. B: Shown are representative contour maps of epicardial Ca2+ ALT magnitude induced by rapid pacing (120 ms) in an MI heart before and after ALLO, overlaid on a schematic of the whole heart. The size of the mapping field corresponds to 14.2 × 14.2 mm. Ca2+ ALT is present in the region of the scar (dashed line) of the MI heart and is attenuated by ALLO. C: ROS plotted against Ca2+ ALT for MI (filled circles) and MI+ALLO (open circles) in BZ and REM regions (connected by dashed line).
Previous studies have shown that ROS decreases SERCA2a function12, which may explain why Ca2+ transients are longer in MI. If so, inhibiting XO ROS with ALLO should shorten Ca2+ transient duration. Shown in Figure 6 are Ca2+ transients measured before and after ALLO treatment. The representative traces show that ALLO decreases Ca2+ transient duration at 85% from 249 to 195 ms. In paired comparisons of average data (right, n=5), ALLO significantly shortened average Ca2+ transient duration over all animals tested (p < 0.05). In addition, the decay phase of the Ca2+ transient was significantly faster for ALLO+MI (108±7 ms) compared to MI (119±7 ms, p < 0.01) in the BZ; however, there was no difference in the REM region. It is also possible that phospholamban (PLB) phosphorylation is playing a role independent of SERCA2a. No difference in total PLB was observed (Panel C, left). However, while MI was associated with a slight decrease in PLB phosphorylation, ALLO had no effect (Panel C, right). Thus, the reduction of alternans by ALLO is independent of PLB phosphorylation.
Figure 6.

A: Ca2+ transients normalized peak-to-peak that were recorded at a slow pacing rate (300 ms) from the same location of an MI heart before (MI) and after ALLO treatment (MI+ALLO). Ca2+ transient duration, as measured at 85% of diastole, decreased from 249 to 195 ms following ALLO. Over all experiments ALLO significantly shortened the average Ca2+ transient duration in the setting of MI (n=5). B: Ca2+ decay (tau) from the BZ (left) and REM (right) in MI hearts before (MI) and after ALLO treatment (MI+ALLO). C: Total (left) and phospholamban (PLB) phosphorylation (right) determined in sham (n=3), MI (n=3) and MI+ALLO (n=3).
To determine if blocking XO with ALLO decreased SERCA2a-SOx, free thiols were measured in separate sham hearts (n=4) and in MI hearts treated with (n=5) or without ALLO (n=5). Shown in Figure 7 (top) is a representative, continuous blot of mBB fluorescence in freshly prepared tissue samples from a sham, MI, and MI heart treated with ALLO. Shown below are protein levels for SERCA2a (110 KD), which were used to normalize mBB fluorescence. For each group, the untreated sample was normalized to that treated with DTT (reducer, maximum free thiols) and NEM (oxidizer, minimum free thiols). The higher free thiol level in MI+ALLO and SHAM compared to MI indicates that ALLO increased free thiols (reduced SERCA2a-SOx) to normal levels (Figure 7, bottom). These results suggest that cardiac alternans observed in chronic MI is due, in part, to oxidation of SERCA2a by XO.
Figure 7.

Representative, continuous monobromobimane (mBB) fluorescence image of SERCA2a free thiols (inverse of SERCA2a-SOx) and SERCA2a protein (110 KD) from a sham, untreated (MI) and ALLO treated MI heart (MI+ALLO). DTT (fully reduced) and NEM (fully oxidized) were used to normalize fluorescence intensities. Greater fluorescence intensity indicates more free (less oxidized) thiols. Pooled data (bottom) demonstrate that treatment with ALLO (n=5) significantly increases the number of SERCA2a free thiols (decreases oxidation) compared to MI (n=5, p < 0.05) but not compared to sham (n=4, p = ns).
