

Abstract
Hepatitis B virus (HBV) remains a major human pathogen despite the development of both antiviral drugs and a vaccine, in part because the current therapies do not suppress HBV replication far enough to eradicate the virus. Here, we screened 51 troponoid compounds for their ability to suppress HBV RNaseH activity and HBV replication based on the activities of α-hydroxytropolones against HIV RNaseH, with the goal of determining whether the tropolone pharmacophore may be a promising scaffold for anti-HBV drug development. Thirteen compounds inhibited HBV RNaseH, with the best 50% inhibitory concentration (IC50) being 2.3 μM. Similar inhibition patterns were observed against HBV genotype D and C RNaseHs, implying limited genotype specificity. Six of 10 compounds tested against HBV replication in culture suppressed replication via blocking of viral RNaseH activity, with the best 50% effective concentration (EC50) being 0.34 μM. Eighteen compounds inhibited recombinant human RNaseH1, and moderate cytotoxicity was observed for all compounds (50% cytotoxic concentration [CC50] = 25 to 79 μM). Therapeutic indexes ranged from 3.8 to 94. Efficient inhibition required an intact α-hydroxytropolone moiety plus one or more short appendages on the tropolone ring, but a wide variety of constituents were permissible. These data indicate that troponoids and specifically α-hydroxytropolones are promising lead candidates for development as anti-HBV drugs, providing that toxicity can be minimized. Potential anti-RNaseH drugs are envisioned to be employed in combination with the existing nucleos(t)ide analogs to suppress HBV replication far enough to block genomic maintenance, with the goal of eradicating infection.
INTRODUCTION
More than 2 billion people have been infected with hepatitis B virus (HBV) at some time in their lives and up to 350 million remain chronically infected as carriers of HBV (1, 2). Approximately 20% of chronic hepatitis B patients develop liver cirrhosis, leading to hepatic insufficiency and portal hypertension (3). Furthermore, there is a 100-fold higher risk of development of hepatocellular carcinoma in chronic HBV patients than in noncarriers (4). Every year, HBV infection kills more than 500,000 people from cirrhosis, liver failure, and hepatocellular carcinoma (5).
The global level of chronic HBV infection still mandates development of new drugs despite the development of excellent vaccines and drugs against the virus. Seven drugs have been approved by the U.S. Food and Drug Administration for treating HBV infection. Interferon alpha and pegylated interferon alpha are immunomodulatory agents. However, the need for subcutaneous administration, the poor long-term responses, the very low cure rates, and the high frequency of adverse side effects make interferon far from an ideal drug (6). The nucleos(t)ide analog drugs lamivudine, adefovir, entecavir, telbivudine, and tenofovir are phosphorylated to their triphosphate derivatives by cellular enzymes and become chain-terminating substrates of the HBV reverse transcriptase (7). While these drugs profoundly suppress HBV replication in most patients, often to below the clinical limit of detection (8, 9), they have a number of limitations. The emergence of resistant HBV variants with mutations in the reverse transcriptase is a major problem for the nucleos(t)ide analogs except entecavir and tenofovir, and multidrug resistance has increased the risk of exacerbation of liver disease (5, 10). Furthermore, viral replication almost always resurges following termination of therapy. Thus, although viral replication can be controlled, the drugs must be given essentially indefinitely. This therapy is very expensive and may cause unpredictable adverse effects after long-term drug administration (11, 12).
HBV encodes two enzymes essential for its replication that are attractive drug targets (13). The reverse transcriptase domain of the viral polymerase protein contains the DNA polymerase activity that synthesizes new DNA. The RNaseH domain is encoded at the carboxy terminus of the viral polymerase and degrades the viral RNA template after it is copied into minus-polarity DNA, allowing the subsequent synthesis of the plus-polarity DNA strand. No drugs against HBV RNaseH are available, primarily due to difficulties in establishing screening assays. We recently developed a low-throughput screening pipeline for HBV RNaseH inhibitors (14–16), opening the door to anti-RNaseH drug discovery.
Both the HBV and HIV RNaseH enzymes belong to the nucleotidyl transferase superfamily (17, 18). We previously demonstrated that many compounds selected for the ability to suppress HIV RNaseH activity also inhibit HBV RNaseH, and some can also block HBV replication in cell cultures via suppression of the viral RNaseH activity (15, 16). One of these compounds was the natural product β-thujaplicinol (14). β-Thujaplicinol is a member of the most widely studied class of troponoids, the α-hydroxytropolones, that have been identified as anticancer agents (19, 20) as well as lead therapeutic agents for a number of infections, including HIV (21–25), herpes simplex virus (26), malaria (27), and many bacteria (28). This antimicrobial activity is often attributed to the ability of the compounds to sequester the divalent cations in the active sites of dinuclear metalloenzymes, a feature arising from the three contiguous oxygen atoms on the troponoid ring (29–31). Here, 51 troponoids, including 36 α-hydroxytropolones, were screened for their ability to inhibit HBV RNaseH activity and viral replication to assess whether tropolones and specifically α-hydroxytropolones may be attractive candidates for development as anti-HBV drugs.
MATERIALS AND METHODS
Compound acquisition and synthesis.
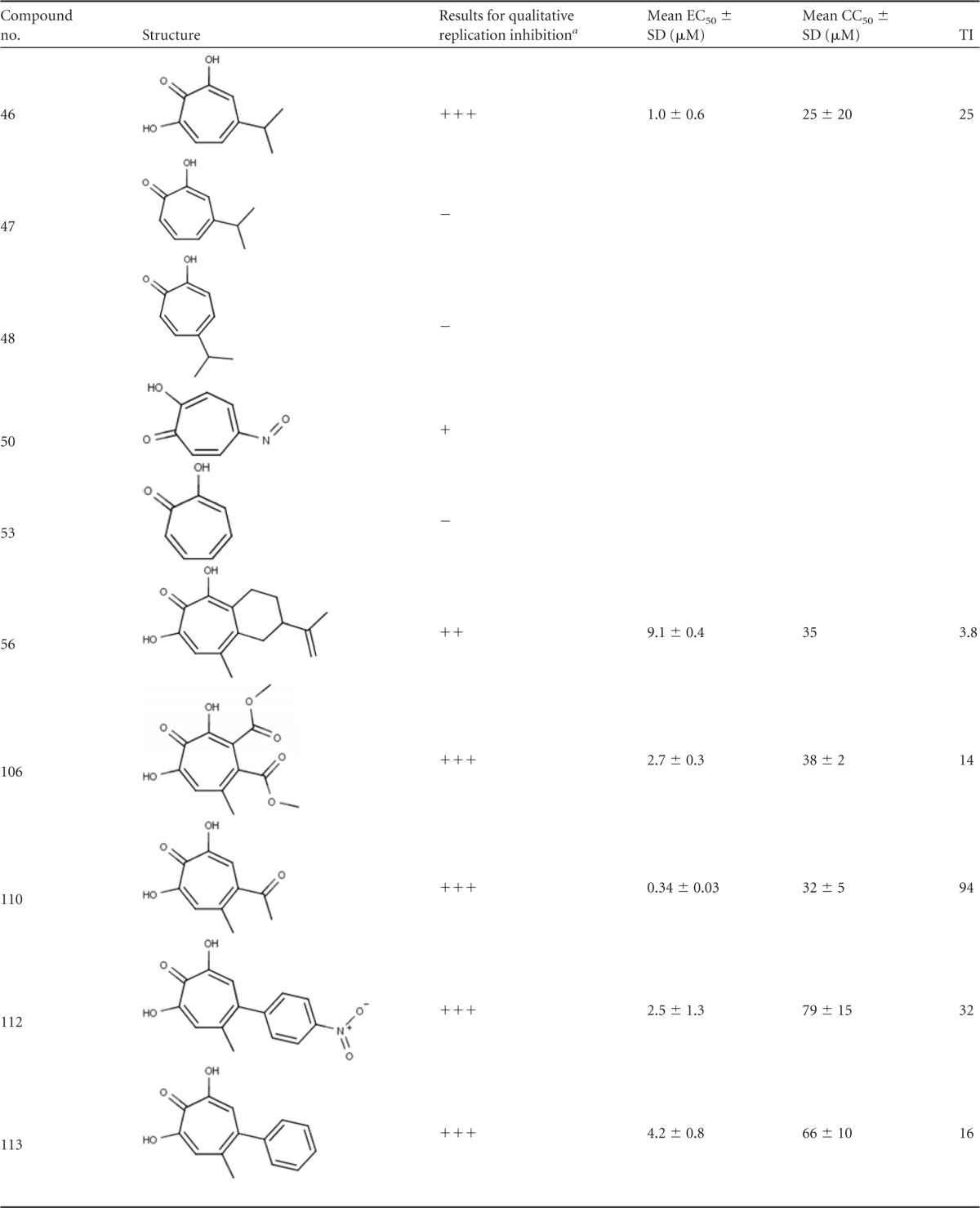
The compounds employed are listed in Table 1, and their structures are in Fig. S1 in the supplemental material. Compounds 46 to 57 were acquired from the National Cancer Institute (NCI) Developmental Therapeutics Program, and compounds 59 to 63 were purchased. α-Hydroxytropolone (compound 172) was synthesized in 3 steps from tropolone based on the procedure of Takeshita et al. (32). Compounds 92 to 105 were synthesized from manicol as previously described (21), and compounds 106 to 120 and 143 to 147 were synthesized in 5 to 7 steps from kojic acid as previously described (33–35). All compounds were dissolved in dimethyl sulfoxide (DMSO) and stored at −80°C.
