

Abstract
Dengue fever is a severe, widespread, and neglected disease with more than 2 million diagnosed infections per year. The dengue virus NS2B/NS3 protease (PR) represents a prime target for rational drug design. At the moment, there are no clinical PR inhibitors (PIs) available. We have identified diaryl (thio)ethers as candidates for a novel class of PIs. Here, we report the selective and noncompetitive inhibition of the serotype 2 and 3 dengue virus PR in vitro and in cells by benzothiazole derivatives exhibiting 50% inhibitory concentrations (IC50s) in the low-micromolar range. Inhibition of replication of DENV serotypes 1 to 3 was specific, since all substances influenced neither hepatitis C virus (HCV) nor HIV-1 replication. Molecular docking suggests binding at a specific allosteric binding site. In addition to the in vitro assays, a cell-based PR assay was developed to test these substances in a replication-independent way. The new compounds inhibited the DENV PR with IC50s in the low-micromolar or submicromolar range in cells. Furthermore, these novel PIs inhibit viral replication at submicromolar concentrations.
INTRODUCTION
Dengue viruses (DENVs) are enveloped positive-strand RNA viruses and belong to the family Flaviviridae. DENV is the most important arthropod-borne viral infection. Over one-third of the world population lives in areas of DENV endemicity, and an estimated 390 million infections occur every year. In addition, the number of countries having experienced DENV epidemics has risen from 9 in 1970 to more than 100 today (1, 2). Furthermore, the number of diagnosed infections across America, Southeast Asia, and the Western Pacific nearly doubled from 1.2 million in 2008 to over 2.3 million in 2010 (2). Four different DENV serotypes have been identified so far. Recently, evidence for an additional subtype has been presented (3). Serotypes 1 to 4 are now prevalent in Asia, Africa, and America, and the regions where dengue is endemic are still increasing (4–6), with dengue endangering even Europe and the United States due to vector spread. DENV infections can be associated with dengue fever, but up to 88% of the infections remain inapparent (7). These nonpersistent infected patients serve besides persistently infected mosquitoes as a virus reservoir. Severe DENV infections and especially reinfections may lead to dengue hemorrhagic fever and dengue shock syndrome, with lethality up to 5% (2, 8, 9). There is neither a vaccination nor a specific treatment for DENV infections.
The DENV genome contains a single open reading frame, which encodes the structural proteins capsid, membrane precursor (prM), and envelope and the nonstructural proteins NS1, NS2, NS3, NS4, and NS5 (10). Cellular proteases and the viral serine protease (PR) are responsible for cleaving the viral precursor polyprotein into functional proteins. The DENV PR consists of the amino-terminal domain of the NS3 protein and requires NS2B, a 14-kDa protein, as a cofactor to form a stable complex. This heterodimeric PR cleaves at the capsid-prM, NS2A/NS2B, NS2B/NS3, NS3/NS4A, NS4B/NS5, internal NS2A, NS3, and NS4A cleavage sites (10, 11). PR inhibitors (PIs) have been shown to be valuable antiviral drugs, especially in the treatment of HIV-1 and hepatitis C virus (HCV) (12–17). In the case of DENV, some already published substances showed inhibition of 50% of DENV replication in a minireplicon system at a submicromolar range (18) or inhibition to 10% of DENV replication with wild-type virus at micromolar concentrations (19). Despite these promising developments, antiviral DENV drugs are still not available.
In this report, we describe the specific and selective inhibition of the DENV PR by diaryl (thio)ethers. Evidence is provided for inhibition of the DENV-2 and -3 PR in vitro and for DENV-2 PR in cell culture. Furthermore, we show suppression of DENV-2 replication at submicromolar PI concentrations.
MATERIALS AND METHODS
DENV-2/DENV-3 PR expression and purification.
Competent BL21 Star Escherichia coli (GE Life Technologies) or BL21Gold(DE3) cells were transformed with either the pET-28C plasmid encoding DENV-2 PR (for the DENV-2 enzyme used for fluorometric enzyme assays) (20), pET15b vector (for the DENV-2 enzyme used for microscale thermophoresis) (21), or a pGEX6P1 vector encoding DENV-3 PR (22). PR expression was induced by addition of 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 2 to 7 h at 37°C. Bacteria were lysed by sonication in lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole [pH 8.0], 100 μM phenylmethylsulfonyl fluoride [PMSF], 3.08 μM aprotinin). The DENV-2 PR was purified using either Ni-nitrilotriacetic acid (NTA) beads (Qiagen) and native wash buffer (lysis buffer with 20 mM imidazole) or elution buffer (lysis buffer with 250 mM imidazole) or loaded onto a HisTrap FF (DENV-2) or GSTrap FF (DENV-3) (GE Healthcare) column and eluted using the particular lysis buffers (see the supplemental material) containing either 300 mM imidazole (DENV-2) or 10 mM reduced glutathione (DENV-3). The PR was further purified by size exclusion chromatography. Protein amounts were determined by Bradford assay and verified by Coomassie-stained PAGE. DENV PR purity was analyzed by silver-stained PAGE.
Chemistry.
The synthesis of the compounds is described in the supplemental material (see supplemental information and Fig. S1 and S2).
Quantum mechanics computations.
Single-point calculations as well as full geometry optimizations with several different structures and corresponding frequency calculations were performed on the B3LYP-D3/cc-pVDZ (23–28) level of theory as implemented in Turbomole (Turbomole GmbH, Germany). Additional constrained geometry optimizations of the docking structures were performed with the B3LYP functional and the cc-pVDZ basis sets using the Gaussian 09 program package (Gaussian, Inc.). To obtain the relative energies of all conformers, we performed single-point B3LYP-D3/cc-pVDZ calculation on the geometries obtained from the constrained optimizations.
Molecular docking.
For molecular docking of compounds 1 to 8, the recently solved crystal structure of DENV-3 PR in complex with the aldehyde inhibitor Bz-nKKR-H (PDB accession code 3U1I) was used (22). Possible docking modes between ligands and the PR were studied using the FlexX docking approach of the LeadIT 2.1.6 suite (BioSolveIt, Germany). Energies of compound structures were minimized using the MOE software (Molecular Operating Environment, 2012.10). All water and ligand molecules were deleted from the PR structure. The binding site was defined on a proper protein pocket, which was shown to be a specific allosteric binding site for other noncompetitive inhibitors near the catalytic site (29).
Fluorometric DENV PR assays.
For a preliminary screening of the substance library, DENV-2 PR assays using 100 μM the fluorogenic substrate Boc-Gly-Arg-Arg-7-amino-4-methylcoumarin (AMC) (20, 30) were performed. The assay conditions were taken from reference 20. For the first screening approach, substrate hydrolysis was measured in the absence (100% enzyme activity) or presence of 50 μM inhibitor. Fifty percent inhibitory concentration (IC50) values for selected compounds were determined measuring the increase of fluorescence at 10 different PI concentrations ranging from 0 to 500 μM, 0 to 100 μM, or 0 to 10 μM, depending on their inhibitory effects. Fluorescence increase resulting from the product AMC by substrate hydrolysis was measured for 10 min after starting the reaction using an Infinite 200 PRO (Tecan, Männedorf, Switzerland) (DENV-2) or a Saphire2 plate reader (Tecan) (DENV-3) at room temperature with 380-nm excitation and 460-nm emission wavelengths. For DENV-3 PR assays, 50 mM Tris-HCl, 1 mM CHAPS {3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate} at pH 9.0, and 40 μM substrate (PhAc-Lys-Arg-Arg-AMC) were used. The final assay volume was 200 μl. The reactions were started by adding either DENV-2 or DENV-3 PR to a final concentration of 50 nM. Each assay was performed in triplicate at room temperature, and IC50 values were calculated using GraFit (Erithacus Software Limited). IC50 values were obtained by fitting the residual enzyme activities to the four-parameter IC50 equation
| (1) |
where y [ΔF/min] is the substrate hydrolysis rate, ymax is the maximum value of the dose-response curve that is observed at very low inhibitor concentrations, ymin is the minimum value that is obtained at high inhibitor concentrations, and s denotes the Hill coefficient (31).