It is also possible that in MI, ROS activates CaMKII, which has been previously associated with increased alternans29. However, as shown in Figure 8, when hearts were treated with KN-93 or its inactive analog KN-92, no significant change in Ca2+ ALT was observed. In comparison, treatment with ALLO (ΔCa2+ ALT calculated from Figure 5) caused a significant decrease in Ca2+ ALT. Similarly, acute treatment with the general antioxidant, NAC, had no effect on alternans. In addition, we have previously shown that ALLO has no effect on ICa,L 20 and in the present study we found no change in peak ICa,L or ICa,L inactivation (inset) from MI hearts compared to shams (p=ns). However, the reversal potential was increased in MI (64±2 mV) compared to sham (55±1 mV, p<0.01). Nevertheless, changes in ICa,L cannot explain the occurrence of repolarization alternans in our model of MI or the effects of ALLO. It is also possible that ALLO decreased APD, which could also decrease Ca2+ ALT independent of any changes in Ca2+ regulation. However, QT interval if anything, was slightly prolonged by 16±7 ms with ALLO, however this did not reach statistical significance. Finally, in control animals (n=4), ALLO had no effect on ECG QT interval (1±1%, p= ns), ECG QRS width (0±4%, p=ns), CaD (1±1% p=ns), or the decay of the calcium transient (1±11%, p=ns). Taken together, these data demonstrate that cardiac alternans associated with MI is due to, in large part, XO mediated oxidation of SERCA2a.
Figure 8.

A: Summary data showing that unlike ALLO (n=5), neither the CaMKII inhibitor (KN-93, n=6), its inactive analogue (KN-92, n=5), nor the general antioxidant N-acetylcysteine (NAC, n=4) significantly reduces Ca2+ transient alternans (Ca2+ ALT). B: Current-voltage curves show that the L-type Ca2+ current (ICa,L) and current inactivation (inset) are unchanged in MI compared to Sham (5–7 cells for 2 animals in each group).
Discussion
The primary findings of this report are: (1) ECG T-wave and Ca2+ transient alternans in chronic MI are associated with elevated ROS and reduced SR Ca2+ cycling, (2) in chronic MI, decreased SERCA2a-SOx enhanced SR Ca2+ cycling and reduced ECG T-wave and Ca2+ transient alternans, and (3) increased ROS from XO is an important mechanism of SERCA2a-SOx and, thus, cardiac alternans in chronic MI. Importantly, this study is the first to report that XO mediated oxidation of SERCA2a plays a significant role in repolarization alternans associated with MI. These results increase the growing body of evidence that suggests arrhythmogenic ROS production is ubiquitous in cardiovascular disease, and that targeting specific oxidant sources may be effective at preventing sudden cardiac death.
Alternans Mediated by Oxidative Stress in Chronic MI
Like acute ischemia 30, 31, chronic MI has been previously associated with cardiac alternans in experimental models4, 5 and in humans3. However, much less in known about the mechanisms of alternans in the setting of chronic MI. Previous studies have shown that oxidative stress is increased in MI32, and that increased oxidative stress can disrupt Ca2+ cycling. For example, Belevytch et al. have shown that increased ROS in chronic MI causes alternans by increasing RyR oxidation9. Similarly, isolated myocyte and whole heart studies have demonstrated that acute oxidative stress, in the form of H2O2 administration, slows SERCA2a mediated Ca2+ cycling12, 33 and causes action potential duration alternans and arrhythmias13. This is consistent with previous reports showing that reduced SERCA2a is a well-known mechanism of Ca2+ transient and repolarization alternans8, 10, 34, 35. In the present study we show that alternans associated with chronic MI is secondary to increased ROS and resultant SERCA2a-SOx. Our results could have also been explained by decreased SERCA2a expression, however, this is not what we found, and decreased SERCA2a expression is not a universal finding in chronic MI36.
CaMKII activity is increased in heart disease and has been previously shown to significantly modulate Ca2+ regulatory proteins and cause arrhythmias37. In addition, modeling studies have shown that increased CaMKII activity is linked to increased cardiac alternans29. Since ROS can activate CaMKII38, it is possible that the increase in alternans we observed is due to increased CaMKII activity. Surprisingly, we saw no significant effect of the CaMKII inhibitor KN-93 or its inactive analogue KN-92 on alternans activity. This does not necessarily mean that CaMKII is unrelated to alternans. Rather, it is possible that CaMKII is not significantly increased in the rat model of chronic MI, as previously shown39.