TABLE 1.
Anti-RNaseH profiles of hydroxytropolones and related compounds
| Compound no. | Qualitative biochemical screening results fora: |
Mean IC50 ± SD (μM) for: |
||||
|---|---|---|---|---|---|---|
| HBV gtD | HBV gtC | Human RNaseH1 | HBV gtD | HBV gtC | Human RNaseH1 | |
| 46 | +++ | +++ | ++ | 5.9 ± 0.7 | 2.3 ± 1.7 | 58 ± 14.4 |
| 47 | − | − | − | |||
| 48 | − | − | − | |||
| 49 | − | − | − | |||
| 50 | − | − | − | |||
| 51 | + | − | − | |||
| 52 | − | − | ||||
| 53 | − | − | − | |||
| 54 | − | − | ||||
| 55 | + | − | + | |||
| 56 | +/− | +/− | − | |||
| 57 | − | + | ||||
| 59 | − | + | ||||
| 60 | − | + | ||||
| 61 | − | ++ | ||||
| 62 | + | − | − | |||
| 63 | − | + | ||||
| 92 | − | − | − | |||
| 93 | − | − | − | |||
| 94 | − | − | ||||
| 95 | − | − | ||||
| 96 | − | + | ||||
| 97 | − | − | ||||
| 98 | − | − | + | |||
| 99 | − | − | − | |||
| 100 | − | − | ||||
| 101 | − | − | ||||
| 102 | − | |||||
| 103 | − | |||||
| 104 | − | |||||
| 105 | − | |||||
| 106 | +++ | +++ | +++ | 29.6 ± 1.4 | 29.0 ± 0.4 | 47.8 ± 3.5 |
| 107 | ++ | + | ++ | 40 ± 18 | 65 ± 38 | icab |
| 108 | + | + | ++ | |||
| 109 | + | + | ++ | |||
| 110 | ++ | ++ | +++ | 34.6 ± 25 | 22.9 ± 2 | 29.3 ± 3.3 |
| 111 | − | |||||
| 112 | + | + | +++ | >100 | >100 | 91.2 ± 23 |
| 113 | ++ | + | ++ | >100 | >100 | 27.4 ± 8.9 |
| 114 | − | |||||
| 115 | − | |||||
| 117 | − | |||||
| 118 | − | |||||
| 119 | − | |||||
| 120 | + | + | +++ | |||
| 143 | + | + | + | |||
| 144 | − | |||||
| 145 | − | |||||
| 146 | − | |||||
| 147 | − | |||||
| 172 | − | − | − | |||
+++, inhibition at 10 μM; ++, inhibition at 20 μM; +, detectable inhibition at 60 μM; +/−, moderate inhibition in some but not all assays; −, no inhibition. gtD, genotype D; gtC, genotype C.
ica, insufficient compound available.
RNaseH expression and purification.
Recombinant HBV RNaseH and human RNaseH1 were expressed in Escherichia coli and partially purified by nickel-affinity chromatography as previously described (15). The enriched extracts were dialyzed into 50 mM HEPES (pH 7.3), 300 mM NaCl, 20% glycerol, and 5 mM dithiothreitol (DTT) and stored in liquid nitrogen.
Oligonucleotide-directed RNA cleavage assay.
The RNaseH activity was measured using an oligonucleotide-directed RNA cleavage assay as previously described (14–16). Briefly, 6 μl protein extract was mixed on ice with an internally 32P-labeled 264-nucleotide (nt) RNA derived from the duck hepatitis B virus genome (DRF+ RNA) plus 3 μg oligonucleotide D2507− or its inverse-complement oligonucleotide D2526+ as a negative control. This mixture was incubated with test compounds in 50 mM Tris (pH 8.0), 190 mM NaCl, 5 mM MgCl2, 3.5 mM DTT, 0.05% NP-40, 6% glycerol, and 1% DMSO at 42°C for 90 min. The cleavage products were resolved by denaturing polyacrylamide gel electrophoresis, detected by autoradiography, and quantified using ImageJ. The nonspecific background values were determined from the incorrect oligonucleotide negative-control lane and subtracted from all experimental values. The 50% inhibitory concentrations (IC50s) were then calculated with GraphPad Prism using three-parameter log(inhibitor) versus response nonlinear curve fitting with the curve minimum set to zero to reflect background subtraction.
HBV replication assay.
Inhibition of the HBV replication was measured in HepDES19 cells as previously described (16). Cells were seeded in 6-well plates and incubated in Dulbecco's modified Eagle's medium (DMEM)/F12, 10% fetal bovine serum (FBS), and 1% penicillin-streptomycin (P/S) with 1 μg/ml tetracycline. The tetracycline was withdrawn after 24 h. The test compound was applied to duplicate wells 48 h later in medium containing a final DMSO concentration of 1%, and medium containing the compound was refreshed daily for the following 2 days. Cells were harvested, and nonencapsidated nucleic acids were digested with micrococcal nuclease (New England BioLabs). HBV DNA was purified from capsids using a QIAamp cador pathogen minikit (Qiagen) with proteinase K incubation overnight at 37°C. TaqMan PCR was performed for 40 cycles at an annealing temperature of 60°C. The primers and probe (IDT Inc.) for the plus-polarity strand were 5′CATGAACAAGAGATGATTAGGCAGAG3′, 5′GGAGGCTGTAGGCATAAATTGG3′, and 5′/56-FAM/CTGCGCACC/ZEN/AGCACCATGCA/3IABkFQ. The primers and probe for the minus-polarity strand were 5′GCAGATGAGAAGGCACAGA3′, 5′CTTCTCCGTCTGCCGTT3′, and 5′/56-FAM/AGTCCGCGT/ZEN/AAAGAGAGGTGCG/3IABkFQ.
