

Abstract
Due to the profound extent to which natural products inspire medicinal chemists in drug discovery, there is demand for innovative syntheses of these often complex materials. This article describes the synthesis of tricarbocyclic natural product architectures through an extension of the enantioselective Birch-Cope sequence with intramolecular Friedel-Crafts alkylation reactions. Additionally, palladium-catalyzed enol silane cycloalkenylation of the tricarbocyclic structures afforded the challenging bicyclo[3.2.1]octane C/D ring system found in the gibberellins and the ent-kauranes, two natural products with diverse medicinal value. In the case of the ent-kaurane derivative, an unprecedented alkene rearrangement converted four alkene isomers to one final product.
Introduction
Natural product structures continue to inspire synthetic chemists and drug developers alike with their complex molecular architecture that frequently exceeds human imagination. More importantly, these fascinating structures often possess useful and unique biological activities that inspire new therapeutic approaches1–5. Gibberellin6 and ent-kaurane derivatives7, 8 are two diterpene natural products9 with a complex tetracyclic molecular architecture (Figure 1) featuring, most prominently, a bicyclo[3.2.1]octane system, which has been the focus of significant recent synthetic efforts10, 11. Both gibberellins and ent-kauranes have noteworthy biological activity; the gibberellins as plant hormones and growth regulators12–14, and the ent-kauranes in a wide array of therapies. In fact, recent therapeutic applications of ent-kauranes include anti-inflammation15, anti-HIV16, antibacterial17, anti-tuberculosis18, and anti-cancer19–23. Clearly these molecular architectures have promising potential value.
Figure 1.

Structures of ent-kaurane skeleton and a gibberellin.
Although both gibberellins and ent-kauranes have members that are commercially available, the de novo construction of their structures can facilitate derivatization and potentially more active analogs. Since the 1970’s, there has been considerable synthetic effort aimed at the de novo construction of both frameworks, although there has been much less enantioselective work. Enantioselective de novo syntheses of gibberellins include two impressive works in 1981 by Takano24, 25 and one by Corey in 199126. Enantioselective de novo syntheses of the ent-kauranes include two works by Corey in 199727, 28, Toyota in 200029, and Reisman in 201330, 31.
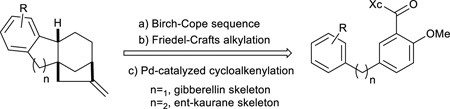
At the core of both the gibberellins and the ent-kauranes is a tetracarbocyclic ring system that creates significant synthetic challenges, including an all-carbon quaternary stereocenter. We hypothesized that the tetracyclic ring system might be accessible from a properly designed product of our previously reported Birch-Cope sequence32. We have illustrated applications of the Birch-Cope sequence products in the enantioselective synthesis of two alkaloids, (−)-lycoramine33 and (+)-mesembrine34. An enantioselective approach to the diterpene frameworks of gibberellins or ent-kauranes would expand the potential applications in natural product-like molecule synthesis. To that end, we fashioned the retrosynthetic plan shown in Scheme 1, in which the tetracarbocyclic core of these valuable compounds is generated with just two additional steps after the Birch-Cope sequence: a Friedel-Crafts alkylation and a palladium-catalyzed cycloalkenylation. The final product would contain the carbon skeleton of the gibberellins or ent-kauranes depending on the length of the linker between the two diaryl rings of the Birch-Cope sequence starting material. In the process, the strategy would create ring D and the bicyclo[3.2.1]octane at a later stage, after rings A-C have been generated. To the best of our knowledge, this approach has not previously been used with the palladium-catalyzed cycloalkenylation35–38. The Friedel-Crafts alkylation would rely on precedent39–43 and would be the second example of an intramolecular carbon nucleophile conjugate addition to the enone system of the Birch-Cope sequence product; along with the Rauhut-Currier reaction that we have recently communicated44, 45. In combination, the two steps would be a relatively short enantioselective entry to two natural product-like scaffolds.
Scheme 1.

Retrosynthetic Plan for the Construction of the Gibberellin and ent-Kaurane Skeletons
Results and Discussion
Construction of the Diaryl Starting Materials
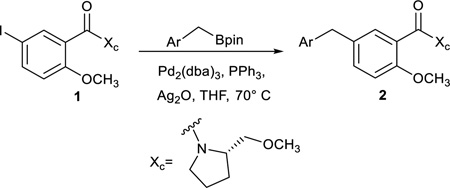
The first stage of the process to generate gibberellin or ent-kaurane carbon skeletons involved synthesizing the Birch-Cope sequence reactants. We previously used cross-coupling chemistry in our synthesis of (−)-lycoramine and (+)-mesembrine to make biaryl reactants, but the diaryl materials with methane or ethane linkers required for this work would, at first glance, seem to necessitate the much less common cross-coupling reaction of an sp2 carbon with an sp3 carbon. Therefore, we initially explored the synthesis of the diarylmethane substrates through Grignard reagent nucleophilic additions to benzaldehyde derivatives followed by reduction of the resulting diaryl alcohol. These efforts were successful, but not without some complications (e.g. magnesium-Oppenhauer oxidation side reactions46) and eventually proved less expedient than a cross-coupling approach. In particular, Suzuki reaction of the 5-iodo-salicylate derivative 134 (Table 1) with benzyl boronic acid derivatives47 in the presence of catalytic Pd2(dba)3 rapidly afforded the diaryl substrates 2 with a methane linker, albeit with some modest yields. Benzylboronic pinacol ester is commercially available and the four other aryl substituted derivatives were synthesized following literature protocols48 in one step from the appropriately substituted benzyl bromide, stoichiometric magnesium and pinacolborane.
Table 1.
Synthesis of Diarylmethane Starting Materials
 | ||
|---|---|---|
| Ar | Product | % Yield |
| C6H5- | 2a | 84 |
| 3-MeO-C6H5- | 2b | 58 |
| 4-MeO-C6H5- | 2c | 61 |
| 3,4-(MeO)2C6H5- | 2d | 56a |
| 3-Me-C6H5- | 2e | 47 |
Coupling conditions: Pd(OAc)2, SPhos, K2CO3, DMF, 60° C
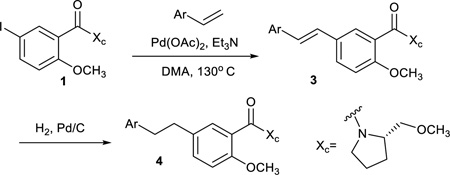
To leverage the facility of sp2-sp2 cross-coupling reactions and avoid the beta-hydride elimination complications introduced by a phenylethane cross-coupling partner, the ethane linked diaryl substrates were synthesized through a two-step process: Heck reaction of 1 with an appropriate styrene analog and subsequent hydrogenation of the resulting stilbene derivative (Table 2). Styrene worked efficiently with N,N-dimethyl-β-alanine as the ligand, but the same alanine ligand afforded low yields with the more electron rich styrene derivative in the synthesis of 3b. After some experimentation, the optimal Heck reaction conditions were found with the ligandless palladium conditions of Botella49. As can be seen in Table 2, both steps of the process were quite efficient for a range of substrates.
Table 2.
Synthesis of Diarylethane Starting Materials
 | ||||
|---|---|---|---|---|
| Ar | Product | % Yield | Product | % Yield |
| C6H5- | 3a | 76a | 4a | 90 |
| 3-MeO-C6H5- | 3b | 95 | 4b | 83 |
| 4-MeO-C6H5- | 3c | 86 | 4c | 87 |
| 3,4-(MeO)2C6H5- | 3d | 88 | 4d | 88 |
| 3-Me-C6H5- | 3e | 88 | 4e | 85 |
Conditions: Pd(OAc)2, K2CO3, N,N-dimethyl-β-alanine, NMP50
Birch-Cope sequence
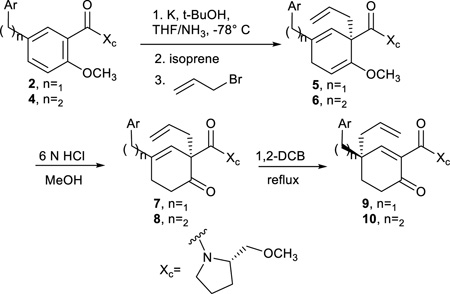
Subjecting the methane- and ethane-linked diaryl substrates, 2 and 4, to the Birch-Cope sequence began with the enantioselective Birch reduction-allylation, which afforded products 5 and 6 in moderate to very good yields (Table 3). As has been demonstrated before32, 33, the Birch reduction is selective for the more electron deficient aryl ring. The enantioselectivity of the Birch reduction-alkylation has been demonstrated on many previous occasions32–34, 51, 52 and was confirmed to afford an enantiomeric ratio of 24:1 for 4d in the current work (see Supporting Information). Note that the natural enantiomers of the gibberellins and ent-kauranes would be generated from the use of D-prolinol as a chiral auxiliary (Xc), however L-prolinol was used for this exploratory work to reduce costs while still illustrating the feasibility of the process. The Birch products, 5 and 6, were purified, but they decomposed over time so they were immediately taken through the next two steps of the Birch-Cope sequence. In that event, hydrolysis of the enol ether and stereoselective Cope rearrangement provided 9 and 10. Like prior analogs32, 34, the Cope rearrangement favors the more thermodynamically stable conjugated enone and the reduction in steric congestion around the C-2 position of the cyclohex-3-enones 7 and 8. Although all of these substrates were new examples in the Birch-Cope sequence, they demonstrated similar efficiency as previously reported examples with aryl or alkyl groups in the C-4 position.
Table 3.
Birch-Cope Sequence
 | ||||
|---|---|---|---|---|
| Reactant | Ar/n | Product / % Yield | ||
| 2a | C6H5-/1 | 5a/48 | 7a/87 | 9a/59 |
| 2b | 3-MeO-C6H5-/1 | 5b/46 | 7b/75 | 9b/59 |
| 2c | 4-MeO-C6H5-/1 | 5c/70 | 7c/90 | 9c/80 |
| 2d | 3,4-(MeO)2C6H5-/1 | 5d/72 | 7d/85 | 9d/73 |
| 2e | 3-Me-C6H5-/1 | 5e/61 | 7e/62 | 9e/69 |
| 4a | C6H5-/2 | 6a/97 | 8a/90 | 10a/75 |
| 4b | 3-MeO-C6H5-/2 | 6b/73 | 8b/70 | 10b/76 |
| 4c | 4-MeO-C6H5-/2 | 6c/67 | 8c/81 | 10c/63 |
| 4d | 3,4-(MeO)2C6H5-/2 | 6d/72 | 8d/89 | 10d/72 |
| 4e | 3-Me-C6H5-/2 | 6e/78 | 8e/94 | 10e/84 |
Intramolecular Friedel-Crafts alkylation
Elaboration of these Birch-Cope sequence products, 9 and 10, into the tetracarbocyclic framework of the gibberellins or the ent-kauranes began by the construction of the central B ring through a Friedel-Crafts alkylation. A standard selection of Lewis acids were screened including AlCl3, TiCl4, and SnCl4, but BF3·Et2O was clearly the best at coaxing the conjugate addition of the aromatic nucleophile to add to the cyclohexenone electrophile40–43. The cyclization occurred to form the cis isomers 11 and 12, but most of the products were isolated as C-2 epimers, typically a 1:1 mixture. Subsequent cleavage of the chiral auxiliary (vide infra) confirmed this observation by affording one enantiomerically pure product, thus dispelling the possibility of epimers at the C-3 bridgehead position. As anticipated, the intramolecular Friedel-Crafts alkylation reactions were most facile when a strong electron donating group was located para to the aromatic carbon that attacks the conjugated enone system (e.g. 9b, 9d, 9e, 10b, 10d, and 10e). Substrates without this characteristic suffered from lower conversion. For example, electron releasing groups meta to the nucleophilic aromatic carbon (9c and 10c) exerted a stronger inductive effect and the reaction suffered accordingly. Where regioisomeric ortho/para products could result (i.e. 9b, 9e, 10b, and 10e), complete selectivity for the para addition product was observed. Following chiral auxiliary removal, two-dimensional NMR experiments (see Supporting Information) confirmed both the structrure of the Friedel-Crafts products and the formation of the expected cis isomers.
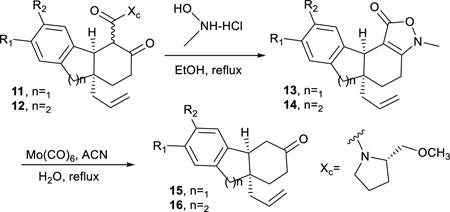
L-Prolinol chiral auxiliary removal occurred efficiently through a previously reported procedure33, 34 involving formation of an oxime and intramolecular isoxazolidinone formation (Table 5). Reduction of the N-O bond in 13/14 by Mo(CO)6 facilitates hydrolysis and decarboxylation to provide 15/16. Compound 12a failed to undergo isoxazolidinone formation and therefore the chiral auxiliary could not be removed.
Table 5.
Chiral Auxiliary Removal
 | |||||
|---|---|---|---|---|---|
| Reactant | N | R1 | R2 | Product / % Yield | Product / % Yield |
| 11b | 1 | OCH3 | H | 13b/77 | 15b/86 |
| 11d | 1 | OCH3 | OCH3 | 13d/72 | 15d/82 |
| 11e | 1 | CH3 | H | 13e/60 | 15e/80 |
| 12b | 2 | OCH3 | H | 14b/83 | 16b/89 |
| 12d | 2 | OCH3 | OCH3 | 14d/84 | 16d/82 |
| 12e | 2 | CH3 | H | 14e/77 | 16e/70 |
Palladium-catalyzed cycloalkenylation
A variety of approaches to form ring D and the bicyclo[3.2.1]octane with the palladium-catalyzed cycloalkenylation were explored. Initially, these included reactions with earlier structures, i.e. 10a, 10d, 12d, and 14b, where greater regiocontrol could be expected in the palladium-catalyzed cycloalkenylation. Consistent with literature reports on similar compounds36–38, cyclohexenones 10a and 10d afforded a 75 and 66% yield of the cycloalkenylation product upon conversion to an enol silane and exposure to the standard cyclization conditions (10 mol% Pd(OAc)2, O2, DMSO, 45°C). However, both cycloalkenylation products failed to undergo the Friedel-Crafts alkylation. Attempted cycloalkenylation with 12d was stymied by regioisomeric mixtures in the silylation step and with 14b, by the absence of any enol silane formation.
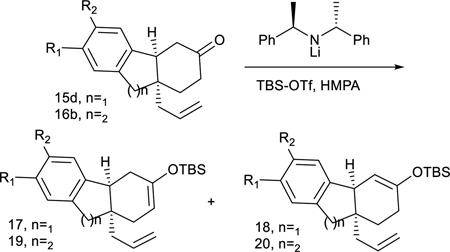
With the failure of these approaches using earlier intermediates, attention turned to compounds 15 and 16 with the hope that a cycloalkenylation could be realized despite the rigidifying effect of the tricyclic structures and the regioselectivity challenges created by removal of the chiral auxiliary. A variety of silylation procedures were explored with 15d and 16b, the representative substrates chosen from the 6-5-6 and 6-6-6 tricyclic category, respectively. In both cases, standard enol silane formation conditions (TBS-OTf, Et3N) afforded a 2:1 mixture of desired (17 and 19, Table 6) to undesired enol silane (18 and 20). Although trimethylsilyl (TMS) and triethylsilyl (TES) were also tried, tert-butyldimethylsilyl (TBS) enol ethers proved the most reproducible and stable for purification of the enol silane prior to the cycloalkenylation reaction. In addition, there is evidence from the Toyota lab that TBS enol silanes work best in palladium-catalyzed cycloalkenylation reactions53. The use of a bulkier base (iPr2NEt), kinetic conditions (LiHMDS at −78°C) or a bromomagnesium diisopropylamide54 failed to improve the yield or the ratio of desired to undesired enol silane. A small improvement in the regioisomeric distribution was achieved by using a chiral lithium amide base50, 55, which provided moderately better regioselectivity for 17 (2.5:1 versus 18) from 15d and even better results for the synthesis of 19 (4:1 versus 20) from 16b. Not unexpectedly, these silyl ether regioisomers could not be separated chromatographically; therefore they were subjected to the cycloalkenylation conditions as a mixture.
Table 6.
Enol Silane Formation
 | |||||
|---|---|---|---|---|---|
| Reactant | n | R1 | R2 | Product | % Yield (regioselectivity) |
| 15d | 1 | OCH3 | OCH3 | 17, 18 | 91 (2.5:1) |
| 16b | 2 | OCH3 | H | 19, 20 | 95 (4:1) |
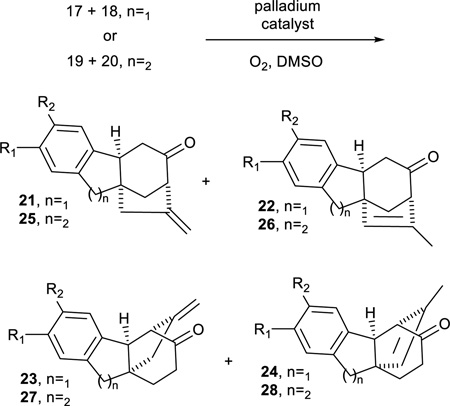
With the TBS enol silanes in hand, we were poised to attempt the palladium-catalyzed cycloalkenylation with the tricyclic substrates. For enol silane mixture 17/18, treatment with Pd(OAc)2 in DMSO under an oxygen atmosphere at 45°C resulted in selective cyclization of 17 to the desired tetracyclic structure 21 in a modest 41% yield (Table 7). The product was slightly contaminated by the undesired cycloalkenylation product 23 (5%) and unreacted enol silane 18 (6%). The use of different palladium catalysts (Pd(OCOCF3)2, [Pd(CH3CN)4BF4]), or solvents (CH3CN) failed to improve the outcome. A similar array of reaction conditions were also surveyed for cycloalkenylation of the enol silane mixture 19/20. In the end, the best results were obtained with the slightly more electrophilic catalyst Pd(OCOCF3)2, however it afforded a mixture of four inseparable products: the two cycloalkenylation products, 25 and 27, along with their alkene regioisomers, 26 and 28. The overall yield of the four isomers was 60%, but the product was a 46:31:15:8 mixture of 25:26:27:28. It should be noted that this distribution roughly parallels the 4:1 enol silane composition with 25 and 26 arising from 19, and 27 and 28 derived from 20. Running the reaction at a lower temperature (room temp.) failed to reduce the formation of alkene isomers. The richer mixture of products resulting from the 6-6-6 tricyclic system versus the 6-5-6 system is likely the result of slightly greater conformational flexibility which permits alternative reaction paths. In contrast, there was a greater proportion of the undesired silane 18 in the 6-5-6 tricycle, but very little of the corresponding cycloalkenylation products 23/24 actually formed. Nevertheless, the overall mixed results in the cycloalkenylation transformation of the 6-5-6 and 6-6-6 systems highlights the challenges of conducting the reaction on the more rigid tricyclic systems.
Table 7.
Palladium-catalyzed Cycloalkenylation
 | |||
|---|---|---|---|
| Reactant | Palladium catalyst | Product | % Yield |
| 17, 18 | Pd(OAc)2 | 21 | 41 |
| 19, 20 | Pd(OCOCF3)2 | 25–28 | 60 (46:31:15:8 ratio) |
In an attempt to isomerize the alkene mixture 25–28 to the most thermodynamically stable regioisomers, presumably 26 and 28, the mixture of 25–28 was exposed to pTsOH under benzene reflux conditions56. Unexpectedly and quite fortuitously, this resulted in complete conversion of the entire mixture to 26 in a 74% yield. We hypothesize that this rearrangement and isomerization occurs along the path shown in Figure 2. Protonation of the alkenes to form a tertiary carbocation and formation of the enol begins the process. Preliminary computational modeling suggests the empty tertiary carbocation p orbital is not far from the enol pi system. Consequently, a four-member transition state can permit 1,3-migration of the alkyl carbocation piece to the opposite alpha position generating the isomeric tertiary carbocation and enol. Elimination and isomerization affords the product 26. Thermodynamic calculations support compound 26 as the most stable constitutional isomer among 25–28. A similar attempt to isomerize the 6-5-6 tricyclic mixture of 21 and 23 noted above, failed to afford a similar transformation despite an analogous calculated thermodynamic preference for compound 22.