Role of Xanthine Oxidase (XO) in Alternans
Cardiac alternans is highly dependent on SR Ca2+ regulatory proteins and their redox status. XO, an important source of ROS, is expressed in the SR where it is inhibited by neuronal nitric oxide synthase (nNOS)18. In ischemic heart failure, nNOS can translocate to the sarcolemma, relieving its inhibition of XO, which may explain increased ROS local to the SR21,22. We have previously shown that ALLO, a specific blocker of XO, can normalize RyR oxidation (RyR-SOx) secondary to nNOS inhibition20. Similarly, Gonzalez et al.18, have shown that ALLO reduces myocardial ROS and improves RyR function in a non-ischemic rat heart failure model. In addition to the effect ALLO has on RyR, we found in the present study that ALLO reduced alternans secondary to increased rate of Ca2+ cycling and reduced SERCA2a-SOx. Previous studies have shown that oxidation of SERCA2a is associated with reduced function, which is consistent with our findings33. ALLO can also reduce NCX,40 but this would slow Ca2+ cycling and promote Ca2+alternans, which is not what we observed. Thus, our results suggest that SERCA2a-SOx, along with RyR oxidation shown by others9, are important mechanisms of arrhythmia and cardiac alternans associated with chronic MI.
Interestingly, we found that acute treatment with the general antioxidant, NAC, had no effect on alternans activity (Figure 8). This has two important implications. First, this result suggests limited ROS-induced ROS-release between mitochondria and XO in our model, because previous studies have shown that NAC can suppress ROS-induced ROS-release41. Second, this result suggests that targeted inhibition of XO rather than a general antioxidant can reduce SERCA2a-SOx and, thus, alternans. This idea is consistent with a recent study by Sovari et al.17, who showed that targeted mitochondrial antioxidant therapy, as opposed to non-specific antioxidants, can decrease reentrant arrhythmias in a sudden cardiac death mouse model by preventing ROS mediated connexin 43 destabilization. The present study and that by Sovari et al.17 may explain why clinical trials have shown that non-specific antioxidants, such as Vitamin C and E, are ineffective at improving cardiac outcomes14, 15. Importantly, the present study adds further evidence in support of the idea that targeted antioxidant therapies may be an effective approach to reduce cardiac events associated with heart disease.
Clinical Implications
Our results suggest that treatment with ALLO may be effective at ameliorating cardiac alternans in patients with chronic MI. Recent clinical trials have shown that ALLO improves myocardial relaxation in patients with ischemic heart disease, as evidenced by a trend towards reduced left ventricular end-diastolic volume (LVEDV)42. Moreover, overexpression of SERCA2a in humans may also decrease LVEDV43. Thus, our result demonstrating that ALLO treatment increases SERCA2a function is consistent with these findings in patients with heart disease. However, such hemodynamic parameters must be compared cautiously with SERCA2a function. It is important to note that oxypurinol (active ALLO metabolite) has been shown to be ineffective at improving outcomes in a moderate to severe heart failure population, but there was a trend for reduced mortality in patients with high uric acid levels (XO activity)44. The EXACT-HF trial is currently underway to determine the benefit of ALLO for heart failure patients with high serum uric acid levels (ClinicalTrials.gov, NCT00987415).
Limitations
We did not measure alternans throughout the whole heart; however, we observed significant ECG T-wave ALT, suggesting alternans over large portions of the heart. ALLO treatment did not completely ameliorate alternans, suggesting the involvement of other molecular mechanisms. For example, RyR can influence alternans in chronic MI independent of SERCA2a activity9, and ATP depletion associated with chronic MI can have similar action45. In addition, we cannot exclude the influence of other ROS sources beyond XO, such as the mitochondria46 or NADPH oxidases47. While we cannot exclude these possibilities, they are independent of and, thus, not mutually exclusive with SERCA2a-SOx by XO as a cause of repolarization alternans in chronic MI.
Acknowledgments
Funding Sources: This material is based upon work supported by NIH grants HL-084142 and HL-100105 (KRL), and NIH grants HL-094450 and HL-096962 (ID).