HPLC-based enzyme assay.
For details on the high-performance liquid chromatography (HPLC)-based enzyme assay, see the supplemental material.
Binding assay based on MST.
For compound 6, binding of the inhibitor to the enzyme was quantified in a substrate-free assay using microscale thermophoresis (MST) technology (32). For MST, purified DENV-2/3 PR was labeled with the Monolith NTTM protein labeling kit Blue according to the supplied labeling protocol, and 10 μl of a 95 nM stock solution of DENV-2/3 PR was mixed with 10 μl of a serial dilution of compound 6 starting at 625 μM for DENV-2 and 250 μM for DENV-3. Samples were diluted with buffers (for DENV-2, 200 mM Tris-HCl buffer [pH 9.5] containing 30% glycerol; for DENV-3, 50 mM Tris-HCl buffer [pH 9.0] containing 1 mM CHAPS) supplemented with dimethyl sulfoxide (DMSO) at a final concentration of 5% (vol/vol) to ensure equal DMSO concentrations. After incubation for 10 min, samples were measured at an MST power of 40% and a light-emitting diode (LED) power of 20%, with a laser-on time of 30 s and a laser-off time 5 s on a NanoTemper Monolith NT.115 instrument using standard capillaries.
DENV-2 replication, qRT-PCR, and cell culture-based PR assays.
The DENV was obtained from J. Schneider-Schaulies (Virology, Würzburg, Germany). The PR-encoding region was amplified and sequenced in order to confirm the serotype. In order to obtain high-titer DENV, the virus was amplified by coculture techniques as described previously for HIV-1 (33). Vero cells were infected and incubated for 3 days. One-fifth of the infected cells and one-fifth of the cell culture supernatants were used to infect Vero cells. This process was repeated 10 times, and the resulting supernatant was stored at −80°C. To analyze inhibition of viral replication, Vero cells were preincubated with the components at decreasing concentrations below the cell toxicity. The cells were subsequently infected with DENV-2 (multiplicity of infection [MOI] of 0.5). Cellular supernatants were collected after 4 days, centrifuged at 2,000 rpm for 5 min to remove detached cells, and titrated on Vero cells. These cells were fixed with 4% paraformaldehyde 3 days after infection. To visualize infected cells, immunofluorescence staining was performed with monoclonal anti-DENV-2 E antibodies. Bound antibodies were visualized with a Cy3-conjugated donkey anti-mouse antibody (1:500) (Jackson ImmunoResearch). Stained infected cells were counted using a fluorescence microscope, and viral titers were calculated. All experiments were performed in three independent triplicates. The significances of differences in infectivity compared to wild-type maturation efficiencies were determined by the paired two-sample t test.
To analyze the serotype specificity of the PIs, DENV replication was monitored using a commercial DENV quantitative reverse transcription-PCR (qRT-PCR) kit (Genesig, Primerdesign, Ltd., Southampton, United Kingdom). Cells were infected with either DENV-1 or DENV-2 (one genome copy per cell). The compounds were added subsequently, and the cells were incubated for 4 days. Cell culture supernatants were collected and centrifuged in order to remove detached cells. Viral RNA was isolated using the QIAamp viral RNA minikit, reverse transcribed, and amplified according to the manufacturer's instructions.
A reporter system was constructed to analyze inhibition of the DENV-2 PR in cell culture. First, the PR coding sequence fused to an N-terminal FLAG tag was introduced into a eukaryotic expression vector, giving rise to the pDENV2-PR plasmid. Second, the DsRed2 open reading frame was amplified with a sense primer encoding the DENV-2 NS2A/2B cleavage site. The amplicon was introduced into the green fluorescent protein gene (gfp)-containing plasmid peGFP-C1 (Clontech) 3′ of the gfp gene. The resulting peGFPCSDsRed reporter plasmid encodes a GFP-NS2A/2B-DsRed fusion protein. HEK 293T cells were cotransfected with both the PR expression plasmid pDENV2-PR and the peGFPCSDsRed reporter. PIs were added directly after transfection. Cells were harvested 2 days posttransfection and lysed in radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS, 50 mM Tris [pH 8.0]). Three independent Western blots of each of the triplicate samples were used to calculate relative substrate and product amounts using the AIDA software package (GE Healthcare). All experiments were repeated at least three times. The significances of differences in processing compared to wild-type maturation efficiencies were determined by the paired two-sample t test. Expression of the viral PR was monitored by anti-FLAG tag Western blotting using monoclonal anti-FLAG M2 antibodies (Sigma-Aldrich).
HIV-1 replication assay.
Viruses were produced by transfection of HEK 293T cells with proviral plasmid pNL4-3 encoding infectious HIV-1 subtype B as described before (34). Viral titers were determined on TZM-bl indicator cells (CD4+ CCR5+ CXCR4+ HeLa cells) in single-cycle infections by serial dilutions of the cell culture supernatants as previously described (33, 34). Briefly, cell culture supernatants were collected, centrifuged (1,500 rpm, 5 min) to remove transfected HEK 293T cells, and titrated in serial dilutions on TZM-bl cells (96-well plate, 2 × 104 cells per well). TZM-bl cells carry a stably integrated copy of an HIV long terminal repeat (LTR) promoter driving a lacZ gene. Two days postinfection, TZM-bl cells were fixed with ice-cold methanol/acetone for 5 min followed by a β-galactosidase stain, using X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) as the substrate. Stained cells were counted, and the viral titers were calculated.
HCV replication assay.
Huh7.5 cells stably expressing the firefly luciferase reporter gene (Huh7.5-Fluc) were transfected by electroporation with in vitro-transcribed RNA of the fully functional Renilla luciferase-encoding reporter virus genome JcR2a (35). DMSO or different concentrations of the compounds were added to the cells immediately after transfection. After 72 h of incubation, virus replication and cell viability were determined by measuring Renilla luciferase (Rluc) and Firefly luciferase (Fluc) activity from cell lysates, respectively. Production of infectious particles from transfected cells was determined by infection of naive Huh7.5 cells with supernatants collected from transfected cells and quantification of Rluc activity 72 h later. Experiments were performed three times in triplicate (or sextuplicate in the case of DMSO-treated cells). The HCV protease inhibitor boceprevir served as a positive control (36).
Cell toxicity test.
To exclude toxic side effects of the PIs, the cell survival and metabolism were measured by a cell proliferation assay (Promega, Germany). Vero and HEK 293T cells (2 × 104) were incubated with decreasing amounts of compounds solubilized in DMSO or with DMSO alone as a control. The assays were performed in triplicate according to the manufacturer's instructions. After 4 days, 20 μl of the MTS substrate was added, the cells were further incubated for 90 min, and the optical density at 490 nm (OD490) was measured. Concentrations of substances inhibiting the MTS substrate conversation were excluded from further analyses.
RESULTS AND DISCUSSION
Inhibitor design, docking, and PR inhibition.
