

Abstract
Oritavancin is a semisynthetic derivative of the glycopeptide antibiotic chloroeremomycin with activity against Gram-positive pathogens, including vancomycin-resistant staphylococci and enterococci. Compared to vancomycin, oritavancin is characterized by the presence of two additional residues, a hydrophobic 4′-chlorobiphenyl methyl moiety and a 4-epi-vancosamine substituent, which is also present in chloroeremomycin. Here, we show that oritavancin and its des-N-methylleucyl variant (des-oritavancin) effectively inhibit lipid I- and lipid II-consuming peptidoglycan biosynthesis reactions in vitro. In contrast to that for vancomycin, the binding affinity of oritavancin to the cell wall precursor lipid II appears to involve, in addition to the d-Ala-d-Ala terminus, other species-specific binding sites of the lipid II molecule, i.e., the crossbridge and d-isoglutamine in position 2 of the lipid II stem peptide, both characteristic for a number of Gram-positive pathogens, including staphylococci and enterococci. Using purified lipid II and modified lipid II variants, we studied the impact of these modifications on the binding of oritavancin and compared it to those of vancomycin, chloroeremomycin, and des-oritavancin. Analysis of the binding parameters revealed that additional intramolecular interactions of oritavancin with the peptidoglycan precursor appear to compensate for the loss of a crucial hydrogen bond in vancomycin-resistant strains, resulting in enhanced binding affinity. Augmenting previous findings, we show that amidation of the lipid II stem peptide predominantly accounts for the increased binding of oritavancin to the modified intermediates ending in d-Ala-d-Lac. Corroborating our conclusions, we further provide biochemical evidence for the phenomenon of the antagonistic effects of mecA and vanA resistance determinants in Staphylococcus aureus, thus partially explaining the low frequency of methicillin-resistant S. aureus (MRSA) acquiring high-level vancomycin resistance.
INTRODUCTION
Glycopeptide antibiotics constitute a diverse group of natural products (1). The largest and most prominent class among the glycopeptide antibiotics is represented by the vancomycin group. The members of this class are characterized by an aglycone core consisting of seven amino acids forming a trimacrocyclic or tetramacrocyclic structure, synthesized by nonribosomal peptide synthetases. Additional structural heterogeneity within this class arises from the sugar moieties attached to the heptapeptide core, which vary in number, position, and chemical structure.
The glycopeptide antibiotic vancomycin was first isolated and described in the early 1950s (2). As a consequence of the worldwide emergence of methicillin-resistant Staphylococcus aureus (MRSA) in the 1970s, vancomycin was increasingly considered the last resort for treatment of MRSA infections. For more than a decade, vancomycin was used successfully without a significant rise in resistance, until enterococcal strains with acquired resistance to vancomycin emerged in 1986 (3–5). Today, vancomycin-resistant enterococci (VRE) represent >30% of all clinical enterococcal isolates in the United States (6). In Europe, surveillance data show large variabilities between various countries with VRE ranging from <2% (Finland and Holland) to >25% (Ireland, Greece, Portugal, and United Kingdom) (7). Although high-level resistance to vancomycin in staphylococci is rare, the increased occurrence of vancomycin-intermediate S. aureus (VISA) and heterogeneous VISA (hVISA) in the hospital setting is increasingly limiting treatment options.
The mode of action of vancomycin and other glycopeptide antibiotics relies on binding to the d-Ala-d-Ala terminus of the peptidoglycan (PG) cell wall precursor lipid II (8–10). Lipid II represents the central cell wall building block of PG biosynthesis. The precursor consists of the bactoprenol carrier, which is linked to the disaccharide unit N-acetylmuramyl-pentapeptide-N-acetylglucosamine via a pyrophosphate bridge (10). Binding of vancomycin to lipid II, which is mediated by formation of five hydrogen bonds between the glycopeptide and the d-Ala-d-Ala terminus of lipid II, locks the cell wall precursor in a stable complex, thereby blocking the entire peptidoglycan synthesis cycle. More precisely, binding to lipid II sterically shields the cell wall precursor substrate from the penicillin binding proteins (PBPs) that catalyze transglycosylation and transpeptidation (9, 11). In VRE and vancomycin-resistant S. aureus (VRSA), the mechanism of resistance results from an alteration of the molecular target, i.e., the replacement of the d-Ala-d-Ala terminus by d-Ala-d-Lac (VanA/VanB) or d-Ala-d-Ser (VanC). The incorporation of d-lactate into peptidoglycan precursors results in the loss of one of the five hydrogen bonds, leading to a 1,000-fold decrease in vancomycin antibiotic activity, while decreased binding of vancomycin to the d-Ala-d-Ser termini of lipid II is due to steric hindrance (12–14). The emergence of vancomycin-resistant strains has revived the development of second-generation glycopeptides with improved activities against these pathogens (15–20). Oritavancin is the N-substituted 4′-chlorobiphenyl methyl synthetic derivative of the naturally occurring glycopeptide chloroeremomycin (Fig. 1A) (21–23). Chloroeremomycin (Fig. 1B) itself belongs to the eremomycin class, which differs from vancomycin by an additional monosaccharide moiety (4-epi-vancosamine) attached to the ring 6-amino acid residue and the substitution of the existing vancosamine at position 4 by 4-epi-vancosamine (24). In contrast to chloroeremomycin, oritavancin is rapidly bactericidal (25, 26) and displays antibacterial activity against MRSA, VISA, VRSA, daptomycin-nonsusceptible S. aureus, and VRE (22, 27, 28). Oritavancin was approved in 2014 for the treatment of complicated skin and skin structure infections caused by Gram-positive pathogens, including MRSA (29). The reasons for the enhanced antimicrobial activity of oritavancin even against vancomycin-resistant strains and its detailed mode of action are still a matter of debate. Like other glycopeptides, oritavancin inhibits the late steps of peptidoglycan biosynthesis (transglycosylation) by binding to the d-Ala-d-Ala terminus of lipid II. Oritavancin was proposed to additionally promote membrane depolarization and permeabilization, leading to rapid cell death (30, 31). It has further been suggested that the hydrophobic substituent promotes dimerization of the antibiotic, resulting in increased binding affinity (32, 33). Moreover, membrane anchoring is thought to direct the antibiotic in close proximity to its membrane-standing target and to further stabilize the glycopeptide-lipid II interaction (34). The additional carbohydrate moiety present in oritavancin and chloroeremomycin has been reported to directly interfere with penicillin binding proteins, resulting in inhibition of peptidoglycan biosynthesis independent of d-Ala-d-Ala binding (35). Recent studies suggested that binding of oritavancin involves multiple interactions with its molecular target lipid II. Solid-state nuclear magnetic resonance (NMR) and computational modeling predicted secondary interactions of the 4′-chlorobiphenyl methyl side chain with the S. aureus pentaglycine crossbridge and the d-aspartate/d-asparagine (d-Asx) crossbridge in Enterococcus faecium (36–40). To better understand the subtleties of the mechanism of action of oritavancin, we characterized the binding to its target lipid II in vivo and in vitro using purified lipid II variants. A comprehensive comparison of oritavancin-binding parameters to variants of the natural occurring cell wall building block and its depsipeptide counterparts, ending in d-Ala-d-Lac, revealed that amidation of the lipid II stem peptide accounts for increased binding affinity of oritavancin in VRSA, rather than interaction with the pentaglycine crossbridge, which is almost absent in these strains (41). Corroborating these muropeptide analyses of VRSA (41), we provide the first biochemical evidence that lipid II terminating in d-Ala-d-Lac is a poor substrate for the Fem peptidyl transferases of S. aureus.
FIG 1.

Chemical structure of oritavancin (A), chloroeremomycin (B), and the glycopeptide backbone (C). The hydrophobic 4′-chlorobiphenyl methyl moiety and the 4-epi-vancosamine substituent are highlighted. The leucyl residue missing in des-N-methylleucyl-oritavancin is marked by a dashed circle. Hydrogen bonds are marked by dashed lines.
MATERIALS AND METHODS
Susceptibility testing.
MICs were determined by standard broth microdilution methods (CLSI) in a polypropylene microtiter plate using cation-adjusted Mueller-Hinton broth (MHB) (Oxoid) for S. aureus ATCC 29213 and brain heart infusion broth (BHIB) (Oxoid) for E. faecium BM4147. Bacteria in the exponential growth phase were diluted to give a final inoculum of 105 CFU. The MICs were read after 16 h at 37°C. Oritavancin and des-oritavancin (23, 42) were kindly provided by The Medicines Company and dissolved in 0.002% Tween 80 (vol/vol). A final concentration of 0.002% Tween 80 (vol/vol) was present in all dilution steps and assays performed in this study.