Footnotes
Conflict of Interest Disclosures: None.
References
- 1.Adabag AS, Luepker RV, Roger VL, Gersh BJ. Sudden cardiac death: Epidemiology and risk factors. Nat Rev Cardiol. 2010;7:216–225. doi: 10.1038/nrcardio.2010.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yeh RW, Sidney S, Chandra M, Sorel M, Selby JV, Go AS. Population trends in the incidence and outcomes of acute myocardial infarction. N Engl J Med. 2010;362:2155–2165. doi: 10.1056/NEJMoa0908610. [DOI] [PubMed] [Google Scholar]
- 3.Chow T, Keriakes D, Bartone C, Booth T, Schloss E, Waller T, Chung E, Menon S, Nallamothu B, Chan P. Prognostic utility of microvolt t-wave alternans in risk stratification of patients with ischemic cardiomyopathy. J Am Coll Cardiol. 2006;47:1820–1827. doi: 10.1016/j.jacc.2005.11.079. [DOI] [PubMed] [Google Scholar]
- 4.Lou Q, Efimov IR. Enhanced susceptibility to alternans in a rabbit model of chronic myocardial infarction. Conf Proc IEEE Eng Med Biol Soc. 2009;2009:4527–4530. doi: 10.1109/IEMBS.2009.5334102. [DOI] [PubMed] [Google Scholar]
- 5.Chou CC, Zhou S, Hayashi H, Nihei M, Liu YB, Wen MS, Yeh SJ, Fishbein MC, Weiss JN, Lin SF, Wu D, Chen PS. Remodelling of action potential and intracellular calcium cycling dynamics during subacute myocardial infarction promotes ventricular arrhythmias in langendorff-perfused rabbit hearts. J Physiol. 2007;580:895–906. doi: 10.1113/jphysiol.2006.120659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pruvot EJ, Katra RP, Rosenbaum DS, Laurita KR. Role of calcium cycling versus restitution in the mechanism of repolarization alternans. Circ Res. 2004;94:1083–1090. doi: 10.1161/01.RES.0000125629.72053.95. [DOI] [PubMed] [Google Scholar]
- 7.Wilson LD, Jeyaraj D, Wan X, Hoeker GS, Said TH, Gittinger M, Laurita KR, Rosenbaum DS. Heart failure enhances susceptibility to arrhythmogenic cardiac alternans. Heart Rhythm. 2009;6:251–259. doi: 10.1016/j.hrthm.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cutler MJ, Wan X, Laurita KR, Hajjar RJ, Rosenbaum DS. Targeted serca2a gene expression identifies molecular mechanism and therapeutic target for arrhythmogenic cardiac alternans. Circ Arrhythm Electrophysiol. 2009;2:686–694. doi: 10.1161/CIRCEP.109.863118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belevych AE, Terentyev D, Viatchenko-Karpinski S, Terentyeva R, Sridhar A, Nishijima Y, Wilson LD, Cardounel AJ, Laurita KR, Carnes CA, Billman GE, Gyorke S. Redox modification of ryanodine receptors underlies calcium alternans in a canine model of sudden cardiac death. Cardiovasc Res. 2009;84:387–395. doi: 10.1093/cvr/cvp246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xie LH, Sato D, Garfinkel A, Qu Z, Weiss JN. Intracellular ca alternans: Coordinated regulation by sarcoplasmic reticulum release, uptake, and leak. Biophys J. 2008;95:3100–3110. doi: 10.1529/biophysj.108.130955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McMurray J, McLay J, Chopra M, Bridges A, Belch JJ. Evidence for enhanced free radical activity in chronic congestive heart failure secondary to coronary artery disease. Am J Cardiol. 1990;65:1261–1262. doi: 10.1016/0002-9149(90)90985-a. [DOI] [PubMed] [Google Scholar]