Figure 2.

Possible pathway for rearrangement of 27/28 into 26
Conclusion
An enantioselective procedure for the synthesis of the tetracarbocyclic skeleton of the gibberellins or the ent-kauranes has been illustrated. The reported results demonstrate another extension of the Birch-Cope sequence products, which in this case affords a diterpene natural product-like structure. An intramolecular Friedel-Crafts alkylation of the enone system demonstrates the ability of aromatic carbon nucleophiles to be used in conjugate addition reactions with the Birch-Cope sequence products. The tricarbocyclic products were subsequently subjected to palladium-catalyzed cycloalkenylation to create the challenging bicyclo[3.2.1]octane C/D ring system. The cycloalkenylation reaction was modestly successful and illustrated the challenges of conducting this reaction on more complex and rigid substrates. An unprecedented isomerization of the ent-kaurane skeleton under acid conditions provided a good yield of the final ent-kaurane-like structure from a mixture of alkene isomers. The overall process illustrates a new enantioselective procedure towards a complex and important natural product-like carbon skeleton.
Experimental
General Procedures
All reactants and reagents were commercially available and were used without further purification unless otherwise indicated. Anhydrous THF was obtained by distillation from benzophenone-sodium under nitrogen. All reactions were carried out under an inert atmosphere of argon or nitrogen unless otherwise indicated. Concentrated refers to the removal of solvent with a rotary evaporator at normal water aspirator pressure followed by further evacuation with a direct-drive rotary vane vacuum pump. Yields refer to chromatographically and spectroscopically pure (>95%) compounds, except as otherwise indicated. All new compounds were determined to be >95% pure by NMR and/or GC as indicated. Thin layer chromatography was performed using silica gel 60 Å precoated aluminum backed plates (0.25 mm thickness) with fluorescent indicator, which were cut. Developed TLC plates were visualized with UV light (254 nm), iodine, and p-anisaldehyde staining. Flash column chromatography was conducted with the indicated solvent system using normal phase silica gel 60 Å, 230–400 mesh. Optical rotation measurements were taken on a Perkin-Elmer 341 polarimeter.1H and 13C NMR spectra were recorded at 400 MHz. Chemical shifts are reported in δ values (ppm) relative to an internal reference (0.05% v/v) of tetramethylsilane (TMS) or residual CHCl3 for 1H NMR and CDCl3in 13C NMR. Peak splitting patterns in the 1H NMR are reported as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad. 13C NMR experiments were conducted with the attached proton test (APT) pulse sequence. 13C multiplicities are reported as δu(up) for methyl and methine, and δd(down) for methylene and quaternary carbons. IR data was obtained with an FT-IR spectrometer. GC analyses were performed with an EI-MS detector fitted with a 30 m × 0.25 mm column filled with cross-linked 5% PH ME siloxane (0.25 µm film thickness); gas pressure 7.63 psi He. One method for analysis of samples involved heating from 70 to 250°C (10°C/min) and finally holding at 250°C for 7 min. HRMS were determined by electrospray ionization (ESI) using an infusion pump on a Thermo-Electron LTQ-FT 7T Fourier transform ion cyclotron resonance (FT-ICR) spectrometer. Samples were dissolved in neat methanol and then diluted to a 90/10/0.01 solution of CH3OH/H2O/formic acid. Alternatively, for lower molecular weight samples, determinations were by solid probe desorption, chemical ionization (CI) at 35 eV, using methane as the ionizing gas, on a Micromasss (now Waters) AutoSpec-Ultima M high-resolution triple sector (EBE) mass spectrometer. Thermodynamic computational calculations were conducted with Spartan ‘14 v. 1.1.8 using density functional theory with EDF2 functional, basis set 6–31G* and toluene as the solvent.
General Procedures for Diarylmethylene Synthesis
Benzylboronic Pinacol Ester Coupling
Method A
Aryl iodide (1.0 eq.), Pd2(dba)3 (8 mol%), PPh3 (1.0 eq.), Ag2O (1.5 eq.) and benzylboronic acid pinacol ester (1.5 eq.) were dissolved in THF and heated overnight at 70°C. The next day the reaction mixture was passed through a plug of silica using EtOAc. The resulting solution was washed with five times with saturated sodium bicarbonate and brine, dried over Na2SO4, filtered and concentrated under reduced pressure to yield the diaryl methylene product. Pure product was obtained through column chromatography (EtOAc in heptanes)47.
Method B
DMF (3 mL/mmol aryl halide) was added to a flask containing Pd(OAc)2 (3 mol%) and SPhos (6 mol%). Next finely crushed K3PO4 (3.0 eq.) was added, followed by aryl halide (1.0 eq.) and boronic ester (2.0 eq.). This was heated to 60°C overnight. The next day the reaction mixture was diluted with EtOAc and this was washed two times with 20% NaOH, then brine. The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure to yield the diaryl methylene product. Pure product was obtained through column chromatography (7:3 EtOAc in heptanes).57
Diarylmethylene 2a, Method A
Use of the general procedure with aryl iodide 1 (207.4 mg, 0.553 mmol) provided 130.2 mg of the diarylmethylene product, an 84% yield. 1H NMR (CDCl3, a mixture of rotamers) δ 7.31-7.24 (m, 2H), 7.23-7.05 (m, 5H), 6.88-6.79 (m, 1H), 4.44-4.34 (m, 1H), 3.93 (s, 2H), 3.83-3.79 (2s, 3H), 3.78-3.70 (m, 1H), 3.60-3.51 (m, 1H), 3.41 (s, 2H), 3.27-3.14 (m, 1H), 3.00 (s, 2H), 2.07-1.98 (m, 1H), 1.97-1.86 (m, 2H), 1.82-1.69 (m, 1H). 13C NMR (CDCl3, a mixture of rotamers) δu 130.5, 128.85, 128.80, 128.2, 126.1, 111.35, 111.28, 59.1, 58.7, 56.3, 55.8, 55.7; δd 168.0, 153.7, 141.2, 133.5, 127.7, 127.2, 73.4, 72.4, 48.4, 45.8, 40.9, 28.5, 27.8, 24.2, 22.2. GC tR = 11.77 min. EI-MS m/z (%): 339 (M+, 1), 307 (9), 294 (15), 225 (100). HRMS (CI) (m/z): [M+H]+ calcd for C21H26NO3:340.1913, found: 340.1926.
Diarylmethylene 2b, Method A
Use of the general procedure with aryl iodide 1 (1.53 g, 4.08 mmol) provided 874 mg of the diarylmethylene product, a 58% yield. 1H NMR (CDCl3, a mixture of rotamers) δ 7.24-7.6 (m, 3H), 6.88-6.69 (m, 4H), 4.42-4.38 (m, 1H), 3.91 (s, 2H), 3.84-3.80 (2s, 3H), 3.80-3.77 (2s, 3H), 3.77-3.72 (m, 1H), 3.60-3.52 (m, 1H), 3.42 (s, 2H), 3.29-3.17 (m, 1H), 3.01 (s, 2H), 2.07-1.98 (m, 1H), 1.97-1.87 (m, 2H), 1.82-1.72 (m, 1H). 13C NMR (CDCl3, a mixture of rotamers) δu130.51, 130.46, 129.4, 128.1, 121.2, 121.16, 114.6, 111.37, 111.33, 59.0, 58.5, 57.2, 56.3, 55.67, 55.61, 55.02, 55.00 ; δd 167.9, 159.7, 153.18, 153.6, 142.69, 142.56, 133.3, 127.5, 127.2, 73.4, 72.35, 48.4, 45.7, 40.8, 28.5, 27.6, 23.96, 22.2. GC tR = 13.51 min. EI-MS m/z (%): 369 (M+, 1), 337 (7), 324 (10), 255 (100). HRMS (CI) (m/z): [M+1]+ calcd for C22H28NO4: 370.2018, found: 370.2022.
Diarylmethylene 2c, Method A
Use of the general procedure with aryl iodide 1(531.0 mg, 1.42 mmol) provided 320.7 mg of the diarylmethylene product, a 61% yield based on GCMS. 1H NMR (CDCl3, a mixture of rotamers) δ 7.17-7.03 (m, 4H), 6.87-6.74 (m, 3H), 4.49-4.32, (m, 1H), 3.73 (s, 2H), 3.85-3.76 (m, 6H), 3.76-3.71 (m, 1H), 3.61-3.51 (m, 1H), 3.40 (s, 2H), 3.29-3.13 (m, 1H), 3.10-2.95 (m, 1H), 2.08-1.84 (m, 4H), 1.81-1.69 (m, 1H). 13C NMR (CDCl3, a mixture of rotamers) δu 130.41, 130.38, 130.3, 130.2, 129.79, 129.75, 128.0, 127.7, 120.79, 120.73. 113.9, 111.3, 111.2, 111.15, 59.1, 58.7, 57.2, 56.3, 55.74, 55.67, 55.61, 55.2; δd 168.1, 157.97, 153.6, 133.99, 133.97, 133.26, 133.14, 127.5, 127.1, 73.4, 72.3, 48.4, 45.8, 40.0, 28.4, 28.9, 24.2, 22.2. GC tR = 13.91 min. EI-MS m/z (%): 369 (M+, 1), 337 (7), 324 (10), 255 (100). HRMS (CI) (m/z): [M+1]+ calcd for C22H28NO4: 370.2018, found: 370.2018.
Diarylmethylene 2d, Method B
Use of the general procedure with aryl iodide 1 (191.6 mg, 0.511 mmol) provided 114.2 mg of the diarylmethylene product, a 56% yield. 1H NMR (CDCl3, a mixture of rotamers) δ 7.19-7.02 (m, 2H), 6.88-6.74(m, 2H), 6.74-6.62 (m, 2H), 4.42-4.34 (m, 1H), 3.86 (s, 2H), 3.86-3.84 (2s, 3H), 3.82 (s, 3H), 3.81-3.78 (2s, 3H), 3.77-3.70 (m, 1H), 3.60-3.50 (m, 1H), 3.41 (s, 2H), 3.28-3.14 (m, 1H), 3.01 (s, 2H), 2.06-1.97 (m, 1H), 1.97-1.85 (m, 2H), 1.81-1.69 (m, 1H). 13C NMR (CDCl3, a mixture of rotamers) δu 130.38, 130.35, 128.0, 120.87, 120.83, 112.17, 112.12, 111.27, 111.22, 59.1, 58.7, 57.2, 56.3, 55.9, 55.8, 55.7, 55.66; δd 168.1, 153.6, 148.9, 147.4, 133.78, 133.60, 127.5, 127.1, 73.4, 72.3, 48.4, 45.7, 40.4, 28.4, 28.77, 24.2, 22.2.GC tR = 15.64 min. EI-MS m/z (%): 399 (M+, 1), 367 (3), 354 (17), 285 (100). HRMS (CI) (m/z): [M+1]+ calcd for C23H30NO5:400.2124, found: 400.2113.
Diarylmethylene 2e, Method A
Use of the general procedure with aryl iodide 1 (2.11 g, 5.62 mmol) provided 930 mg of the diarylmethylene product, a 47% yield. 1H NMR (CDCl3, a mixture of rotamers) δ 7.22-7.06 (m, 3H), 7.05-6.92 (m, 3H), 6.89-6.78 (m, 1H), 4.50-4.35, (m, 1H), 3.89 (s, 2H), 3.85-3.79 (2s, 3H), 3.79-3.71 (m, 1H), 3.62-3.52 (m, 1H), 3.42 (s, 2H), 3.29-3.15 (m, 1H), 3.06-2.92 (m, 1H), 2.34-2.29 (2s, 3H), 2.08-1.86 (m, 4H), 1.83-1.70 (m, 1H). 13C NMR (CDCl3, a mixture of rotamers) δu 130.52, 130.49, 129.66, 129.60, 128.3, 128.2, 126.8, 125.90, 125.84, 111.3, 59.1, 58.6, 57.2, 56.3, 55.76, 55.68, 21.4; δd 167.98, 153.7, 141.0, 140.96, 138.02, 133.6, 127.6, 127.2, 73.4, 72.4, 48.4, 45.8, 40.8, 28.5, 27.8, 24.8, 22.2. GC tR = 12.28 min. EI-MS m/z (%): 353 (M+, 1), 331 (7), 308 (10), 239 (10). HRMS (CI) (m/z): [M+1]+ calcd for C22H28NO3: 354.2069, found: 354.2070.
General Procedures for the Heck Cross-Coupling Reaction
Method A, for electron-poor styrenes
A flame-dried flask was charged with Pd(OAc)2 (0.15 eq.), K2CO3 (2.0 eq.), N,N-dimethyl-β-alanine (0.15 eq.), and styrene (1.5 eq.). Next aryl iodide (1.0 eq.) dissolved in NMP was added and this was heated to 130°C overnight. The next day the reaction was concentrated directly onto silica under reduced pressure and purified via column chromatography (1:1 EtOAc in heptanes)58.
Method B, for electron-rich styrenes
To a flask containing aryl iodide (1.0 eq.) was added triethylamine (1.5 eq.), Pd(OAc)2 (0.10 eq.), and DMA. This was heated overnight at 120°C. The next day the reaction was concentrated directly onto silica under reduced pressure and purified via column chromatography (1:1 EtOAc in heptanes)49.
Stilbene 3a, Method A
Use of the general procedure with aryl iodide 1 (2.81 g, 7.48 mmol) provided 2.02 g of the cross-coupled product in a 77% yield (as the desired product and a Heck regioisomer). 1H NMR (CDCl3, a mixture of regioisomers and rotamers) δ 7.54-7.42 (m, 3H), 7.41-7.31 (t, 2H, J=7.84 Hz), 7.31-7.22 (m, 2H), 7.12-6.97 (m, 2H), 6.96-6.86 (m, 1H), 4.50-4.39 (m, 1H), 3.87 (s, 3H), 3.83-3.73 (m, 1H), 3.67-3.54 (m, 1H), 3.45 (s, 2H), 3.34-3.19 (m, 1H), 3.16-3.07 (m, 1H), 2.10-1.88 (m, 4H), 1.85-1.73 (m, 1H). 13C NMR (CDCl3, a mixture of regioisomers and rotamers) δu 128.5, 128.47, 128.41, 128.0, 127.9, 127.3, 127.28, 127.25, 127.21, 126.1, 125.4, 111.3, 58.9, 58.6, 56.2, 55.6, 55.5, 29.3, 24.4; δd 177.1, 167.45, 167.41, 154.7, 137.2, 137.17, 130.1, 130.0, 127.8, 127.4, 73.4, 72.2, 49.1, 48.2, 45.7, 45.6, 30.4, 28.4, 28.0, 27.7, 24.0, 22.1, 17.4. GC tR = 12.347 min. (Minor Heck regioisomer, 4.38%). EI-MS m/z (%): 351 (M+, 2), 306 (11), 237 (100), 178 (11), 165 (11). tR = 15.29 min (Major Heck regioisomer, 94.97%). EI-MS m/z (%): 351 (M+, 6), 306 (11), 237 (100), 178 (14), 165 (15). HRMS (CI) (m/z): [M+1]+ calcd for C22H26NO3: 352.1913, found: 352.1910.
Stilbene 3b, Method B
Use of the general procedure with aryl iodide 1 (4.79 g, 12.8 mmol) provided 4.88 g of the cross-coupled product in a 96% yield (as the desired product and a Heck regioisomer). 1H NMR (CDCl3, a mixture of regioisomers and rotamers) δ 7.52-7.43 (m, 2H), 7.31-7.25 (t, 2H, J=7.68 Hz), 7.12-7.06 (t, 1H, J=7.36 Hz), 7.05-6.98 (d, 2H, J=11.28 Hz), 6.96-6.86 (m, 1H), 6.84-6.80 (m, 1H), 4.50-4.40 (m, 1H), 3.89-3.83 (m, 6H), 3.83-3.53 (m, 2H), 3.48-3.40 (m, 2H), 3.33-3.20 (m, 1H), 3.15-3.06 (m, 1H), 2.10-1.88 (m, 4H), 1.86-1.73 (m, 1H). 13C NMR (CDCl3, a mixture of regioisomers and rotamers) δu 130.1, 130.02, 129.6, 129.2, 128.63, 128.60, 127.79, 127.7, 127.46, 127.4, 125.7, 120.86, 120.81, 120.79, 119.0, 113.9, 113.3, 113.13, 113.09, 111.58, 111.41, 111.18, 110.9, 59.1, 58.8, 57.3, 56.3, 56.2, 55.8, 55.75, 55.2; δd 167.74, 167.67, 159.46, 155.0, 154.89, 148.9, 138.8, 134.1, 130.36, 130.24, 128.2, 127.55, 113.64, 73.5, 72.34, 48.5, 45.8, 28.52, 28.47, 27.8, 24.2, 22.2, 20.3. GC tR = 14.30 min. (Minor Heck regioisomer, 4.72%). EI-MS m/z (%): 381 (M+, 1), 336 (8), 267 (100), 133 (8). tR = 19.99 min. (Major Heck regioisomer, 92.79%). EI-MS m/z (%): 381 (M+, 9), 336 (5), 267 (100), 133 (10). HRMS (CI) (m/z): [M+1]+ calcd for C23H28NO4: 382.2018, found: 382.2007.
Stilbene 3c, Method B
Use of the general procedure with aryl iodide 1 (5.016 g, 13.4 mmol) provided 4.39 g of the cross-coupled product in an 86 % yield (as the desired product and a Heck regioisomer). 1H NMR (CDCl3, a mixture of regioisomers and rotamers) δ 7.42-7.35 (m, 3H), 7.29-7.19 (m, 1H), 6.96-6.78 (m, 5H), 4.46-4.37 (m, 1H), 3.83-3.76 (m, 6H), 3.76-3.70 (dd, 1H, 9.4 Hz) 3.61-3.47 (m, 1H), 3.40 (s, 2H), 3.31-3.15 (m, 1H), 3.14-3.01 (m, 1H), 2.05-1.82 (m, 4H), 1.81-1.66 (m, 1H). 13C NMR (CDCl3, a mixture of regioisomers and rotamers) δu 132.8, 132.29, 132.26, 132.74, 132.67, 130.19, 130.14, 129.4, 128.29, 128.25, 127.5, 127.06, 125.40, 125.33, 114.12, 114.04, 113.62, 113.57, 111.38, 59.1, 58.9, 56.3, 55.8, 55.3; δd 167.91, 167.83. 159.19, 154.5, 127.9, 73.5, 72.36, 72.25, 48.57, 48.58, 45.8, 28.5, 27.81, 27.78, 24.2, 22.2. GC tR = 14.89 min. (Minor Heck regioisomer, 5.87%). EI-MS m/z (%): 381 (M+, 1), 336 (10), 267 (100), 133 (8). tR = 20.68 min. (Major Heck regioisomer, 92.92%). EI-MS m/z (%): 381 (M+, 21), 336 (3), 267 (100), 133 (8). HRMS (CI) (m/z): [M+1]+ calcd for C23H28NO4: 382.2018, found: 382.2031.