Analysis of the cytoplasmic peptidoglycan nucleotide precursor pool.
E. faecium BM4147 was grown in BHI broth (0.002% Tween 80) to an optical density at 600 nm (OD600) of 0.5 and supplemented with 80 mg/liter vancomycin to induce vanA expression. After 10 min, oritavancin or des-N-methylleucyl-oritavancin (des-oritavancin) was added at 10× MIC and incubated for 60 min. Cells were harvested and extracted with boiling water. The suspension was then centrifuged, and the supernatant was lyophilized. UDP-linked cell wall precursors were analyzed by high-performance liquid chromatography (HPLC) (43), and the corresponding fractions were confirmed by matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometry.
Cloning, overexpression, and purification of Enterococcus faecium Asl.
E. faecium BM4147 asl was amplified using forward and reverse primers (Asl_for 5′-TAGGCTAGCATGAACAGTATTGAAAATG-3′ and Asl_rev 5′-TTACTCGAGGCCTTCTTTCACATGAAAATA-3′) and cloned into a pET21b vector (Novagen) using NdeI and XhoI restriction sites to generate C-terminal His6 fusion proteins. Escherichia coli BL21(DE3) (Promega) cells transformed with the appropriate recombinant plasmid were grown in LB medium (50 μg/ml ampicillin) at 37°C. At an OD600 of 0.6, isopropyl-β-d-thiogalactopyranoside (IPTG) was added at a concentration of 0.75 mM to induce expression of the recombinant proteins. After 2 h, cells were harvested and resuspended in lysis buffer (50 mM Tris-HCl [pH 7.5], 300 mM NaCl, 10 mM imidazole). Aliquots of 200 mg/ml lysozyme, 100 mg/ml DNase, and 10 mg/ml RNase were added; cells were incubated for 30 min on ice and sonicated. Cell debris was spun down, and purification was performed as described for the MurG enzyme (44).
Analysis of enzyme activities.
MurG activity assays were performed in a final volume of 30 μl containing 2.5 nmol purified lipid I, 25 nmol UDP-GlcNAc, or [14C]UDP-GlcNAc in 200 mM Tris-HCl, 5.7 mM MgCl2 [pH 7.5], and 0.8% Triton X-100 in the presence of 0.45 μg of purified MurG-His6 enzyme. Reaction mixtures were incubated for 30 min at 30°C (44). The assay for synthesis of lipid II-Gly1 catalyzed by FemX and lipid II-Gly3 and lipid II-Gly5 catalyzed by FemXA and FemXAB, respectively, was performed as described previously without any modifications (45); 5 nmol of lipid II or lipid II-d-Lac was used as a substrate. The enzymatic activity of PBP2 was determined by incubating 2.5 nmol [14C]lipid II in 100 mM morpholineethanesulfonic acid (MES), 10 mM MgCl2 [pH 5.5], and 0.1% Triton X-100 in a total volume of 50 μl. The reaction was initiated by the addition of 7.5 μg PBP2-His6 and incubated for 1.5 h at 30°C (44). The Asl-catalyzed addition of d-[14C]aspartate to lipid II was performed in a total volume of 50 μl containing 2.5 nmol lipid II, 25 nmol d-[14C]aspartate in 120 mM Tris-HCl, 40 mM MgCl2 [pH 8], 8 mM ATP, 0.4% Triton X-100, and 2 μg Asl-His6 (37, 38). The synthesized lipid intermediates were extracted from the reaction mixtures with n-butanol-pyridine acetate (pH 4.2) (2:1, vol/vol) and analyzed by thin-layer chromatography (TLC) as described earlier (45). The analysis of lipid II conversion catalyzed by PBP2 was carried out by applying reaction mixtures directly onto TLC plates developed in solvent B (butanol-acetic acid-water-pyridine [15:3:12:10, vol/vol/vol/vol]). The quantitative analysis was performed by phosphorimaging. Antibiotics were added at increasing molar ratios of 0.25 to 4 with respect to the amount of lipid precursor, respectively. The lantibiotic nisin was used as a control.
In vitro synthesis and purification of lipid II variants.
Large-scale synthesis and purification of the peptidoglycan precursors lipid I, lipid II, lipid II-Gly1−5 and amidated lipid II were performed as described previously (45, 46). Lipid II-Asp was synthesized with purified lipid II and purified recombinant Asl-His6 as described above using nonradioactive d-aspartate (37, 38). Purification was performed as described for unmodified lipid II (45, 47).
For synthesis of the lipid II variant containing the depsipeptide terminus (d-Ala-d-Lac), UDP-MurNAc-depsipeptide was purified from Lactobacillus casei ATCC 393. L. casei was grown in MRS broth to an OD600 of 0.6 and incubated with 65 μg/ml of chloramphenicol for 15 min. Intracellular accumulation was achieved by incubation with bacitracin (10× MIC, 40 μg/ml) in the presence of 1.25 mM zinc for another 60 min. The assay for synthesizing lipid II-d-Lac was performed in a final volume of 60 μl using 3 μg recombinant MraY-His6 and 1.25 μg recombinant MurG-His6 by incubating the UDP-MurNAc-depsipeptide, 5 nmol C55P, and 150 nmol UDP-GlcNAc in 83.3 mM Tris-HCl (pH 7.5), 6.3 mM MgCl2 and 0.5% Triton X-100. The reaction mixtures were incubated for 4 h at 30°C. Lipid II-d-Lac was extracted from the reaction mixtures with n-butanol-pyridine acetate (pH 4.2) (2:1, vol/vol), analyzed by TLC, and purified by HPLC.
Amidated lipid II and lipid II-d-Lac, containing d-isoglutamine in position 2 of the stem peptide, were synthesized and purified as described previously (46). Lipid II or amidated lipid II was used as a substrate in a Fem peptidyl transferase assay (45). Vancomycin, chloroeremomycin, or oritavancin was added in molar ratio with respect to the lipid precursors. The reaction products were extracted with BuOH-PyrAc (2:1) and separated by TLC in solvent Rick (chloroform, methanol, water, ammonium hydroxide [88:48:10:1, vol/vol]). The detection and quantification of radiolabeled products were carried out using a Storm PhosphorImager.
Complex formation of oritavancin and des-oritavancin with purified lipid II and lipid II variants.
Complex formation of oritavancin and des-oritavancin with lipid II and lipid II variants was analyzed by incubating 2 nmol of each purified cell wall precursor with increasing concentrations of oritavancin or des-oritavancin in 50 mM Tris-HCl (pH 7.5) for 20 min at room temperature. The free precursors were extracted with n-butanol-pyridine acetate (pH 4.2) (2:1, vol/vol) and analyzed by TLC and subsequent phosphomolybdic acid (PMA) staining. The quantitative analysis of the lipid-containing precursors in the butanol phase was carried out using phosphorimaging.
Quartz crystal microbalance.
To apply this mass-sensitive biosensor technique, a LiquiLab21 quartz crystal microbalance (QCM) device (ifake.V., Germany) was employed. Changes in the oscillation frequency of quartz sensors, representing binding events at the sensor surface equipped in a flow chamber, were followed in real time and used to calculate binding kinetic constants with appropriate software. The quartz sensors were cleaned and prepared with the covalent immobilization of a hexadecanthiol monolayer as described previously (48). The supported bilayers were completed by transferring a monolayer consisting of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC)-0.1 mol% lipid intermediate onto a covalently fixed C16H33SH monolayer by the Langmuir-Blodgett technique. After equilibration of the quartz crystals under flow conditions until they reached a constant frequency, 1 μM peptide solution was added. The frequency curves allow the calculation of the kinetic binding constants as described previously (49, 50).
Statistics.
All experiments were performed at least in triplicate, and the values are expressed as the means ± standard deviations (SD).
RESULTS
Oritavancin blocks individual cell wall biosynthesis reactions in vitro.