- 12.Greensmith DJ, Eisner DA, Nirmalan M. The effects of hydrogen peroxide on intracellular calcium handling and contractility in the rat ventricular myocyte. Cell Calcium. 2010;48:341–351. doi: 10.1016/j.ceca.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 13.Morita N, Sovari AA, Xie Y, Fishbein MC, Mandel WJ, Garfinkel A, Lin SF, Chen PS, Xie LH, Chen F, Qu Z, Weiss JN, Karagueuzian HS. Increased susceptibility of aged hearts to ventricular fibrillation during oxidative stress. Am J Physiol Heart Circ Physiol. 2009;297:H1594–1605. doi: 10.1152/ajpheart.00579.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sesso HD, Buring JE, Christen WG, Kurth T, Belanger C, MacFadyen J, Bubes V, Manson JE, Glynn RJ, Gaziano JM. Vitamins e and c in the prevention of cardiovascular disease in men: The physicians’ health study ii randomized controlled trial. JAMA. 2008;300:2123–2133. doi: 10.1001/jama.2008.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yusuf S, Dagenais G, Pogue J, Bosch J, Sleight P. Vitamin e supplementation and cardiovascular events in high-risk patients. The heart outcomes prevention evaluation study investigators. N Engl J Med. 2000;342:154–160. doi: 10.1056/NEJM200001203420302. [DOI] [PubMed] [Google Scholar]
- 16.Vivekananthan DP, Penn MS, Sapp SK, Hsu A, Topol EJ. Use of antioxidant vitamins for the prevention of cardiovascular disease: Meta-analysis of randomised trials. Lancet. 2003;361:2017–2023. doi: 10.1016/S0140-6736(03)13637-9. [DOI] [PubMed] [Google Scholar]
- 17.Sovari AA, Rutledge CA, Jeong EM, Dolmatova E, Arasu D, Liu H, Vahdani N, Gu L, Zandieh S, Xiao L, Bonini MG, Duffy HS, Dudley SC., Jr Mitochondria oxidative stress, connexin43 remodeling, and sudden arrhythmic death. Circ Arrhythm Electrophysiol. 2013;6:623–631. doi: 10.1161/CIRCEP.112.976787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gonzalez DR, Treuer AV, Castellanos J, Dulce RA, Hare JM. Impaired s-nitrosylation of the ryanodine receptor caused by xanthine oxidase activity contributes to calcium leak in heart failure. J Biol Chem. 2010;285:28938–28945. doi: 10.1074/jbc.M110.154948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khan SA, Lee K, Minhas KM, Gonzalez DR, Raju SV, Tejani AD, Li D, Berkowitz DE, Hare JM. Neuronal nitric oxide synthase negatively regulates xanthine oxidoreductase inhibition of cardiac excitation-contraction coupling. Proc Natl Acad Sci U S A. 2004;101:15944–15948. doi: 10.1073/pnas.0404136101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cutler MJ, Plummer BN, Wan X, Sun QA, Hess D, Liu H, Deschenes I, Rosenbaum DS, Stamler JS, Laurita KR. Aberrant s-nitrosylation mediates calcium-triggered ventricular arrhythmia in the intact heart. Proc Natl Acad Sci U S A. 2012;109:18186–18191. doi: 10.1073/pnas.1210565109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bendall JK, Damy T, Ratajczak P, Loyer X, Monceau V, Marty I, Milliez P, Robidel E, Marotte F, Samuel JL, Heymes C. Role of myocardial neuronal nitric oxide synthase-derived nitric oxide in beta-adrenergic hyporesponsiveness after myocardial infarction-induced heart failure in rat. Circulation. 2004;110:2368–2375. doi: 10.1161/01.CIR.0000145160.04084.AC. [DOI] [PubMed] [Google Scholar]
- 22.Damy T, Ratajczak P, Shah AM, Camors E, Marty I, Hasenfuss G, Marotte F, Samuel JL, Heymes C. Increased neuronal nitric oxide synthase-derived no production in the failing human heart. Lancet. 2004;363:1365–1367. doi: 10.1016/S0140-6736(04)16048-0. [DOI] [PubMed] [Google Scholar]