Stilbene 3d, Method B
Use of the general procedure with aryl iodide 1 (3.65 g, 9.73 mmol) provided 3.51 g of the cross-coupled product in an 88% yield (as the desired product and a Heck regioisomer). 1H NMR (CDCl3, a mixture of regioisomers and rotamers) δ 7.46-7.36 (m, 2H), 7.07-6.94 (m, 2H), 6.93-6.78 (m, 4H), 4.47-4.36 (m, 1H), 3.90 (s, 3H), 3.85 (s, 3H), 3.81 (m, 3H), 3.78-3.70 (m, 1H), 3.64-3.50 (m, 1H), 3.41 (s, 2H), 3.30-3.16 (m, 1H), 3.12-3.02 (m, 1H), 2.06-1.84 (m, 4H), 1.81-1.68 (m, 1H). 13C NMR (CDCl3, a mixture of regioisomers and rotamers) δu 130.05, 130.04, 128.25, 127.2, 125.6, 125.3, 120.9, 119.65, 119.61, 111.48, 111.39, 111.31, 110.88, 110.82, 108.7, 59.02, 59.01, 56.3, 55.86, 55.78, 55.70, 55. 64; δd 167.7, 154.5, 149.1, 148.6, 134.2, 130.52, 130.49, 130.44, 127.9, 127.46, 127.43, 112.3, 73.5, 72.3, 72.27, 48.3, 45.8, 28.5, 27.8, 24.1, 22.2. GC tR = 16.66 min. (Minor Heck regioisomer, 8.57%). EI-MS m/z (%): 411 (M+, 4), 366 (8), 297 (100), 148 (11). tR = 25.84 min. (Major Heck regioisomer, 87.67%). EI-MS m/z (%): 411 (M+, 13), 366 (1), 297 (100), 148 (13). HRMS (CI) (m/z): [M]+ calcd for C24H29NO5: 411.2046, found: 411.2039.
Stilbene 3e, Method B
Use of the general procedure with aryl iodide 1 (3.13 g, 8.57 mmol) provided 2.78 g of the cross-coupled product in an 89% yield (as the desired product and a Heck regioisomer). 1H NMR (CDCl3, a mixture of regioisomers and rotamers) δ 7.47-7.41 (m, 2H), 7.33-7.18 (m, 3H), 7.17-6.82 (m, 4H), 4.49-4.36 (m, 1H), 3.82 (s, 3H), 3.79-3.72 (dd, 1H, 9.4 Hz) 3.65-3.47 (m, 1H), 3.43 (s, 2H), 3.31-3.16 (m, 1H), 3.17-3.02 (m, 1H), 2.36 (s, 3H), 2.13-1.84 (m, 4H), 1.81-1.68 (m, 1H). 13C NMR (CDCl3, a mixture of regioisomers and rotamers) δu 128.95, 128.93, 128.55, 128.52, 128.28, 128.24, 128.09, 127.60, 127.45, 127.28, 127.02, 125.56, 125.41, 125.36, 123.52, 111.4, 110.9, 59.1, 58.8, 57.4, 56.4, 55.8, 55.7, 21.4; δd 167.7, 154.97, 138.14, 138.12, 137.32, 137.27, 130.51, 130.4, 128.02, 113.28, 73.5, 72.40, 72.27, 48.5, 45.82, 45.76, 28.54, 27.8, 24.2, 22.2. GC tR = 12.89 min. (Minor Heck regioisomer, 5.78%). EI-MS m/z (%): 365 (M+, 1), 333 (10), 320 (14), 251 (100), 178 (14). tR = 16.83 min. (Major Heck regioisomer, 94.21%). EI-MS m/z (%): 365 (M+, 9), 333 (3), 320 (7), 251 (100), 178 (19). HRMS (CI) (m/z): [M+1]+ calcd for C23H28NO3: 366.2069, found: 366.2072.
General Procedure for Alkene Hydrogenation
A flask containing alkene(1.0 eq.) was reduced under a hydrogen atmosphere in a Parr shaker hydrogenator with Pd/C (0.20 g/g alkene) and a 1:1 mixture of EtOAc and EtOH. After 4 h the reaction mixture was filtered through celite and concentrated under reduced pressure. The reduced product was then purified via column chromatography (1:1 EtOAc in heptanes).
Diaryl 4a
Use of the general procedure with alkene 3a (2.32 g, 8.11 mmol) provided 2.09 g of the hydrogenated product in an 90% yield. 1H NMR (CDCl3, a mixture of regioisomers and rotamers) δ 7.30-7.21 (m, 2H), 7.22-6.99 (m, 5H), 6.83-6.78 (d, 1H, J=8.56 Hz), 4.44-4.35, (m, 1H), 3.85-3.77 (m, 3H), 3.77-3.70 (m, 1H), 3.60-3.50 (m, 1H), 3.41 (s, 2H), 3.25-3.10 (m, 1H), 3.09-2.95 (m, 1H), 2.87 (s, 4H), 2.07-1.84 (m, 4H),1.80-1.68 (m, 1H). 13C NMR (CDCl3, a mixture of regioisomers and rotamers) δu 130.06, 129.98, 128.47, 128.43, 128.30, 128. 25, 127.67, 125.89, 125.83, 111.2, 59.07, 58.71, 57.2, 56.2, 55.71, 55.62, 24.7; δd 177.3, 167.99. 153.5, 151.54, 151.45, 133.99, 133.90, 127.4, 127.03, 73.5, 72.4, 48.4, 45.7, 37.91, 37.88, 36.81, 36.72, 28.47, 28.17, 27.78, 24.2, 22.2. GC tR = 11.98 min. (Minor Heck regioisomer, 4.66%). EI-MS m/z (%): 353 (M+, 1), 321 (8), 308 (13), 239 (100), 164 (8). tR = 12.57 min. (Major Heck regioisomer, 95.09%). EI-MS m/z (%): 353 (M+, 1), 321 (7), 308 (11), 239 (100), 91 (8). HRMS (CI) (m/z): [M+1]+ calcd for C22H28NO3: 354.2069, found: 354.2069.
Diaryl 4b
Use of the general procedure with alkene 3b (4.67 g, 12.26 mmol) provided 3.91 g of the hydrogenated product in an 83% yield. 1H NMR (CDCl3, a mixture of regioisomers and rotamers) δ 7.24-7.16 (t, 1H, J=7.36 Hz), 7.16-7.11 (dd, 1H, J=8.40, 2.20 Hz), 7.07-7.03 (d, 1H, J=2.12 Hz), 6.87-6.80 (d, 1H, 8.44 Hz), 6.80-6.69 (m, 3H), 4.47-4.38, (m, 1H), 3.86-3.71 (m, 7H), 3.64-3.52 (m, 1H), 3.44 (s, 2H), 3.27-3.12 (m, 1H), 3.11-2.98 (m, 1H), 2.88 (s, 4H), 2.08-1.84 (m, 4H), 1.83-1.64 (m, 1H). 13C NMR (CDCl3, a mixture of regioisomers and rotamers) δu 130.07, 130.01, 129.29, 129.25, 127.7, 120.91, 120.85, 114.25, 114.19, 111.27, 111.22, 111.18, 111.04, 59.1, 58.7, 57.3, 56.2, 55.76, 55.65, 55.62, 55.1, 43.88, 43.82; δd 168.13, 168.06, 159.65, 159.60, 153.5, 143.2, 143.1, 134.0, 133.95, 127.06, 73.5, 72.4, 48.45, 48.41, 37.99, 37.95, 36.73, 36.62, 28.5, 27.8, 24.2, 22.2.GC tR = 13.73 min. (Minor Heck regioisomer, 3.59%). EI-MS m/z (%): 383 (M+, 1), 351 (5), 338 (8), 269 (100), 134 (7). tR = 15.27 min. (Major Heck regioisomer, 95.30%). EI-MS m/z (%): 383 (M+, 1), 351 (10), 338 (14), 269 (100), 134 (7). HRMS (CI) (m/z): [M+1]+ calcd for C23H30NO4: 384.2175, found: 384.2191.
Diaryl 4c
Use of the general procedure with alkene 3c (4.44 g, 11.7 mmol) provided 3.90 g of the hydrogenated product in an 87% yield. 1H NMR (CDCl3, a mixture of regioisomers and rotamers) δ 7.16-7.00 (m, 4H), 6.85-6.78 (m, 3H), 4.46-4.37 (m, 1H), 3.80 (s, 3H), 3.78 (s, 3H), 3.76-3.73 (m, 1H), 3.59-3.51 (m, 1H), 3.42 (s, 2H), 3.24-3.11 (m, 1H), 3.10-2.98 (m, 1H), 2.83 (s, 4H), 2.06-1.98 (m, 2H), 1.98-1.86 (m, 2H), 1.80-1.70 (m, 1H). 13C NMR (CDCl3, a mixture of regioisomers and rotamers) δu 130.1, 130.0, 129.4, 129.36, 127.7, 113.74, 113.69, 111.15, 59.1, 58.8, 57.3, 56.26, 56.21, 55.74, 55.63, 55.23;δd 168.1, 157.8, 134.1, 134.0, 133.7 133.6, 127.4, 73.5, 72.4, 48.4, 45.8, 37.12, 37.04, 37.03, 28.5, 27.8, 24.2, 22.26, 22.22.GC tR = 13.99 min. (Minor Heck regioisomer, 10.20%). EI-MS m/z (%): 383 (M+, not shown due to loss of two methyl groups in the GC-MS), 351 (7), 338 (10), 269 (100), 126 (10). tR = 14.76 min. (Major Heck regioisomer, 89.49%). EI-MS m/z (%): 383 (M+, 1), 351 (7), 338 (12), 269 (100), 121 (15). HRMS (CI) (m/z): [M+1]+ calcd for C23H30NO4: 384.2175, found: 384.2162.
Diaryl 4d
Use of the general procedure with alkene 3d (3.51 g, 8.54 mmol) provided 3.12 g of the hydrogenated product in an 88% yield. 1H NMR (CDCl3, a mixture of regioisomers and rotamers) δ 7.13-7.03 (td, 2H, J=8.08, 2.48 Hz), 6.83-6.74 (m, 2H), 6.72-6.63 (m, 2H), 4.45-4.36, (m, 1H), 3.85 (s, 4H), 2.83 (s, 2H), 3.82-3.78 (m, 3H), 3.77-3.70 (m, 1H), 3.61-3.5 (m, 1H), 3.41 (s, 2H), 3.27-3.11 (m, 1H), 3.10-2.96 (m, 1H), 2.83 (s, 4H), 2.06-1.85 (m, 4H), 1.82-1.67 (m, 1H). 13C NMR (CDCl3, a mixture of regioisomers and rotamers) δu 130.12, 130.6, 127.7, 120.31, 120.28, 111.94, 111.87, 111.25, 111.23, 111.16, 59.1, 59.7, 58.6, 57.3, 56.26, 56.22, 55.89, 55.83, 55.80, 55.79, 55.78, 55.74, 55.63, 43.4; δd 168.1, 148.8, 147.33, 147.26, 134.23, 134.14, 134.05, 133.96, 127.5, 127.07, 73.5, 72.4, 48.4, 45.7, 37.5, 37.0, 36.95, 28.45, 27.8, 24.2, 22.2. GC tR = 15.59 min. (Minor Heck regioisomer, 5.82%). EI-MS m/z (%): 413 (M+, 2), 381 (3), 368 (7), 299 (100), 149 (8). tR = 16.86 min. (Major Heck regioisomer, 91.76%). EI-MS m/z (%): 413 (M+, 5), 381 (3), 368 (8), 299 (100), 151 (18). HRMS (CI) (m/z): [M+1]+ calcd for C23H32NO5: 414.2280, found: 414.2279.
Diaryl 4e
Use of the general procedure with alkene 3e (3.10 g, 8.49 mmol) provided 3.12 g of the hydrogenated product in an 85% yield. 1H NMR (CDCl3, a mixture of regioisomers and rotamers) δ 7.19-7.05 (m, 2H), 7.01-6.87 (m, 4H), 6.84-6.73 (d, 1H, J=7.2 Hz), 4.46-4.32 (m, 1H), 3.80-3.64 (m, 4H), 3.60-3.47 (m, 1H), 3.41-3.35 (s, 2H), 3.22-2.93 (m, 2H), 2.88-2.76 (s, 4H), 2.32-2.26 (s, 3H), 2.05-1.80 (m, 2H). 13C NMR (CDCl3, a mixture of regioisomers and rotamers) δu 130.07, 130.00, 129.30, 129.26, 128.25, 127.7, 126.7, 126.6, 125.50, 125.46, 111.2, 59.1, 58.7, 57.30, 56.2, 55.78, 55.67, 43.8, 21.4; δd 168.2, 153.6, 141.5, 141.48, 137.90, 137.88, 134.27, 134.13, 73.55, 72.45, 48.4, 45.8, 37.91, 36.90, 36.81, 28.5, 27.8, 22.2. GC tR = 12.42 min. (Minor Heck regioisomer, 5.74%). EI-MS m/z (%): 367 (M+, 1), 335 (10), 322 (13), 253 (100). tR = 13.14 min. (Major Heck regioisomer, 93.26%). EI-MS m/z (%): 367 (M+, 2), 335 (8), 322 (10), 253 (100), 105 (24). HRMS (CI) (m/z): [M+1]+ calcd for C23H30NO3: 368.2226, found:368.2215.
General Procedure for Birch Reduction-Allylation
To a solution of benzamide (1.0 eq.) and tert-butyl alcohol (1.0 eq.) in THF (10 mL/mmol amide) and NH3 (130 mL/mmol amide) at −78°C was added potassium in small pieces until blue coloration was maintained for 20 min. Isoprene was added dropwise to consume the excess metal, and then allyl bromide (2.5 eq.) was added. The solution was stirred at −78°C and allowed to slowly warm to room temperature to allow the NH3 to evaporate. Water was then added, and the mixture was extracted three times with DCM. The combined organics were washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure to yield the 2,5-cyclohexadiene product. Pure product was obtained through column chromatography (3:7 EtOAc in heptanes). Note: Birch reduction-allylation products were not stable enough to obtain an HRMS or optical rotations.
Cyclohexadiene 5a
Use of the general procedure with benzamide 2a (1.1710 g, 3.45 mmol) provided 0.640 g of the 2,5-cyclohexadiene product in a 49% yield. 1H NMR (CDCl3, a mixture of rotamers) δ 7.28 (t, 2H, J=7.80 Hz), 7.22-7.12 (m, 3H), 5.73-5.55 (m, 1H), 5.25 (s, 1H), 5.07-4.90 (m, 2H), 4.68 (t, 1H, J=3.44 Hz), 4.39-4.26 (m, 1H), 3.67-3.55 (m, 1H), 3.47 (s, 3H), 3.38-3.25 (m, 6H), 3.23-3.11 (m, 1H), 2.87-2.47 (m, 3H), 1.91-1.74 (m, 3H), 1.74-1.59 (m, 1H). 13C NMR (CDCl3, a mixture of rotamers) δu 135.1, 128.8, 128.3, 126.3, 122.8, 92.6, 58.9, 58.1, 54.2; δd 170.7, 152.5, 139.3, 136.9, 116.8, 72.1, 52.8, 46.0, 43.3, 41.3, 29.3, 26.6, 24.8.
Cyclohexadiene 5b
Use of the general procedure with benzamide 2b (0.971 g, 2.63 mmol) provided 0.497 g of the 2,5-cyclohexadiene product in a 46% yield. 1H NMR (CDCl3, a mixture of rotamers) δ 7.18 (t, 1H, J=7.72 Hz), 6.79-6.65 (m, 3H), 5.70-5.56 (m, 1H), 5.24 (s, 1H), 5.04-4.89 (m, 2H), 4.68 (t, 1H, J=3.64 Hz), 4.36-4.24 (m, 1H), 3.78 (s, 3H), 3.68-3.56 (m, 1H), 3.49 (t, 2H, J=2.12 Hz), 3.46 (s, 2H), 3.37-3.26 (m, 6H), 2.91-2.28 (m, 3H), 1.93-1.76 (m, 3H), 1.74-1.54 (m, 1H). 13C NMR (CDCl3, a mixture of rotamers) δu 135.1, 129.3, 122.9, 121.2, 114.7, 111.4, 92.6, 58.9, 58.1, 55.1, 54.23; δd 170.7, 159.7, 153.0, 140.9, 136.8, 116.8, 72.0, 52.8, 46.1, 43.3, 41.1, 29.4, 26.5, 24.9.
Cyclohexadiene 5c
Use of the general procedure with benzamide 2c (1.07 g, 2.89 mmol) provided 0.8359 g of the 2,5-cyclohexadiene product in a 70% yield. 1H NMR (CDCl3, a mixture of rotamers) δ 7.13-7.00 (t, 2H, J=7.24 Hz), 6.87-6.76 (d, 2H, J=8.60 Hz), 5.70-5.54 (m, 1H), 5.24-5.17 (d, 1H, J=7.32 Hz), 5.03-4.89 (m, 2H), 4.67 (s, 1H), 4.38-4.26, (m, 1H), 3.78 (s, 3H), 3.66-3.50 (m, 2H), 3.46 (s, 3H), 3.39-3.30 (m, 4H), 3.30-3.12 (m, 3H), 2.89-2.44 (m, 4H), 1.93-1.73 (m, 3H), 1.74-1.59 (m, 1H). 13C NMR (CDCl3, a mixture of rotamers) δu 135.2, 129.7, 123.1, 122.4, 113.74, 113.71, 92.76, 92.65, 58.92, 58.88, 58.04, 57.89, 55.23, 54.25, 54.21; δd 170.8, 170.3, 158.2, 152.9, 152.5, 137.3, 136.9, 131.4, 131.3, 116.7, 72.17, 72.07, 52.8, 52.77, 46.2, 46.0, 42.3, 41.28, 41.27, 29.31, 29.26, 26.6, 26.4, 25.0, 24.8, 22.7.
Cyclohexadiene 5d
Use of the general procedure with benzamide 2d (2.62 g, 6.34 mmol) provided 2.07 g of the 2,5-cyclohexadiene product in a 72% yield. 1H NMR (CDCl3, a mixture of rotamers) δ 6.78-6.71 (d, 1H, J=8.0 Hz), 6.71-6.59 (m, 2H), 5.67-5.53 (m, 1H), 5.19 (s, 1H), 5.00-4.84 (m, 2H), 4.64 (s, 1H), 4.33-4.21, (m, 1H), 3.84-3.75 (m, 7H), 3.61-3.46 (m, 2H), 3.43 (s, 3H), 3.37-3.10 (m, 6H), 2.84-2.68 (m, 1H), 2.68-2.60 (m, 1H), 2.60-2.43 (m, 2H), 1.91-1.70 (m, 3H), 1.69-1.54 (m, 1H). 13C NMR (CDCl3, a mixture of rotamers) δu 135.2, 123.2, 122.5, 120.9, 112.0, 111.1, 92.7, 58.9, 58.1, 57.9, 55.9, 55.8, 54.3, 54.2; δd 170.3, 152.9, 148.9, 147.5, 137.1, 136.9, 131.9, 116.1, 72.6, 72.02, 72.0, 51.3, 47.7, 46.3, 46.0, 43.0, 41.3, 29.7, 29.3, 29.3, 26.9, 26.5, 26.4, 25.2, 25.0, 24.9.
Cyclohexadiene 5e
Use of the general procedure with benzamide 2e (0.880 g, 2.49 mmol) provided 0.60 g of the 2,5-cyclohexadiene product in a 61% yield. 1H NMR (CDCl3, a mixture of rotamers) δ 7.17 (t, 1H, J=7.48 Hz), 7.08-6.91 (m, 3H), 5.73-5.53 (m, 1H), 5.27 (s, 1H), 5.09-4.88 (m, 2H), 4.68 (t, 1H, J=3.52 Hz), 4.41-4.27, (m, 1H), 3.70-3.60 (m, 1H), 3.60-3.52 (m, 1H), 3.48 (s, 3H), 3.41-3.24 (m, 6H), 3.23-3.14 (m, 1H), 2.87-2.52 (m, 4H), 2.33 (2s, 3H), 1.75-1.57 (m, 2H). 13C NMR (CDCl3, a mixture of rotamers) δu 135.5, 129.7, 129.66, 128.2, 127.0, 125.9, 123.3, 92.7, 59.0, 57.9, 54.3, 21.4; δd 170.8, 170.3, 152.9, 152.5, 139.2, 137.9, 136.9, 116.7, 72.1, 52.8, 46.1, 43.2, 41.3, 29.4, 26.6.