To identify new chemical scaffolds for inhibitors of the DENV PR, an in-house library consisting of approximately 250 compounds was screened at 50 μM. A previously described DENV-2 PR assay was used to analyze the substance library using a fluorogenic Boc-Gly-Arg-Arg-AMC substrate (20, 30). By this assay, an active key substance (compound 1) was discovered to inhibit the DENV-2 PR by about 35% at 50 μM (Table 1). In a first approach to improve affinity, the thiophene moiety was replaced by alkyl chains or other (hetero)aromatic ring systems, and the amide moiety was replaced by an ester or amine function. The diaryl thioethers were prepared in a two-step synthesis (see Table S1 in the supplemental material). HBTU [N,N,N′,N′-tetramethyl-O-(1-H-benzotriazol-1-yl)uronium hexafluorophosphate] served as a coupling reagent for condensation of the respective phenyl thiobenzoic acid with the amine parts of the inhibitors. Phenyl thiobenzoic acids were prepared by a nucleophilic aromatic substitution reaction from corresponding arylmercaptans and arylhalides (37, 38).
TABLE 1.
Structures and activities of the NS2B/NS3 protease inhibitors
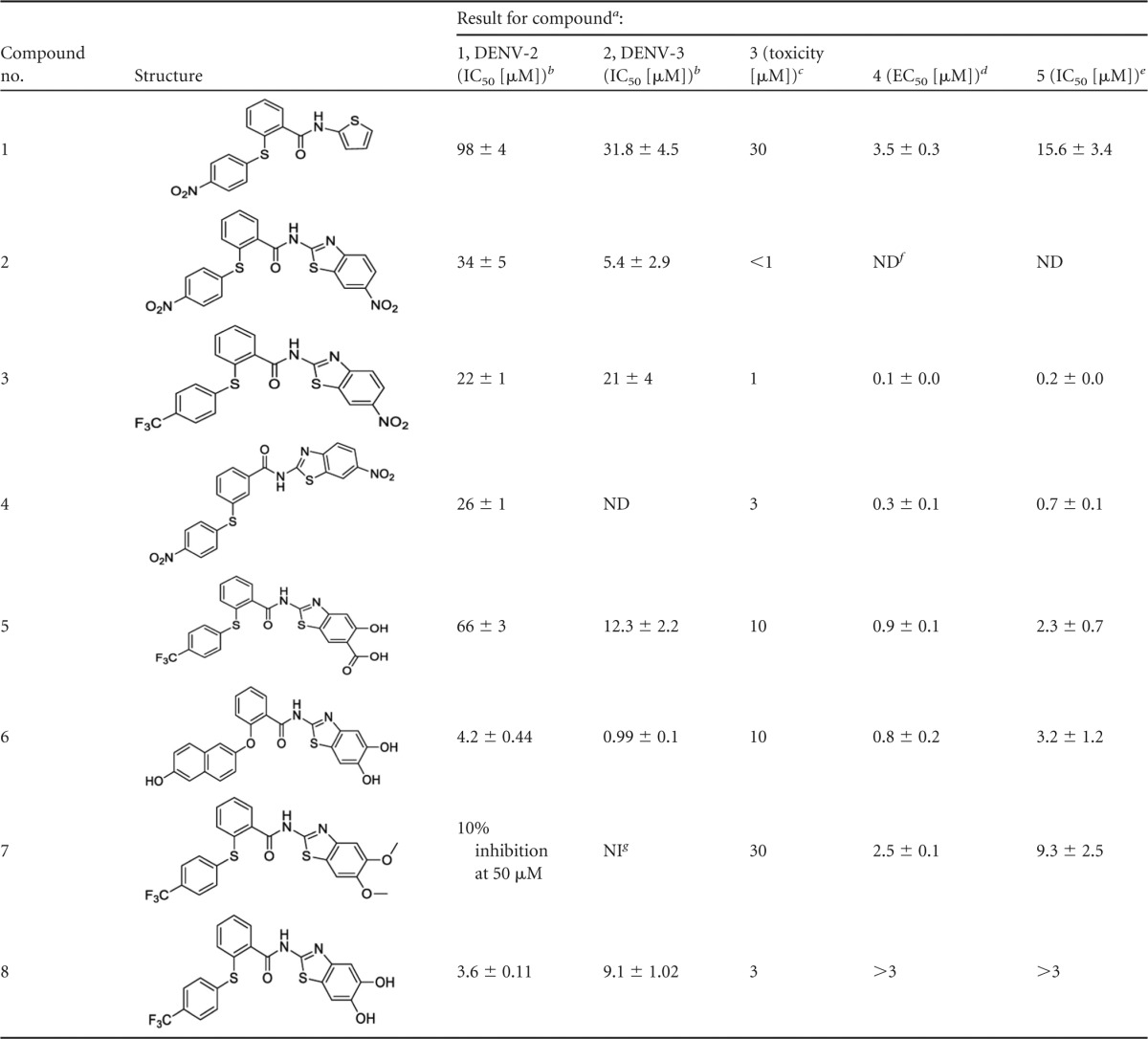
Values are indicated as means ± standard deviations from 3 independent experiments performed in triplicate.
Inhibition of isolated proteases determined by fluorometric enzyme assays with an AMC-derived substrate.
Concentration at which no influence on cellular metabolism was observed.
Antiviral activity, inhibition of viral replication.
Biochemical inhibition of PR in cell culture.
ND, not done.
NI, no inhibition at 50 μM.
Thus, we obtained a small series of new compounds (see the supporting information in the supplemental material). Replacement of the amide linker by an amine or ester group led to loss of activity, and also compounds with alkyl chains instead of the thiophene moiety turned out to be inactive. Among the new compounds with other heteroaromatic moieties, PI 2 with a nitro-substituted benzothiazol fragment was identified as the most potent (Table 1; see the supporting information and Fig. S1 in the supplemental material). In the next step, the position of the thioether fragment was changed from the ortho- to the para- or meta-position. Furthermore, the thioether moiety was replaced by a methylene group, and the nitro group at the phenyl ring was replaced by hydrogen, primary amine, or a trifluoromethyl group. Among these new compounds, the meta-substituted dinitro derivative 4 and the trifluoromethyl derivative 3 exhibited an improved inhibition (see Fig. S1 in the supplemental material). In order to explain these structure-activity relationships and to propel optimization forward on a rational basis, we determined the inhibition mechanisms of the original lead compound, compound 1, and, to that point, the most active inhibitor, compound 3. Both compounds were found to be noncompetitive with respect to the substrate. This was shown by determination of IC50s at different substrate concentrations (20, 50, 100, and 200 mM) and, the other way round, by determination of Km and Vmax values at various inhibitor concentrations. The first assays provided no significant differences of IC50s, and plots from the second assays showed typical noncompetitive graphs with decreasing Vmax values and nearly constant Km values at increasing concentrations [I] of inhibitor 3: at [I] = 0 μM, Vmax = 52 ΔF/min and Km = 75 μM; at [I] = 5 μM, Vmax = 56 ΔF/min and Km = 112 μM; at [I] = 25 μM, Vmax = 32 ΔF/min and Km = 108 μM; at [I] = 50 μM, Vmax = 21 ΔF/min and Km = 128 μM; and at [I] = 75 μM, Vmax = 14 ΔF/min and Km = 95 μM. Additionally, to exclude false-positive results due to quenching of the fluorescence of the hydrolysis product AMC by the nitro aromatics, we determined inhibition of the DENV-2 PR by compounds 2 and 3 using a semiquantitative HPLC assay (see the supplemental material), which confirmed the inhibition of substrate hydrolysis observed with compounds 2 and 3 (∼70 to 75% inhibition in the HPLC assay versus ∼70% inhibition in the fluorescence assay).
The noncompetitive inhibition raised the question of the binding region within the enzyme. Previously a specific allosteric binding site for noncompetitive and nonpeptidic inhibitors had been proposed. This deep binding pocket is located behind the active site (Ser135, Asp175, and His51) and is mainly formed by the amino acids Trp89, Thr120, Gly121, Glu122, Ile123, Gly124, Gly164, Ile165, Ala166, and Gln167 on one side and Lys73, Lys74, Asn152, Val78, Gly82, and Met84 on the other, the last three of which are NS2B derived (Fig. 1).