- 23.de Jong JW, Schoemaker RG, de Jonge R, Bernocchi P, Keijzer E, Harrison R, Sharma HS, Ceconi C. Enhanced expression and activity of xanthine oxidoreductase in the failing heart. J Mol Cell Cardiol. 2000;32:2083–2089. doi: 10.1006/jmcc.2000.1240. [DOI] [PubMed] [Google Scholar]
- 24.Mills WR, Mal N, Forudi F, Popovic ZB, Penn MS, Laurita KR. Optical mapping of late myocardial infarction in rats. Am J Physiol Heart Circ Physiol. 2006;290:H1298–H1306. doi: 10.1152/ajpheart.00437.2005. [DOI] [PubMed] [Google Scholar]
- 25.Dikalov S, Griendling KK, Harrison DG. Measurement of reactive oxygen species in cardiovascular studies. Hypertension. 2007;49:717–727. doi: 10.1161/01.HYP.0000258594.87211.6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gonzalez DR, Beigi F, Treuer AV, Hare JM. Deficient ryanodine receptor s-nitrosylation increases sarcoplasmic reticulum calcium leak and arrhythmogenesis in cardiomyocytes. Proc Natl Acad Sci U S A. 2007;104:20612–20617. doi: 10.1073/pnas.0706796104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walker ML, Wan X, Kirsch GE, Rosenbaum DS. Hysteresis effect implicates calcium cycling as a mechanism of repolarization alternans. Circulation. 2003;108:2704–2709. doi: 10.1161/01.CIR.0000093276.10885.5B. [DOI] [PubMed] [Google Scholar]
- 28.Pastore JM, Girouard SD, Laurita KR, Akar FG, Rosenbaum DS. Mechanism linking t-wave alternans to the genesis of cardiac fibrillation. Circulation. 1999;99:1385–1394. doi: 10.1161/01.cir.99.10.1385. [DOI] [PubMed] [Google Scholar]
- 29.Livshitz LM, Rudy Y. Regulation of ca2+ and electrical alternans in cardiac myocytes: Role of camkii and repolarizing currents. Am J Physiol Heart Circ Physiol. 2007;292:H2854–2866. doi: 10.1152/ajpheart.01347.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee HC, Mohabir R, Smith N, Franz MR, Clusin WT. Effect of ischemia on calcium-dependent fluorescence transients in rabbit hearts containing indo-1: Correlation with monophasic action potentials and contraction. Circulation. 1988;78:1047–1059. doi: 10.1161/01.cir.78.4.1047. [DOI] [PubMed] [Google Scholar]
- 31.Kurz RW, Mohabir R, Ren XL, Franz MR. Ischaemia induced alternans of action potential duration in the intact-heart: Dependence on coronary flow, preload and cycle length. Eur Heart J. 1993;14:1410–1420. doi: 10.1093/eurheartj/14.10.1410. [DOI] [PubMed] [Google Scholar]
- 32.Belch JJ, Bridges AB, Scott N, Chopra M. Oxygen free radicals and congestive heart failure. Br Heart J. 1991;65:245–248. doi: 10.1136/hrt.65.5.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kukreja RC, Kearns AA, Zweier JL, Kuppusamy P, Hess ML. Singlet oxygen interaction with ca(2+)-atpase of cardiac sarcoplasmic reticulum. Circ Res. 1991;69:1003–1014. doi: 10.1161/01.res.69.4.1003. [DOI] [PubMed] [Google Scholar]
- 34.Laurita KR, Katra R, Wible B, Wan X, Koo MH. Transmural heterogeneity of calcium handling in canine. Circ Res. 2003;92:668–675. doi: 10.1161/01.RES.0000062468.25308.27. [DOI] [PubMed] [Google Scholar]
- 35.Wan X, Laurita KR, Pruvot E, Rosenbaum DS. Molecular correlates of repolarization alternans in cardiac myocytes. J Mol Cell Cardiol. 2005;39:419–428. doi: 10.1016/j.yjmcc.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 36.Houser SR, Piacentino V, III, Weisser J. Abnormalities of calcium cycling in the hypertrophied and failing heart. J Mol Cell Cardiol. 2000;32:1595–1607. doi: 10.1006/jmcc.2000.1206. [DOI] [PubMed] [Google Scholar]