MTT cytotoxicity assays.
HepDES19 cells (1.0 × 104 cells per well) were seeded in 96-well plates and incubated in DMEM with 10% FBS plus 1% P/S, 1% nonessential amino acids, and 1% glutamine. The compounds were diluted in the medium to the indicated concentrations plus 1% DMSO and added to the cells 24 h after plating, with each concentration tested in triplicate. The medium containing the compound was refreshed daily for the next 2 days. Thiazolyl blue tetrazolium bromide (MTT) (Sigma-Aldrich) was added to 0.25 mg/ml, the cultures were incubated for 60 min, the metabolites were solubilized in acidic isopropanol, and the absorbance was read at 570 nm.
RESULTS
Biochemical screening against HBV RNaseH.
Fifty-one troponoids (Table 1; see also Fig. S1 in the supplemental material) were screened for anti-HBV RNaseH activity. The screening employed an oligonucleotide-directed RNA cleavage assay (Fig. 1A) in which an internally 32P-labeled RNA is annealed to a cDNA oligonucleotide, and the cleavage at the heteroduplex at the annealing site is detected by resolving the RNA fragments by denaturing polyacrylamide gel electrophoresis followed by autoradiography. A qualitative primary screening assay was initially performed with compounds at concentrations of 60, 20, and 10 μM. The screening was first performed against a recombinant genotype D HBV RNaseH isolate and subsequently against a genotype C HBV RNaseH isolate to ensure that screening hits were not specific for only one of HBV's 8 major genotypes. Finally, IC50s were determined for select compounds against the genotype D and C enzymes.
FIG 1.
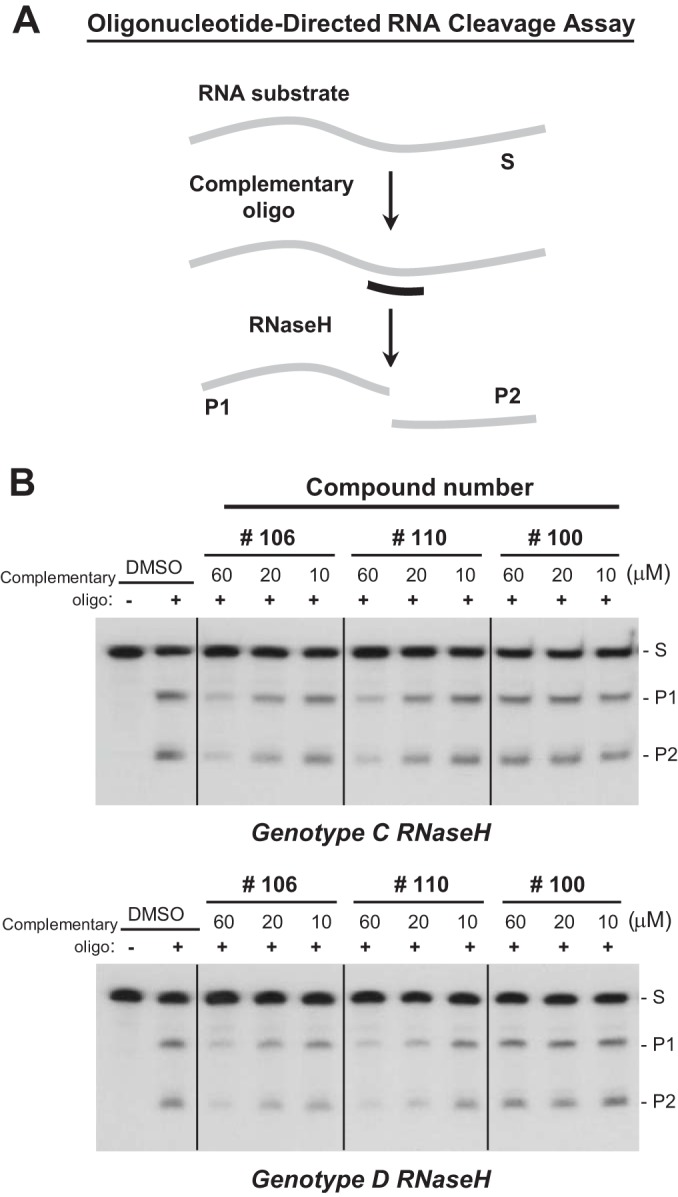
Primary screening of hydroxytropolones against HBV RNaseH. (A) Oligonucleotide-directed RNA cleavage assay. Reaction products were resolved by denaturing polyacrylamide gel electrophoresis and detected by autoradiography. Gray line, 32P-labeled RNA; black line, DNA oligonucleotide; S, substrate; P1, product 1; P2, product 2. (B) Examples of primary screening assays against HBV genotype C and D RNaseH enzymes. DMSO, vehicle control; +, complementary oligo, −, inclusion of a non-complementary DNA oligonucleotide as a specificity control.
Eight compounds had activity at only 60 μM against the genotype D and/or C enzymes (Table 1 and Fig. 1B). Three additional compounds inhibited the genotype D and/or C enzymes at 20 μM, and the two most active compounds (compounds 46 and 106) inhibited RNaseHs from both genotypes at 10 μM. The results with compound 46 (β-thujaplicinol) were consistent with those from our previous studies (14). Compound 56 gave inconsistent results in these assays. In some assays, it inhibited RNaseH at 20 μM, but it was inactive in the large majority of assays. The IC50s were determined for the six most active compounds against both genotype D and C enzymes. Compounds 46, 106, 107, and 110 had IC50s of ≤40 μM against both the genotype D and/or C enzymes (Fig. 2 and Table 1).
FIG 2.

IC50s for hydroxylated tropolone inhibitors of HBV RNaseH. IC50s were measured using the oligonucleotide-directed RNA cleavage assay. (A) IC50 curves for compound 106 against genotype C and D HBV RNaseHs. (B) IC50 curves for compound 110 against genotype C and D HBV RNaseHs. The IC50 curves are from representative assays, and the IC50s are the averages ± 1 standard deviation from two or three assays.
Counterscreening against human RNaseH1.
Thirty-five of the 51 compounds were counterscreened against recombinant human RNaseH1 in an initial effort to evaluate the specificity of the inhibition. Four compounds were active against the human enzyme at 10 μM, and 14 had activity at 20 μM or 60 μM. The inhibition patterns differed between HBV and human RNaseH1, with compounds generally having greater efficacy against the human enzyme than against HBV RNaseH. However, three compounds, compounds 46, 51, and 62, were more active against the HBV enzyme in the primary screening assays (Table 1). IC50s were determined against human RNaseH1 for the five compounds selected for full replication analyses (Table 1). These data confirmed the different patterns of the inhibition between the HBV and human enzymes, with compound 46 being 10- to 20-fold more active against the HBV enzyme, compound 106 being almost 2-fold more active against the HBV enzyme, compounds 110 and 112 having similar IC50s, and compound 113 being about 4-fold more active against human RNaseH1.
HBV replication inhibition and cytotoxicity.