We investigated the effect of oritavancin in various individual in vitro peptidoglycan biosynthesis assays using purified substrates and enzymes and included the lantibiotic nisin, which is known to target lipid II and other bactoprenol-bound precursors, as a control (Fig. 2A) (51). In the presence of increasing concentrations of oritavancin, inhibition of all enzymatic steps (MurG, FemX, PBP2) using lipid I or lipid II as a substrate was observed. Since the crossbridge has been proposed to be an additional binding site for oritavancin (36, 40), we further analyzed the impact of oritavancin on both pentaglycine (S. aureus) and d-aspartate (E. faecium) crossbridge formation in vitro, as catalyzed by the purified enzymes FemXAB and Asl (37, 38, 45), respectively (Fig. 2B). Compared to the control reactions in the absence of an antibiotic, increasing concentrations of oritavancin in the reaction mixtures resulted in successive decreases of the respective products. An almost complete inhibition of each reaction was observed when oritavancin was added in 2-fold molar excess with respect to the lipid intermediate (Fig. 2).
FIG 2.
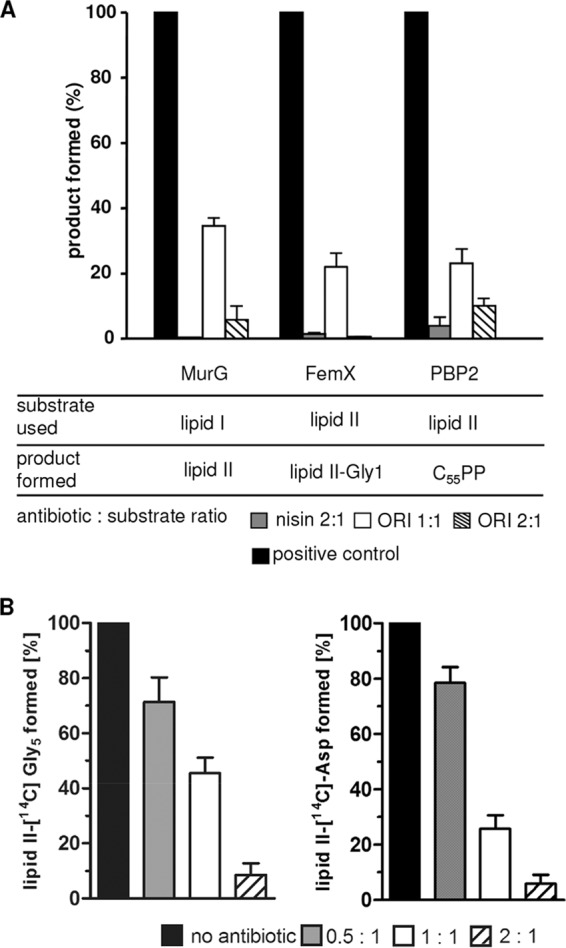
Inhibition of membrane-associated cell wall biosynthesis steps by oritavancin. (A) Synthesis and analysis of the individual biosynthesis steps were performed as described in Materials and Methods. The amount of reaction products synthesized in the absence of antibiotics was taken as 100%. The bactoprenol-containing products were analyzed by TLC. Radiolabeling was based on 3H-labeled C55P (for lipid I), [14C]GlcNAc for lipid II, and [14C]glycine for lipid II-Gly1. Antibiotics were added in molar ratios with respect to the bactoprenol-bound substrates as indicated. The lantibiotic nisin was used as a control. (B) Impact of oritavancin on the staphylococcal pentaglycine and enterococcal d-aspartate crossbridge formation. The addition of [14C]glycine by purified FemXAB-His6 (S. aureus) or the formation of the [14C]aspartate crossbridge by Asl-His6 (E. faecium) was assayed in the presence of increasing oritavancin concentrations (0.5 to 2 molar ratio with respect to lipid II). The reaction products synthesized in the absence of oritavancin were taken as the 100% level. The lipid intermediates were extracted from the reaction mixture and analyzed by TLC and subsequent phosphorimaging. The mean values from three independent experiments are shown. All experiments were performed at least in triplicate, and data are presented as means ± standard deviations (SD). ORI, oritavancin.
Influence of lipid II modifications on the binding of oritavancin.
For numerous Gram-positive pathogens, the amidation of the α-carbonyl group of d-glutamate in position 2 of the lipid II stem peptide is essential and has been shown to affect the interaction with antimicrobial compounds, such as plectasin (46). To investigate the impact of lipid II amidation on the interaction with selected glycopeptide antibiotics, either lipid II or amidated lipid II was used as a substrate in a FemX-catalyzed lipid II-Gly1 synthesis assay (Fig. 3). The glycopeptides tested (vancomycin, oritavancin, and chloroeremomycin) were added at a 1:1 molar ratio with respect to the lipid intermediate. Compared to the negative control, vancomycin inhibited the FemX-catalyzed addition of [14C]glycine to unmodified lipid II by 30%, whereas no significant inhibitory effect was observed when amidated lipid II was used as a substrate. Oritavancin, in contrast, showed a markedly increased affinity for the amidated lipid II variant, resulting in an almost complete inhibition (>95%) of the FemX-catalyzed reaction at an equimolar ratio. The inhibitory effect was reduced when unmodified lipid II was used as a substrate (62%), suggesting enhanced binding of oritavancin to amidated lipid II. Although the overall inhibitory effect was less pronounced with chloroeremomycin, enhanced binding to amidated lipid II was also observed (Fig. 3).
FIG 3.
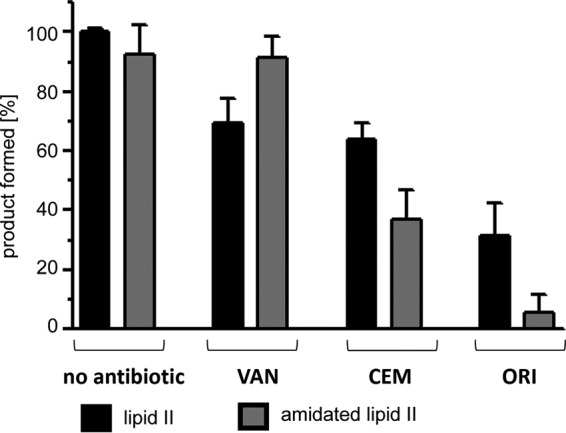
Impact of lipid II amidation on the inhibitory effect of different glycopeptide antibiotics in vitro. Purified lipid II or amidated lipid II (d-isoglutamine instead of d-glutamate in position 2 of the stem peptide) was used as a substrate in a FemX in vitro assay (see Materials und Methods). The addition of [14C]glycine to the lipid precursor in the absence and presence of vancomycin, chloroeremomycin, or oritavancin was quantified using a Storm PhosphorImager. Antibiotics were added in equimolar ratio (1:1) with respect to the lipid precursor. The mean values from three independent experiments are shown. All experiments were performed at least in triplicate, and data are presented as means ± standard deviations (SD). VAN, vancomycin; CEM, chloroeremomycin; ORI, oritavancin.
To further validate the data obtained from the in vitro assays, the binding parameters of oritavancin and vancomycin to selected lipid II variants (lipid II, lipid II-Gly5, amidated lipid II, amidated lipid II-Gly5, lipid II-d-Lac, and amidated lipid II-d-Lac) were determined using a quartz crystal microbalance (QCM) technique (48–50).
QCM is a versatile mass-sensitive biosensor system based on the piezoelectric effect of quartz crystals. QCM inverses this effect, enabling time-resolved measurements of mass changes at the sensor surface, capable of measuring subnanogram levels. Covering the quartz surface by using supported lipid bilayers allows for determination of the membrane interaction of analytes, whereas covering by embedding specific targets into the lipid bilayer allows for detection of kinetic binding constants. Consequently, this approach combines the advantage of analyzing the interaction of the antibiotic with its membrane-bound target (lipid II) embedded in a lipid bilayer, mimicking its natural membrane environment. In agreement with the observations obtained from the in vitro assays, determination of the binding parameters revealed that vancomycin binds to unmodified lipid II (equilibrium constant [KD] = 5.37 × 10−7 ± 0.41 × 10−7 M), and this was unchanged for lipid II-Gly5, while no binding to the amidated lipid II variant was detected. Oritavancin, in contrast, efficiently bound to lipid II (KD = 2.13 × 10−7 ± 0.60 × 10−7 M), and the binding affinity is further increased by 1 order of magnitude (KD = 6.64 × 10−8 ± 1.62 × 10−8 M) when the amidated lipid II variant is used for the binding approach. The association rates (Kass) of oritavancin binding to the various lipid variants are meaningful indicators of successively increased affinity of oritavancin for its target, depending on the extent of lipid II modification (lipid II < amidated lipid II < lipid II-Gly5 < amidated lipid II-Gly5) (Fig. 4). A 3-fold increase in the association rate of oritavancin was observed for the fully modified cell wall precursor (amidated lipid II-Gly5) (Kass = 2,896.0 ± 994.0 M−1 s−1) compared to that for lipid II lacking modifications (Kass = 890.11 ± 275.16 M−1 s−1) (Fig. 4).