Cyclohexadiene 6a
Use of the general procedure with benzamide 4a (3.45 g, 9.77 mmol) as 2 separate Birch reductions which were combined for purification provided 3.74 g of the 2,5-cyclohexadiene product in a 97% yield. 1H NMR (CDCl3, a mixture of rotamers) δ 7.31-7.22 (t, 2H, J=5.28 Hz), 7.22-7.13 (d, 3H, J=6.52 Hz), 5.60-5.45 (m, 1H), 5.15 (s, 1H), 4.99-4.85, (m, 2H), 4.73-4.68 (t, 1H, 3.56 Hz), 4.31-4.24 (m, 1H), 3.62-3.57 (dd, 1H, J=9.6, 3.2 Hz), 3.47 (s, 3H), 3.33 (s, 3H), 3.31-3.15 (m, 3H), 2.89-2.63 (m, 5H), 2.53- 2.45 (m, 1H), 2.43-2.35 (t, 2H, J=8.00 Hz), 1.87-1.56 (m, 5H). 13C NMR (CDCl3, a mixture of rotamers) δu 135.0, 128.3, 128.2, 125.9, 121.6, 92.7, 58.8, 58.1, 54.2;δd170.3, 152.99, 141.45, 136.6, 116.5, 71.9, 52.6, 45.8, 45.3, 37.5, 33.9, 29.9, 26.3, 24.9.
Cyclohexadiene 6b
Use of the general procedure with benzamide 4b (8.52 g, 22.2 mmol) as 3 separate Birch reductions which were combined for purification to provide 6.96 g of the 2,5-cyclohexadiene product in a 73% yield. 1H NMR (CDCl3, a mixture of rotamers) δ 7.24-7.17 (m, 1H), 6.83-6.70 (m, 3H), 5.63-5.47 (m, 1H), 5.17 (s, 1H), 5.00-4.88 (m, 2H), 4.73 (t, 1H, J=3.64 Hz), 4.36-4.24 (m, 1H), 3.81 (s, 3H), 3.66-3.59 (m, 1H), 3.50 (s, 3H), 3.39-3.33 (m, 3H), 3.33-3.17 (m, 2H), 3.89-2.65 (m, 5H), 2.56-2.46 (m, 1H), 2.40 (t, 2H, J=8.04 Hz), 1.90-1.70 (m, 3H), 1.68-1.59 (m, 2H). 13C NMR (CDCl3, a mixture of rotamers) δu 135.1, 129.3, 121.6, 120.7, 114.1, 111.1, 92.6, 58.9, 58.1, 55.1, 54.2; δd 170.5, 159.7, 153.0, 143.1, 136.6, 116.6, 71.9, 45.9, 41.3, 37.4, 34.1, 29.9, 26.3, 24.8.
Cyclohexadiene 6c
Use of the general procedure with benzamide 4c (0.500 g, 1.30 mmol) provided 0.290 g of the 2,5-cyclohexadiene product in a 52% yield. 1H NMR (CDCl3, a mixture of rotamers) δ 7.14-7.04 (d, 2H, J=8.52 Hz), 6.85-6.76 (d, 2H, J=10.7 Hz) 5.66-5.44 (m, 1H), 5.13 (s, 1H), 5.00-4.85 (m, 2H), 4.74-4.67 (t, 1H, J=3.56 Hz), 4.36-4.22 (m, 1H), 3.75 (s, 3H), 3.66-3.54 (m, 1H), 3.47 (s, 3H), 3.33 (s, 3H), 3.30-3.13 (m, 3H), 2.91-2.61 (m, 4H), 2.56-2.41 (m, 1H), 2.40-2.31 (t, 2H, J=3.96 Hz), 2.02-1.53 (m, 5H). 13C NMR (CDCl3, a mixture of rotamers) δu 135.1, 129.24, 129.14, 128.23, 128.18, 121.5, 113.73, 113.69, 113.62, 92.6, 58.9, 58.0, 55.2, 54.2 ;δd 170.8, 157.8, 136.7, 133.5, 116.54, 72.0, 52.6, 45.9, 41.29, 41.06, 37.7, 33.1, 29.9, 26.3, 24.9.
Cyclohexadiene 6d
Use of the general procedure with benzamide 4d (2.62 g, 6.34 mmol) provided 2.07 g of the 2,5-cyclohexadiene product in a 72% yield. 1H NMR (CDCl3, a mixture of rotamers) δ 6.80-6.67 (m, 3H), 5.64-5.46 (m, 1H), 5.19-5.12 (d, 1H, J=9.56 Hz), 4.98-4.87 (m, 2H), 4.74-4.69 (t, 1H, J=3.92 Hz), 4.36-4.23 (m, 1H), 3.89-3.86 (d, 3H, J= 2.44 Hz), 3.86-3.82 (d, 3H, J=2.28 Hz), 3.65-3.52 (m, 1H), 3.49 (s, 3H), 3.37-3.31 (d, 3H, J=4.00 Hz), 3.32-3.16 (m, 3H), 2.60-2.29 (m, 3H), 2.90-2.62 (m, 4H), 1.89-1.57 (m, 5H). 13C NMR (CDCl3, a mixture of rotamers) δu 134.33, 134.30, 126.22, 126.19, 112.2, 110.61, 110.68, 55.4, 44.2, 38.4;δd171.3, 164.2, 159.1, 159.07, 142.0, 136.4, 118.1, 118.06, 102.7, 45.7, 42.9, 41.1, 27.7, 18.9.
Cyclohexadiene 6e
Use of the general procedure with benzamide 4e (1.6299 g, 4.44 mmol) provided 1.43 g of the 2,5-cyclohexadiene product in a 78% yield. 1H NMR (CDCl3, a mixture of rotamers) δ 7.14 (t, 1H, J=7.36 Hz), 7.02-6.91 (m, 3H), 5.66-5.46 (m, 1H), 5.15 (s, 1H), 5.00-4.86 (m, 2H), 4.71 (t, 1H, J=3.48 Hz), 4.35-4.21 (m, 1H), 3.61-3.57 (m, 1H), 3.33 (s, 3H), 3.27-3.13 (m, 1H), 2.90-2.62 (m, 4H), 2.56-2.44 (m, 1H), 2.35 (t, 2H, J=3.96 Hz), 2.31 (2, 3H), 1.88-1.53 (m, 4H), 1.42-1.30 (m, 1H).13C NMR (CDCl3, a mixture of rotamers) δu 135.1, 129.0, 128.2, 126.6, 125.2, 121.5, 92.6, 58.8, 58.1, 54.2, 21.4; δd 170.3, 153.0, 141.4, 137.8, 136.7, 116.5, 71.97, 52.6, 45.9, 41.3, 37.6, 34.1, 29.9, 26.3, 24.8.
General Procedure for Enol Ether Hydrolysis
Enol ether (1.0 eq.) was dissolved in MeOH (6 mL/mmol enol ether), cooled to 0°C, and treated with 6 N HCl (2.5 mL/mmol enol ether). After stirring overnight, the reaction solution was diluted with water and extracted three times with DCM. The combined organics were washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure. Column chromatography (1:1 EtOAc in heptanes) afforded pure ketone.
Ketone 7a
Use of the general procedure with enol ether 5a (0.64 g, 1.68 mmol) provided 0.5411 g of the ketone product in an 88% yield. [α]D24 = −16.5 (c=0.4, CH2Cl2). 1H NMR (CDCl3, a mixture of rotamers) δ 7.33-7.26 (m, 2H), 7.26-7.20 (m, 1H), 7.19-7.23 (m, 2H), 5.90-5.73 (m, 1H), 5.49 (s, 1H), 5.09-4.99 (m, 2H), 4.34-4.21 (m, 1H), 3.66-3.59 (m, 1H), 3.42 (s, 2H), 3.39 (s, 3H), 3.33 (m, 1H), 3.28-3.12 (m, 1H), 3.07-2.96 (m, 1H), 2.73-2.65 (d, 2H, J=7.36 Hz), 2.60-2.29 (m, 4H), 1.96-1.76 (m, 3H), 1.76-1.61 (m, 1H). 13C NMR (CDCl3, a mixture of rotamers) δu134.3, 134.2, 128.73, 128.70, 128.55, 126.6, 124.9, 124.4, 59.0, 57.9; δd 208.2, 207.7, 169.0, 168.5, 139.3, 138.6, 118.2, 72.1, 71.9, 61.2, 46.7, 46.4, 43.7, 41.9, 41.6, 37.0, 36.9, 28.4, 28.2, 26.8, 26.7, 24.6. HRMS (CI) (m/z): [M+1]+ calcd for C23H30NO3: 368.2226, found: 368.2235.
Ketone 7b
Use of the general procedure with enol ether 5b (0.497 g, 1.21 mmol) provided 0.362 g of the ketone product in a 75% yield. [α]D24 = −14.8 (c=0.73, CH2Cl2). 1H NMR (CDCl3, a mixture of rotamers) δ 7.25-7.17 (t, 1H, J=7.80 Hz), 6.81-6.67 (m, 3H), 5.89-5.74 (m, 1H), 5.48 (s, 1H), 5.10-4.98 (m, 2H), 4.34-4.21 (m, 1H), 3.78 (m, 2H), 3.67-3.58 (m, 1H), 3.43-3.29 (m, 6H), 3.30-3.18 (m, 1H), 3.10-2.96 (m, 1H), 2.72-2.64 (d, 2H, J=7.32 Hz), 2.62-2.26 (m, 4H), 1.93-1.79 (m, 4H), 1.80-1.58 (m, 1H). 13C NMR (CDCl3, a mixture of rotamers) δu 134.3, 129.5, 124.5, 121.1, 114.8, 111.6, 59.0, 57.9, 55.1; δd 168.5, 159.7, 140.2, 139.1, 118.2, 118.19, 71.8, 61.2, 46.5, 46.4, 43.7, 41.6, 37.3, 36.9, 28.2, 26.9, 24.5. HRMS (CI) (m/z): [M+1]+ calcd for C24H32NO4: 398.2331, found: 398.2333.
Ketone 7c
Use of the general procedure with enol ether 5c (0.8359 g, 2.24 mmol) provided 0.7329 g of the ketone product in a 91% yield. 1H NMR (CDCl3, a mixture of rotamers) δ 7.07-6.95 (dd, 2H, J=8.64, 2.36 Hz), 6.80-6.75 (d, 2H, J=8.52), 5.87-5.68 (m, 1H), 5.41-4.34 (d, 1H, J=8.04 Hz), 5.05-4.92 (m, 2H), 4.30-4.15 (m, 1H), 3.73 (s, 3H), 3.6-3.40 (m, 1H), 3.39-3.28 (m, 6H), 3.25-3.08 (m, 1H), 3.04-2.88 (m, 1H), 2.70-2.55 (d, 2H), 2.55-2.22 (m, 4H), 1.91-1.73 (m, 3H), 1.73-1.56 (m, 1H). 13C NMR (CDCl3, a mixture of rotamers) δu 134.3, 134.25, 129.7, 129.66, 124.5, 124.1, 114.0, 113.9, 59.02, 59.0, 57.8, 55.3; δd 208.3, 207.8, 169.0, 168.5, 158.4, 139.6, 139.2, 130.63, 130.60, 118.15, 118.13, 72.1, 71.9, 61.3, 61.2, 46.8, 46.4, 42.9, 41.9, 41.6, 37.0, 36.9, 28.4, 28.2, 26.8, 26.7, 24.6. A sample of compound 7c decomposed before an HRMS or optical rotation could be obtained.
Ketone 7d
Use of the general procedure with enol ether 5d (0.990 g, 2.24 mmol) provided 0.959 g of the ketone product in an 85% yield. [α]D24 = −20.6 (c=0.47, CH2Cl2). 1H NMR (CDCl3, a mixture of rotamers) δ 6.74-6.69 (d, 1H, J=7.84 Hz), 6.67-6.60 (m, 2H), 5.83-5.70 (m, 1H), 5.40 (s, 1H), 5.04-4.92 (m, 2H), 4.28-4.14 (m, 1H), 3.80 (s, 6H), 3.61-3.39 (m, 1H), 3.38-3.08 (m, 7H), 3.05-2.93 (m, 1H), 2.65-2.55 (d, 2H, J=7.72 Hz), 2.53-2.42 (m, 1H), 2.42-2.25 (m, 3H), 1.92-1.72 (m, 3H), 1.72-1.55 (m, 1H). 13C NMR (CDCl3, a mixture of rotamers) δu 134.3, 134.2, 124.6, 124.2, 119.3, 112.0, 111.3, 58.95, 57.9; δd 208.2, 207.8, 168.5, 149.0, 147.8, 138.5, 139.1, 131.16, 131.13, 118.1, 72.2, 71.8, 61.3, 61.2, 55.9, 55.8, 46.8, 46.5, 43.2, 41.7, 41.4, 37.0, 36.9, 28.3, 28.2, 26.8, 26.7, 24.7, 24.6. HRMS (CI) (m/z): [M+1]+ calcd for C25H34NO5: 428.2437, found: 428.2448.
Ketone 7e
Use of the general procedure with enol ether 5e (0.60 g, 1.52 mmol) provided 0.360 g of the ketone product in a 62% yield. [α]D24 = −14.2 (c=0.33, CH2Cl2). 1H NMR (CDCl3, a mixture of rotamers) δ 7.19 (t, 1H, J=7.44 Hz), 7.08-7.03 (d, 1H, J=7.56), 7.01-6.92 (m, 2H), 5.92-5.76 (m, 1H), 5.48 (s, 1H), 5.11-4.99 (m, 2H), 4.39-4.19 (m, 1H), 3.69-3.47 (m, 1H), 3.43-3.30 (m, 6H), 3.30-3.18 (m, 1H), 3.08-2.93 (m, 1H), 2.74-2.64 (d, 2H, J=7.36 Hz), 2.59-2.35 (m, 4H), 2.34 (s, 3H), 1.95-1.78 (m, 3H), 1.77-1.60 (m, 1H). 13C NMR (CDCl3, a mixture of rotamers) δu 134.3, 134.27, 129.5, 128.4, 127.3, 125.7, 124.7, 124.3, 59.00, 58.98, 57.9, 21.4; δd 208.3, 169.0, 168.5, 139.3, 138.6, 137.9, 118.0, 72.1, 71.9, 61.2, 46.8, 46.4, 43.4, 41.9, 41.7, 37.0, 36.7, 28.4, 28.2, 26.83, 26.81, 24.9. HRMS (CI) (m/z): [M+1]+ calcd for C24H32NO3: 382.2382, found: 382.2391.
Ketone 8a
Use of the general procedure with enol ether 6a (1.69 g, 4.28 mmol) provided 1.48 g of the ketone product in a 91 % yield. [α]D24 = −7.8 (c=0.27, CH2Cl2). 1H NMR (CDCl3, a mixture of rotamers) δ 7.35-7.25 (m, 2H), 7.24-7.12 (m, 3H), 5.82-5.62 (m, 1H), 5.37 (s, 1H), 5.11-4.93 (m, 2H), 4.35-4.17 (m, 1H), 3.65-3.56 (dd, 1H, J=9.32 Hz, J=3.04 Hz), 3.34 (s, 3H), 3.33-3.26 (m, 1H), 3.10-2.99 (m, 1H), 2.97-2.85 (m, 1H), 2.83-2.74 (t, 2H, J=6.52 Hz), 2.65-2.53 (m, 3H), 2.51-2.38 (m, 5H), 1.92-1.71 (m, 3H), 1.70-1.56 (m, 1H). 13C NMR (CDCl3, a mixture of rotamers) δu134.3, 128.3, 128.2, 126.0, 123.3, 58.8, 57.8; δd207.5, 168.4, 141.0, 139.1, 117.8, 71.7, 60.95, 46.3, 41.3, 38.2, 36.7, 22.6, 28.3, 26.5, 24.4. HRMS (CI) (m/z): [M+1]+ calcd for C24H32NO3: 382.2382, found: 382.2377.
Ketone 8b
Use of the general procedure with enol ether 6b (2.52 g, 5.93mmol) provided 1.72 g of the ketone product in a 72% yield.[α]D24= −7.6 (c=0.67, CH2Cl2). 1H NMR (CDCl3, a mixture of rotamers) δ 7.26-7.17 (t, 1H, J=7.80 Hz), 6.82-6.70 (m, 3H), 5.82-5.64 (m, 1H), 5.37 (s, 1H), 5.12-4.95 (m, 2H), 4.35-4.17 (m, 1H), 3.80 (s, 3H), 3.62 (dd, 1H, J=9.32, 3.20 Hz), 3.36 (s, 3H), 3.34-3.26 (m, 1H), 3.12-3.01 (m, 1H), 2.97-2.88 (m, 1H), 2.83-2.74 (t, 2H, J=8.00 Hz), 2.65-2.53 (m, 3H), 2.52-2.39 (m, 5H), 1.98-1.73 (m, 3H), 1.71-1.57 (m, 1H). 13C NMR (CDCl3, a mixture of rotamers) δu134.3, 129.4, 123.3, 120.6, 114.2, 111.1, 58.9, 57.8, 55.1; δd207.9, 168.4, 159.7, 142.6, 138.9, 118.1, 71.9, 61.0, 46.6, 41.7. 38.1, 36.9, 33.7, 28.4, 26.6, 24.5. HRMS (CI) (m/z): [M+1]+ calcd for C25H34NO4: 412.2488, found: 412.2474.
Ketone 8c
Use of the general procedure with enol ether 6c (1.40 g, 3.29 mmol) provided 0.880 g of the ketone product in a 65% yield. [α]D24 = −12.8 (c=0.53, CH2Cl2). 1H NMR (CDCl3, a mixture of rotamers) δ 7.07-6.95 (d, 2H, J=8.48 Hz), 6.79-6.70 (d, 2H, J=8.48 Hz), 5.82-5.56 (m, 1H), 5.26 (s, 1H), 5.04-4.86 (m, 2H), 4.26-4.09 (m, 1H), 3.75-3.36 (m, 3H), 3.58-3.50 (dd, 1H, J=9.24, 3.20 Hz), 3.27 (s, 3H), 3.26-3.19 (m, 2H), 3.00-2.90 (m, 1H), 2.85-2.75 (m, 1H), 2.71-2.61 (m, 2H), 2.56-2.44 (m, 3H), 2.43-2.31 (m, 5H), 1.88-1.62 (m, 3H), 1.61-1.49 (m, 1H). 13C NMR (CDCl3, a mixture of rotamers) δu134.3, 129.2, 123.4, 113.8, 113.8, 59.03, 58.98, 55.26, 55.23; δd 168.5, 157.9, 139.1, 133.0, 128.2, 128.12, 118.0, 71.99, 71.7, 61.16, 61.1, 46.3, 41.6, 41.5, 38.5, 36.9, 32.9, 28.5, 26.6, 24.6, 24.5. HRMS (CI) (m/z): [M+1]+ calcd for C25H33NO4: 412.2488, found: 412.2471.
Ketone 8d
Use of the general procedure with enol ether 6d (1.82 g, 4.00 mmol) provided 1.58 g of the ketone product in a 90% yield. [α]D24 = −17.7 (c=0.6, CH2Cl2). 1H NMR (CDCl3, a mixture of rotamers) δ 6.82-6.75 (d, 1H, J=7.92 Hz), 6.72 (s, 1H), 6.69 (s, 1H), 5.84-5.67 (m, 1H), 5.41-5.34 (d, 1H, J=5.00 Hz), 5.11-4.94 (m, 2H), 4.34-4.16 (m, 1H), 3.86 (s, 3H), 3.85-3.83 (d, 3H, J=1.28 Hz), 3.63-3.41 (m, 1H), 3.34 (s, 2H), 3.31 (s, 2H), 3.14-2.98 (m, 1H), 2.97-2.87 (m, 1H), 2.79-2.66 (t, 2H, J=7.68 Hz), 2.65-2.53 (m, 3H), 2.52-2.38 (m, 5H), 1.93-1.72 (m, 3H), 1.71-1.59 (m, 1H). 13C NMR (CDCl3, a mixture of rotamers) δu 155.1, 132.8, 128.5, 128.2, 126.1, 59.1, 58.9, 57.9, 56.4; δd 195.51, 195.53, 165.5, 142.6, 142.3, 137.81, 137.76, 119.4, 74.0, 72.1, 48.3, 46.1, 41.9, 41.6, 39.8, 39.75, 38.7, 34.0, 33.9, 30.8, 30.7, 30.5, 30.4, 28.5, 27.7, 24.3, 22.1. HRMS (CI) (m/z): [M+1]+ calcd for C26H36NO5: 442.2593, found: 442.2576.