The inhibition of viral replication was measured in HepDES19 cells (HepG2 cells stably transfected with an HBV genotype D genome under the control of a tetracycline-repressible promoter [36]) using a protocol we recently developed (16). Tetracycline was withdrawn from the medium, cells were incubated with the compound for 3 days with daily replenishment of the medium and compound, and cytoplasmic lysates containing viral capsid particles were prepared. The lysates were treated with micrococcal nuclease to destroy contaminating chromosomal DNA, and the capsids were treated with proteinase K to release the viral DNAs. Synthesis of the HBV plus-polarity DNA strand is suppressed by RNaseH inhibitors, and many minus-polarity DNA strands are truncated prematurely (14–16, 37). Therefore, we employed a strand-preferential quantitative PCR to measure accumulation of the amount of plus- and minus-polarity HBV DNAs recovered from the capsids (Fig. 3A).
FIG 3.

Inhibition of HBV replication by hydroxylated tropolones. The inhibition of replication by hydroxylated tropolones was measured against an HBV genotype D isolate in HepDES19 cells. (A) Principle underlying the strand-preferential quantitative PCR (qPCR) assay. Plus-polarity DNA is measured by amplifying HBV DNA across the gap in the minus-polarity DNA strand. Minus-polarity DNA strands are measured by amplifying sequences downstream of the 3′ end of the vast majority of plus-polarity DNA strands in viral capsids. (B to D) Select EC50 curves. EC50s were calculated based on the decline of the plus-polarity DNA strand. The curves and the EC50s are the means ± 1 standard deviation from two or three independent experiments, each conducted in duplicate.
Ten compounds were tested in the initial semiquantitative HBV replication inhibition assays at 60, 20, or 6.7 μM (Table 2) based on their efficacy in biochemical screening. Five compounds that inhibited RNaseH (compounds 46, 106, 110, 112, and 113) were selected; compound 107 was not tested because insufficient compound was available. We also selected five compounds that lacked activity against HBV RNaseH (compounds 47, 48, 50, 53, and 56) as specificity controls to evaluate the accuracy of our biochemical assay in identifying the inhibitors of viral replication. All five RNaseH inhibitors selectively inhibited accumulation of plus-polarity HBV DNA at 6.7 μM. Three of the five compounds that were negative in the biochemical assays were also negative in the replication inhibition assays (compounds 47, 48, and 53). Compound 50 was negative in the biochemical assays but suppressed accumulation of both DNA strands at 60 μM, consistent with inhibition being due to cytotoxicity and/or other mechanisms not involving RNaseH. Compound 56 occasionally had low activity in the biochemical assays. It suppressed accumulation of both DNA strands at 60 μM but had a weak preferential effect against the plus-polarity DNA at 20 μM. The cytotoxicity of the six compounds that selectively suppressed the plus-polarity DNA strand was assessed in HepDES19 cells using an MTT assay under the culture conditions employed for the replication inhibition assays. Moderate cytotoxicity was observed for all compounds, with 50% cytotoxic concentration (CC50) values ranging from 25 to 79 μM (Table 2 and Fig. 4).
TABLE 2.
Inhibition of HBV replication by tropolones
+++, inhibition at 6.7 μM; ++, inhibition at 20 μM; +, detectable inhibition at 60 μM; −, no inhibition.
FIG 4.
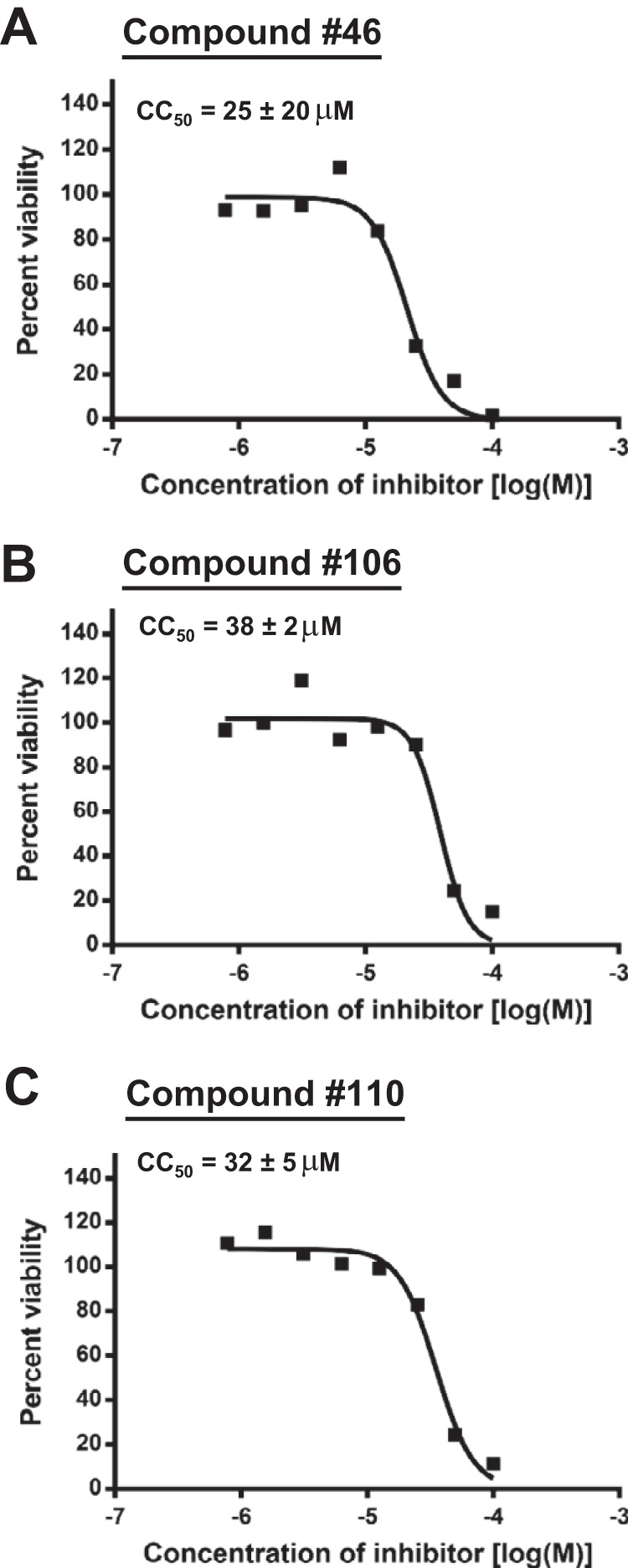
Representative cytotoxicity measurements. Representative MTT cytotoxicity assay results are shown. The curves are from representative experiments conducted in triplicate, and the CC50 values are the averages ± 1 standard deviation from two or three independent experiments, each conducted in triplicate.
The 50% effective concentrations (EC50s) were then determined for the six compounds which preferentially suppressed the plus-polarity DNA strand (compounds 46, 56, 106, 110, 112, and 113) (Table 2 and Fig. 3). All EC50s were <10 μM, with the best value being 0.34 μM for compound 110. The minus-polarity DNA strand in the same samples was either not suppressed by treatment with the compounds or was suppressed at much higher compound concentrations for all compounds except compound 56, confirming that synthesis of mature HBV genomes was suppressed by inhibition of the viral RNaseH activity. This led to therapeutic indexes (TI) (CC50/EC50) from 3.8 for compound 56 up to 94 for compound 110. For compound 56, minus-polarity DNA accumulation was suppressed in parallel with plus-polarity DNA (data not shown) over a concentration range in which significant cytotoxicity was evident in the MTT assays. This implies that compound 56 inhibited HBV replication in large part by inducing cytotoxicity.