FIG 4.
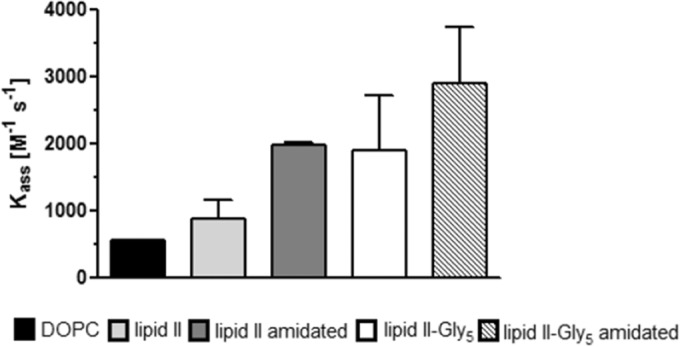
Association rate (Kass) of oritavancin binding to different lipid II variants embedded in DOPC model membranes or pure DOPC as determined by QCM biosensor measurements.
Taken together, these results demonstrate that the binding of oritavancin to the cell wall precursor involves, in addition to the d-Ala-d-Ala terminus of lipid II, the interaction with the pentaglycine crossbridge. Furthermore, a tertiary binding site, d-isoglutamine in position 2 of the stem peptide, significantly contributes to binding affinity.
In VRSA, the terminal d-Ala residue is exchanged to d-Lac, and it has been proposed that oritavancin compensates for the resulting loss of one critical hydrogen bond to this target through the interaction with additional binding sites on the lipid II molecule, i.e., the crossbridge (36, 39). However, vanA-type vancomycin-resistant S. aureus strains are characterized by a complete lack of or an incomplete chain length of the pentaglycine crossbridge (41), suggesting that in these strains yet another binding site on the lipid II molecule contributes to the high-affinity binding. Since the amidation of d-Glu of the lipid II stem peptide proved to significantly enhance binding affinity by 1 order of magnitude, oritavancin binding to the unmodified and amidated depsipeptide counterparts, as present in vanA-type VRSA, was investigated (Fig. 5). Indeed, the binding of oritavancin to lipid II-d-Lac is comparatively low (Fig. 5D) compared to that of the other lipid II derivatives tested. Although the overall binding affinity (KD = 1.05 × 10−7 M) is in the range of that of unmodified lipid II, tracking of frequency changes provides an interesting insight into the binding process. While target recognition is obviously represented by a low dissociation rate (koff) (afferent part of the curve) in Fig. 5B, C, and D for lipid II, amidated lipid II, and amidated lipid II-d-Lac, respectively, the koff rate of the nonamidated lipid II-d-Lac variant is evidently higher and comparable to that of oritavancin binding to the plain DOPC membrane (Fig. 5A). The data show that amidation of the cell wall precursor is a crucial factor for oritavancin target recognition of both lipid II and its depsipeptide counterpart (lipid II-d-Lac) as represented by decreased dissociation rates and increased binding affinity compared to those for the respective nonamidated variants (Fig. 5C and D).
FIG 5.

Frequency shifts of the QCM measurements of oritavancin binding to pure DOPC (A) or DOPC containing 0.1 mol% of lipid II derivative (B to E). The binding affinity KD results from the ratio of koff (afferent shift) and Kass (slope). (A) Intrinsic affinity of oritavancin to membranes; (B, C, and E) target binding by a much slower dissociation tendency, which is superimposed on membrane affinity.
Lipid II-d-Lac is a poor substrate for Fem peptidyl transferases of S. aureus.
In S. aureus, the cell wall building block lipid II is characteristically modified by a pentaglycine crossbridge attached to the l-Lys of the stem peptide, which is catalyzed by FemXAB peptidyl transferases (45, 52).
Severin et al. hypothesized that Fem peptidyl transferases of S. aureus do not efficiently recognize the d-Lac-containing depsipeptide precursors as a substrate (41) to explain the pentaglycine bridge deficiency in vanA-type VRSA. Here we provide the first biochemical evidence that the Fem-catalyzed addition of glycine residues to lipid II-d-Lac is strongly hampered. Compared to the control reaction using lipid II-d-Ala, the addition of the first glycine catalyzed by FemX is strongly decreased (34%) when lipid II-d-Lac was used as a substrate (Fig. 6). This effect was even more pronounced in a coupled FemXA synthesis assay (17%), providing biochemical evidence that the depsipeptide precursor is a poor substrate for the Fem enzymes of S. aureus. Most likely, the modification of the stem peptide terminus interferes with substrate recognition.
FIG 6.
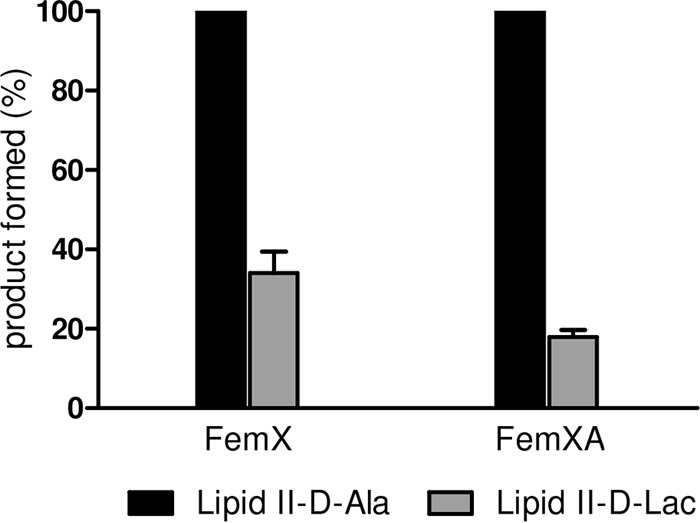
Enzymatic activities of FemX and FemXA. The addition of [14C]glycine to the substrate lipid II-d-Ala or lipid II-d-Lac by FemX and FemXA was analyzed by TLC and quantified by phosphorimaging. Reaction products using unmodified lipid II were taken as the 100% level.
Interactions of des-oritavancin with lipid II and lipid II variants.
The chemical removal of the N-terminal amino acid of the glycopeptide aglycone core results in damage to the binding pocket, crucial for the tight binding of d-Ala-d-Ala residues of the lipid II molecule. Damage to the carboxylate binding pocket leads to a complete loss of vancomycin antibacterial activity (36). In contrast, des-N-methylleucyl-oritavancin (des-oritavancin), lacking the terminal leucyl residue (Fig. 1A), still retains significant antibiotic activity against vancomycin-resistant isolates (Table 1) (36). To further investigate the activity of des-oritavancin in vivo, we determined the level of the UDP-MurNAc-depsipeptide in the cytoplasm of VanA-type E. faecium BM4147 after treatment with oritavancin and des-oritavancin (Fig. 7A). Antibiotics that interfere with the late steps of peptidoglycan synthesis block the recycling of the undecaprenol lipid carrier and lead to accumulation of the soluble cell wall precursor (UDP-MurNAc-penta-/depsipeptide) in the cytoplasm of treated cells (42). As previously reported, no accumulation of UDP-MurNAc-depsipeptide was detected after treatment of VRE with vancomycin (53). Oritavancin, in contrast, led to a significant accumulation of UDP-MurNAc-depsipeptide at 10× MIC, in either the VanA-induced (80 mg/liter vancomycin) or uninduced cells, comparable to that for chloroeremomycin (53). In line with its antibiotic activity against VRE, treatment with des-oritavancin (10× MIC) also led to accumulation of the d-Lac (UDP-MurNAc-depsipeptide)-containing soluble precursor, although the overall level of accumulation was markedly reduced compared to that in oritavancin-treated cells (Fig. 7A). Additionally, the antibiotic activities of both oritavancin and des-N-methylleucyl-oritavancin were antagonized by full-length lipid I and lipid II, as well as lipid II-Gly5 and lipid II-Asp variants (data not shown). Since Ge et al. proposed a direct interaction of the 4-epi-vancosamine sugar attached to ring 6 of the peptide core with penicillin binding proteins, thereby inhibiting enzyme activity without binding to the d-Ala-d-Ala of the cell wall precursor (35), we investigated the effect of des-oritavancin on the PBP2-catalyzed transglycosylation in vitro. In this test system, the addition of des-methylleucyl-oritavancin was found to inhibit the polymerization of lipid II-Gly5 in a dose-dependent manner (Fig. 7B), and an almost complete inhibition was observed at a molar ratio of 4:1 (antibiotic to lipid), suggesting substrate sequestration rather than a direct inhibition of enzyme activity. To further verify the binding of des-oritavancin to lipid II, the glycopeptide was incubated with the purified cell wall precursor at various molar ratios. In line with the in vitro assay (Fig. 7B), only traces of unbound lipid II (10%) were detectable at a 4:1 (antibiotic to lipid) molar ratio after extraction from the reaction mixtures and subsequent TLC analysis (Fig. 7C). In contrast to des-N-methylleucyl-vancomycin, which has no measurable affinity for lipid II (36), des-oritavancin is able to trap the unmodified lipid II in a stable complex, thereby preventing its extraction. Analysis of oritavancin binding to lipid II revealed a 2:1 binding stoichiometry (Fig. 7C), supporting dimerization of the heptapeptide (32).
