

Abstract
Objective:
We investigated system-level corticostriatal changes in a human model of premotor Parkinson disease (PD), i.e., healthy carriers of the G2019S LRRK2 mutation that is associated with a markedly increased, age-dependent risk of developing PD.
Methods:
We compared 37 asymptomatic LRRK2 G2019S mutation carriers (age range 30–78 years) with 32 matched, asymptomatic nonmutation carriers (age range 30–74 years). Using fMRI, we tested the hypothesis that corticostriatal connectivity in premotor PD shifts from severely affected to less affected striatal subregions, as shown previously in symptomatic PD. Specifically, we predicted that in premotor PD, the shift in corticostriatal connectivity would follow the same gradient of striatal dopamine depletion known from overt PD, with the dorsoposterior putamen being more affected than the ventroanterior putamen.
Results:
The known parallel topology of corticostriatal loops was preserved in each group, but the topography of putamen connectivity shifted. In LRRK2 G2019S mutation carriers, the right inferior parietal cortex had reduced functional connectivity with the dorsoposterior putamen but increased connectivity with the ventroanterior putamen, as compared with noncarriers. This shift in functional connectivity increased with age in LRRK2 G2019S mutation carriers.
Conclusions:
Asymptomatic LRRK2 G2019S mutation carriers show a reorganization of corticostriatal circuits that mirrors findings in idiopathic PD. These changes may reflect premotor basal ganglia dysfunction or circuit-level compensatory changes.
Parkinson disease (PD) is a progressive, neurodegenerative disorder characterized by nigrostriatal dopamine depletion. The motor symptoms of PD appear only when dopaminergic cell death reaches a critical threshold of 50% to 80%.1 This suggests that the nervous system has a marked potential to compensate for these changes.2 However, human studies of cerebral compensation in premotor PD are lacking, given the difficulty of predicting who will develop PD in the future.
Previous work has shown that compensatory mechanisms in idiopathic PD depend on brain regions relatively unaffected by dopamine depletion, such as the anterior striatum.3,4 Using fMRI in early-stage PD, we recently showed a shift in corticostriatal connectivity from severely affected striatal regions (posterior putamen) to less affected striatal regions (anterior putamen).5 If these changes reflect compensation, they should also occur in the premotor phase of PD, when functional reorganization of cerebral circuits prevents overt clinical symptoms.2
We tested this prediction in a genetic model of premotor PD, comparing corticostriatal functional connectivity between healthy carriers and noncarriers of the G2019S mutation in the LRRK2 gene. LRRK2 parkinsonism is associated with Lewy body pathology6 and with similar clinical signs as idiopathic PD.7,8 Healthy carriers of the LRRK2 G2019S mutation therefore form a unique population at risk of PD.9,10 Penetrance estimations range from 10%–17% at the age 50 years to 25%–85% at the age of 70 years,7,11–13 suggesting that the risk of developing PD is age-dependent.7 Thus, we investigated age-related changes in corticostriatal connectivity in healthy LRRK2 G2019S mutation carriers.
METHODS
Subjects.
We recruited 75 healthy first-degree relatives of leucine-rich repeat kinase 2 (LRRK2) Ashkenazi Jewish patients with PD into this study through their affected PD family members. All patients with PD were recruited from the Movement Disorder Unit of Tel Aviv Sourasky Medical Center, as part of a larger study (the Michael J. Fox Ashkenazi Jewish LRRK2 Consortium) aimed to characterize the G2019S mutation phenotype. Participants were not aware of their genetic status throughout the study. Exclusion criteria were clinical diagnosis of PD according to the Queen Square Brain Bank criteria, the presence of a mutation in the β-glucocerebrosidase (GBA) gene (an independent risk factor for developing PD14), other neuropsychiatric diseases, and general exclusion criteria for MRI scanning (e.g., claustrophobia, pacemaker, implanted metal parts). Of 75 tested subjects, 6 were excluded. Reasons for exclusion were a panic attack in the MRI scanner (n = 1), technical problems (storage failure; n = 2), and the presence of a GBA mutation (n = 3). Of 69 included subjects, 37 (20 women) were carriers of the LRRK2 G2019S mutation and 32 (16 women) were noncarriers. The 69 subjects came from 43 families, 18 of which had ≥2 included subjects (range 2–5 members, average ± standard error = 2.4 ± 0.86 members).
Standard protocol approvals, registrations, and patient consents.
Before the beginning of the study, all subjects signed an informed consent form approved by Tel Aviv Sourasky Medical Center institutional review board. Genomic DNA was isolated from peripheral blood leukocytes, and the 6055G_A (G2019S) mutation in exon 41 of the LRRK2 gene was determined as previously described.14 All subjects were administered the Unified Parkinson's Disease Rating Scale, Part III, the Beck Depression Inventory II, the State-Trait Anxiety Inventory, and the University of Pennsylvania Smell Identification Test. We compared LRRK2 G2019S mutation carriers and noncarriers using 2-sample t tests.