DISCUSSION
The HBV and HIV RNaseH enzymes both belong to the nucleotidyl transferase superfamily (17, 18), and they share about 23% identity and 33% similarity in the core catalytic domain (15, 38). Previously, we identified the α-hydroxytropolone natural product β-thujaplicinol (compound 46) as an inhibitor of HBV RNaseH (14) based on its efficacy against HIV RNaseH (21–25). Here, 50 additional troponoids, including 36 α-hydroxytropolones, were screened against HBV RNaseH based on our previous results and the ability of this compound class to inhibit the HIV enzyme and to cross-inhibit other viruses such as the herpes simplex viruses (26) and foamy virus (39). Our goal was to determine whether tropolones are candidates for development as anti-HBV drugs and, if so, to derive initial structure-activity relationships (SAR) that could help guide their chemical optimization. Screening against recombinant HBV RNaseH identified five compounds that were active at ≤20 μM. These compounds inhibited HBV DNA synthesis with EC50s of <5 μM, and their selective suppression of the HBV plus-polarity DNA strand accumulation confirmed that inhibition of viral replication was due to blocking of the RNaseH activity.
The compounds were screened against HBV RNaseH enzymes from both genotype C and D isolates to maximize the chances of finding hits with efficacy against genetically divergent HBV isolates. Ten of the 13 compounds active against the genotype D RNaseH at ≤60 μM were also active against the genotype C enzyme (Table 1), indicating that hydroxylated tropolones can inhibit the RNaseH enzymes from multiple HBV genotypes. Five of these compounds had somewhat different efficacies against the two genotypes in primary screening, suggesting a potential for the HBV genotype to affect compound efficacy. However, the small magnitude of these differences implies that any potential genotype specificity is unlikely to present an insurmountable hurdle during drug development.
Two major observations regarding the SAR for the tropolones against HBV RNaseH can be drawn from the biochemical studies (Fig. 5). First, the α-hydroxytropolone moiety is needed for strong inhibition (compare compound 46 to compounds 47 and 48). The four most active inhibitors (compounds 46, 106, 107, and 110) and five of the six less efficient inhibitors (compounds 108, 109, 112, 113, and 120) are all α-hydroxytropolones. Almost every other free tropolone tested, including the close analogs of compound 46 (compounds 47 and 48), was inactive. Interestingly, two of the four benzoylated tropolones, compounds 62 and 51, showed minor activity, and broader follow-up of this class of molecules may be warranted. This need for three adjacent Lewis basic moieties on the tropolone ring for efficient inhibition of RNaseH activity is consistent with mechanistic studies indicating that the α-hydroxytropolones bind to the active site and sequester the divalent cations via interactions with the Lewis basic moieties (21–25). Most reported HIV RNaseH inhibitors from other chemical classes also act by the same mechanism (40–43), but compounds that inhibit HIV RNaseH by altering its conformation or its interaction with nucleic acids have also been reported (44, 45).
FIG 5.
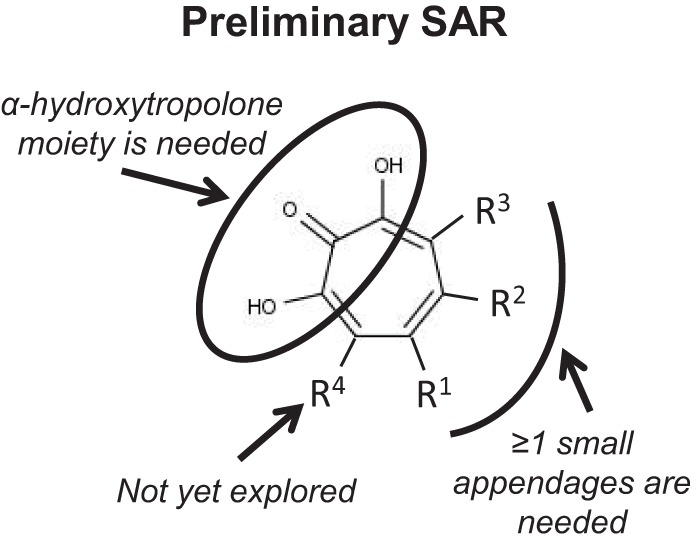
Preliminary SAR for hydroxylated tropolones against HBV RNaseH.
Second, there appears to be a size limit for substitutions on the hydroxytropolone ring above which the compounds fail to inhibit RNaseH, although the lack of activity of the parent α-hydroxytropolone (compound 172) demonstrates that some substitution is necessary. The most active α-hydroxytropolone was the least-substituted molecule, compound 46, which has a single isopropyl group, and the other most active inhibitors (compounds 106, 107, and 110) all have similarly small appendages on the hydroxytropolone ring (methyl ester, methyl ketone, methyl, and chloromethyl). As these appendages lengthen, however, the activity diminishes. For example, compound 108 differs from compounds 106 and 107 only at R1, where it has a larger methoxymethylene group, and this change makes it inactive. The trend continues with compounds 109, 143, 120, 111, and 118, which differ from compound 110 only at R2, where they have an ethyl ester, an isopropyl ketone, a cyclohexyl ketone, a phenyl ketone, and a biphenyl ketone, respectively. An ethyl ester and an isopropyl ketone are only moderately larger than the methyl ketone, and in this case the activities of compounds 109 and 143 decrease but are not ablated. On the other hand, the biphenyl ketone is the longest of the series, and compound 118 is inactive. The phenyl ketone and cyclohexyl ketone are similar in size and are each considerably longer then the methyl ketone. Interestingly, while the phenyl ketone-containing compound 111 is inactive, the cyclohexyl ketone-containing compound 120 retains minor activity. This could be due to the relative flexibility of the cyclohexane ring to adopt chair-like conformations that would shorten the side chain. Similarly, other larger molecules, such as manicol (compound 56) and its derivatives (compounds 92 to 105), lacked activity. This is in contrast to their enzymatic inhibition of HIV RNaseH, where both compounds 46 and 56 are highly active (21). This size limitation suggests that the binding site of HBV RNaseH may be less open than that of HIV RNaseH, providing an opportunity for exploitation during inhibitor development.
Several compounds that differ from compound 110 with an aryl substitution at R2 were also tested. The smallest of these, compound 113, has a phenyl group at R2 and retained some activity. However, even a minor change to this structure, such as introducing a halogen (compounds 114, 119, and 144), resulted in no inhibition (see also compound 115 and compounds 145 to 147). The only close derivative of compound 113 with activity was compound 112, which has a nitroaryl at R2. It is unclear why this substitution would give greater activity, but it contains the most electronically poor aryl group of the series. This suggests that the favorable interactions between the electronically poor aryl group of the ligand and the electronically rich aromatic residues in the binding cavity may offset any negative effects, as we have seen in other projects (46).
Overall, the relationship between the longer appendage length and lack of activity at R1 and R2 suggests unfavorable interactions due to steric strain either within the binding site or perhaps with the substrate. It is unclear whether similar effects would be seen with substitution at the R3 position. However, the inactivity of compound 117, which differs from the moderately active compound 109 by only an added methyl group at R3, implies that substantial changes in activity will be observed.