Ketone 8e
Use of the general procedure with enol ether 6e (1.43 g, 3.50 mmol) provided 1.30 g of the ketone product in a 94% yield. [α]D24 = −6.1 (c=0.67, CH2Cl2). 1H NMR (CDCl3, a mixture of rotamers) δ 7.18 (t, 1H, J=7.48 Hz), 7.05-6.94 (m, 3H), 5.86-5.64 (m, 1H), 5.38 (s, 1H), 5.13-4.94 (m, 2H), 4.34-4.17 (m, 1H), 3.69-3.42 (m, 1H), 3.36 (s, 3H), 3.34-3.26 (m, 1H), 3.14-3.03 (m, 1H), 2.98-2.87 (m, 1H), 2.76 (t, 2H, J=7.80 Hz), 2.66-2.51 (m, 3H), 2.51-2.41 (m, 5H), 2.33 (s, 3H), 1.95-1.74 (m, 3H), 1.73-1.56 (m, 1H). 13C NMR (CDCl3, a mixture of rotamers) δu 134.4, 129.0, 128.2, 126.7, 125.2, 123.2, 58.8, 57.8, 21.3; δd 207.5, 207.46, 168.9, 168.4, 168.35, 140.9, 139.2, 137.8, 117.8, 71.95, 71.7, 60.97, 46.5, 46.3, 41.6, 41.4, 38.2, 36.7, 33.7, 28.5, 28.4, 26.7, 26.6, 24.5. HRMS (CI) (m/z): [M+1]+ calcd for C25H34NO3: 396.2539, found: 396.2534.
General Procedure for the Cope Rearrangement
The 1,5-diene (1.0 eq.) was dissolved in 1,2-dichlorobenzene (2 mL/mmol diene) and refluxed overnight. The solvent was removed in vacuo and the crude enone was purified by chromatography (EtOAc).
Enone 9a
Use of the general procedure with 1,5-diene 7a (0.541 g, 1.47 mmol) provided 0.320 g of the pure enone product in a 59% yield. [α]D24 = −44.2 (c=1.0, CH2Cl2). 1H NMR (CDCl3, a mixture of rotamers) δ 7.35-7.21 (m, 3H), 7.21-7.12 (m, 2H), 6.82 (s, 1H), 5.94-5.78 (m, 1H), 5.25-5.11 (m, 2H), 4.34-4.25 (m, 1H), 3.73-3.61 (m, 1H), 3.51-3.40 (m, 1H), 3.37 (s, 2H), 3.22 (s, 1H), 3.15-3.03 (m, 1H), 3.03-2.91 (m, 1H), 2.90-2.79 (d, 2H, J=10.4 Hz), 2.57-2.21 (m, 4H), 2.04-1.79 (m, 5H), 1.79-1.68 (m, 1H). 13C NMR (CDCl3, a mixture of rotamers) δu 155.09, 155.0 133.2, 133.0, 130.4, 128.4, 128.3, 126.9, 126.8, 59.1, 58.9, 57.8, 56.4; δd 195.0, 165.9, 165.8, 138.5, 136.5, 119.6, 119.5, 74.1, 72.2, 48.3, 45.7, 44.4, 44.3, 43.9, 42.9, 42.8, 41.3, 39.9, 34.1, 34.0, 30.5, 30.3, 30.2, 28.5, 27.7, 24.3, 24.2, 22.0.GC tR = 13.15 min. EI-MS m/z (%): 367 (M+, 6), 322 (99), 276 (16), 253 (39), 91 (100). HRMS (CI) (m/z): [M+1]+ calcd for C23H30NO3: 368.2226, found: 368.2218.
Enone 9b
Use of the general procedure with 1,5-diene 7b (0.4004 g, 1.01 mmol) provided 0.235 g of the pure enone product in a 59% yield. [α]D24 = −31.3 (c=0.53, CH2Cl2). 1H NMR (CDCl3, a mixture of rotamers) δ 7.23-7.15 (t, 1H, J=8.00 Hz), 6.84-6.70 (M, 3H), 6.68 (s, 1H), 5.93-5.76 (m, 1H), 5.22-5.11 (t, 2H, J=9.96 Hz), 4.33-4.23 (m, 1H), 3.80-3.75 (d, 3H, J=4.00 Hz), 3.67-3.62 (dd, 1H, J=9.44, 3.40 Hz), 3.48-3.40 (m, 1H), 3.36 (s, 2H), 3.20 (s, 1H), 3.14-3.07 (m, 1H), 3.05-2.96 (m, 1H), 2.81 (s, 1H), 2.53-2.23 (m, 4H), 2.03-1.81 (m, 6H), 1.79-1.68 (m, 1H). 13C NMR (CDCl3, a mixture of rotamers) δu 155.0, 133.2, 133.0, 129.3, 129.2, 122.8, 122.78, 119.6, 119.5, 116.4, 112.0, 111.9, 59.1, 58.9, 57.8, 56.4, 55.2, 55.19; δd 195.1, 165.8, 159.5, 138.5, 138.2, 138.0, 119.6, 119.5, 74.1, 72.2, 48.3, 45.7, 44.5, 43.9, 43.0, 42.4, 39.9, 39.7, 34.1, 34.0, 30.5, 30.3, 29.7, 28.5, 27.7, 24.2, 22.0.GC tR = 15.31 min. (Major isomer, 94.45%). EI-MS m/z (%): 397 (M+, 5), 352 (73), 283 (20), 121 (100). HRMS (CI) (m/z): [M+1]+ calcd for C24H32NO4: 398.2331, found: 398.2332.
Enone 9c
Use of the general procedure with 1,5-diene 7c (0.7329 g, 1.85 mmol) provided 0.5869 g of the pure enone product, a mixture of rotamers in an 80% yield. [α]D24 = −60.8 (c=0.13, CH2Cl2). 1H NMR (CDCl3) δ 7.08-6.96 (m, 2H), 6.82-6.69 (m, 3H), 5.85-5.70 (m, 1H), 5.16-5.02 (m, 2H), 4.26-4.17 (m, 1H), 3.71 (s, 3H), 3.64-3.55 (m, 1H), 3.43-3.33 (m, 1H), 3.33-3.28 (m, 2H), 3.17-3.13 (m, 1H), 3.10-3.29 (m, 2H), 2.78-2.67 (m, 2H), 2.46-2.30 (m, 2H), 2.30-2.13 (m, 2H), 1.94-1.77 (m, 5H), 1.74-1.61 (m, 1H). 13C NMR (CDCl3, a mixture of rotamers) δu 134.3, 134.2, 133.25, 133.17, 133.14, 131.4, 131.37, 131.3, 129.7, 113.96, 113.94, 113.81, 113.76, 113.72, 113.68, 59.11, 58.90, 58.55, 57.84, 57.74, 57.56, 56.37, 56.34, 55.26, 55.22; δd 195.6, 195.5, 195.1, 195.7, 166.3, 165.92, 165.86, 165.83, 158.5, 158.3, 155.3, 155.2, 154.7, 138.5, 138.0, 137.9, 128.55, 128.43, 128.38, 128.30, 119.45, 119.41, 119.38, 118.1, 74.04, 74.03, 72.21, 72.16, 72.11, 48.33, 48.30, 46.75, 46.43, 45.66, 45.48, 43.5, 43.4, 42.99, 42.90, 42.85, 42.80, 42.3, 39.95, 39.90, 39.82, 37.02, 36.92, 34.10, 34.04, 34.01, 33.98, 30.5, 3.4, 30.24, 30.18, 28.5, 28.3, 27.7, 24.2, 24.2, 21.99, 21.95. GC tR = 16.01 min. EI-MS m/z (%): 397 (M+, 3), 352 (6), 121 (100). HRMS (CI) (m/z): [M+1]+ calcd for C24H32NO4: 398.2331, found: 398.2343.
Enone 9d
Use of the general procedure with 1,5-diene 7d (0.8188 g, 1.92 mmol) provided 0.6026 g of the pure enone product in a 73% yield. [α]D24 = −22.2 (c=0.33, CH2Cl2). 1H NMR (CDCl3, a mixture of rotamers) δ 6.83-6.75 (m, 2H), 6.75-6.70 (m, 1H), 6.70-6.66 (m, 1H), 5.92-5.76 (m, 1H), 5.23-5.10 (m, 2H), 4.33-4.23 (m, 1H), 3.89-3.84 (m, 6H), 3.67-3.59 (dd, 1H, J=9.36, 3.32 Hz), 3.48-3.41 (m, 1H), 3.36 (s, 2H), 3.22 (s, 1H), 3.17-3.09 (m, 1H), 3.09-3.00 (m, 1H), 2.88-2.72 (m, 1H), 2.53-2.37 (m, 2H), 2.37-2.21 (m, 2H), 2.07-1.84 (m, 5H), 1.79-1.67 (m, 1H).13C NMR (CDCl3, a mixture of rotamers) δu 133.25,133.2, 133.1122.5, 122.5, 113.75, 113.70, 113.6, 111.08, 111.01, 110.98, 59.1, 58.9, 57.9, 57.5, 56.34, 56.32, 56.01, 55.99, 55.87; δd 195.5, 195.4, 195.08, 195.98, 165.9, 165.8, 155.3, 155.2, 154.3, 148.7, 148.0, 138.4, 137.8, 129.0, 128.98, 128.81, 119.5, 119.4, 74.1, 72.3, 72.2, 53.4, 48.33, 48.26, 45.7, 45.5, 44.03, 43.6, 43.5, 43.0, 42.5, 39.9, 39.8, 39.77, 39.69, 34.1, 33.98, 30.8, 30.6, 30.5, 28.6, 28.3, 27.7, 27.6, 24.2, 24.1, 22.0, 21.9. GC tR = 18.69 min. EI-MS m/z (%): 427 (M+, 4), 382 (4), 151 (100). HRMS (CI) (m/z): [M]+ calcd for C25H34NO5: 428.2437, found: 428.2420.
Enone 9e
Use of the general procedure with 1,5-diene 7e (0.360 g, 0.945 mmol) provided 0.25 g of the pure enone product in a 69% yield. [α]D24 = −22.8 (c=1.2, CH2Cl2). 1H NMR (CDCl3, a mixture of rotamers) δ 7.23-7.13 (m, 1H), 7.10-7.02 (m, 1H), 7.01-6.93 (m, 2H), 6.81 (s, 1H), 5.95-5.78 (m, 1H), 5.25-5.10 (m, 2H), 4.35-4.24 (m, 1H), 3.71-3.60 (m, 1H), 3.50-3.40 (m, 1H), 3.40 (s, 2H), 3.20 (s, 1H), 3.17-2.93 (m, 2H), 2.87-2.76 (m, 2H), 2.54-2.36 (m, 2H), 2.37-2.25 (m, 5H), 2.20-1.83 (m, 5H), 1.81-1.67 (m, 1H). 13C NMR (CDCl3, a mixture of rotamers) δu 134.27, 134.21, 134.03, 129.5, 128.8, 127.6, 125.0, 124.4, 121.1, 114.8, 111.6, 59.0, 57.97, 57.88, 55.1; δd 168.5, 159.8, 140.2, 139.1, 118.24, 118.20, 77.0, 71.8, 61.6, 61.2, 46.5, 46.4, 43.7, 41.6, 41.5, 37.2, 36.9, 28.4, 28.2, 26.7, 25.9, 24.5. GC tR = 18.69 min. EI-MS m/z (%): 381 (M+, 3), 336 (46), 267 (14), 105 (100). HRMS (CI) (m/z): [M+1]+ calcd for C24H32NO3: 382.2382, found: 382.2374.
Enone 10a
Use of the general procedure with 1,5-diene 8a (2.60 g, 6.82 mmol) provided 1.96 g of the pure enone product in a 75% yield. [α]D24 = −31.8 (c=0.67, CH2Cl2). 1H NMR (CDCl3, a mixture of rotamers) δ 7.34-7.24 (m, 2H), 7.23-7.12 (m, 3H), 6.89-6.83 (m, 1H), 5.91-5.75 (m, 1H), 5.24-5.13 (m, 2H), 4.36-4.26 (m, 1H), 3.71-3.62 (m, 1H), 3.51-3.43 (m, 1H), 3.38 (s, 2H), 3.30-3.22 (m, 1H), 3.21-3.12 (m, 2H), 2.75-2.59 (m, 2H), 2.57-2.51 (t, 2H, J=6.96 Hz), 2.42-2.34 (t, 2H, J=7.80 Hz), 2.06-1.87 (m, 5H), 1.87-1.75 (m, 3H). 13C NMR (CDCl3, a mixture of rotamers) δu155.1, 154.7132.9, 132.8, 128.5, 128.2, 128.1, 126.1, 59.1, 58.9, 57.9, 56.4; δd 195.5, 194.0, 166.0, 165.6, 141.6, 138.2, 137.8, 119.3, 74.0, 72.0, 48.3, 45.5, 42.0, 41.7, 39.8, 39.7, 38.7, 33.97, 33.93, 39.77, 30.69, 30.44, 30.38, 28.5, 27.6, 24.2, 22.0. GC tR = 14.98 min. (Major isomer, 93.62%). EI-MS m/z (%): 381 (M+, 3), 336 (56), 267 (60), 135 (34), 91 (100). tR = 15.18 min (Minor isomer, 6.12%). EI-MS m/z (%): 381 (M+, 5), 336 (82), 267 (61), 91 (100). HRMS (CI) (m/z): [M+1]+ calcd for C24H32NO3: 382.2382, found: 382.2386.
Enone 10b
Use of the general procedure with 1,5-diene 8b (1.72 g, 4.20 mmol) provided 1.32 g of the pure enone product in a 76% yield. [α]D24 = −30.3 (c=0.73, CH2Cl2). 1H NMR (CDCl3, a mixture of rotamers) δ 7.34-7.24 (m, 2H), 7.08-6.98 (t, 1H, J=8.40 Hz), 6.69 (s, 1H), 6.65-6.50 (m, 3H), 5.74-5.59 (m, 1H), 5.09-4.96 (m, 2H), 4.18-4.08 (m, 1H), 3.60 (s, 3H), 3.54-3.45 (dd, 1H, J=9.32, 3.20 Hz), 3.35-3.25 (t, 1H, J=8.04 Hz), 3.20 (s, 2H), 3.15-2.98 (m, 3H), 2.53-2.41 (m, 2H), 2.41-2.31 (t, 2H, J=6.48 Hz), 2.26-2.14 (t, 2H, J=7.28 Hz), 1.88-1.70 (m, 5H), 1.70-1.57 (m, 3H). 13C NMR (CDCl3, a mixture of rotamers) δu 155.151.1, 154.4, 132.8, 132.77, 129.5, 120.54, 120.49, 114.1, 111.2, 111.18, 59.06, 58.94, 57.9, 56.4, 55.1,14.2; δd 195.6, 195.1, 166.0, 165.8, 159.7, 143.2, 143.2, 138.2, 137.8, 137.7, 119.4, 74.2, 72.1, 48.5, 45.5, 41.9, 41.6, 39.61, 39.4, 38.6, 33.93, 33.90, 30.72, 30.64, 30.46, 30.40, 28.5, 27.6, 24.2, 22.0. GC tR = 18.02 min. (Major isomer, 92.69%). EI-MS m/z (%): 411 (M+, 10), 366 (100), 297 (53), 135 (48), 121 (94). tR = 18.51 min (Minor isomer, 6.77%). EI-MS m/z (%): 411 (M+, 10), 366 (73), 297 (100), 121 (55). HRMS (CI) (m/z): [M+1]+ calcd for C25H34NO4: 412.2488, found: 412.2482.
Enone 10c
Use of the general procedure with 1,5-diene 8c (1.00 g, 2.43 mmol) provided 0.630 g of the pure enone product in a 63% yield. [α]D24 = −22.1 (c=1.2, CH2Cl2). 1H NMR (CDCl3, a mixture of rotamers) δ 7.14-7.05 (d, 2H, J=8.56 Hz), 6.91-6.81 (t, 3H, J=8.56 Hz), 5.93-5.76 (m, 1H), 5.25-5.11 (m, 2H), 4.37-4.27 (m, 1H), 3.80 (s, 4H), 3.73-3.63 (dd, 1H, J=9.48, 3.32 Hz), 3.54-3.43 (m, 1H), 3.39 (s, 2H), 3.31-3.23 (m, 1H), 3.19 (m, 2H), 3.69-3.51 (m, 4H), 2.43-2.35 (m, 2H), 2.05-1.88 (m, 6H), 1.86-1.73 (m, 1H). 13C NMR (CDCl3, a mixture of rotamers) δu 155.38, 154.8, 132.9, 132.8, 129.1, 129.05, 114.0, 59.1, 59.0, 57.9, 56.4, 55.3; δd 195.65, 195.15, 166.0, 165.9, 158.0, 138.14, 137.7, 133.7, 74.2, 72.2, 48.4, 45.5, 41.9, 41.6, 40.0, 38.7, 34.0, 33.95, 30.79, 30.71, 29.54, 29.49, 28.5, 27.8, 24.3. GC tR = 18.69 min. (Major isomer, 94.45%). EI-MS m/z (%): 411 (M+, 3), 366 (22), 297 (22), 277 (15), 121 (100). tR = 19.06 min. (Minor isomer, 5.49%). 411 (M+, 4), 366 (36), 297 (17), 121 (100). HRMS (CI) (m/z): [M+1]+ calcd for C25H34NO4: 412.2488, found: 412.2476.
Enone 10d
Use of the general procedure with 1,5-diene 8d (1.58 g, 3.58 mmol) provided 1.14 g of the pure enone product in a 72% yield. [α]D24 = −33.2 (c=1.7, CH2Cl2). 1H NMR (CDCl3, a mixture of rotamers) δ 6.89-6.85 (d, 1H, J=2.24 Hz), 6.83-6.77 (d, 1H, J=8.04 Hz), 6.74-6.66 (dd, 2H, J=10.08, 1.92 Hz), 5.92-5.76 (m, 1H), 5.25-5.13 (m, 2H), 4.36-4.25 (m, 1H), 3.87 (s, 3H), 3.84 (s, 3H), 3.71-3.61 (m, 1H), 3.51-3.41 (m, 1H), 3.37 (s, 2H), 3.30-3.15 (m, 3H), 2.67-2.58 (m, 2H), 2.57-2.51 (t, 2H, J=7.32 Hz), 2.42-2.34 (t, 2H, J=7.48 Hz), 2.06-1.81 (m, 5H), 1.87-1.75 (m, 3H). 13C NMR (CDCl3, a mixture of rotamers) δu 155.1, 132.8, 120.0, 111.6, 111.4, 59.0, 58.9, 57.85, 57.73, 56.3, 55.87, 55.82, 14.1; δd 295.5, 195.0, 166.0, 165.3, 148.9, 147.4, 138.1, 137.78, 137.65, 134.2, 119.2, 74.2, 72.14, 72.09, 60.24, 48.4, 48.39, 45.5, 45.4, 42.1, 42., 41.9, 41.6, 39.8, 39.7, 38.6, 33.93, 33.89, 30.8, 30.7, 29.97, 28.4, 27.6, 24.2, 21.95. GC tR = 21.85 min. (Major isomer, 95.49%). EI-MS m/z (%): 441 (M+, 3), 396 (17), 327 (22), 277 (13), 151 (100). tR = 22.58 min. (Minor isomer, 4.5%). EI-MS m/z (%): 441 (M+, 8), 396 (40), 327 (45), 277 (36), 151 (100). HRMS (CI) (m/z): [M+1]+ calcd for C26H36NO5: 442.2593, found:442.2572.