FIG 1.
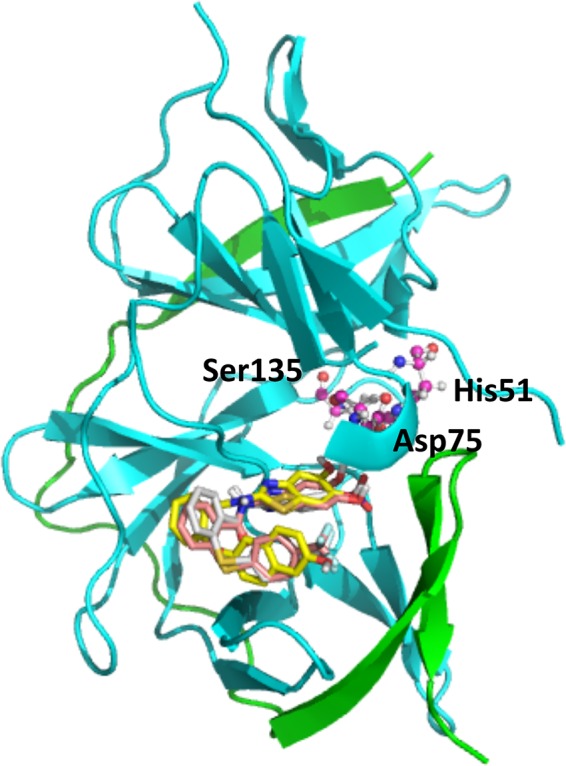
Allosteric site of DENV-3 PR with the docked compounds 3, 6, and 8. The NS2B unit is shown in green and the NS3 unit in cyan. Ligands are rendered as sticks colored according to the Corey, Pauling, and Koltun (CPK) color scheme, with the exception of carbon atoms (compound 3 in white, compound 6 in yellow, and compound 8 in pink). Catalytic triad residues His51, Asp75, and Ser135 in the active site, which is behind the allosteric site, are represented as balls and sticks.
This binding pocket was taken for docking studies with compound 3 using the FlexX docking approach of the LeadIT 2.1.6 suite with the recently solved crystal structure of DENV-3 PR in complex with the aldehyde inhibitor Bz-nKKR-H (PDB accession code 3U1I) (22). The structure of the DENV-3 PR was taken since both PRs show a high degree of similarity and the DENV-3 PR structure used is the only available structure of a DENV PR in complex with a low-molecular-weight inhibitor.
Since diaryl thioethers are flexible compounds with respect to their thioether bond, quantum chemical computations were performed in order to confirm that conformations obtained by docking studies are plausible, and this is indeed the case (see the supporting information in the supplemental material). For compound 3, pose 3 is predicted to be only about 1 to 5 kcal/mol higher than the global minima of the molecule, which is stabilized by intramolecular π-π interactions. This energy difference can easily be compensated for by inhibitor-enzyme interactions. Poses 1 and 2 seem to be higher in energy since the amide unit is not or only partially in conjugation with the other aromatic rings (see supporting information and Fig. S3 in the supplemental material).
The docking studies proposed an interaction of the nitro group of compound 3 with the side chain of Asn152 (Fig. 2). In order to verify this binding mode by addressing additional possible binding partners within the allosteric pocket, we introduced a hydroxyl group adjacent to the nitro group. This should enable an additional hydrogen bond with the oxygen of the backbone carbonyl group in Asn152. Additionally, instead of the nitro group, a carboxylic acid function (compound 5) or a second hydroxyl group (compound 8) were introduced to reduce possible quenching effects in the PR assays but to maintain the possible interaction with Asn152. While introduction of an acid and a hydroxyl function at the benzothiazole moiety did not lead to improved affinity (compound 5), the dihydroxy-substituted inhibitor, compound 8, was found to be highly active, with an IC50 of 3.6 μM. Also this PI was shown to be a noncompetitive inhibitor. It has to be mentioned that despite the catechol structure, the compound is stable and not sensitive to oxidation. The docking studies with this inhibitor propose interactions of one hydroxyl group with the side chain of Asn152 and with the backbone of Lys74 and an interaction of the other hydroxyl group with Lys73 (Fig. 2).
FIG 2.
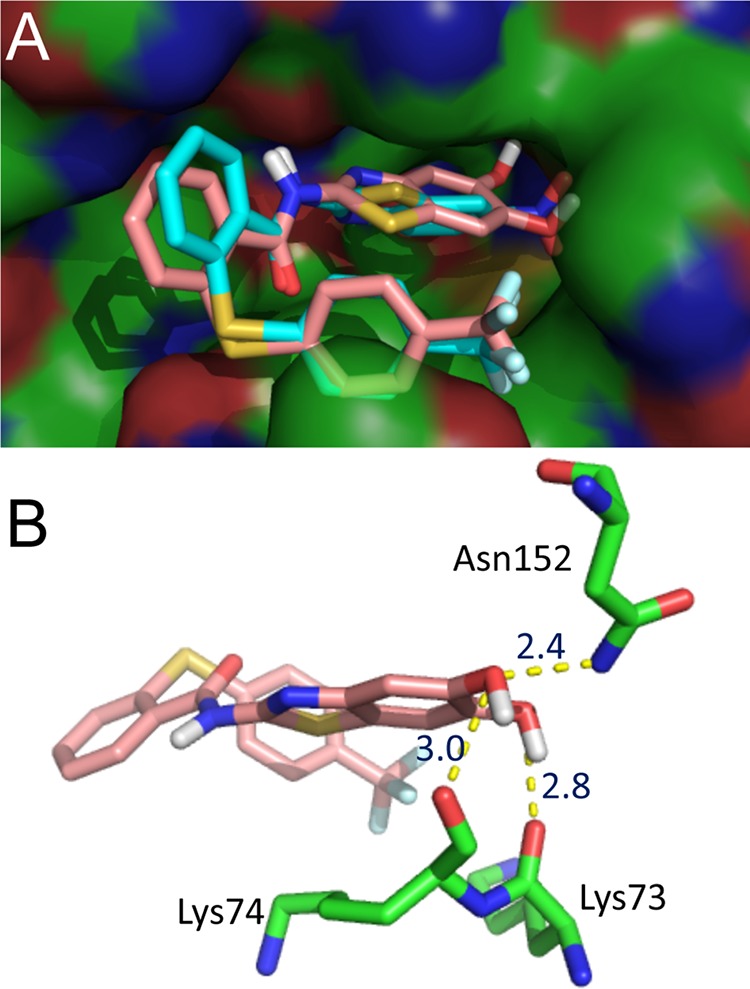
Predicted binding modes of compounds 3 and 8. The PIs were docked into DENV-3 PR by the LeadIT-FlexX program. The image was generated with PyMOL. (A) Surface view of the allosteric site with the docked compounds 3 and 8. Ligands are rendered as sticks colored according to the Corey, Pauling, and Koltun (CPK) color scheme, with the exception of carbon atoms (compound 3 in cyan and compound 8 in pink). (B) Binding mode of compound 8 showing the H bonds between two the hydroxyl groups and the amino acids Lys73, Lys74, and Asn152.