Eighteen of the 35 compounds that were counterscreened against human RNaseH1 inhibited the enzyme, including 11 of the 13 inhibitors of HBV RNaseH. Therefore, the potential exists for cross-inhibition of the human enzyme that should not be overlooked during drug development. However, differences were observed in the sensitivity profiles for HBV and human RNaseHs, implying that selectivity is achievable. For example, compound 46 was 10- to 20-fold more active against HBV RNaseH than against the human enzyme and compound 106 was almost 2-fold more active against the HBV enzyme (Table 1). Furthermore, the human enzyme was modestly sensitive to compounds with substituents attached to one of the two oxygens on the tropolone ring (compounds 59 and 61 to 63), whereas the HBV enzyme was not, and two manicol derivatives (compounds 96 and 98) which were inactive against HBV RNaseH had modest activity against human RNaseH1.
All five of the HBV RNaseH inhibitors that were active at ≤20 μM against HBV RNaseH in biochemical assays (compounds 46, 106, 110, 112, and 113) also preferentially suppressed accumulation of the viral plus-polarity DNA strand in the replication assays. Each compound had an EC50 of <5 μM, with compound 110 having an EC50 of 0.34 μM. The CC50 values for these compounds were 25 to 79 μM by MTT assays (Table 2), leading to TI values from 14 for compound 106 to 94 for 110. Together, these observations indicate that HBV replication was inhibited by these five compounds primarily via suppression of the viral RNaseH activity.
Some of the cytotoxicity (measured as a loss of mitochondrial function in the MTT assays) may be due to inhibition of human RNaseH1 because RNaseH contributes to mitochondrial DNA replication and pre-rRNA processing (47–49). However, the cytotoxicity did not correlate perfectly with inhibition of RNaseH1 (Tables 1 and 2). Compound 56 was inactive against human RNaseH1 yet had a CC50 of 35 μM, whereas compounds 106 and 110 had CC50 values similar to that of compound 56 but inhibited human RNaseH1 well at 10 μM. This discord implies that inhibition of human RNaseH1 is not the sole source of toxicity in our assays; this possibility is consistent with reports of direct toxicity against isolated mitochondria for some troponoids (50).
In all cases for which both EC50s and IC50s were determined, the EC50 was from ∼6-fold (for compound 46) to 102-fold (compound 110) lower. This is reminiscent of the lower EC50 than IC50 that we recently observed with an N-hydroxyisoquinolinedione (4.2 versus 28 μM) (16). Some possible explanations for this difference include cellular retention of the compounds, enzymatic conversion to more active derivatives, or greater efficacy against RNaseH in the context of the native enzyme than the recombinant form used in the biochemical studies. Given that this greater efficacy in cells has been observed with seven compounds from two different chemical classes, we favor the possibility that this reflects differences between the native and recombinant forms of RNaseH. However, the good correlation between inhibition at 20 μM in the biochemical assays and activity against replication in culture (Tables 1 and 2) indicates that the biochemical screening assay provides a valuable screening tool with respect to viral replication in the context of simultaneous cytotoxicity testing.
Our studies indicate that α-hydroxytropolones are promising candidates for development as novel anti-HBV RNaseH drugs. They provide the initial guidance for chemical optimization, and they highlight the particular need for care to exploit differences between HBV RNaseH and human RNaseH1 and to address mitochondrial toxicity to avoid unacceptable side effects during the long treatment period envisioned for curative HBV therapy. RNaseH inhibitors may work additively or synergistically with the nucleos(t)ide analogs because the two classes of inhibitors target physically distinct enzyme active sites (51), but this remains to be determined. If so, RNaseH inhibitors would be good candidates for use in the multidrug regimens that will be needed to suppress viral replication far enough to block HBV genomic maintenance in chronically infected individuals (11, 52, 53).
Supplementary Material
ACKNOWLEDGMENTS
We thank Aisha Uraizee, Kunjan Patel, Deborah Roby, and Monideepa Sengupta for technical assistance. We thank John Beutler from the National Cancer Institute for facilitating collaboration among J.T., S.L.G., and R.M.
The work at Saint Louis University was supported by Saint Louis University funds from the School of Medicine and the Cancer Center and by grants from the Friends of the Saint Louis University Liver Center, NIH/National Center for Advancing Translational Sciences (NCATS) (UL1 TR000448), and R01 AI104494 to J.T. G.L. was partially supported by the Henan Province, China, medical technology fund. R.M. was supported by NIH grant SC2 GM09959, and S.L.G. was funded by the Intramural Research Program of the National Cancer Institute, National Institutes of Health, Department of Health and Human Services.
Potential applications for some of these compounds against HBV are covered by U.S. patent application 13/072201 (pending).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.04617-14.
REFERENCES
- 1.Lavanchy D. 2004. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J Viral Hepat 11:97–107. doi: 10.1046/j.1365-2893.2003.00487.x. [DOI] [PubMed] [Google Scholar]
- 2.Shepard CW, Simard EP, Finelli L, Fiore AE, Bell BP. 2006. Hepatitis B virus infection: epidemiology and vaccination. Epidemiol Rev 28:112–125. doi: 10.1093/epirev/mxj009. [DOI] [PubMed] [Google Scholar]
- 3.Ganem D, Prince AM. 2004. Hepatitis B virus infection—natural history and clinical consequences. N Engl J Med 350:1118–1129. doi: 10.1056/NEJMra031087. [DOI] [PubMed] [Google Scholar]
- 4.Fattovich G, Pantalena M, Zagni I, Realdi G, Schalm SW, Christensen E, European Concerted Action on Viral Hepatitis (EUROHEP) . 2002. Effect of hepatitis B and C virus infections on the natural history of compensated cirrhosis: a cohort study of 297 patients. Am J Gastroenterol 97:2886–2895. doi: 10.1111/j.1572-0241.2002.07057.x. [DOI] [PubMed] [Google Scholar]
- 5.Sorrell MF, Belongia EA, Costa J, Gareen IF, Grem JL, Inadomi JM, Kern ER, McHugh JA, Petersen GM, Rein MF, Strader DB, Trotter HT. 2009. National Institutes of Health Consensus Development Conference Statement: management of hepatitis B. Ann Intern Med 150:104–110. doi: 10.7326/0003-4819-150-2-200901200-00100. [DOI] [PubMed] [Google Scholar]
- 6.Lok AS, McMahon BJ. 2009. Chronic hepatitis B: update 2009. Hepatology 50:661–662. doi: 10.1002/hep.23190. [DOI] [PubMed] [Google Scholar]