Enone 10e
Use of the general procedure with 1,5-diene 8e (1.30 g, 3.29 mmol) provided 1.10 g of the pure enone product in a 84% yield. [α]D24 = −27.5 (c=0.4, CH2Cl2). 1H NMR (CDCl3, a mixture of rotamers) δ 7.14 (t, 1H, J=7.44 Hz), 7.04-6.89 (m, 3H), 6.85 (s, 1H), 5.90-5.72 (m, 1H), 5.22-5.09 (m, 2H), 4.34-4.22 (m, 1H), 3.70-3.60 (m, 1H), 3.50-3.40 (m, 1H), 3.36 (s, 2H), 3.29-3.11 (m, 3H), 2.55-2.55 (m, 2H), 2.52 (t, 2H, J=6.83 Hz), 2.40-2.32 (m, 2H), 2.30 (s, 3H), 2.00-1.84 (m, 5H), 1.85-1.70 (m, 3H). 13C NMR (CDCl3, a mixture of rotamers) δu 155.2, 154.7, 132.9, 132.8, 129.0, 129.97, 128.4, 126.8, 125.2, 125.18, 59.1, 58.9, 57.8, 56.4, 21.3, 14.2; δd 195.5, 195.2, 166.0, 165.9, 141.6, 138.2, 138.1, 137.8, 119.3, 74.2, 72.2, 60.3, 48.3, 46.4, 46.3, 41.9, 41.7, 39.8, 38.7, 34.0, 33.9, 30.77, 30.67, 30.38, 30.33, 28.5, 27.6, 24.2, 22.0. GC tR = 15.66 min. (Major isomer, 93.34%). EI-MS m/z (%): 395 (M+, 6), 350 (77), 281 (43), 105 (100). tR = 16.00 min. (Minor isomer, 5.13%). 395 (M+, 10), 350 (100), 281 (78), 105 (77). HRMS (CI) (m/z): [M+1]+ calcd for C25H34NO3: 396.2539, found: 396.2536.
General Procedure for Friedel-Crafts Conjugate Addition
Under an argon atmosphere, enone (1.0 eq.) was dissolved in DCM (8 mL/mmol enone). The mixture was cooled to 0°C and BF3·Et2O (1.2 eq.) was added dropwise. The reaction was then left to warm to room temperature and stirred overnight. The next day the reaction was quenched with saturated NH4Cl and water. The aqueous layer was extracted 3× with DCM and the combined organics were washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified using column chromatography (1:1 EtOAc in heptanes).
Tricyclic Compound 11b
Use of the general procedure with enone 9b (92.3 mg, 0.232 mmol) provided 90.5 mg of the tricyclic product in as a mixture of inseparable epimers in a 98% yield. [α]D24 = −201 (c=0.17, CH2Cl2). 1H NMR (CDCl3, a mixture of rotamers and epimers) δ 7.13-6.94 (m, 1H), 6.80-6.70 (m, 1H), 6.70-6.59 (m, 1H), 5.98-5.73 (m, 1H), 5.22-4.96 (m, 2H), 4.77-4.30 (m, 1H), 4.00 (s, 1H), 3.96-3.87 (m, 1H), 3.77 (s, 3H), 3.74-3.67 (m, 1H), 3.60-3.51 (dd, 1H, J=9.84, 2.44 Hz), 3.50-3.42 (m, 2H), 3.42-3.33 (m, 1H), 3.33-3.25 (m, 1H), 3.06-2.79 (m, 2H), 2.66-2.37 (m, 3H), 2.37-2.20 (m, 2H), 2.21-1.80 (m, 5H), 1.72-1.44 (m, 2H). 13C NMR (CDCl3, a mixture of rotamers and epimers) δu 134.3, 134.2, 134.1, 124.6, 124.4, 123.7, 112.3, 112.2, 111.15, 111.96, 111.89, 111.70, 111.64, 111.18, 110.2, 61.6, 59.3, 59.18, 59.4, 57.4, 55.4, 47.2, 46.7; δd 178.0, 177.2, 166.7, 159.23, 159.18, 142.9, 142.5, 138.0, 137.97, 118.37, 118.20, 118.15, 117.98, 96.4, 74.8, 73.7, 72.4, 71.7, 50.3, 49.6, 48.8, 47.0, 43.9, 43.3, 4.4, 4.3, 36.5, 29.7, 29.2, 28.8, 28.6, 26.4, 25.8, 25.3, 25.0, 19.8. HRMS (ESI) (m/z): [M+1]+ calcd for C24H32NO4: 398.2326, found: 398.2323.
Tricyclic Compound 11d
Use of the general procedure with enone 9d (233.7 mg, 0.548 mmol) provided 227.6 mg of the tricyclic product in as a mixture of epimers in a 97% yield. [α]D24 = +8.6 (c=0.27, CH2Cl2). Less polar epimer: 1H NMR (CDCl3, a mixture of rotamers) δ 6.80-6.74 (d, 1H, J=7.76 Hz), 6.58 (s, 0.65H), 6.39 (s, 0.14H), 6.32 (s, 0.14H), 5.96-5.81 (m, 1H), 5.22-5.09 (m, 2H), 4.81-4.71 (m, 1H), 4.12-3.96 (m, 2H), 3.96-3.89 (m, 1H), 4.12-3.96 (m, 1H), 3.96-3.89 (m, 1H), 3.85 (s, 1H), 3.82 (s, 2H), 3.75-3.62 (m, 1H), 3.58-3.50 (dd, 1H, J=10.0, 3.16 Hz), 3.42-3.30 (m, 3H), 3.05-2.91 (m, 1H), 2.61-2.39 (m, 3H), 2.36-2.23 (m, 2H), 2.03-1.89 (m, 4H), 1.70-1.44 (m, 3H). More polar epimer: (fractions 9–35) 1H NMR (CDCl3, a mixture of rotamers) δ 6.74 (s, 0.5H), 6.73 (d, 1H, J=5.12 Hz), 6.67 (s, 0.5H), 5.88-5.73 (m, 1H), 5.15-5.00 (m, 2H), 4.51-4.28 (m, 1H), 3.90-3.82 (m, 3H), 3.81-3.75 (m, 3H), 3.51-3.13 (m, 4H), 3.09-2.71 (m, 4H), 2.66-2.57 (m, 1H), 2.55-2.36 (m, 2H), 2.33-2.20 (m, 2H), 2.19-2.12 (m, 1H), 2.06-1.87 (m, 4H), 1.88-1.68 (m, 2H). Less polar epimer: 13C NMR (CDCl3, a mixture of rotamers) δu134.5, 134.4, 109.3, 109.2, 109.0, 108.3, 107.7, 107.3, 61.7, 61.6, 59.26, 59.21, 59.18, 57.4, 56.43, 56.39, 56.13, 56.11, 47.4, 47.3, 21.0, 14.2, 14.1; δd 178.6, 178.3, 177.8, 166.7, 148.72, 148.62, 148.49, 148.32, 138.0, 137.8, 137.5, 133.3, 133.2, 132.9, 118.14, 118.08, 117.96, 95.8, 73.6, 73.0, 72.4, 60.3, 50.3, 49.6, 49.0, 47.7, 47.3, 47.2, 43.6, 43.2, 40.4, 40.3, 39.9, 31.9, 29.7, 29.4, 28.9, 28.8, 26.7, 26.1, 25.9, 25.3, 25.1, 22.7, 20.0. More polar epimer: 13C NMR (CDCl3, a mixture of rotamers) δu 134.2, 134.1, 134.05, 108.7, 108.3, 108.0, 107.73, 107.71, 107.57, 107.33, 59.1, 58.9, 58.7, 58.4, 57.8, 57.7, 57.3, 57.2, 57.0, 56.6, 56.4, 56., 55.9, 53.1, 52.0, 51.75, 51.71, 31.6, 14.1; δd 209.1, 208.5, 208.2, 168.4, 167.9, 148.8, 148.7, 148.2, 148.1, 136.4, 136.3, 136.0, 135.8, 132.9, 132.8, 118.4, 118.3, 114.0, 75.1, 74.9, 72.7, 71.7, 47.3, 47.2, 46.3, 45.9, 45.0, 44.8, 44.66, 44.61, 44.0, 43.89, 43.85, 43.73, 43.68, 36.5, 36.4, 33.8, 31.9, 31.2, 31.01, 30.97, 30.89, 29.7, 29.5, 29.3, 29.2, 29.1, 28.9, 27.5, 27.4, 24.0, 23.8, 22.7, 22.3. HRMS (ESI) (m/z): [M+1]+ calcd for C25H34NO5: 428.2432, found: 428.2428.
Tricyclic Compound 11e
Use of the general procedure with enone 9e (211.8 mg, 0.534 mmol) provided 189.2 mg of an inseparable mixture of 2 epimers, a 76% yield. [α]D24 = −154 (c=0.53, CH2Cl2). 1H NMR (CDCl3, a mixture of rotamers and epimers) δ 7.09-6.86 (m, 3H), 6.03-5.81 (m, 1H), 5.24-5.07 (m, 2H), 4.81-4.49 (m, 1H), 4.05 (s, 1H), 3.98-3.87 (m, 1H), 3.81-3.65 (m, 1H), 3.62-3.53 (m, 1H), 3.47 (s, 2H), 3.35-3.26 (m, 1H), 3.06-2.90 (m, 1H), 2.66-2.41 (m, 3H), 2.49-2.22 (m, 5H), 2.21-1.82 (m, 5H), 1.76-1.45 (m, 3H). 13C NMR (CDCl3) δu134.5, 134.4, 127.6, 127.5, 126.3, 126.0, 123.7, 122.9, 61.7, 59.3, 59.0, 47.1, 21.2; δd178.3, 143.0, 141.0, 136.8, 118.1, 73.7, 72.4, 49.6, 46.8, 43.1, 40.4, 29.2, 28.6, 26.5, 25.8, 25.3, 24.9. HRMS (ESI) (m/z): [M+1]+ calcd for C24H32NO3: 382.2377, found: 382.2374.
Tricyclic Compound 12a
Use of the general procedure with enone 10a (71.5 mg, 0.188 mmol) provided 45.1 mg of the pure tricyclic product in a 63% yield (as a mixture of epimers and rotomers). [α]D24 = −78.3 (c=0.33, CH2Cl2). 1H NMR (CDCl3, a mixture of rotamers) δ 7.34-7.25 (m, 2H), 7.25-7.11 (m, 2H), 6.03-5.75 (m, 1H), 5.24-5.10 (m, 2H), 4.76-4.65, 4.49-4.31 (m, 1H), 3.78-3.49 (m, 2H), 3.45-3.25 (m, 2H), 3.12-2.96 (m, 1H), 2.86-2.30 (m, 4H), 2.30-2.14 (m, 2H), 2.14-1.91 (m, 5H), 1.91-1.76 (m, 1H), 1.74-1.51 (m, 3H), 1.48-1.36 (m, 3H). 13C NMR (CDCl3, a mixture of rotamers) δu 134.3, 133.9, 128.5, 128.4, 126.5, 125.6, 84.2, 62.4, 61.6, 60.8, 59.1, 58.3, 41.1; δd 208.9, 172.9, 142.7, 118.6, 118.1, 73.6, 72.2, 71.9, 64.0, 49.2, 44.9, 42.2, 42.0, 36.7, 35.6, 35.5, 34.2, 33.8, 30.5, 30.3, 27.1, 27.0, 26.8, 26.3, 25.4, 25.1. HRMS (ESI) (m/z): [M+1]+ calcd for C24H32NO3: 382.2377, found: 382.2372.
Tricyclic Compound 12b
Use of the general procedure with enone 10b (750 mg, 1.82 mmol) provided 662.2 mg of the pure tricyclic product as a 1:1 mixture of two epimers in an 88% yield. [α]D24 = −128 (c=0.2, CH2Cl2). Less polar epimer: 1H NMR (CDCl3, a mixture of rotamers) δ 6.88-6.79 (d, 1H, J=9.24 Hz), 6.76-6.62 (m, 2H), 6.03-5.85 (m, 1H), 5.24-5.05 (m, 2H), 4.77-4.61 (m, 1H), 3.99-3.82 (m, 1H), 3.78 (s, 3H), 3.74-3.59 (m, 2H), 3.59-3.48 (m, 1H), 3.48-3.40 (m, 1H), 3.38 (s, 2H), 3.35-3.27 (m, 1H), 2.82-2.62 (m, 2H), 2.58-2.39 (m, 1H), 2.39-2.16 (m, 3H), 2.11-1.52 (m, 6H), 1.50-1.32 (m, 3H). More polar epimer: 1H NMR (CDCl3, a mixture of rotamers) δ 7.13-6.95 (dd, 1H, J=37.6, 8.52 Hz), 6.67-6.61 (dd, 1H, J=9.48, 2.72 Hz), 6.60-6.64 (dd, 1H, J=8.48, 2.72 Hz), 5.89-5.74 (m, 1H), 5.11-4.91 (m, 2H), 4.25-4.15 (m, 1H), 3.74 (s, 3H), 3.70-3.49 (m, 2H), 3.42-3.27 (m, 3H), 3.19-3.02 (m, 1H), 2.96-2.87 (m, 3H), 2.52-2.37 (m, 2H), 2.28-2.10 (m, 2H), 2.00-1.80 (m, 5H), 1.80-1.70 (m, 2H), 1.65-1.54 (m, 2H), 1.54-1.41 (m, 1H). Less polar epimer: 13C NMR (CDCl3, a mixture of rotamers) δu 134.3, 134.16, 134.07, 133.89, 128.5, 128.4, 127.9, 127.7, 124.04, 113.5, 113.4, 111.3, 113.2, 113.1, 111.7, 111.6, 111.5, 111.4, 111.2, 61.7, 61.6, 61.5, 59.2, 59.1, .5, 55.4, 55.2, 41.3, 41.5, 37.0; δd 178.59, 178.57, 177.9, 166.6, 158.46, 158.29, 158.17, 146.8, 139.7, 138.6, 138.3, 138.5, 131.73, 131.66, 118.7, 118.5, 118.3, 118.1, 97.0, 95.8, 74.0, 73.8, 72.4, 72.34, 72.27, 72.18, 50.3, 48.9, 48.8, 42.1, 41.9, 37.2, 36.8, 36.6, 36.4, 35.8, 34.4, 34.3, 34.1, 33.9, 29.7, 29.3, 29.0, 28.8, 28.4, 28.0, 27.54, 27.48, 27.32, 27.14, 26.6, 26.5, 26.3, 24.7, 25.4, 24.2, 25.05, 24.9, 22.7, 21.8, 19.5. More polar epimer: 13C NMR (CDCl3, a mixture of rotamers) δu 133.7, 133.6, 131.4, 130.9, 113.7, 113.6, 111.6, 111.5, 61.3, 60.9, 60.8, 60.4, 59.2, 59.0, 58.7, 57.0, 56.6, 56.3, 55.2, 55.17, 48.0, 46.9, 46.7, 46.5; δd 207.1, 206.6, 168.4, 158.3, 135.6, 135.4, 130.0, 129.3, 118.5, 118.4, 75.1, 73.7, 71.9, 47.3, 45.9, 45.7, 42.0, 36.6, 34.5, 34.4, 34.3, 34.19, 34.15, 33.9, 28.7, 27.2, 25.64, 25.56, 25.3, 25.1, 25.0, 23.6, 22.7, 22.3, 21.9. HRMS (ESI) (m/z): [M+1]+ calcd for C25H34NO4: 412.2482, found: 412.2479.
Tricyclic Compound 12d
Use of the general procedure with enone 10d (358.4 mg, 0.813 mmol) provided 299.6 mg of the pure tricyclic product as a 1:1 mixture of two epimers in an 84% yield. [α]D24 = −37 (c=0.4, CH2Cl2).Less polar epimer: 1H NMR (CDCl3, a mixture of rotamers) δ 6.71-6.60 (m, 1H), 6.51-6.35 (m, 2H), 6.03-5.84 (m, 1H), 5.24-5.04 (m, 2H), 4.78-4.64 (m, 1H), 3.85 (s, 3H), 3.83-3.71 (m, 3H), 3.70-3.59 (m, 2H), 3.59-3.47 (m, 2H), 3.39-3.28 (m, 3H), 2.76-2.58 (m, 2H), 2.58-2.41 (m, 1H), 2.40-2.28 (m, 2H), 2.09-1.78 (m, 5H), 1.53-1.15 (m, 6H).More polar epimer: 1H NMR (CDCl3, a mixture of rotamers) δ 6.82-6.54 (m, 2H), 5.94-5.76 (m, 1H), 5.14-4.93 (m, 2H), 4.42-4.17 (m, 1H), 3.90-3.79 (m, 4H), 3.79-3.73 (m, 2H), 3.68-3.23 (m, 4H), 3.22-2.96 (m, 3H), 2.96-2.82 (m, 3H), 2.53-2.40 (m, 2H), 2.33-2.06 (m, 4H), 2.02-1.84 (m, 4H), 1.83-1.44 (m, 3H). Less polar epimer: 13C NMR (CDCl3, a mixture of rotamers) δu 134.1, 134.0, 111.2, 111.1, 111.0, 110.4, 61.6, 61.56, 59.2, 59.1, 57.5, 56.4, 56.0, 55.9, 55.8, 40.9, 40.2, 21.0, 14.13, 14.1; δd 179.2, 166.8, 147.6, 147.5, 131.7, 129.0, 128.7, 118.7, 118.5, 97.5, 72.6, 72.4, 49.4, 49., 42.3, 42.1, 36.6, 36.3, 34.2, 33.9, 28.8, 27.3, 26.7, 26.6, 26.4, 26.3, 25.8, 25.5, 25.1. More polar epimer: 13C NMR (CDCl3, a mixture of rotamers) δu133.76, 133.69, 133.63, 113.6, 113.2, 112.5, 112.3, 111.67, 111.59, 111.52, 111.37, 61.3, 61.0, 60.94, 60.3, 59.2, 59.0, 58.8, 58.7, 57.0, 56.7, 56.5, 56.0, 55.89, 55.86, 55.82, 55.79, 55.73, 48.1, 46.8, 46.8, 46.7, 14.2; δd 206.91, 206.84, 26.42, 206.3, 169.1, 168.6, 168.5, 168.4, 147.78, 147.67, 147.64, 146.94, 146.90, 146.79, 146.71, 129.9, 129.7, 129.4, 128.9, 125.98, 125.95, 125.75, 125.70, 118.4, 118.3, 75.1, 73.8, 72.3, 71.5, 47.4, 47.2, 46.0, 45.9, 42.0, 41.9, 41.86, 36.7, 36.68, 36.58, 34.43, 34.36, 34.30, 34.12, 34.09, 34.0, 33.93, 33.89, 28.9, 27.3, 27.2, 25.5, 25.4, 25.3, 25.2, 25.11, 25.04, 25.01, 24.99, 23.37, 23.61, 22.3, 22.0. HRMS (ESI) (m/z): [M+1]+ calcd for C26H36NO5: 442.2588, found: 442.2584.