- 37.Wu YJ, Temple J, Zhang R, Dzhura I, Zhang W, Trimble R, Roden DM, Passier R, Olson EN, Colbran RJ, Anderson ME. Calmodulin kinase ii and arrhythmias in a mouse model of cardiac hypertrophy. Circulation. 2002;106:1288–1293. doi: 10.1161/01.cir.0000027583.73268.e7. [DOI] [PubMed] [Google Scholar]
- 38.Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O’Donnell SE, Aykin-Burns N, Zimmerman MC, Zimmerman K, Ham AJ, Weiss RM, Spitz DR, Shea MA, Colbran RJ, Mohler PJ, Anderson ME. A dynamic pathway for calcium-independent activation of camkii by methionine oxidation. Cell. 2008;133:462–474. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Respress JL, van Oort RJ, Li N, Rolim N, Dixit SS, deAlmeida A, Voigt N, Lawrence WS, Skapura DG, Skardal K, Wisloff U, Wieland T, Ai X, Pogwizd SM, Dobrev D, Wehrens XH. Role of ryr2 phosphorylation at s2814 during heart failure progression. Circ Res. 2012;110:1474–1483. doi: 10.1161/CIRCRESAHA.112.268094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saliaris AP, Amado LC, Minhas KM, Schuleri KH, Lehrke S, St John M, Fitton T, Barreiro C, Berry C, Zheng M, Kozielski K, Eneboe V, Brawn J, Hare JM. Chronic allopurinol administration ameliorates maladaptive alterations in ca2+ cycling proteins and beta-adrenergic hyporesponsiveness in heart failure. Am J Physiol Heart Circ Physiol. 2007;292:H1328–1335. doi: 10.1152/ajpheart.00461.2006. [DOI] [PubMed] [Google Scholar]
- 41.Aon MA, Cortassa S, O’Rourke B. Percolation and criticality in a mitochondrial network. Proc Natl Acad Sci U S A. 2004;101:4447–4452. doi: 10.1073/pnas.0307156101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rekhraj S, Gandy SJ, Szwejkowski BR, Nadir MA, Noman A, Houston JG, Lang CC, George J, Struthers AD. High-dose allopurinol reduces left ventricular mass in patients with ischemic heart disease. J Am Coll Cardiol. 2013;61:926–932. doi: 10.1016/j.jacc.2012.09.066. [DOI] [PubMed] [Google Scholar]
- 43.Jessup M, Greenberg B, Mancini D, Cappola T, Pauly DF, Jaski B, Yaroshinsky A, Zsebo KM, Dittrich H, Hajjar RJ. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (cupid): A phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum ca2+-atpase in patients with advanced heart failure. Circulation. 2011;124:304–313. doi: 10.1161/CIRCULATIONAHA.111.022889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hare JM, Mangal B, Brown J, Fisher C, Jr, Freudenberger R, Colucci WS, Mann DL, Liu P, Givertz MM, Schwarz RP. Impact of oxypurinol in patients with symptomatic heart failure. Results of the opt-chf study. J Am Coll Cardiol. 2008;51:2301–2309. doi: 10.1016/j.jacc.2008.01.068. [DOI] [PubMed] [Google Scholar]
- 45.Hüser J, Wang YG, Sheehan KA, Cifuentes F, Lipsius SL, Blatter LA. Functional coupling between glycolysis and excitation-contraction coupling underlies alternans in cat heart cells. J Physiol (Lond) 2000;524:795–806. doi: 10.1111/j.1469-7793.2000.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsutsui H, Ide T, Kinugawa S. Mitochondrial oxidative stress, DNA damage, and heart failure. Antioxid Redox Signal. 2006;8:1737–1744. doi: 10.1089/ars.2006.8.1737. [DOI] [PubMed] [Google Scholar]
- 47.Krijnen PA, Meischl C, Hack CE, Meijer CJ, Visser CA, Roos D, Niessen HW. Increased nox2 expression in human cardiomyocytes after acute myocardial infarction. J Clin Pathol. 2003;56:194–199. doi: 10.1136/jcp.56.3.194. [DOI] [PMC free article] [PubMed] [Google Scholar]