- 7.Grimm D, Thimme R, Blum HE. 2011. HBV life cycle and novel drug targets. Hepatol Int 5:644–653. doi: 10.1007/s12072-011-9261-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Bömmel F, De Man RA, Wedemeyer H, Deterding K, Petersen J, Buggisch P, Erhardt A, Huppe D, Stein K, Trojan J, Sarrazin C, Bocher WO, Spengler U, Wasmuth HE, Reinders JG, Moller B, Rhode P, Feucht HH, Wiedenmann B, Berg T. 2010. Long-term efficacy of tenofovir monotherapy for hepatitis B virus-monoinfected patients after failure of nucleoside/nucleotide analogues. Hepatology 51:73–80. doi: 10.1002/hep.23246. [DOI] [PubMed] [Google Scholar]
- 9.Berg T, Zoulim F, Moeller B, Trinh H, Marcellin P, Chan S, Kitrinos KM, Dinh P, Flaherty JF Jr, McHutchison JG, Manns M. 2014. Long-term efficacy and safety of emtricitabine plus tenofovir DF vs. tenofovir DF monotherapy in adefovir-experienced chronic hepatitis B patients. J Hepatol 60:715–722. doi: 10.1016/j.jhep.2013.11.024. [DOI] [PubMed] [Google Scholar]
- 10.Iwamoto M, Watashi K, Tsukuda S, Aly HH, Fukasawa M, Fujimoto A, Suzuki R, Aizaki H, Ito T, Koiwai O, Kusuhara H, Wakita T. 2014. Evaluation and identification of hepatitis B virus entry inhibitors using HepG2 cells overexpressing a membrane transporter NTCP. Biochem Biophys Res Commun 443:808–813. doi: 10.1016/j.bbrc.2013.12.052. [DOI] [PubMed] [Google Scholar]
- 11.Tavis JE, Gehring AJ, Hu Y. 2013. How further suppression of virus replication could improve current HBV treatment. Expert Rev Anti Infect Ther 11:755–757. doi: 10.1586/14787210.2013.814846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strasser SI. 2012. Drugs in development for the treatment of chronic hepatitis B. Curr Hepatitis Rep 11:111–118. doi: 10.1007/s11901-012-0131-9. [DOI] [Google Scholar]
- 13.De Clercq E, Ferir G, Kaptein S, Neyts J. 2010. Antiviral treatment of chronic hepatitis B virus (HBV) infections. Viruses 2:1279–1305. doi: 10.3390/v2061279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu Y, Cheng X, Cao F, Huang A, Tavis JE. 2013. Beta-thujaplicinol inhibits hepatitis B virus replication by blocking the viral ribonuclease H activity. Antiviral Res 99:221–229. doi: 10.1016/j.antiviral.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 15.Tavis JE, Cheng X, Hu Y, Totten M, Cao F, Michailidis E, Aurora R, Meyers MJ, Jacobsen EJ, Parniak MA, Sarafianos SG. 2013. The hepatitis B virus ribonuclease H is sensitive to inhibitors of the human immunodeficiency virus ribonuclease H and integrase enzymes. PLoS Pathog 9:e1003125. doi: 10.1371/journal.ppat.1003125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cai CW, Lomonosova E, Moran EA, Cheng X, Patel KB, Bailly F, Cotelle P, Meyers MJ, Tavis JE. 2014. Hepatitis B virus replication is blocked by a 2-hydroxyisoquinoline-1,3(2H,4H)-dione (HID) inhibitor of the viral ribonuclease H activity. Antiviral Res 108:48–55. doi: 10.1016/j.antiviral.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nowotny M. 2009. Retroviral integrase superfamily: the structural perspective. EMBO Rep 10:144–151. doi: 10.1038/embor.2008.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang W, Steitz TA. 1995. Recombining the structures of HIV integrase, RuvC and RNase H. Structure 3:131–134. doi: 10.1016/S0969-2126(01)00142-3. [DOI] [PubMed] [Google Scholar]
- 19.Sugawara K, Ohbayashi M, Shimizu K, Hatori M, Kamei H, Konishi M, Oki T, Kawaguchi H. 1988. BMY-28438 (3,7-dihydroxytropolone), a new antitumor antibiotic active against B16 melanoma. I. Production, isolation, structure and biological activity. J Antibiot (Tokyo) 41:862–868. [DOI] [PubMed] [Google Scholar]
- 20.Meck C, D'Erasmo MP, Hirsch DR, Murelli RP. 2014. The biology and synthesis of alpha-hydroxytropolones. Medchemcomm 5:842–852. doi: 10.1039/c4md00055b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chung S, Himmel DM, Jiang JK, Wojtak K, Bauman JD, Rausch JW, Wilson JA, Beutler JA, Thomas CJ, Arnold E, Le Grice SF. 2011. Synthesis, activity, and structural analysis of novel alpha-hydroxytropolone inhibitors of human immunodeficiency virus reverse transcriptase-associated ribonuclease H. J Med Chem 54:4462–4473. doi: 10.1021/jm2000757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Didierjean J, Isel C, Querre F, Mouscadet JF, Aubertin AM, Valnot JY, Piettre SR, Marquet R. 2005. Inhibition of human immunodeficiency virus type 1 reverse transcriptase, RNase H, and integrase activities by hydroxytropolones. Antimicrob Agents Chemother 49:4884–4894. doi: 10.1128/AAC.49.12.4884-4894.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beilhartz GL, Wendeler M, Baichoo N, Rausch J, Le Grice S, Gotte M. 2009. HIV-1 reverse transcriptase can simultaneously engage its DNA/RNA substrate at both DNA polymerase and RNase H active sites: implications for RNase H inhibition. J Mol Biol 388:462–474. doi: 10.1016/j.jmb.2009.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Budihas SR, Gorshkova I, Gaidamakov S, Wamiru A, Bona MK, Parniak MA, Crouch RJ, McMahon JB, Beutler JA, Le Grice SF. 2005. Selective inhibition of HIV-1 reverse transcriptase-associated ribonuclease H activity by hydroxylated tropolones. Nucleic Acids Res 33:1249–1256. doi: 10.1093/nar/gki268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Himmel DM, Maegley KA, Pauly TA, Bauman JD, Das K, Dharia C, Clark AD Jr, Ryan K, Hickey MJ, Love RA, Hughes SH, Bergqvist S, Arnold E. 2009. Structure of HIV-1 reverse transcriptase with the inhibitor beta-thujaplicinol bound at the RNase H active site. Structure 17:1625–1635. doi: 10.1016/j.str.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tavis JE, Wang H, Tollefson AE, Ying B, Korom M, Cheng X, Cao F, Davis KL, Wold WS, Morrison LA. 2014. Inhibitors of nucleotidyl transferase superfamily enzymes suppress herpes simplex virus replication. Antimicrob Agents Chemother 58:7451–7461. doi: 10.1128/AAC.03875-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iwatsuki M, Takada S, Mori M, Ishiyama A, Namatame M, Nishihara-Tsukashima A, Nonaka K, Masuma R, Otoguro K, Shiomi K, Omura S. 2011. In vitro and in vivo antimalarial activity of puberulic acid and its new analogs, viticolins A-C, produced by Penicillium sp. FKI-4410. J Antibiot (Tokyo) 64:183–188. doi: 10.1038/ja.2010.124. [DOI] [PubMed] [Google Scholar]
- 28.Allen NE, Alborn WE Jr, Hobbs JN Jr, Kirst HA. 1982. 7-Hydroxytropolone: an inhibitor of aminoglycoside-2″-O-adenylyltransferase. Antimicrob Agents Chemother 22:824–831. doi: 10.1128/AAC.22.5.824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Piettre SR, Ganzhorn A, Hoflack J, Islam K, Hornsperger J-M. 1997. Alpha-hydroxytropolones: a new class of potent inhibitors of inositol monophosphatase and other bimetallic enzymes. J Am Chem Soc 119:3201–3204. doi: 10.1021/ja9634278. [DOI] [Google Scholar]