Tricyclic Compound 12e
Use of the general procedure with enone 10e (356.6 mg, 0.903 mmol) provided 251.0 mg of the pure tricyclic product as a 1:1 mixture of two epimers in a 70% yield. [α]D24 = −48 (c=0.87, CH2Cl2). Less polar epimer: 1H NMR (CDCl3, a mixture of rotamers) δ 7.08-6.89 (m, 2H), 6.89-6.76 (m, 1H), 6.00-5.74 (m, 1H), 5.25-4.97 (m, 2H), 4.78-4.65 (m, 1H), 3.81-3.50 (m, 3H), 3.50-3.27 (m, 4H), 2.83-2.62 (m, 2H), 2.62-2.42 (m, 2H), 2.40-2.14 (m, 6H), 2.14-1.75 (m, 6H), 1.73-1.14 (m, 3H). More polar epimer: 1H NMR (CDCl3, a mixture of rotamers) δ 6.11-6.78 (m, 3H), 5.99-5.73 (m, 1H), 5.17-4.93 (m, 2H), 4.35-4.14 (m, 1H), 3.75-3.50 (m, 2H), 3.50-3.31 (m, 3H), 3.17-3.06 (m, 1H), 2.97-2.88 (m, 3H), 2.54-2.24 (m, 2H), 2.28 (s, 3H), 2.26-2.09 (m, 2H), 2.08-1.80 (m, 5H), 1.83-1.56 (m, 4H), 1.52-1.41 (m, 1H).Less polar epimer: 13C NMR (CDCl3, a mixture of rotamers) δu134.3, 133.7, 133.6, 130.4, 129.7, 129.6, 129.5, 129.3, 129.1, 128.7,128.5, 128.3, 127.4, 127.3, 127.2, 126.9, 126.6, 126.5, 61.6, 61.5, 61.1, 60.9, 60.7, 59.1, 59.0, 58.6, 57.0, 56.6, 47.15, 47.04, 46.8, 40.9, 40.3, 21.0, 20.9; δd 206.6, 178.7, 136.6, 136.5, 136.3, 136.1, 134.8, 134.0, 133.8, 118.5, 118.4, 118.3, 118.1, 73.9, 73.5, 72.4, 72.2, 71.9, 50.3, 49.2, 48.9, 47.3, 45.7, 42.2, 42.1, 42.0, 36.6, 36.3, 35.9, 34.12, 34.06, 33.8, 28.8, 28.7, 28.0, 27.3, 27.2, 27.1, 26.9, 26.6, 26.5, 26.3, 25.5, 25.4, 25.3, 25.2,25.1, 24.9, 23.6, 21.9, 19.6.More polar epimer: 13C NMR (CDCl3, a mixture of rotamers) δu133.7, 133.6, 130.3, 129.7, 129.6, 129.5, 126.7, 126.5, 60.9, 60.7, 59.0, 58.6, 57.0, 56.6, 47.1, 46.8, 21.0, 20.9; δd 206.98, 206.54, 168.6, 168.3, 136.2, 136.0, 134.7, 134.1, 133.94, 133.8, 118.4, 118.3, 73.5, 73.3, 47.3, 45.7, 42.1, 42.0,36.6, 34.17, 34.11, 34.05, 33.8, 28.7, 27.3, 25.5, 25.3, 25.2, 23.6, 21.9. HRMS (ESI) (m/z): [M+1]+ calcd for C25H34NO3: 396.2533, found: 396.2530.
General Procedure for Chiral Auxiliary Removal
β-ketoamide (1.0 eq.) and N-methylhydroxylamine hydrochloride (2.0 eq.) were dissolved in EtOH (10 mL/mmol β-ketoamide). This was refluxed overnight. The next day EtOH was removed in vacuo, and the resulting residue was re-dissolved in EtOAc and water. The organic layer was separated and the aqueous layer was extracted two times more with EtOAc. The combined organics were washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The crude isoxazolone was purified using column chromatography (EtOAc in heptanes).
Isoxazolidinone 13b
Use of the general procedure with β-ketoamide 11b (122.0 mg, 0.307 mmol) provided 74.1 mg of the pure isoxazolidinone in a 77% yield. [α]D24 = −113 (c=0.4, CH2Cl2). 1H NMR (CDCl3) δ 7.51-7.43 (d, 1H, J=3.02 Hz), 6.75-6.69 (m, 2H), 5.95-5.81 (m, 1H), 5.18-5.09 (m, 2H), 3.78 (s, 3H), 3.75 (s, 1H), 3.22 (s, 3H), 3.05-2.66 (dd, 2H, J=123.2, 15.76 Hz), 2.50-2.21 (m, 4H), 1.84-1.62 (m, 2H). 13C NMR (CDCl3) δu134.3, 126.2, 112.3, 110.6, 55.4, 44.2, 38.4; δd 171.3, 164.2, 159.1, 142.0, 136.4, 118.1, 102.7, 45.7, 42.9, 41.1, 29.7, 18.9. GC tR = 15.23 min. EI-MS m/z (%): 311 (M+, 9), 269 (100), 224 (32), 152 (20). HRMS (ESI) (m/z): [M+1]+ calcd for C19H22NO3: 312.1594, found: 312.1591.
Isoxazolidinone 13d
Use of the general procedure with β-ketoamide 11d (259 mg, 0.607 mmol) provided 150.2 mg of the pure isoxazolidinone in a 72% yield. [α]D24 = −156 (c=0.53, CH2Cl2). 1H NMR (CDCl3) δ 7.16, 6.68 (two s, 2H), 5.93-5.77 (m, 1H), 5.18-5.07 (m, 2H), 3.84 (2s, 6H), 3.75 (s, 1H), 3.22 (s, 3H), 3.00-2.60 (dd, 2H, J=128, 19.36), 2.50-2.24 (m, 4H), 1.84-1.65 (m, 2H).13C NMR (CDCl3) δu134.4, 108.6, 108.0, 56.04, 55.98, 44.98, 38.4; δd 171.4, 164.3, 148.43, 148.37, 136.0, 131.8, 118.0, 102.5, 45.7, 42.8, 41.3, 28.0, 18.9. GC tR = 13.49 min. EI-MS m/z (%): 341 (M+, 52), 299 (97), 254 (79), 224 (100). HRMS (ESI) (m/z): [M+1]+ calcd for C20H24NO4: 342.1700, found: 342.1696.
Isoxazolidinone 13e
Use of the general procedure with β-ketoamide 11e (112.5 mg, 0.295 mmol) provided 52.6 mg of the pure isoxazolidinone in a 60% yield. [α]D24 = −200 (c=0.27, CH2Cl2). 1H NMR (CDCl3) δ 7.52-7.41 (d, 1H, J=8.08 Hz), 6.99 (s, 2H), 5.98-5.80 (m, 1H), 5.20-5.06 (m, 2H), 3.78 (s, 1H), 3.23 (s, 3H), 3.05-2.95 (m, 1H), 2.74-2.64 (m, 1H), 2.50-2.36 (m, 1H), 2.36-2.26 (m, 6H), 1.84-1.62 (m, 2H). 13C NMR (CDCl3) δu 134.4, 127.7, 125.5, 125.3, 44.6, 38.4, 21.3; δd 171.3, 164.3, 141.2, 140.6, 136.6, 118.0, 102.7, 45.5, 42.7, 41.1, 27.6, 18.9. GC tR = 11.21 min. EI-MS m/z (%): 295 (M+, 17), 253 (100), 208 (42), 193 (36), 164 (27).HRMS (ESI) (m/z): [M+1]+ calcd for C19H22NO2: 296.1645, found: 296.1642.
Isoxazolidinone 14b
Use of the general procedure with β-ketoamide 12b (344.1 mg, 0.837 mmol) provided 225.6 mg of the pure isoxazolidinone in an 83% yield. [α]D24 = −135 (c=0.2, CH2Cl2). 1H NMR (CDCl3) δ 7.60-7.51 (d, 1H, J=8.80 Hz), 6.78-6.66 (dd, 1H, J=8.68, 2.76 Hz), 6.63-6.58 (d, 1H, J=2.72 Hz), 5.95-5.81 (m, 1H), 5.20-5.03 (m, 2H), 3.76 (s, 3H), 3.47 (s, 1H), 3.23 (s, 3H), 2.87-2.68 (m, 2H), 2.48-2.28 (m, 2H), 2.24-2.01 (m, 2H), 1.88-1.73 (m, 3H), 1.60-1.51 (m, 1H). 13C NMR (CDCl3) δu 133.7, 131.0, 113.0, 112.3, 55.2, 38.4, 38.1; δd 172.1, 164.4, 157.8, 136.2, 129.9, 118.2, 103.9, 41.6, 35.4, 31.8, 26.0, 25.5, 19.0. GC tR = 13.89 min. EI-MS m/z (%): 325 (M+, 24), 297 (8), 283 (100), 171 (18). HRMS (ESI) (m/z): [M+1]+ calcd for C20H24NO3: 326.1751, found: 326.1747.
Isoxazolidinone 14d
Use of the general procedure with β-ketoamide 12d (283.6 mg, 0.643 mmol) provided 192.0 mg of the pure isoxazolidinone in an 84% yield. [α]D24 = +4.4 (c=0.93, CH2Cl2). 1H NMR (CDCl3) δ 7.43 (s, 1H), 6.53 (s, 1H), 6.63-6.58, 5.96-5.75 (m, 1H), 5.21-4.98 (m, 2H), 3.92-3.73 (m, 6H), 3.51 (s, 1H), 3.31-3.15 (m, 3H), 2.85-2.52 (m, 2H), 2.48-2.29 (m, 2H), 2.22-2.11 (m, 1H), 2.08-1.98 (m, 1H), 1.95-1.82 (m, 2H), 1.79-1.66 (m, 1H), 1.60-1.49 (m, 1H). 13C NMR (CDCl3) δu 133.6, 112.7, 110.9, 55.9, 55.8, 38.4, 38.3; δd 172.4, 164.4, 147.7, 147.2, 130.0, 126.1, 118.5, 103.9, 41.6, 34.9, 32.0, 25.0, 24.8, 18.9. GC tR = 15.23 min. EI-MS m/z (%): 355 (M+, 70), 313 (100), 296 (49), 268 (50), 201 (55). HRMS (ESI) (m/z): [M+1]+ calcd for C21H26NO4: 356.1856, found:356.1852.
Isoxazolidinone 14e
Use of the general procedure with β-ketoamide 12e (246.3 mg, 0.624 mmol) provided 150.0 mg of the pure isoxazolidinone in a 78% yield. [α]D24 = −149 (c=0.13, CH2Cl2). 1H NMR (CDCl3) δ 7.59-7.60 (d, 1H, J=7.96 Hz), 7.03-6.95 (d, 1H, J=7.36 Hz), 6.91 (s, 1H), 5.99-5.80 (m, 1H), 5.21-5.00 (m, 2H), 3.52 (s, 1H), 3.24 (s, 3H), 2.86-2.65 (m, 2H), 2.47-2.32 (m, 2H), 2.30 (s, 3H), 2.23-2.01 (m, 3H), 1.89-1.75 (m, 3H). 13C NMR (CDCl3) δu 133.7, 129.8, 128.9, 127.2, 38.42, 38.40, 20.9; δd 172.1, 164.6, 135.6, 134.9, 134.7, 118.5, 104.0, 41.6, 35.2, 31.9, 25.6, 25.4, 19.0. GC tR = 12.33 min. EI-MS m/z (%): 309 (M+, 42), 267 (100), 222 (39), 165 (42), 155 (60). HRMS (ESI) (m/z): [M+1]+ calcd for C20H24NO2: 310.1802, found:310.1798.
General Procedure for Isoxazolidinone Cleavage
Isoxazolidinone (1.0 eq.) and Mo(CO)6 (1.2 eq.) were dissolved in a 15:1 ratio of ACN:H2O (15 mL ACN/mmol isoxazolidinone). This was refluxed overnight. The next day the reaction mixture was concentrated directly onto silica and purified via column chromatography (EtOAc in heptanes).
Ketone 15b
Use of the general procedure with isoxazolidinone 13b (39.5 mg, 0.127 mmol) provided 28.2 mg of the pure product in an 87% yield. [α]D24 = −68 (c=0.13, CH2Cl2). 1H NMR (CDCl3) δ 7.05-6.97 (d, 1H, J=9.08 Hz), 6.74-6.68 (m, 2H), 5.94-5.81 (m, 1H), 5.18-5.09 (m, 2H), 3.76 (s, 3H), 3.33-3.28 (t, 1H, J=5.64 Hz), 3.08-2.99 (d, 1H, J=16.32 Hz), 2.78-2.60 (m, 3H), 2.50-2.42 (m, 1H), 2.42-2.47 (m, 2H), 2.10-2.02 (m, 1H), 1.98-1.88 (m, 1H), 1.84-1.75 (m, 1H). 13C NMR (CDCl3) δu134.5, 124.2, 112.5, 110.3, 55.4, 48.7; δd 212.3, 159.3, 143.2, 136.6, 118.2, 44.7, 44.4, 44.0, 41.8, 36.4, 32.2.GC tR = 8.55 min. EI-MS m/z (%): 256 (M+, 10), 214 (100), 172 (30), 115 (26). HRMS (ESI) (m/z): [M-1]+ calcd for C17H19O2: 255.1380, found: 255.1377.
Ketone 15d
Use of the general procedure with isoxazolidinone 13d (150.2 mg, 0.440 mmol) provided 104.1 mg of the pure product in an 82% yield. [α]D24 = −12 (c=0.4, CH2Cl2). 1H NMR (CDCl3) δ 6.69, 6.61 (two s, 2H), 5.94-5.77 (m, 1H), 5.20-5.07 (m, 2H), 3.84 (s, 6H), 3.36-3.28 (m, 1H), 3.07-2.97 (d, 1H, J=16.04 Hz), 2.81-2.68 (m, 2H), 2.69-2.59 (m, 1H), 2.50-2.23 (m, 3H), 2.13-2.02 (m, 1H), 2.01-1.89 (m, 1H), 1.89-1.75 (m, 1H). 13C NMR (CDCl3) δu134.4, 107.8, 106.7, 56.06, 56.03, 49.2; δd 212.4, 148.7, 148.6, 136.1, 133.1, 118.2, 44.55, 44.53, 44.3, 42.0, 36.2, 32.3.GC tR = 9.52 min. EI-MS m/z (%): 286 (M+, 35), 244 (100), 188 (35), 115 (26). HRMS (ESI) (m/z): [M]+ calcd for C18H22O3: 286.1564, found: 286.1559.
Ketone 15e
Use of the general procedure with isoxazolidinone 13e (42.7 mg, 0.145 mmol) provided 34.8 mg of the pure product in an 80% yield. [α]D24 = −27.6 (c=0.47, CH2Cl2). 1H NMR (CDCl3) δ 7.09-6.94 (m, 3H), 6.01-5.81 (m, 1H), 5.25-5.09 (m, 2H), 3.36 (t, 1H, J=5.52 Hz), 3.10-3.00 (m, 1H), 2.81-2.65 (m, 3H), 2.54-2.36 (m, 2H), 2.32 (s, 4H), 2.15-2.02 (m, 1H), 2.02-1.89 (m, 1H), 1.89-1.76 (m, 1H). 13C NMR (CDCl3) δu134.5, 127.6, 125.5, 123.4, 49.1, 21.2; δd 212.2, 141.8, 141.6, 136.8, 118.1, 44.5, 44.2, 43.9, 21.7, 36.5, 32.1. GC tR = 7.44 min. EI-MS m/z (%): 240 (M+, 33), 197 (100), 155 (56), 127 (36). HRMS (CI) (m/z): [M+1]+ calcd for C17H21O: 241.1592, found: 241.1589.
Ketone 16b
Use of the general procedure with isoxazolidinone 14b (225.6 mg, 0.694 mmol) provided 167.6 mg of the pure product in a 90% yield. [α]D24 = +21.5 (c=0.87, CH2Cl2). 1H NMR (CDCl3) δ 6.99-6.88 (d, 1H, J=8.36 Hz), 6.76-7.70 (dd, 1H, J=8.40, 2.72 Hz), 6.69-6.66 (d, 1H, J=2.60 Hz), 5.95-5.75 (m, 1H), 5.18-4.95 (m, 2H), 3.79 (m, 3H), 2.98-2.87 (m, 2H), 2.85-2.76 (m, 1H), 2.52-2.40 (m, 3H), 2.37-2.28 (m, 1H), 2.28-2.20 (m, 1H) 2.00-1.79 (m, 3H), 1.65-1.54 (m, 2H). 13C NMR (CDCl3) δu 133.8, 130.0, 113.8, 112.4, 55.2, 45.3; δd 211.5, 158.0, 135.6, 130.9, 118.1, 47.7, 41.9, 37.1, 33.3, 34.4, 25.8, 25.1. GC tR = 9.43 min. EI-MS m/z (%): 270 (M+, 32), 228 (100), 199 (18), 171 (27). HRMS (CI) (m/z): [M+1]+ calcd for C18H23O2: 271.1698, found: 271.1698.
Ketone 16d
Use of the general procedure with isoxazolidinone 14d (192.0 mg, 0.541 mmol) provided 133.9 mg of the pure product in an 82% yield. [α]D24 = +73 (c=0.2, CH2Cl2). 1H NMR (CDCl3) δ 6.61 (s, 1H), 6.49 (s, 1H), 5.96-5.72 (m, 1H), 5.17-4.96 (m, 2H), 3.93-3.76 (m, 6H), 2.93-2.80 (m, 2H), 2.80-2.72 (m, 1H), 2.56-2.40 (m, 3H), 2.39-2.11 (m, 3H), 1.98-1.83 (m, 3H), 1.63-1.54 (m, 1H). 13C NMR (CDCl3) δu 133.8, 111.7, 111.6, 55.9, 55.8, 45.8; δd 211.6, 147.6, 147.5, 130.6, 126.0, 118.3, 47.9, 41.8, 37.1, 35.3, 34.3, 25.2, 25.0. GC tR = 10.38 min. EI-MS m/z (%): 300 (M+, 100), 258 (87), 201 (63), 151 (57). HRMS (CI) (m/z): [M]+ calcd for C19H24O3: 300.1725, found: 300.1713.
Ketone 16e
Use of the general procedure with isoxazolidinone 14e (138.1 mg, 0.447 mmol) provided 138.1 mg of the pure product in a 70% yield. [α]D24 = +78.5 (c=0.2, CH2Cl2). 1H NMR (CDCl3) δ 7.03-6.87 (m, 3H), 5.97-5.75 (m, 1H), 5.20-4.97 (m, 2H), 3.97-3.78 (m, 3H), 2.53-2.40 (m, 3H), 2.39-2.33 (m, 1H), 2.32 (s, 3H), 2.30-2.15 (m, 2H), 2.00-1.81 (m, 3H), 1.68-1.57 (m, 1H). 13C NMR (CDCl3) δu 133.9, 129.8, 129.0, 127.1, 45.6, 21.0; δd 211.5, 135.8, 135.7, 134.1, 118.1, 48.1, 42.0, 37.0, 35.2, 34.3, 25.26, 25.19. GC tR = 8.45 min. EI-MS m/z (%): 254 (M+, 9), 212 (100), 155 (65), 141 (45), 128 (44). HRMS (CI) (m/z): [M+1]+ calcd for C18H23O: 255.1749, found: 255.1737.
General Procedure for Regioselective Enol Silane Formation
Chiral amine, (+)-bis[(R)-1-phenethyl]amine (2.0 eq.), was dissolved in toluene and the mixture was cooled to −78°C. Next n-BuLi (2.0 eq.) was added, and the reaction was stirred at this temperature for 30 min. HMPA (2.0 eq.) was added, and at this point the reaction turned a pink/peach color. The reaction was warmed to room temperature, recooled to −78°C, and TBS-OTf (5.0 eq.) was added followed by the addition of ketone (1.0 eq.) dropwise. The reaction then became a cloudy opaque white color, and was stirred overnight while warming to room temperature. The next day the reaction was quenched with Et3N and saturated NaHCO3. The aqueous layer was extracted 3× with ether and the combined organics were washed with brine, dried over Na2SO4, filtered and concentrated. The crude product was purified via column chromatography (EtOAc in heptanes). HRMS and optical rotations were not determined as the enol silanes were susceptible to decomposition and therefore were used soon after isolation.
Enol Silanes 17 and 18
Use of the general procedure with ketone 15d (64.6 mg, 0.226 mmol) provided 82.0 mg of pure product as a mixture of regioisomers in ~2.5:1 ratio by 1H NMR, a 91% yield. 1H NMR (CDCl3) δ 6.76, 6.72, 6.68 (3s, 2H), 5.98-5.82 (m, 1H), 5.21, 5.19 (d, 0.30H, J = 5.08 Hz), 5.14-5.05 (m, 2H), 4.78-4.73 (br, 0.67H), 3.88 (s, 3H), 3.86 (s, 3H), 3.38-3.05 (m, 1H), 2.90-2.70 (m, 1H), 2.59-2.11 (m, 5H), 2.00-1.80 (m, 2H), 0.97-0.83 (m, 9H), 0.22-0.054 (m, 6H). GC tR = 10.54 min. (Major regioisomer, 66%) EI-MS m/z (%): 400 (M+, 19), 358 (16), 269 (100), 216 (34). tR = 10.57 min. (Major regioisomer, 33%) EI-MS m/z (%): 400 (M+, 19), 358 (100), 327 (16), 269 (10), 227 (16).