In agreement with the proposed binding mode, the dimethoxy-substituted compound 7 is not active (10% inhibition at 50 μM). Also the derivatives with only one hydroxyl group are less active, corroborating the docking studies (see the supporting information and Table S1 in the supplemental material). Further docking studies proposed replacement of the trifluoromethyl-substituted phenyl ring by a hydroxyl-substituted naphthyl moiety and replacement of the sulfur of the thioether bridge by oxygen (Fig. 3). This yielded compound 6, which was shown to be a noncompetitive inhibitor with an IC50 of ca. 4 μM. For that compound, binding studies using microscale thermophoresis were performed (see Fig. S4 in the supplemental material). Since these studies need labeling of the protein, we used the more stable mutant of the DENV-2 PR. With this substrate-independent binding assay, we found a dissociation constant (KD) of 15.2 μM, which confirms the fluorometric enzyme assays (see Fig. S4).
FIG 3.
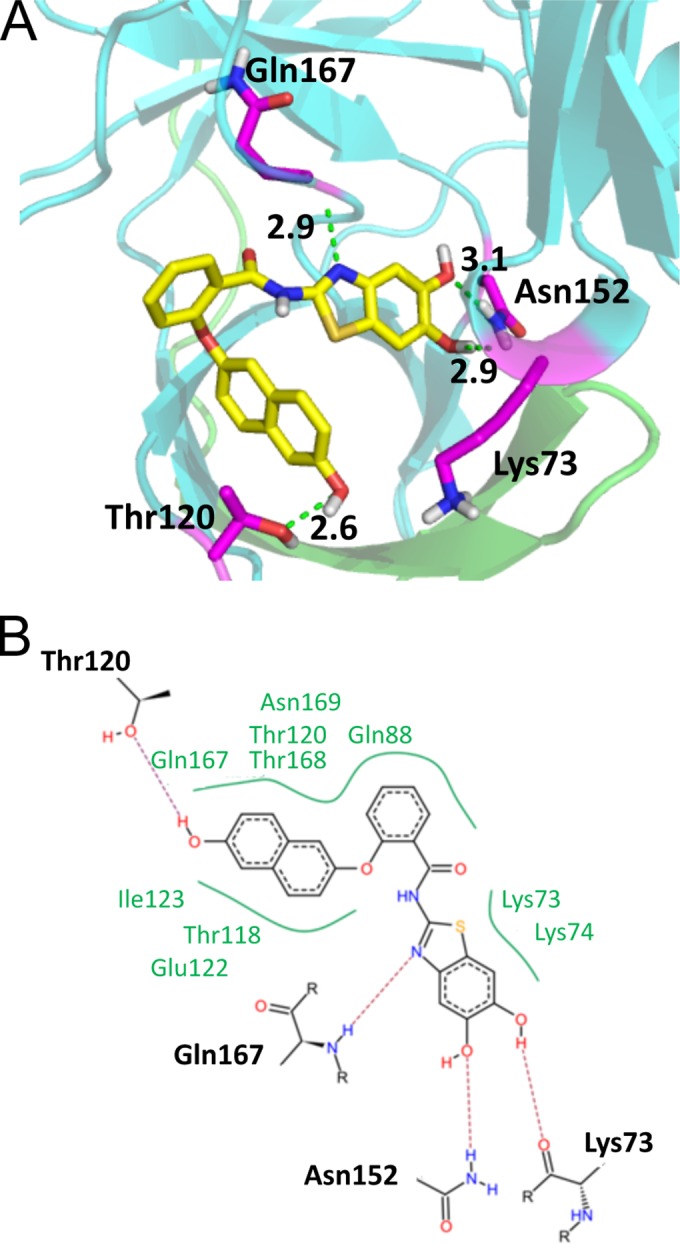
Docking results for PI 6 within the DENV-3 PR. (A) Three-dimensional interaction diagram of compound 6 with the amino acids Lys73, Thr120, Asn152, and Gln167. (B) Schematic view of all interactions (H bonds and hydrophobic interactions) of compound 6.
For selected compounds, namely, 1, 2, 3, 5, 6, 7, and 8, the inhibition of the PR of the serotype 3 virus was tested. With the exception of compound 8, DENV-3 PR is more sensitive to the inhibitors with derivative 6, which is the most active one against DENV-2 PR, being also the most potent DENV-3 PI (IC50, 1 μM). This also underlines the results of the docking studies, which were done with the structure of DENV-3 PR.
Toxicity and antiviral activities.
Next, we analyzed inhibition of the viral replication by the PIs. In order to exclude toxic side effects of the substances, the cell survival and metabolism were measured by a cell proliferation assay. Vero cells were incubated with decreasing amounts of the PIs solubilized in DMSO or with DMSO alone as a control (Table 1). Substance concentrations inhibiting the MTS substrate conversation were excluded from further analyses. Substance 2 was not tested in cellular assays, since it exhibited toxicity at concentrations below 1 μM (Table 1; see Fig. S5 in the supplemental material). Cell death due to cytotoxicity of the compounds was not observed at concentrations of 30 μM. Higher concentrations were not analyzed in cell culture.
To analyze inhibition of viral replication, Vero cells were preincubated with the PIs at decreasing concentrations below the cell toxicity (Table 1; see Fig. S5 in the supplemental material) and subsequently infected with DENV-2. Cellular supernatants were collected after 4 days and titrated on Vero cells. Virus-infected cells were visualized by immunofluorescence staining, and viral titers were calculated (Table 1). All PIs did significantly decrease viral replication. Furthermore, substances 3 and 4 inhibited viral replication more than 3 orders of magnitude at 1 μM and 3 μM, respectively (Fig. 4). This indicates that the PIs described here, except compounds 2 and 8, suppressed viral replication quite efficiently at low-micromolar or even submicromolar concentrations. Especially the low 50% effective concentrations (EC50s) for compounds 3, 4, 5, 6, and 8 showed that we have identified inhibitors of DENV-2 replication.
FIG 4.

Inhibition of DENV-1 and DENV-2 replication. (A) Vero cells were preincubated with decreasing amounts of the compounds. DMSO served as a control. The cells were subsequently infected with DENV-2. Cell culture supernatants were collected and viruses titrated on Vero cells. All experiments were performed in triplicate assays. Error bars represent the standard deviation. Significances of differences in viral titers compared to the DMSO control were determined by the paired two-sample t test and are indicated by one (P < 0.05) or two (P < 0.01) asterisks above the bars. (B) Subtype specificity inhibition of replication by the PIs. Vero cells were preincubated with the compounds and infected with DENV-1 or DENV-2 (one genome copy [cp] per cell). Cell culture supernatants were collected, and viral genome amounts were determined by qRT-PCR. All experiments were performed in independent triplicate assays. Error bars represent the standard deviation.
In order to analyze whether the compounds are able to inhibit the PRs of other DENV serotypes, effects on replication were determined with DENV-1. Cells were infected with DENV-1 or DENV-2 in the presence of the most effective compounds, 3 and 4. Viral replication was determined by quantification of viral RNA genomes in the cellular supernatants by qRT-PCR. Both compounds 3 and 4 decreased DENV-2 genome copies to 27% and 3.1% of the DMSO control and DENV-1 genome copies to 10% and 3.5%, indicating that DENV-1 exhibits a similar sensitivity to both substances (Fig. 4). These results together with the results of the in vitro PR analyses provide evidence that compounds 3 and 4 inhibit at least replication or PRs of DENV-1, DENV-2, and DENV-3.
Interestingly, the dimethoxy derivative 7 also exhibits antiviral activity against DENV-2, while it is inactive against DENV-2 PR in the fluorometric enzyme assay (Table 1 and Fig. 4). Vice versa, the dihydroxy derivative 8 displays good inhibition of PR of serotypes 2 and 3 but is not active in cells at concentrations below 3 μM. This observation leads to the hypotheses that compound 7 may act like a prodrug being demethylated in cells yielding to the more potent dihydroxy derivative. This hypothesis is supported by the fact that compound 7 inhibits DENV-1 replication as well (Fig. 4) and the PR in the cell-based assay (described below).