TABLE 1.
MICs of test strains
| Test strain | MIC (μg/ml) of test strain fora: |
|||
|---|---|---|---|---|
| ORI | des-ORI | CEM | VAN | |
| S. aureus ATCC 29213 | 0.08 | 2 | 0.15 | 0.5 |
| E. faecium BM 4147 | 1.4 | 4 | 16 | >512 |
ORI, oritavancin; des-ORI, des-N-methylleucyl-oritavancin; CEM, chloroeremomycin; VAN, vancomycin.
FIG 7.

Intracellular accumulation of the soluble cell wall precursor UDP-MurNAc-depsipeptide in VanA-type E. faecium. (A) E. faecium BM4147 was grown in HHD broth (0.002% Tween 80) to an OD600 of 0.5 and supplemented with 80 mg/liter vancomycin (VAN) to induce vanA expression. After 10 min, oritavancin (ORI) or des-N-methylleucyl-oritavancin (des-ORI) was added at 10× MIC and incubated for 60 min. Cells were harvested and extracted with boiling water, and the intracellular nucleotide pool was analyzed by reversed-phase HPLC. UDP-MurNAc-depsipeptide (UDP-MurNAc-l-Ala-d-Gln-l-Lys-d-Ala-d-Lac) was identified by MALDI-TOF mass spectrometry. (B) Inhibition of the PBP2-catalyzed reaction by des-ORI in vitro. The conversion of [14C]lipid II-Gly5 into polymeric peptidoglycan in the presence of increasing concentrations of des-ORI was quantitatively analyzed by applying the reaction mixtures directly onto TLC plates and subsequent separation in solvent B (butanol-acetic acid-water-pyridine [15:3:12:10, vol/vol/vol/vol]) followed by the detection and quantification of residual [14C]lipid II-Gly5 using phosphorimaging. (C) Purified lipid II was incubated with increasing concentrations of ORI or des-ORI at molar ratios of 0.5 to 4:1 with respect to the substrate lipid II. The reaction mixtures were extracted, and unbound lipid was analyzed by TLC and visualized by PMA staining.
DISCUSSION
Semisynthetic glycopeptide antibiotics, like oritavancin, represent promising agents for the treatment of infections caused by multidrug-resistant Gram-positive bacteria. Like vancomycin, the lipoglycopeptide antibiotics such as oritavancin and telavancin inhibit transglycosylation via binding to the d-Ala-d-Ala terminus of lipid II. In addition, structural modifications, like the 4-epi-vancosamine and the 4′-chlorobiphenyl methyl side chain of oritavancin, account for improved antibacterial activity against MRSA, VISA, VRSA, and VRE strains (22, 40, 54).
Here, we provide evidence at the molecular level that these structural modifications enable oritavancin, in contrast to vancomycin, to bind to further target sites of the lipid II peptidoglycan precursor other than the d-Ala-d-Ala terminus. With use of solid-state nuclear magnetic resonance (NMR) and rotational-echo double resonance (REDOR), a model for the oritavancin-PG complex has been proposed in which the crossbridge is bound in a cleft between the epi-vancosamine residue and the core of the oritavancin molecule (36, 39, 40). We now provide biochemical evidence that the crossbridge and the d-iso-glutamine in position 2 of the lipid II stem peptide represent crucial binding sites for oritavancin. In vitro assays and determination of binding parameters using a QCM-based approach (Fig. 4 and 5) with different purified lipid II variants embedded in a lipid bilayer revealed that a progressive increase in oritavancin binding affinity to lipid II involves both lipid II modifications of S. aureus: the crossbridge and the amidation of the glutamate residue of the stem peptide (45, 46). In contrast, this increase in binding affinity was not observed for vancomycin. Rather, lipid II amidation appeared to strongly decrease vancomycin binding affinity, both in the in vitro system and as revealed by the determination of binding parameters. Earlier studies suggested a correlation between vancomycin resistance and the reduced degree of peptidoglycan amidation in S. aureus strain Mu50, and inducible amidation of d-iso-Glu has been reported in Clostridium difficile and Streptomyces coelicolor A3(2), but not in glycopeptide-resistant enterococci (55–61). Moreover, it was shown that the antimicrobial activity of vancomycin is antagonized more efficiently by short synthetic peptides (d-Glu/Gln-l-Lys-d-Ala-d-Ala) containing d-iso-Glu instead of d-iso-Gln (55, 62). It was concluded that the increase in the glutamine nonamidated muropeptide components in the peptidoglycan of Mu50 may efficiently trap a larger amount of vancomycin in the thickened cell wall.
In contrast to observations with vancomycin, enhanced binding affinity to amidated lipid II was also observed with chloroeremomycin, suggesting that the 4-epi-vancosamine residue, present in both oritavancin and chloroeremomycin but absent in vancomycin, is involved in the interaction with the amidated precursor.
Oritavancin exhibits antimicrobial activity against VRSA (27, 28), in which resistance to vancomycin is mediated by the exchange of the terminal d-Ala to d-Lac. Muropeptide analyses of vanA-type VRSA revealed that these strains are characterized by a complete lack of or at least severely shortened pentaglycine crossbridges (41). Thus, the interaction of oritavancin with the crossbridge appears less relevant in VRSA. Rather, as revealed by comparative analysis of binding parameters, enhanced binding affinity to lipid II-d-Lac relies on additional interactions of oritavancin with d-iso-Gln in position 2 of the stem peptide (Fig. 5) and thereby compensates for the loss of one hydrogen bond.
Up to now, the vanA-mediated mechanism of resistance has not been evolving or spreading rapidly in MRSA (63). It has been shown that the simultaneous expressions of chromosomally located mecA and plasmid-borne vanA resistance determinants are mutually antagonistic, and it was concluded that this phenomenon is based on the inefficient recognition of depsipeptide precursors by the alternative penicillin binding protein PBP2a (41, 64).
Biochemical analysis of the FemX- and FemXA-catalyzed reactions in this study clearly demonstrated that the d-Lac-containing cell wall precursors are poor substrates for the staphylococcal Fem peptidyl transferases, presumably due to interference with substrate recognition. In staphylococci, FemX, which initiates pentaglycine bridge formation by adding the first glycine residue, is essential (52). Although characterized by abnormal cell morphology, reduced cell wall turnover, and retarded cell separation, cells expressing at least monoglycine crossbridges are generally viable, but hypersusceptible to oxacillin (65).
Since pentaglycine bridge formation is severely hampered in VRSA, a reduction in FemX activity (34%) (Fig. 6) results in a synthetically lethal phenotype in the presence of oxacillin, because PBP2a′ is unable to cross-link monoglycyl-containing precursors (65, 66).
Interestingly, Streptomyces coelicolor produces an alternative Fem homologue, VanK, which is essential under conditions of vanHAX induction (67) to ensure the formation of the crossbridge that consists of a single glycine residue. This indicates that FemX of S. coelicolor is also unable to efficiently recognize lipid II-d-Lac, although biochemical evidence is lacking so far. FemX of enterococci, in contrast, appears to efficiently recognize depsipeptide precursors (68), which might partially explain the enhanced prevalence of VRE compared to that of VRSA.