Enol Silanes 19 and 20
Use of the general procedure with ketone 16b (73.2 mg, 0.271 mmol) provided 98.4 mg of pure product as a mixture of regioisomers in ~4:1 ratio by 1H NMR, a 95% yield. 1H NMR (CDCl3) δ 7.22-7.05 (m, 1H), 7.03-6.94 (m, 1H), 6.77-6.63 (m, 1H), 5.97-5.75 (m, 1H), 5.14-5.92 (m, 2H), 5.92-4.77 (m, 1H), 3.80 (s, 3H), 2.93-2.60 (m, 2H), 2.32-1.79 (m, 7H), 1.49-1.36 (m, 1H), 1.00-0.82 (m, 9H), 0.19-0.057 (m, 6H). GC tR = 10.43 min. (Minor regioisomer, 18.5%) EI-MS m/z (%): 384 (M+, 12), 342 (71), 327 (18), 253 (23), 210 (75), 72 (100). tR = 10.51 min. (Major regioisomer, 66.3%) EI-MS m/z (%): 384 (M+, 6), 253 (100), 210 (51), 72 (24).
Bicyclo[3.2.1]octane 21
Enol silanes 17 and 18 (58.9 mg, 0.147 mmol) were dissolved in DMSO (3 mL) and Pd(OAc)2 (3 mg, 0.013 mmol) was added. The reaction mixture was then put under an oxygen atmosphere and heated to 45°C overnight. Upon completion, as determined by TLC, the reaction was concentrated under reduced pressure, loaded directly onto silica gel and purified via column chromatography (1:10 EtOAc/heptanes). The desired cyclized product 21 was isolated as a 15.4 mg sample, which was a mixture of 21:23:18 in an 88:5:6 ratio (see GC analysis), which amounts to a 41% yield of 21. Further chromatographic purification afforded a pure 2.5 mg (8% yield) sample which was used for characterization purposes. [α]D24 = +44.5 (c=0.4, CH2Cl2). 1H NMR (CDCl3) 6.74 (s, 1H), 6.60 (s, 1H) 5.22 (s, 1H), 5.09 (s, 1H), 3.86 (s, 3H), 3.85 (s, 3H), 3.34-3.26 (d, 1H, J=4.56 Hz), 3.24-3.08 (m, 2H), 2.98-2.78 (m, 2H), 2.71-2.59 (dt, 1H, J=18.84, 2.92 Hz), 2.57-2.39 (m, 2H), 2.08-2.00 (dd, 1H, J=12.12, 12.16 Hz), 1.80-1.71 (dd, 1H, J=12.12, 12.48 Hz). 13C NMR (CDCl3) δ 209.1, 148.6, 146.3, 137.5, 133.2, 111.1, 107.8, 106.8, 57.6, 56.1, 50.8, 47.4, 44.5, 41.0, 40.2, 37.0. GC tR = 9.49 min. (Minor regioisomer, 5%) EI-MS m/z (%): 284 (M+, 50), 242 (100), 227 (2), 128 (15). GC tR = 9.97 min. (Desired product, 88%) EI-MS m/z (%): 284 (M+, 100), 242 (50), 189 (24), 115 (23). GC tR = 10.73 min. (Residual enol silane, 6%) EI-MS m/z (%): 400 (M+, 15), 358 (100), 327 (11), 227 (17), 73 (25). HRMS (CI) (m/z): [M]+ calcd for C18H20O3: 284.1412, found: 284.1412.
Bicyclo[3.2.1]octane 25–28
Enol silanes 19 and 20 (98.4 mg, 0.256 mmol) were dissolved in DMSO (5 mL) and Pd(OCOCF3)2 (12 mg, 0.036 mmol) was added. The reaction mixture was then put under an oxygen atmosphere and heated to 45°C overnight. The reaction was monitored by TLC and upon completion concentrated under reduced pressure, loaded directly onto silica gel and purified via column chromatography (1:19 EtOAc/heptanes). A 41.1 mg mixture of 25:26:27:28 in a 46:31:15:8 ratio was obtained (60% yield of the four isomers). 1H NMR (CDCl3) δ 7.23-6.91 (3 sets of doublets, 1 H, J=8.6 Hz), 6.81-6.62 (m, 2H), 5.89-5.84 (d, 0.078 H, J=1.64 Hz), 5.67-5.61 (d, 0.32 H, J=1.2 Hz), 5.22-4.99 (4s, 2H), 3.83-3.73 (m, 3H), 3.26-2.46 (m, 5H), 2.10-1.65 (m, 5H). GC tR = 8.87 min. (Minor regioisomer, 6.7%) EI-MS m/z (%): 268 (M+, 64), 253 (24), 240 (83), 212 (100), 197 (46). GC tR = 9.22 min. (Minor regioisomer, 1.2%) EI-MS m/z (%): 268 (M+, 100), 228 (39), 211 (47), 159 (47). GC tR = 9.30 min. (Major regioisomer, 45.7%) EI-MS m/z (%): 268 (M+, 93), 240 (21), 225 (40), 212 (100), 92. GC tR = 9.59 min. (Minor regioisomer, 45.3%) EI-MS m/z (%): 268 (M+, 90), 226 (100), 210 (34), 173 (34), 115 (27).
Bicyclo[3.2.1]octane 26
A mixture of alkene isomers 25–28 (46.9 mg, 0.175 mmol) was refluxed for 3 hours with pTsOH·H2O (4.0 eq.) in benzene (50 mL/mmol alkene isomers). Once GC-MS indicated that a complete reaction had occurred, the reaction was diluted with ether and washed with saturated NaHCO3. The aq. layer was extracted 3× with ether, and the combined organics were washed with brine, dried over Na2SO4, and concentrated under reduced pressure to provide the crude product which was purified via column chromatography (10% EtOAc in heptanes) to afford 34.6 mg 74% yield) of compound 26. [α]D24 = −209 (c=0.27, CH2Cl2). 1H NMR (CDCl3) δ 7.06, 7.04 (d, 1H, J=8.6 Hz), 6.80-6.74 (dd, 1H, J=8.52, 2.64 Hz), 6.66, 6.65 (d, 1H, J=2.56 Hz),5.66-5.61 (br, 1H), 3.80 (s, 3H), 3.21-3.11 (m, 1H), 3.11-3.04 (m, 1H), 2.92, 2.91 (d, 1H, J=4.44 Hz), 2.90-2.81 (m, 1H), 2.77-2.68 (m, 1H), 2.64-2.55 (dd, 1H, J=17.5, 4.8 Hz), 2.21, 2.18 (d, 1H, J=11.2 Hz), 2.02-1.85 (m, 2H), 1.85-1.81 (m, 1H), 1.804, 1.801 (d, 3H, J=1.44 Hz ). 13C NMR (CDCl3) δu137.5, 128.7, 113.3, 112.7, 59.6, 55.2, 39.8, 15.1; δd 206.8, 157.4, 139.8, 137.3, 132.4, 47.0, 43.8, 38.5, 32.0, 27.0. GC tR = 9.316 min. EI-MS m/z (%): 268 (M+, 100), 240 (39), 225 (82), 211 (45), 93 (27). HRMS (CI) (m/z): [M]+ calcd for C18H21O2: 269.1542, found: 269.1539.
Supplementary Material
Table 4.
Intramolecular Friedel-Crafts Alkylation
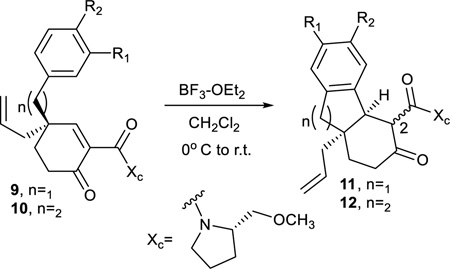 | |||||
|---|---|---|---|---|---|
| Reactant | n | R1 | R2 | Product | % Yield |
| 9a | 1 | H | H | 11a | dec. |
| 9b | 1 | OCH3 | H | 11b | 98* |
| 9c | 1 | H | OCH3 | 11c | dec. |
| 9d | 1 | OCH3 | OCH3 | 11d | 97* |
| 9e | 1 | CH3 | H | 11e | 76* |
| 10a | 2 | H | H | 12a | 63 |
| 10b | 2 | OCH3 | H | 12b | 88* |
| 10c | 2 | H | OCH3 | 12c | dec. |
| 10d | 2 | OCH3 | OCH3 | 12d | 84* |
| 10e | 2 | CH3 | H | 12e | 70* |
Product was obtained as a mixture of C-2 epimers; dec=decomposed
Acknowledgement
The project described was supported by Award Number R15GM087291 from the National Institute of General Medical Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health. A generous award (CHE-0958996) from the National Science Foundation enabling acquisition of the 400 MHz NMR spectrometer used in these studies is gratefully acknowledged. The authors would also like to thank Bryn Mawr College for financial support of this work. Terry Huh is acknowledged for her assistance with the synthesis of an early intermediate in the project. Tim Wade and the Drexel University Mass Spectrometry Facility are acknowledged for acquiring all HRMS data.
Footnotes
Electronic supplementary information available. Copies of 1H and 13C NMR spectra, gas chromatographs and mass spectra for all new compounds.
The authors declare no competing financial interest.
References
- 1.Newman DJ, Cragg GM. Journal of Natural Products. 2012;75:311–335. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mishra BB, Tiwari VK. European Journal of Medicinal Chemistry. 2011;46:4769–4807. doi: 10.1016/j.ejmech.2011.07.057. [DOI] [PubMed] [Google Scholar]
- 3.Lee K-H. Journal of Natural Products. 2010;73:500–516. doi: 10.1021/np900821e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Newman DJ. Journal of Medicinal Chemistry. 2008;51:2589–2599. doi: 10.1021/jm0704090. [DOI] [PubMed] [Google Scholar]
- 5.Cragg GM, Grothaus PG, Newman DJ. Chemical Reviews. 2009;109:3012–3043. doi: 10.1021/cr900019j. [DOI] [PubMed] [Google Scholar]
- 6.Mander LN. Natural Product Reports. 2003;20:49–69. doi: 10.1039/b007744p. [DOI] [PubMed] [Google Scholar]
- 7.Sun H-D, Huang S-X, Han Q-B. Natural Product Reports. 2006;23:673–698. doi: 10.1039/b604174d. [DOI] [PubMed] [Google Scholar]
- 8.Garcia P, De Oliveira A, Batista R. Molecules. 2007;12:455–483. doi: 10.3390/12030455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanson JR. Natural Product Reports. 2009;26:1156–1171. doi: 10.1039/b807311m. [DOI] [PubMed] [Google Scholar]
- 10.Presset M, Coquerel Y, Rodriguez J. Chemical Reviews. 2012;113:525–595. doi: 10.1021/cr200364p. [DOI] [PubMed] [Google Scholar]
- 11.Zhu L, Huang S-H, Yu J, Hong R. Tetrahedron Letters. 2015;56:23–31. [Google Scholar]
- 12.Colebrook EH, Thomas SG, Phillips AL, Hedden P. The Journal of Experimental Biology. 2014;217:67–75. doi: 10.1242/jeb.089938. [DOI] [PubMed] [Google Scholar]
- 13.Davière J-M, Achard P. Development. 2013;140:1147–1151. doi: 10.1242/dev.087650. [DOI] [PubMed] [Google Scholar]
- 14.Davies PJ. Plant Hormones: Biosynthesis, Signal Transduction, Action! 3rd edn. Dordrecht, The Netherlands: Kluwer Academic Publishers; 2010. [Google Scholar]
- 15.Hueso-Falcón I, Cuadrado I, Cidre F, Amaro-Luis JM, Ravelo AG, Estevez-Braun A, De Las Heras B, Hortelano S. European Journal of Medicinal Chemistry. 2011;46:1291–1305. doi: 10.1016/j.ejmech.2011.01.052. [DOI] [PubMed] [Google Scholar]
- 16.Bruno M, Rosselli S, Pibiri I, Kilgore N, Lee K-H. Journal of Natural Products. 2002;65:1594–1597. doi: 10.1021/np020029b. [DOI] [PubMed] [Google Scholar]
- 17.Brucoli F, Borrello MT, Stapleton P, Parkinson GN, Gibbons S. Journal of Natural Products. 2012;75:1070–1075. doi: 10.1021/np300080w. [DOI] [PubMed] [Google Scholar]
- 18.Khaybullin RN, Strobykina IY, Dobrynin AB, Gubaydullin AT, Chestnova RV, Babaev VM, Kataev VE. Bioorganic & Medicinal Chemistry Letters. 2012;22:6909–6913. doi: 10.1016/j.bmcl.2012.09.023. [DOI] [PubMed] [Google Scholar]
- 19.Cotugno R, Gallotta D, Piaz FD, Apicella I, De Falco S, Rosselli S, Bruno M, Belisario MA. Biochimica et Biophysica Acta (BBA) - General Subjects. 2014;1840:1135–1144. doi: 10.1016/j.bbagen.2013.11.023. [DOI] [PubMed] [Google Scholar]
- 20.Wang D LL, Wang C, Zhang Y, Xu J. Mini-Reviews in Medicinal Chemistry. 2011;11:910–919. doi: 10.2174/138955711796575416. [DOI] [PubMed] [Google Scholar]
- 21.Kuo P-C, Shen Y-C, Yang M-L, Wang S-H, Thang TD, Dung NX, Chiang P-C, Lee K-H, Lee EJ, Wu T-S. Journal of Natural Products. 2007;70:1906–1909. doi: 10.1021/np070383f. [DOI] [PubMed] [Google Scholar]
- 22.Ding C, Zhang Y, Chen H, Yang Z, Wild C, Chu L, Liu H, Shen Q, Zhou J. Journal of Medicinal Chemistry. 2013 doi: 10.1021/jm400367n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen J, Sun Z, Zhang Y, Zeng X, Qing C, Liu J, Li L, Zhang H. Bioorganic & Medicinal Chemistry Letters. 2009;19:5496–5499. doi: 10.1016/j.bmcl.2009.07.090. [DOI] [PubMed] [Google Scholar]
- 24.Takano S, Kasahara C, Ogasawara K. Journal of the Chemical Society, Chemical Communications. 1981:635–637. [Google Scholar]
- 25.Takano S, Kasahara C, Ogasawara K. Journal of the Chemical Society, Chemical Communications. 1981:637–638. [Google Scholar]
- 26.Corey EJ, Kigoshi H. Tetrahedron Letters. 1991;32:5025–5028. [Google Scholar]
- 27.Corey EJ, Guzman-Perez A, Lazerwith SE. Journal of the American Chemical Society. 1997;119:11769–11776. [Google Scholar]
- 28.Corey EJ, Liu K. Journal of the American Chemical Society. 1997;119:9929–9930. [Google Scholar]
- 29.Toyota M, Wada T, Ihara M. The Journal of Organic Chemistry. 2000;65:4565–4570. doi: 10.1021/jo000142b. [DOI] [PubMed] [Google Scholar]
- 30.Yeoman JTS, Cha JY, Mak VW, Reisman SE. Tetrahedron. 2014;70:4070–4088. [Google Scholar]
- 31.Yeoman JTS, Mak VW, Reisman SE. Journal of the American Chemical Society. 2013;135:11764–11767. doi: 10.1021/ja406599a. [DOI] [PubMed] [Google Scholar]
- 32.Paul T, Malachowski WP, Lee J. J Org Chem. 2007;72:930–937. doi: 10.1021/jo0621423. [DOI] [PubMed] [Google Scholar]
- 33.Malachowski WP, Paul T, Phounsavath S. J. Org. Chem. 2007;72:6792–6796. doi: 10.1021/jo070976v. [DOI] [PubMed] [Google Scholar]
- 34.Paul T, Malachowski WP, Lee J. Org Lett. 2006;8:4007–4010. doi: 10.1021/ol0615228. [DOI] [PubMed] [Google Scholar]
- 35.Dénès F, Pérez-Luna A, Chemla F. Chemical Reviews. 2010;110:2366–2447. doi: 10.1021/cr800420x. [DOI] [PubMed] [Google Scholar]
- 36.Kende AS, Roth B, Sanfilippo PJ. Journal of the American Chemical Society. 1982;104:1784–1785. [Google Scholar]
- 37.Ito Y, Aoyama H, Saegusa T. Journal of the American Chemical Society. 1980;102:4519–4521. [Google Scholar]
- 38.Toyota M, Ihara M. Synlett. 2002;2002:1211–1222. [Google Scholar]
- 39.Cook HRaGR. Chapter 21 Friedel-Crafts Type Cyclizations. Weinheim: Wiley-VCH; 2010. [Google Scholar]
- 40.Taber DF, Sheth RB. The Journal of Organic Chemistry. 2008;73:8030–8032. doi: 10.1021/jo801767n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu H-J, Tran DD-P. Tetrahedron Letters. 1999;40:3827–3830. [Google Scholar]
- 42.Rogers DH, Frey B, Roden FS, Russkamp F-W, Willis AC, Mander LN. Australian Journal of Chemistry. 1999;52:1093–1108. [Google Scholar]
- 43.Majetich G, Liu S, Fang J, Siesel D, Zhang Y. The Journal of Organic Chemistry. 1997;62:6928–6951. [Google Scholar]
- 44.Ross TM, Burke SJ, Malachowski WP. Tetrahedron Letters. 2014;55:4616–4618. doi: 10.1016/j.tetlet.2014.06.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qiao Y, Kumar S, Malachowski WP. Tetrahedron Letters. 2010;51:2636–2638. [Google Scholar]
- 46.Kloetzing RJ, Krasovskiy A, Knochel P. Chemistry - A European Journal. 2007;13:215–227. doi: 10.1002/chem.200600738. [DOI] [PubMed] [Google Scholar]
- 47.Imao D, Glasspoole BW, Laberge VS, Crudden CM. Journal of the American Chemical Society. 2009;131:5024–5025. doi: 10.1021/ja8094075. [DOI] [PubMed] [Google Scholar]
- 48.Pintaric C, Olivero S, Gimbert Y, Chavant PY, Duñach E. Journal of the American Chemical Society. 2010;132:11825–11827. doi: 10.1021/ja1052973. [DOI] [PubMed] [Google Scholar]
- 49.Botella L, Nájera C. Tetrahedron. 2004;60:5563–5570. [Google Scholar]
- 50.Hodgson DMS, H MA. Top. Organometallic Chem. 2003;5:1–20. [Google Scholar]
- 51.Schultz AG. Chemical Communications. 1999:1263–1271. [Google Scholar]
- 52.Schultz AG, Macielag M, Sundararaman P, Taveras AG, Welch M. J. Am. Chem. Soc. 1988;110:7828–7841. [Google Scholar]
- 53.Toyota M, Wada T, Fukumoto K, Ihara M. Journal of the American Chemical Society. 1998;120:4916–4925. [Google Scholar]
- 54.Jung ME, Light LA. Journal of the American Chemical Society. 1984;106:7614–7618. [Google Scholar]
- 55.O'Brien P. Journal of the Chemical Society, Perkin Transactions 1. 1998:439–1458. [Google Scholar]
- 56.Birman VB, Danishefsky SJ. Journal of the American Chemical Society. 2002;124:2080–2081. doi: 10.1021/ja012495d. [DOI] [PubMed] [Google Scholar]
- 57.Flaherty A, Trunkfield A, Barton W. Organic Letters. 2005;7:4975–4978. doi: 10.1021/ol051929x. [DOI] [PubMed] [Google Scholar]
- 58.Cui X, Li Z, Tao C-Z, Xu Y, Li J, Liu L, Guo Q-X. Organic Letters. 2006;8:2467–2470. doi: 10.1021/ol060585n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.