To further exclude that effects of our compounds on DENV-2 replication are due to cytotoxicity and to prove the specificity of the observed inhibition of DENV-2 replication, we analyzed influences of the PIs on HIV-1 replication. HIV-1 encodes an aspartate PR, which in neither its structure nor to its substrate specificity is similar to the DENV-PR. Thus, DENV PIs should not influence HIV-1 replication. HEK 293T cells were transfected with the proviral HIV-1 plasmid pNL4-3, and PIs were added at concentrations above the EC50. Viral supernatants were collected 2 days posttransfection and titrated on TZM-bl indicator cells (Fig. 5). TZM-bl indicator cells contained a LacZ gene controlled by the HIV-1 LTR promoter. Infected cells were identified by β-galactosidase stain. None of the PIs influenced HIV-1 replication, excluding cellular toxicity and unspecific inhibition of the HIV-1 PR.
FIG 5.
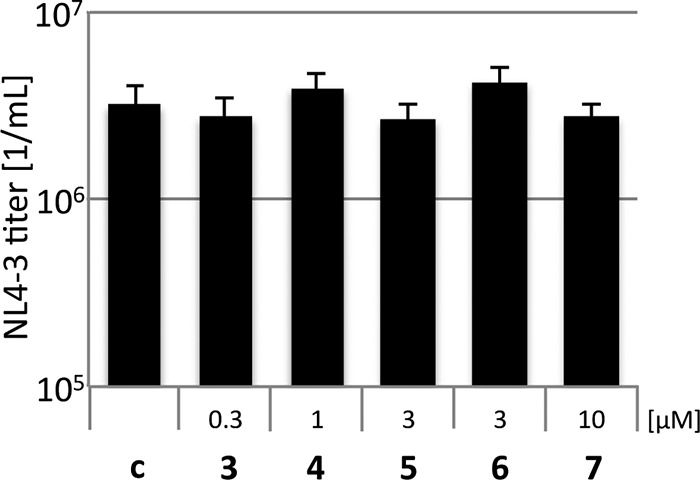
Compounds 3 to 7 do not influence HIV-1. HEK 293T cells were transfected with the proviral HIV-1 plasmid pNL4-3 and subsequently incubated with the PIs. Viral titers were determined on TZM-bl indicator cells, which contain a LacZ gene controlled by the HIV-1 LTR promoter. All experiments were performed in triplicate assays. Error bars represent the standard deviation.
To analyze the specificity of compounds, we determined antiviral activity against HCV, another member of the family Flaviviridae. Human hepatoma cells (Huh7.5-FLuc) expressing a firefly luciferase reporter that was used to measure cytotoxicity (not shown) were transfected with a Renilla luciferase (RLuc)-encoding HCV reporter genome (JcR2a) (Fig. 6A). After 72 h, virus replication was assessed by quantification of RLuc activity in transfected cells (Fig. 6B). To determine virus titers, naive Huh7.5 cells were inoculated with culture supernatants collected 72 h after transfection, and RLuc expression was determined 3 days later (Fig. 6C). None of the compounds influenced HCV replication or production of infectious particles, showing that the PIs are specific for DENV and inactive against the related HCV.
FIG 6.

Components 1 and 3 to 7 do not influence HCV replication. (A) Scheme of the experimental approach. Huh7.5-Fluc cells were transfected with in vitro transcribed HCV RNA of the Renilla luciferase-encoding reporter virus JcR2a. (B) After 72 h, virus replication and cell viability were determined by measuring Renilla luciferase (Rluc) and firefly luciferase (Fluc) activity from cell lysates, respectively. AU, arbitrary fluorescence units. (C) Production of infectious particles was determined by infection of naive Huh7.5 cells with supernatants collected from transfected cells and quantification of Rluc activity after 72 h. Experiments were performed three times in triplicate (or sextuplicate in the case of DMSO-treated cells). The HCV protease inhibitor boceprevir was included as a positive control. A red dashed line indicates the limit of detection. All data represent the mean of three independent experiments, which were performed in triplicate assays. Error bars represent the standard deviation.
Inhibition of the DENV PR in cell culture.
To measure inhibition of the DENV-2 PR in cells quantitatively and without any influences of the viral replication (such as additional replication cycles, etc.), a PR reporter system was constructed. The coding regions of gfp and dsred2 were fused, and an NS2A/NS2B cleavage site was inserted by PCR (Fig. 7). Cleavage of this GFP-NS2A/B cleavage site-DsRed2 fusion protein by the DENV-2 PR would result in separate GFP and DsRed proteins. HEK 293T cells were transfected with the reporter plasmid alone or in combination with a pDENV2-PR expression plasmid. To monitor expression of the DENV PR, an N-terminal M2 FLAG tag was added by PCR. Cleavage of the substrate was analyzed by quantitative Western blotting using anti-GFP antibodies (Fig. 7; see Fig. S6 in the supplemental material). Determination of GAPDH (glyceraldehyde-3-phosphate dehydrogenase) amounts served as loading control. Expression of the substrate alone resulted in the expected 55-kDa protein. Coexpression of the DENV PR led to substrate cleavage of about 75%. Instead of the expected single GFP-derived product band, cleavage of the reporter fusion protein resulted in two product bands (Fig. 7). Therefore, we analyzed whether GFP was cleaved by the DENV-2 PR (see Fig. S7 in the supplemental material) and could show that DENV-2 indeed cleaves the GFP at a potential cleavage site, KRDH. The lead substance, compound 1, inhibited viral PR activity in cell culture already with an IC50 at 15.6 μM. The optimized derivatives resulted in IC50s in the low-micromolar or even submicromolar range (Table 1, Fig. 7; see Fig. S6). The high inhibition of replication in comparison to the IC50s for PR inhibition in cell culture could indicate that an almost complete processing of viral proteins is required for viral infectivity. Similar discrepancies between EC50 and IC50s were observed by others but attributed to the enzyme-based DENV PR assay (18). Furthermore, our result might suggest that the PIs are enriched in cells. Thus, concentrations less than the IC50 in cell culture led to the inhibition of viral replication.
FIG 7.
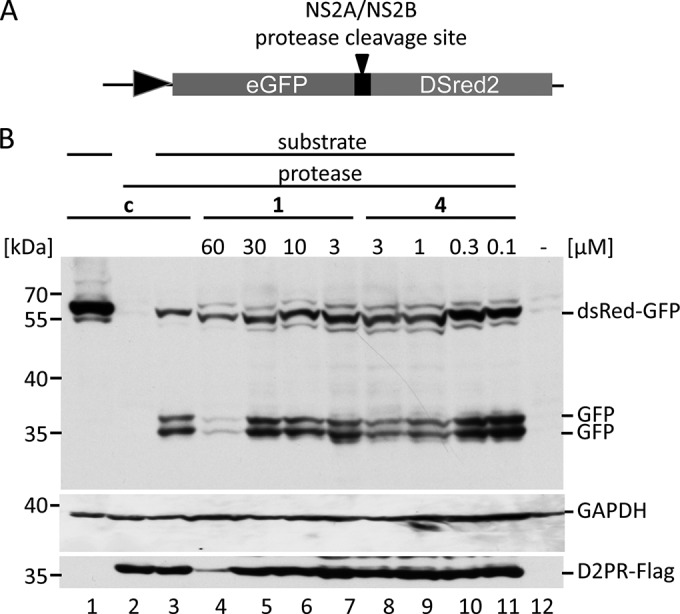
Compounds 1 and 3 to 7 inhibit DENV-2 PR in cells. (A) Scheme of the cleavage reporter construct. (B) Representative quantitative Western blotting analyses of reporter cleavage by the DENV-2 PR using a monoclonal anti-GFP antibody. Cells were transfected with the reporter vector alone (lane 1), with the PR expression vector alone, or with the reporter vector and the PR (lanes 3 to 11). PIs were added after transfection at four nontoxic concentrations (indicated above the panel). Substrate and products were visualized, and the inhibition was calculated as a ratio of fusion protein to fusion protein plus products. Expression of the DENV PR was monitored by Western blotting with monoclonal M2 antibodies. The positions of the molecular mass markers (kDa) are indicated.