- 30.Semenova EA, Johnson AA, Marchand C, Davis DA, Yarchoan R, Pommier Y. 2006. Preferential inhibition of the magnesium-dependent strand transfer reaction of HIV-1 integrase by alpha-hydroxytropolones. Mol Pharmacol 69:1454–1460. doi: 10.1124/mol.105.020321. [DOI] [PubMed] [Google Scholar]
- 31.Martin SF, Follows BC, Hergenrother PJ, Franklin CL. 2000. A novel class of zinc-binding inhibitors for the phosphatidylcholine-preferring phospholipase C from Bacillus cereus. J Org Chem 65:4509–4514. doi: 10.1021/jo9915731. [DOI] [PubMed] [Google Scholar]
- 32.Takeshita H, Mori A, Kusaba T. 1986. An improved synthesis of 2,7-dihydroxytropone (2-hydroxytrolone). Synthesis 7:578–579. [Google Scholar]
- 33.Meck C, Mohd N, Murelli RP. 2012. An oxidopyrylium cyclization/ring-opening route to polysubstituted alpha-hydroxytropolones. Org Lett 14:5988–5991. doi: 10.1021/ol302892g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Williams YD, Meck C, Mohd N, Murelli RP. 2013. Triflic acid-mediated rearrangements of 3-methoxy-8-oxabicyclo[3.2.1]octa-3,6-dien-2-ones: synthesis of methoxytropolones and furans. J Org Chem 78:11707–11713. doi: 10.1021/jo401617r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hirsch DR, Cox G, D'Erasmo MP, Shakya T, Meck C, Mohd N, Wright GD, Murelli RP. 2014. Inhibition of the ANT(2″)-Ia resistance enzyme and rescue of aminoglycoside antibiotic activity by synthetic alpha-hydroxytropolones. Bioorg Med Chem Lett 24:4943–4947. doi: 10.1016/j.bmcl.2014.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guo H, Jiang D, Zhou T, Cuconati A, Block TM, Guo JT. 2007. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J Virol 81:12472–12484. doi: 10.1128/JVI.01123-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gerelsaikhan T, Tavis JE, Bruss V. 1996. Hepatitis B virus nucleocapsid envelopment does not occur without genomic DNA synthesis. J Virol 70:4269–4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li MD, Bronson DL, Lemke TD, Faras AJ. 1995. Phylogenetic analyses of 55 retroelements on the basis of the nucleotide and product amino acid sequences of the pol gene. Mol Biol Evol 12:657–670. [DOI] [PubMed] [Google Scholar]
- 39.Corona A, Schneider A, Schweimer K, Rosch P, Wohrl BM, Tramontano E. 2014. Inhibition of foamy virus reverse transcriptase by human immunodeficiency virus type 1 RNase H inhibitors. Antimicrob Agents Chemother 58:4086–4093. doi: 10.1128/AAC.00056-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fuji H, Urano E, Futahashi Y, Hamatake M, Tatsumi J, Hoshino T, Morikawa Y, Yamamoto N, Komano J. 2009. Derivatives of 5-nitro-furan-2-carboxylic acid carbamoylmethyl ester inhibit RNase H activity associated with HIV-1 reverse transcriptase. J Med Chem 52:1380–1387. doi: 10.1021/jm801071m. [DOI] [PubMed] [Google Scholar]
- 41.Su HP, Yan Y, Prasad GS, Smith RF, Daniels CL, Abeywickrema PD, Reid JC, Loughran HM, Kornienko M, Sharma S, Grobler JA, Xu B, Sardana V, Allison TJ, Williams PD, Darke PL, Hazuda DJ, Munshi S. 2010. Structural basis for the inhibition of RNase H activity of HIV-1 reverse transcriptase by RNase H active site-directed inhibitors. J Virol 84:7625–7633. doi: 10.1128/JVI.00353-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Billamboz M, Bailly F, Lion C, Touati N, Vezin H, Calmels C, Andreola ML, Christ F, Debyser Z, Cotelle P. 2011. Magnesium chelating 2-hydroxyisoquinoline-1,3(2H,4H)-diones, as inhibitors of HIV-1 integrase and/or the HIV-1 reverse transcriptase ribonuclease H domain: discovery of a novel selective inhibitor of the ribonuclease H function. J Med Chem 54:1812–1824. doi: 10.1021/jm1014692. [DOI] [PubMed] [Google Scholar]
- 43.Kirschberg TA, Balakrishnan M, Squires NH, Barnes T, Brendza KM, Chen X, Eisenberg EJ, Jin W, Kutty N, Leavitt S, Liclican A, Liu Q, Liu X, Mak J, Perry JK, Wang M, Watkins WJ, Lansdon EB. 2009. RNase H active site inhibitors of human immunodeficiency virus type 1 reverse transcriptase: design, biochemical activity, and structural information. J Med Chem 52:5781–5784. doi: 10.1021/jm900597q. [DOI] [PubMed] [Google Scholar]
- 44.Wendeler M, Lee HF, Bermingham A, Miller JT, Chertov O, Bona MK, Baichoo NS, Ehteshami M, Beutler J, O'Keefe BR, Gotte M, Kvaratskhelia M, Le GS. 2008. Vinylogous ureas as a novel class of inhibitors of reverse transcriptase-associated ribonuclease H activity. ACS Chem Biol 3:635–644. doi: 10.1021/cb8001039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Himmel DM, Sarafianos SG, Dharmasena S, Hossain MM, Coy-Simandle K, Ilina T, Clark AD Jr, Knight JL, Julias JG, Clark PK, Krogh-Jespersen K, Levy RM, Hughes SH, Parniak MA, Arnold E. 2006. HIV-1 reverse transcriptase structure with RNase H inhibitor dihydroxy benzoyl naphthyl hydrazone bound at a novel site. ACS Chem Biol 1:702–712. doi: 10.1021/cb600303y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang AX, Murelli RP, Barinka C, Michel J, Cocleaza A, Jorgensen WL, Lubkowski J, Spiegel DA. 2010. A remote arene-binding site on prostate specific membrane antigen revealed by antibody-recruiting small molecules. J Am Chem Soc 132:12711–12716. doi: 10.1021/ja104591m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cerritelli SM, Frolova EG, Feng C, Grinberg A, Love PE, Crouch RJ. 2003. Failure to produce mitochondrial DNA results in embryonic lethality in Rnaseh1 null mice. Mol Cell 11:807–815. doi: 10.1016/S1097-2765(03)00088-1. [DOI] [PubMed] [Google Scholar]
- 48.Ruhanen H, Ushakov K, Yasukawa T. 2011. Involvement of DNA ligase III and ribonuclease H1 in mitochondrial DNA replication in cultured human cells. Biochim Biophys Acta 1813:2000–2007. doi: 10.1016/j.bbamcr.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu H, Sun H, Liang X, Lima WF, Crooke ST. 2013. Human RNase H1 is associated with protein P32 and is involved in mitochondrial pre-rRNA processing. PLoS One 8:e71006. doi: 10.1371/journal.pone.0071006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nakagawa Y, Tayama K. 1998. Mechanism of mitochondrial dysfunction and cytotoxicity induced by tropolones in isolated rat hepatocytes. Chem Biol Interact 116:45–60. doi: 10.1016/S0009-2797(98)00078-7. [DOI] [PubMed] [Google Scholar]
- 51.Chang LJ, Hirsch RC, Ganem D, Varmus HE. 1990. Effects of insertional and point mutations on the functions of the duck hepatitis B virus polymerase. J Virol 64:5553–5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gehring A, Bertoletti A, Tavis JE. 2014. Host factor-targeted hepatitis B virus therapies. Intervirology 57:158–162. doi: 10.1159/000360938. [DOI] [PubMed] [Google Scholar]
- 53.Block TM, Gish R, Guo H, Mehta A, Cuconati A, Thomas London W, Guo JT. 2013. Chronic hepatitis B: what should be the goal for new therapies? Antiviral Res 98:27–34. doi: 10.1016/j.antiviral.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.