The differences in the molecular interactions with the PG precursor lipid II between vancomycin and oritavancin are further reflected by the antibiotic activity of their respective Edman degradation products. Chemical removal of the N-terminal N-methylleucyl residue from the aglycone core of vancomycin results in damage to the d-Ala-d-Ala binding cleft, impairing binding to the peptidoglycan precursor, as evidenced by a total loss of its antimicrobial activity (36). Interestingly, the Edman degradation product of oritavancin (des-N-methylleucyl-oritavancin) retains good antimicrobial activity (Table 1) (36). Although species-specific lipid II modifications were shown to significantly contribute to oritavancin binding, its hexapeptide derivative lacking the carboxylate binding pocket is also able to bind to unmodified lipid II (Fig. 7), suggesting that additional features further compensate for the loss of one hydrogen bond. The hydrophobic substitutions on the vancosamine residue have been demonstrated to enhance the antibacterial activity of chloroeremomycin and oritavancin by promoting dimerization of the glycopeptides in solution (32, 33, 55). In contrast, vancomycin lacking these additional substitutions reaches its target site, where its antibacterial activity against susceptible bacteria is assumed to be a sole function of its binding to d-Ala-d-Ala-containing PG residues, predominantly as a monomer (32). Studies on various synthetic covalent dimers of vancomycin, which showed significantly improved antibacterial activity against vancomycin-susceptible and -resistant bacteria, support the crucial role for dimerization as a determinant of antibacterial activity (69).
In the case of oritavancin, enhanced antibacterial activity appears to further rely on the ability to interact with the bacterial membrane, which may contribute to its rapid bactericidal activity (30, 70). Kim et al. have recently shown that the hydrophobic 4′-chlorobiphenyl methyl moiety of oritavancin promotes the interaction with isolated membrane protoplasts and increases membrane permeability in artificial liposomes composed of bacterial membrane phospholipids, when a certain threshold concentration is reached (70). The integration of oritavancin into the membrane might lead to changes in the physicochemical properties of the membrane and affect the organization of highly dynamic biosynthesis machineries and energy-generating systems. One can assume that trapping of the limited central bactoprenol-bound precursor, combined with integration of oritavancin into the cytoplasmic membrane, has a dramatic effect on the tightly controlled membrane-associated steps of cell wall biosynthesis, where proteins are in intimate contact. These pleiotropic effects have not been reported for chloroeremomycin nor for vancomycin, both of which lack alkyl side chains. More recently, it has further been shown that oritavancin prevents vegetative outgrowth and recovery of C. difficile spores probably due to direct binding to the spore surface (71) through the hydrophobic chlorobiphenyl side chain.
In conclusion, we here showed at the molecular level that the binding of oritavancin to its target involves the interaction with additional species-specific binding sites, namely, the crossbridge and d-iso-Gln in position 2 of the lipid II stem peptide. Interactions with d-iso-Gln in position 2 of the lipid II stem peptide in particular facilitate stronger intramolecular interactions with the PG precursor, thereby compensating for the loss of a crucial hydrogen bond in vancomycin-resistant strains.
ACKNOWLEDGMENTS
This work was supported by the German Research Foundation (DFG) (SCHN 1284/1-2) and the German Center for Infection Research (DZIF).
We thank Michaele Josten for performing mass spectrometry and Inge Luhmer-Becker for preparation of the UDP-MurNAc-pentapeptide, Greg Moeck and Adel Rafai Far for discussion and reading of the manuscript, and The Medicines Company for providing oritavancin, des-oritavancin, and chloroeremomycin.
REFERENCES
- 1.Arthur M, Courvalin P. 1993. Genetics and mechanisms of glycopeptide resistance in enterococci. Antimicrob Agents Chemother 37:1563–1571. doi: 10.1128/AAC.37.8.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCormick MH, Stark WM, Pittenger GE, Pittenger RC, McGuire JM. 1956. Vancomycin, a new antibiotic. Chemical and biological properties. Antibiot Annu 1955−1956:606–611. [PubMed] [Google Scholar]
- 3.Courvalin P. 1990. Resistance of enterococci to glycopeptides. Antimicrob Agents Chemother 34:2291–2296. doi: 10.1128/AAC.34.12.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leclercq R, Derlot E, Duval J, Courvalin P. 1988. Plasmid mediated resistance to vancomycin and teicoplanin in Enterococcus faecium. N Engl J Med 319:157–161. doi: 10.1056/NEJM198807213190307. [DOI] [PubMed] [Google Scholar]
- 5.Uttley AHC, Collins CH, Naidoo J, George RC. 1988. Vancomycin-resistant enterococci. Lancet i:57–58. [DOI] [PubMed] [Google Scholar]
- 6.Hidron AI, Edwards JR, Patel J, Horan TC, Sievert DM, Pollock DA, Fridkin SK, National Healthcare Safety Network Team, Participating National Healthcare Safety Network Facilities. 2008. Antimicrobial-resistant pathogens associated with healthcare-associated infections: annual summary of data reported to the National Healthcare Safety Network at Centers for Disease Control and Prevention 2006-2007. Infect Control Hosp Epidemiol. 29:996–1011. doi: 10.1086/591861. [DOI] [PubMed] [Google Scholar]
- 7.Livermore DM. 2012. Fourteen years in resistance. Int J Antimicrob Agents 39:283–294. doi: 10.1016/j.ijantimicag.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 8.Nicas TI, Allen NE. 1994. Resistance and mode of action, p 219–241. In Nagarajan R. (ed), Glycopeptide antibiotics. Marcel Dekker, New York, NY. [Google Scholar]
- 9.Reynolds PE. 1989. Structure, biochemistry and mechanism of action of glycopeptide antibiotics. Eur J Clin Microbiol Infect Dis 8:943–950. doi: 10.1007/BF01967563. [DOI] [PubMed] [Google Scholar]
- 10.Schneider T, Sahl HG. 2010. An oldie but a goodie—cell wall biosynthesis as antibiotic target pathway. Int J Med Microbiol 300:161–169. doi: 10.1016/j.ijmm.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 11.Perkins HR, Nieto M. 1974. The chemical basis for the action of the vancomycin group of antibiotics. Ann N Y Acad Sci 235:348–363. doi: 10.1111/j.1749-6632.1974.tb43276.x. [DOI] [PubMed] [Google Scholar]
- 12.Bugg TDH, Wright GD, Dutka-Malen S, Arthur M, Courvalin P, Walsh CT. 1991. Molecular basis for vancomycin resistance in Enterococcus faecium BM4147: biosynthesis of a depsipeptide peptidoglycan precursor by vancomycin resistance proteins vanH and vanA. Biochemistry 30:10408–10415. doi: 10.1021/bi00107a007. [DOI] [PubMed] [Google Scholar]
- 13.Walsh CT, Fisher SL, Park IS, Prahalad M, Wu Z. 1996. Bacterial resistance to vancomycin: five genes and one missing hydrogen bond tell the story. Chem Biol 3:21–28. doi: 10.1016/S1074-5521(96)90079-4. [DOI] [PubMed] [Google Scholar]
- 14.Reynolds PE, Snaith HA, Maguire AJ, Dutka-Malen S, Courvalin P. 1994. Analysis of peptidoglycan precursors in vancomycin-resistant Enterococcus gallinarum BM4174. Biochem J 301:5–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malabarba A, Nicas TI, Thompson RC. 1997. Structural modifications of glycopeptide antibiotics. Med Res Rev 17:69–137. doi: 10.1002/(SICI)1098-1128(199701)17:1<69::AID-MED3>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 16.Van Bambeke F, Van Laethem Y, Courvalin P, Tulkens PM. 2004. Glycopeptide antibiotics: from conventional molecules to new derivatives. Drugs 64:913–936. doi: 10.2165/00003495-200464090-00001. [DOI] [PubMed] [Google Scholar]