Upon comparison of the inhibition of the isolated proteases (columns 1 and 2 in Table 1) with antiviral activity (column 4) and PR inhibition in cell culture (column 5), no good correlation can be found between PR inhibition (columns 1 and 2) and inhibition of viral replication (column 4) or PR in cell culture (column 5). However, a very good correlation (Fig. 8) can be found between antiviral activity (column 4) and PR inhibition in cell culture (column 5), indicating that indeed the PR inhibition is the main reason for the antiviral activity of the compounds and furthermore substantiating the hypothesis that the dimethoxy-substituted compound 7, which is not active against the isolated proteases, maybe a prodrug for the dihydroxy compound 8. On the other hand, these results show that for rational design of new PR inhibitors, both assays are necessary: the determination of inhibition of isolated proteases in order to verify the docking hypotheses and the determination of PR inhibition in cell culture, which can be used to predict the antiviral activity.
FIG 8.
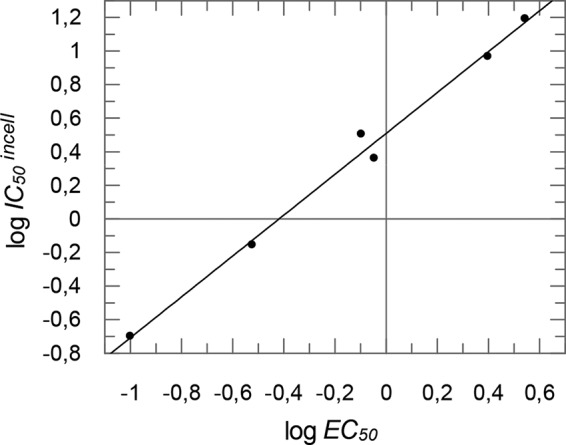
Correlation of antiviral activity (expressed as log EC50 [column 4 in Table 1]) and PR inhibition in cell culture (expressed as log IC50 in the cell [column 5 in Table 1]). For the diagram, results for compounds 1, 3, 4, 5, 6, and 7 were taken.
By analyzing influences of the PR inhibition in cell culture on DENV replication, a high sensitivity of viral replication compared to that of the cell-culture-based cleavage assay was observed (Table 1 and Fig. 7). These results indicate that already partial inhibition of processing is detrimental for DENV, which is similar to partial inhibition of HIV-1 maturation, which results in an almost complete reduction of viral replication (39). Other viruses, such as foamy viruses, tolerate up to 40% of nonprocessed proteins without any influence on viral replication (40). On the other hand, since the cell-culture-based cleavage reporter system uses the NS2B/NS3 cleavage site, which was shown to be the most efficient one, it can be assumed that the PIs will block less-efficient cleavage sites at lower EC50s.
In summary, we describe a novel class of DENV-2/3 PR inhibitors with inhibition of the PR in vitro and in vivo, which are shown to inhibit viral replication. Our substances may serve as promising lead compounds for further studies and optimization.
Supplementary Material
ACKNOWLEDGMENTS
We thank A. Kucharski (University of Würzburg, Institute of Pharmacy and Food Chemistry), S. Mährlein, and U. Nowe (University of Mainz, Institute of Pharmacy and Biochemistry) for technical support. The DENV-2 PR bacterial expression plasmid was kindly provided by C. Klein and C. Steuer (University of Heidelberg), and the anti-DENV-2 E antibodies and dengue viruses were provided by J. Schneider-Schaulies (University of Würzburg).
We thank the University of Mainz for financial support. C.W. was supported by the Studienstiftung des Deutschen Volkes, and T.S. and B.E. were supported by the DFG (SFB630).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.03543-14.
REFERENCES
- 1.Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, Myers MF, George DB, Jaenisch T, Wint GR, Simmons CP, Scott TW, Farrar JJ, Hay SI. 2013. The global distribution and burden of dengue. Nature 496:504–507. doi: 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization. November 2012, posting date Dengue and severe dengue. Fact sheet no. 117 World Health Organization, Geneva, Switzerland. [Google Scholar]
- 3.Normile D. 2013. Tropical medicine. Surprising new dengue virus throws a spanner in disease control efforts. Science 342:415. doi: 10.1126/science.342.6157.415. [DOI] [PubMed] [Google Scholar]
- 4.Messer WB, Gubler DJ, Harris E, Sivananthan K, de Silva AM. 2003. Emergence and global spread of a dengue serotype 3, subtype III virus. Emerg Infect Dis 9:800–809. doi: 10.3201/eid0907.030038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Streit JA, Yang M, Cavanaugh JE, Polgreen PM. 2011. Upward trend in dengue incidence among hospitalized patients, United States. Emerg Infect Dis 17:914–916. doi: 10.3201/eid1705.101023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guzman MG, Halstead SB, Artsob H, Buchy P, Farrar J, Gubler DJ, Hunsperger E, Kroeger A, Margolis HS, Martinez E, Nathan MB, Pelegrino JL, Simmons C, Yoksan S, Peeling RW. 2010. Dengue: a continuing global threat. Nat Rev Microbiol 8:S7–S16. doi: 10.1038/nrmicro2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Endy TP, Anderson KB, Nisalak A, Yoon IK, Green S, Rothman AL, Thomas SJ, Jarman RG, Libraty DH, Gibbons RV. 2011. Determinants of inapparent and symptomatic dengue infection in a prospective study of primary school children in Kamphaeng Phet, Thailand. PLoS Negl Trop Dis 5:e975. doi: 10.1371/journal.pntd.0000975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luz PM, Vanni T, Medlock J, Paltiel AD, Galvani AP. 2011. Dengue vector control strategies in an urban setting: an economic modelling assessment. Lancet 377:1673–1680. doi: 10.1016/S0140-6736(11)60246-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guzman MG, Kouri G. 2002. Dengue: an update. Lancet Infect Dis 2:33–42. doi: 10.1016/S1473-3099(01)00171-2. [DOI] [PubMed] [Google Scholar]
- 10.Lindenbach BD, Thiel H-J, Rice CM. 2007. Flaviviridae: the viruses and their replication, p 1101-1152. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 11.Bera AK, Kuhn RJ, Smith JL. 2007. Functional characterization of cis and trans activity of the flavivirus NS2B-NS3 protease. J Biol Chem 282:12883–12892. doi: 10.1074/jbc.M611318200. [DOI] [PubMed] [Google Scholar]
- 12.Jacobson IM, McHutchison JG, Dusheiko G, Di Bisceglie AM, Reddy KR, Bzowej NH, Marcellin P, Muir AJ, Ferenci P, Flisiak R, George J, Rizzetto M, Shouval D, Sola R, Terg RA, Yoshida EM, Adda N, Bengtsson L, Sankoh AJ, Kieffer TL, George S, Kauffman RS, Zeuzem S, ADVANCE Study Team . 2011. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 364:2405–2416. doi: 10.1056/NEJMoa1012912. [DOI] [PubMed] [Google Scholar]
- 13.Poordad F, McCone J Jr, Bacon BR, Bruno S, Manns MP, Sulkowski MS, Jacobson IM, Reddy KR, Goodman ZD, Boparai N, DiNubile MJ, Sniukiene V, Brass CA, Albrecht JK, Bronowicki JP, SPRINT-2 Investigators . 2011. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med 364:1195–1206. doi: 10.1056/NEJMoa1010494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matthews SJ, Lancaster JW. 2012. Telaprevir: a hepatitis C NS3/4A protease inhibitor. Clin Ther 34:1857–1882. doi: 10.1016/j.clinthera.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 15.Pearlman BL. 2012. Protease inhibitors for the treatment of chronic hepatitis C genotype-1 infection: the new standard of care. Lancet Infect Dis 12:717–728. doi: 10.1016/S1473-3099(12)70060-9. [DOI] [PubMed] [Google Scholar]