- 17.Pace JL, Yang G. 2006. Glycopeptides: update on an old successful antibiotic class. Biochem Pharmacol 71:968–980. doi: 10.1016/j.bcp.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 18.Van Bambeke F, Mingeot-Leclercq MP, Struelens MJ, Tulkens PM. 2008. The bacterial envelope as a target for novel anti-MRSA antibiotics. Trends Pharmacol Sci 29:124–134. doi: 10.1016/j.tips.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 19.Leung SF, Tirado-Rives J, Jorgensen WL. 2009. Vancomycin analogs: seeking improved binding of d-Ala-d-Ala and d-Ala-d-Lac peptides by side-chain and backbone modifications. Bioorg Med Chem 17:5874–5886. doi: 10.1016/j.bmc.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li TL, Liu YC, Lyu SY. 2012. Combining biocatalysis and chemoselective chemistries for glycopeptide antibiotics modification. Curr Opin Chem Biol 16:170–178. doi: 10.1016/j.cbpa.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 21.Allen NE. 2010. From vancomycin to oritavancin: the discovery and development of a novel lipoglycopeptide antibiotic. Antinfect Agents Med Chem 9:23–47. doi: 10.2174/187152110790886745. [DOI] [Google Scholar]
- 22.Arthur M, Depardieu F, Reynolds P, Courvalin P. 1999. Moderate-level resistance to glycopeptide LY333328 mediated by genes of the vanA and vanB clusters in enterococci. Antimicrob Agents Chemother 43:1875–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cooper RD, Snyder NJ, Zweifel MJ. 1996. Reductive alkylation of glycopeptides antibiotics: synthesis and antibacterial activity. J Antibiot (Tokyo) 49:575–581. doi: 10.7164/antibiotics.49.575. [DOI] [PubMed] [Google Scholar]
- 24.Lu W, Oberthur M, Leimkuhler C, Tao J, Kahne D, Walsh CT. 2004. Characterization of a regiospecific epivancosaminyl transferase GtfA and enzymatic reconstitution of the antibiotic chloroeremomycin. Proc Natl Acad Sci U S A 101:4390–4395. doi: 10.1073/pnas.0400277101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Belley A, McKay GA, Arhin FF, Sarmiento I, Beaulieu S, Fadhil I, Parr TR Jr, Moeck G. 2010. Oritavancin disrupts membrane integrity of Staphylococcus aureus and vancomycin-resistant enterococci to effect rapid bacterial killing. Antimicrob Agents Chemother 54:5369–5371. doi: 10.1128/AAC.00760-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McKay GA, Beaulieu S, Arhin FF, Beaulieu S, Sarmiento I, Parr TR Jr, Moeck G. 2009. Time-kill kinetics of oritavancin and comparator agents against Staphylococcus aureus, Enterococcus faecalis and Enterococcus faecium. J Antimicrob Chemother 63:1191–1199. doi: 10.1093/jac/dkp126. [DOI] [PubMed] [Google Scholar]
- 27.Saravolatz LD, Pawlak J, Johnson JB. 2010. In vitro activity of oritavancin against community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA), vancomycin-intermediate S. aureus (VISA), vancomycin resistant S. aureus (VRSA) and daptomycin-non-susceptible S. aureus (DNSSA). Int J Antimicrob Agents 36:69–72. doi: 10.1016/j.ijantimicag.2010.02.023. [DOI] [PubMed] [Google Scholar]
- 28.Arhin FF, Drahi DC, Pillar CM, Parr TR Jr, Moeck G, Sahm DF. 2009. Comparative in vitro activity profile of oritavancin against recent gram-positive clinical isolates. Antimicrob Agents Chemother 53:4762–4771. doi: 10.1128/AAC.00952-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morrow T. 2014. One-shot staph treatment offers many advantages. Manag Care 23:55–56. [PubMed] [Google Scholar]
- 30.Domenech O, Dufrene YF, Van Bambeke F, Tukens PM, Mingeot-Leclercq MP. 2010. Interactions of oritavancin, a new semi-synthetic lipoglycopeptide, with lipids extracted from Staphylococcus aureus. Biochim Biophys Acta 1798:1876−1885. doi: 10.1016/j.bbamem.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 31.Nicolaou KC, Hughes R, Cho SY, Winssinger N, Labischinski H, Endermann R. 2001. Synthesis and biological evaluation of vancomycin dimers with potent activity against vancomycin-resistant bacteria: target-accelerated combinatorial synthesis. Chem Eur J 7:3824–3843. doi: 10.1002/1521-3765(20010903)7:17<3824::AID-CHEM3824>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 32.Mackay JP, Gerhard U, Beauregard DA, Westwell MS, Searle MS, Williams DH. 1994. Glycopeptide antibiotic activity and the possible role of dimerization: a model for biological signalling. J Am Chem Soc 116:4581–4590. doi: 10.1021/ja00090a006. [DOI] [Google Scholar]
- 33.Gerhard U, Mackay JP, Maplestone RA, Williams DH. 1993. The role of the sugar and chlorine substituents in the dimerization of vancomycin antibiotics. J Am Chem Soc 115:232–237. doi: 10.1021/ja00054a033. [DOI] [Google Scholar]
- 34.Beauregard DA, Williams DH, Gwynn MN, Knowles DJ. 1995. Dimerization and membrane anchors in extracellular targeting of vancomycin group antibiotics. Antimicrob Agents Chemother 39:781–778. doi: 10.1128/AAC.39.3.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ge M, Chen Z, Onishi HR, Kohler J, Silver LL, Kerns R, Fukuzawa S, Thompson C, Kahne D. 1999. Vancomycin derivatives that inhibit peptidoglycan biosynthesis without binding d-Ala-d-Ala. Science 284:507–511. doi: 10.1126/science.284.5413.507. [DOI] [PubMed] [Google Scholar]
- 36.Kim SJ, Cegelski L, Stueber D, Singh M, Dietrich E, Tanaka KS, Parr TR Jr, Far AR, Schaefer J. 2008. Oritavancin exhibits dual mode of action to inhibit cell-wall biosynthesis in Staphylococcus aureus. J Mol Biol 377:281–293. doi: 10.1016/j.jmb.2008.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Veiga P, Piquet S, Maisons A, Furlan S, Courtin P, Chapot-Chartier MP, Kulakauskas S. 2006. Identification of an essential gene responsible for d-Asp incorporation in the Lactococcus lactis peptidoglycan crossbridge. Mol Microbiol 62:1713–1724. doi: 10.1111/j.1365-2958.2006.05474.x. [DOI] [PubMed] [Google Scholar]
- 38.Bellais S, Arthur M, Dubost L, Hugonnet JE, Gutmann L, van Heijenoort J, Legrand R, Brouard JP, Rice L, Mainardi JL. 2006. Aslfm, the d-aspartate ligase responsible for the addition of d-aspartic acid onto the peptidoglycan precursor of Enterococcus faecium. J Biol Chem 281:11586–11594. doi: 10.1074/jbc.M600114200. [DOI] [PubMed] [Google Scholar]
- 39.Kim SJ, Cegelski L, Preobrazhenskaya M, Schaefer J. 2006. Structures of Staphylococcus aureus cell-wall complexes with vancomycin, eremomycin, and chloroeremomycin derivatives by 13C{19F} and 15N{19F} rotational-echo double resonance. Biochemistry 45:5235–5250. doi: 10.1021/bi052660s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patti GJ, Kim SJ, Yu TY, Dietrich E, Tanaka KS, Parr TR Jr, Far AR, Schaefer J. 2009. Vancomycin and oritavancin have different modes of action in Enterococcus faecium. J Mol Biol 392:1178–1191. doi: 10.1016/j.jmb.2009.06.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Severin A, Tabei K, Tenover F, Chung M, Clarke N, Tomasz A. 2004. High level oxacillin and vancomycin resistance and altered cell wall composition in Staphylococcus aureus carrying the staphylococcal mecA and the enterococcal vanA gene complex. J Biol Chem 279:3398–3407. doi: 10.1074/jbc.M309593200. [DOI] [PubMed] [Google Scholar]
- 42.Allen NE, LeTourneau DL, Hobbs JN Jr, Thompson RC. 2002. Hexapeptide derivatives of glycopeptide antibiotics: tools for mechanism of action studies. Antimicrob Agents Chemother 46:2344–2348. doi: 10.1128/AAC.46.8.2344-2348.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brötz H, Bierbaum G, Leopold K, Reynolds PE, Sahl HG. 1998. The lantibiotic mersacidin inhibits peptidoglycan synthesis by targeting lipid II. Antimicrob Agents Chemother 42:154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schneider T, Gries K, Josten M, Wiedemann I, Pelzer S, Labischinski H, Sahl HG. 2009. The lipopeptide antibiotic friulimicin B inhibits cell wall biosynthesis through complex formation with bactoprenol phosphate. Antimicrob Agents Chemother 53:1610–1618. doi: 10.1128/AAC.01040-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schneider T, Senn MM, Berger-Bächi B, Tossi A, Sahl HG, Wiedemann I. 2004. In vitro assembly of a complete, pentaglycine interpeptide bridge containing cell wall precursor (lipid II-Gly5) of Staphylococcus aureus. Mol Microbiol 53:675–685. doi: 10.1111/j.1365-2958.2004.04149.x. [DOI] [PubMed] [Google Scholar]
- 46.Münch D, Roemer T, Lee SH, Engeser M, Sahl HG, Schneider T. 2012. Identification and in vitro analysis of the GatD/MurT enzyme-complex catalyzing lipid II amidation in Staphylococcus aureus. PLoS Pathog 8:e1002509. doi: 10.1371/journal.ppat.1002509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Umbreit JN, Strominger JL. 1972. Isolation of the lipid intermediate in peptidoglycan biosynthesis from Escherichia coli. J Bacteriol 112:1306–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Christ K, Rüttinger HH, Höpfner M, Rothe U, Bendas G. 2005. The detection of UV-induced membrane damages by a combination of two biosensor techniques. Photochem Photobiol 81:1417–1423. doi: 10.1562/2005-04-19-RA-493. [DOI] [PubMed] [Google Scholar]
- 49.Christ K, Wiedemann I, Bakowsky U, Sahl HG, Bendas G. 2007. The role of lipid II in membrane binding of and pore formation by nisin analyzed by two combined biosensor techniques. Biochim Biophys Acta 1768:694–704. doi: 10.1016/j.bbamem.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 50.Simonis D, Christ K, Alban S, Bendas G. 2007. Affinity and kinetics of different heparins binding to P- and L-selectin. Semin Thromb Hemost 33:534–539. doi: 10.1055/s-2007-982085. [DOI] [PubMed] [Google Scholar]
- 51.Brötz H, Josten M, Wiedemann I, Schneider U, Götz F, Bierbaum G, Sahl HG. 1998. Role of lipid-bound peptidoglycan precursors in the formation of pores by nisin, epidermin and other lantibiotics. Mol Microbiol 30:317–327. doi: 10.1046/j.1365-2958.1998.01065.x. [DOI] [PubMed] [Google Scholar]
- 52.Rohrer S, Ehlert K, Tschierske M, Labischinski H, Berger-Bächi B. 1999. The essential Staphylococcus aureus gene fmhB is involved in the first step of peptidoglycan pentaglycine interpeptide formation. Proc Natl Acad Sci U S A 96:9351–9356. doi: 10.1073/pnas.96.16.9351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Allen NE, Hobbs JN, Nicas TI. 1996. Inhibition of peptidoglycan biosynthesis in vancomycin-susceptible and -resistant bacteria by a semisynthetic glycopeptide antibiotic. Antimicrob Agents Chemother 40:2356–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Belley A, Harris R, Beveridge T, Parr T Jr, Moeck G. 2009. Ultrastructural effects of oritavancin on methicillin-resistant Staphylococcus aureus and vancomycin-resistant Enterococcus. Antimicrob Agents Chemother 53:800–804. doi: 10.1128/AAC.00603-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hanaki H, Labischinski H, Inaba Y, Kondo N, Murakami H, Hiramatsu K. 1998. Increase in glutamine-non-amidated muropeptides in the peptidoglycan of vancomycin-resistant Staphylococcus aureus strain Mu50. J Antimicrob Chemother 42:315–320. doi: 10.1093/jac/42.3.315. [DOI] [PubMed] [Google Scholar]
- 56.Ammam F, Meziane-Cherif D, Mengin-Lecreulx D, Blanot D, Patin D, Boneca IG, Courvalin P, Lambert T, Candela T. 2013. The functional vanGCd cluster of Clostridium difficile does not confer vancomycin resistance. Mol Microbiol 89:612–625. doi: 10.1111/mmi.12299. [DOI] [PubMed] [Google Scholar]
- 57.Hugonnet JE, Haddache N, Veckerlé C, Dubost L, Marie A, Shikura N, Mainardi JL, Rice LB, Arthur M. 2014. Peptidoglycan cross-linking in glycopeptide-resistant actinomycetales. Antimicrob Agents Chemother 58:1749–1756. doi: 10.1128/AAC.02329-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cegelski L, Steuber D, Mehta AK, Kulp DW, Axelsen PH, Schaefer J. 2006. Conformational and quantitative characterization of oritavancin-peptidoglycan complexes in whole cells of Staphylococcus aureus by in vivo 13C and 15N labeling. J Mol Biol 357:1253–1262. doi: 10.1016/j.jmb.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 59.Boyle-Vavra S, Labischinski H, Ebert Ehlert CCK, Daum RS. 2001. A spectrum of changes occurs in peptidoglycan composition of glycopeptide-intermediate clinical Staphylococcus aureus isolates. Antimicrob Agents Chemother 45:280–287. doi: 10.1128/AAC.45.1.280-287.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schäberle TF, Vollmer W, Frasch HJ, Hüttel S, Kulik A, Röttgen M, Von Thaler AK, Wohlleben W, Stegmann E. 2011. Self-resistance and cell wall composition in the glycopeptide producer Amycolatopsis balhimycina. Antimicrob Agents Chemother 55:4283–4289. doi: 10.1128/AAC.01372-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cremniter J, Mainardi JL, Josseaume N, Quincampoix JC, Dubost L, Hugonnet JE, Marie A, Gutmann L, Rice LB, Arthur M. 2006. Novel mechanisms of resistance to glycopeptide antibiotics in Enterococcus faecium. J Biol Chem 281:32254–32262. doi: 10.1074/jbc.M606920200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nieto M, Perkins HR. 1971. Modification of the acyl-d-alanyl-d-alanine terminus affecting complex-formation with vancomycin. Biochem J 123:789–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Howden BP, Davies JK, Johnson PDR, Stinear TP, Grayson ML. 2010. Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: resistance mechanisms, laboratory detection, and clinical implications. Clin Microbiol Rev 23:99–139. doi: 10.1128/CMR.00042-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Severin A, Wu SW, Tabei K, Tomasz A. 2004. Penicillin-binding protein 2 is essential for expression of high-level vancomycin resistance and cell wall synthesis in vancomycin-resistant Staphylococcus aureus carrying the vanA gene complex. Antimicrob Agents Chemother 48:4566–4573. doi: 10.1128/AAC.48.12.4566-4573.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Strandén AM, Ehlert K, Labischinski H, Berger-Bächi B. 1997. Cell wall monoglycine cross-bridges and methicillin hypersusceptibility in a femAB null mutant of methicillin-resistant Staphylococcus aureus. J Bacteriol 179:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Koyama N, Tokura Y, Münch D, Sahl HG, Schneider T, Shibagaki Y, Ikeda H, Tomoda H. 2012. The nonantibiotic small molecule cyslabdan enhances the potency of β-lactams against MRSA by inhibiting pentaglycine interpeptide bridge synthesis. PLoS One 7:e48981. doi: 10.1371/journal.pone.0048981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hong HJ, Hill MH, Hill LM, Buttner MJ. 2005. The role of the novel Fem protein VanK in vancomycin resistance in Streptomyces coelicolor. J Biol Chem 280:13055–13061. doi: 10.1074/jbc.M413801200. [DOI] [PubMed] [Google Scholar]
- 68.Bouhss A, Josseaume N, Severin Tabei AK, Hugonnet JE, Shlaes D, Mengin-Lecreulx D, van Heijenoort J, Arthur M. 2002. Synthesis of the l-alanyl-l-alanine cross-bridge of Enterococcus faecalis peptidoglycan. J Biol Chem 277:45935–45941. doi: 10.1074/jbc.M207449200. [DOI] [PubMed] [Google Scholar]
- 69.Staroske T, Williams DH. 1998. Synthesis of covalent head-to-tail dimers of vancomycin. Tetrahedron Lett 39:4917–4920. doi: 10.1016/S0040-4039(98)00895-8. [DOI] [Google Scholar]
- 70.Kim SJ, Singh M, Schaefer J. 2009. Oritavancin binds to isolated protoplast membranes but not intact protoplasts of Staphylococcus aureus. J Mol Biol 391:414–425. doi: 10.1016/j.jmb.2009.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chilton CH, Freeman J, Baines SD, Crowther S, Nicholson S, Wilcox MH. 2013. Evaluation of the effect of oritavancin on Clostridium difficile spore germination, outgrowth and recovery. J Antimicrob Chemother 68:2078–2082. doi: 10.1093/jac/dkt160. [DOI] [PubMed] [Google Scholar]