- 16.Kohl NE, Emini EA, Schleif WA, Davis LJ, Heimbach JC, Dixon RA, Scolnick EM, Sigal IS. 1988. Active human immunodeficiency virus protease is required for viral infectivity. Proc Natl Acad Sci U S A 85:4686–4690. doi: 10.1073/pnas.85.13.4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.HHS Panel on Antiretroviral Guidelines for Adults and Adolescents. 2012. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Department of Health and Human Services; Washington, DC: http://aidsinfo.nih.gov. [Google Scholar]
- 18.Yang CC, Hsieh YC, Lee SJ, Wu SH, Liao CL, Tsao CH, Chao YS, Chern JH, Wu CP, Yueh A. 2011. Novel dengue virus-specific NS2B/NS3 protease inhibitor, BP2109, discovered by a high-throughput screening assay. Antimicrob Agents Chemother 55:229–238. doi: 10.1128/AAC.00855-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nitsche C, Schreier VN, Behnam MA, Kumar A, Bartenschlager R, Klein CD. 2013. Thiazolidinone-peptide hybrids as dengue virus protease inhibitors with antiviral activity in cell culture. J Med Chem 56:8389–8403. doi: 10.1021/jm400828u. [DOI] [PubMed] [Google Scholar]
- 20.Steuer C, Heinonen KH, Kattner L, Klein CD. 2009. Optimization of assay conditions for dengue virus protease: effect of various polyols and nonionic detergents. J Biomol Screen 14:1102–1108. doi: 10.1177/1087057109344115. [DOI] [PubMed] [Google Scholar]
- 21.D'Arcy A, Chaillet M, Schiering N, Villard F, Lim SP, Lefeuvre P, Erbel P. 2006. Purification and crystallization of dengue and West Nile virus NS2B-NS3 complexes. Acta Crystallogr Sect F Struct Biol Cryst Commun 62:157–162. doi: 10.1107/S1744309106001199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noble CG, Seh CC, Chao AT, Shi PY. 2012. Ligand-bound structures of the dengue virus protease reveal the active conformation. J Virol 86:438–446. doi: 10.1128/JVI.06225-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grimme S, Antony J, Ehrlich S, Krieg H. 2010. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys 132:154104. doi: 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- 24.Dunning JTH. 1989. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J Chem Phys 90:1007–1023. [Google Scholar]
- 25.Vosko SH, Wilk L, Nusair M. 1980. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Can J Phys 58:1200–1211. doi: 10.1139/p80-159. [DOI] [Google Scholar]
- 26.Becke AD. 1988. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A 38:3098–3100. doi: 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- 27.Lee C, Yang W, Parr RG. 1988. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B Condensed Matter 37:785–789. doi: 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- 28.Becke AD. 1993. Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652. [Google Scholar]
- 29.Othman R, Kiat TS, Khalid N, Yusof R, Newhouse EI, Newhouse JS, Alam M, Rahman NA. 2008. Docking of noncompetitive inhibitors into dengue virus type 2 protease: understanding the interactions with allosteric binding sites. J Chem Infect Model 48:1582–1591. doi: 10.1021/ci700388k. [DOI] [PubMed] [Google Scholar]
- 30.Chanprapaph S, Saparpakorn P, Sangma C, Niyomrattanakit P, Hannongbua S, Angsuthanasombat C, Katzenmeier G. 2005. Competitive inhibition of the dengue virus NS3 serine protease by synthetic peptides representing polyprotein cleavage sites. Biochem Biophys Res Commun 330:1237–1246. doi: 10.1016/j.bbrc.2005.03.107. [DOI] [PubMed] [Google Scholar]
- 31.Ludewig S, Kossner M, Schiller M, Baumann K, Schirmeister T. 2010. Enzyme kinetics and hit validation in fluorimetric protease assays. Curr Top Med Chem 10:368–382. doi: 10.2174/156802610790725498. [DOI] [PubMed] [Google Scholar]
- 32.Jerabek-Willemsen M, Wienken CJ, Braun D, Baaske P, Duhr S. 2011. Molecular interaction studies using microscale thermophoresis. Assay Drug Dev Technol 9:342–353. doi: 10.1089/adt.2011.0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cígler P, Kozísek M, Rezácová P, Brynda J, Otwinowski Z, Pokorná J, Plesek J, Grüner B, Dolecková-Maresová L, Mása M, Sedlácek J, Bodem J, Kräusslich H, Král V, Konvalinka J. 2005. From nonpeptide toward noncarbon protease inhibitors: metallacarboranes as specific and potent inhibitors of HIV protease. Proc Natl Acad Sci U S A 102:15394–15399. doi: 10.1073/pnas.0507577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kozisek M, Henke S, Saskova KG, Jacobs GB, Schuch A, Buchholz B, Müller V, Kräusslich HG, Rezacova P, Konvalinka J, Bodem J. 2012. Mutations in HIV-1 gag and pol compensate for the loss of viral fitness caused by a highly mutated protease. Antimicrob Agents Chemother 56:4320–4330. doi: 10.1128/AAC.00465-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reiss S, Rebhan I, Backes P, Romero-Brey I, Erfle H, Matula P, Kaderali L, Poenisch M, Blankenburg H, Hiet MS, Longerich T, Diehl S, Ramirez F, Balla T, Rohr K, Kaul A, Buhler S, Pepperkok R, Lengauer T, Albrecht M, Eils R, Schirmacher P, Lohmann V, Bartenschlager R. 2011. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 9:32–45. doi: 10.1016/j.chom.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang MH, Gordon LA, Fung HB. 2012. Boceprevir: a protease inhibitor for the treatment of hepatitis C. Clin Ther 34:2021–2038. doi: 10.1016/j.clinthera.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 37.Wu F, Yang X. March 2003. A process for preparing 2-trifluoromethyl-10-oxanone from p-trifluorotoluene and o-mercaptobenzoic acid includes nucleophilic substitution reaction on aryl ring to obtain diaryl thioether, and cyclization reaction with concentrated sulfuric acid. Chinese patent 03115623. [Google Scholar]
- 38.Moon JK, Park JW, Lee WS, Kang YJ, Chung HA, Shin MS, Yoon YJ, Park KH. 1999. Synthesis of some 2-substituted-thioxanthones. J Heterocyclic Chem 36:793–798. doi: 10.1002/jhet.5570360336. [DOI] [Google Scholar]
- 39.Müller B, Anders M, Akiyama H, Welsch S, Glass B, Nikovics K, Clavel F, Tervo HM, Keppler OT, Kräusslich HG. 2009. HIV-1 Gag processing intermediates trans-dominantly interfere with HIV-1 infectivity. J Biol Chem 284:29692–29703. doi: 10.1074/jbc.M109.027144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spannaus R, Bodem J. 2014. Determination of the protease cleavage site repertoire—the RNase H but not the RT domain is essential for foamy viral protease activity. Virology 454-455:145–156. doi: 10.1016/j.virol.2014.02.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.