Image acquisition.
During scanning, all subjects were asked to keep their eyes closed and to avoid repetitive thoughts. Imaging was performed on a GE 3T Signa HDxt scanner (GE Signa EXCITE, Milwaukee, WI) with a resonant gradient echoplanar imaging system, using a standard 8-channel head coil. Each subject received an anatomical scan (spoiled gradient echo sequence: field of view 250 × 250 mm; matrix size 256 × 256 mm; voxel size 0.98 × 0.98 × 1 mm; repetition time = 9 milliseconds; echo time = 3.6 milliseconds) and 266 functional scans (single-shot gradient echoplanar imaging sequence: echo time/repetition time = 35/1,680 milliseconds; 30 axial slices; voxel size = 3.1 × 3.1 × 3.5 mm; no gap; scanning time approximately 7.5 minutes; 266 images).
Preprocessing of imaging data.
Data were preprocessed and analyzed with SPM5 (Statistical Parametric Mapping, www.fil.ion.ucl.ac.uk/spm). Images were spatially realigned, slice-time corrected, normalized to Montreal Neurological Institute (MNI) space using unified segmentation, and smoothed with an isotropic 8-mm full width at half maximum gaussian kernel. We then low-pass filtered the images using a fifth-order Butterworth filter to retain frequencies below 0.1 Hz, because correlations between intrinsic fluctuations are specifically found in this frequency range.15 Anatomical images were spatially coregistered to the mean of the functional image and spatially normalized using the same transformation matrix applied to the functional images.
Striatal seed regions.
We segmented each subject's normalized anatomical MRI scan into left and right putamen, caudate nucleus, and nucleus accumbens (using FIRST v1.1; www.fmrib.ox.ac.uk/fsl). We further subdivided the putamen into a dorsoposterior part, a ventroposterior part, a dorsoanterior part, and a ventroanterior part using the y = 0 and z = 0 axes as the borders between the 4 subregions.16 Finally, we averaged across the left and right striatal regions because none of the subjects had asymmetric signs of PD. We used MarsBaR (http://marsbar.sourceforge.net) to calculate the average BOLD time courses of the bilateral dorsoposterior putamen, ventroposterior putamen, dorsoanterior putamen, ventroposterior putamen, caudate nucleus, and accumbens (figure 1).
Figure 1. Striatal seed regions.
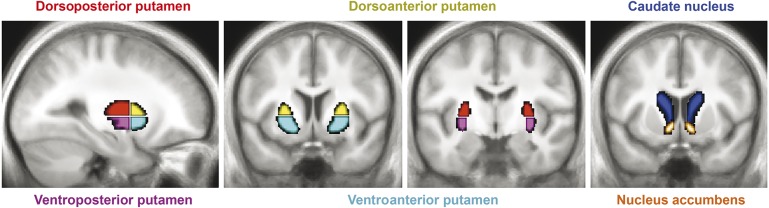
Six striatal seed regions, overlaid onto the average structural MRI of the 37 LRRK2 G2O19S mutation carriers and 32 noncarriers. The putamen is divided into 4 regions, using the lines passing through the anterior commissure (y = 0) and z = 0 (in MNI [Montreal Neurological Institute] space) as the borders, based on previous work.16
Nuisance signals.
We removed nonneuronal fluctuations from the data by adding 3 time courses to our model that describe the average signal intensity in the bilateral lateral ventricles (CSF), a blank portion of the MRIs (out-of-brain signal), and the global gray matter signal. To optimally control for the motion effects, we added 36 motion parameters to our model, as described before.17
Statistical analyses.
First-level analyses.
For each subject, we performed a multiple regression analysis using the general linear model implemented in SPM. This model included the time courses of the 6 striatal seed regions and the 39 nuisance regressors. All regressors and functional images were bandpass-filtered between 0.008 and 0.l Hz. Parameter estimates (β values) for all regressors were obtained by maximum-likelihood estimation, modeling temporal autocorrelation as an AR(1) process. For each seed region, the parameter estimate reflects the influence of the seed region's time course on the time course of that voxel, while controlling for the contribution of all the other regressors in the model.18 Thus, common variance (e.g., global signal fluctuations) is not assigned to any of the regressors, increasing the specificity of our findings.
Second-level analyses.
Group-level analyses were performed using a random-effects model implemented in SPM. For each group, we entered the β images of the 6 striatal seed regions into a 2 × 6 repeated-measures analysis of variance with the factors group (LRRK2 G2019S mutation carriers vs noncarriers) and region (dorsoposterior putamen, ventroposterior putamen, dorsoanterior putamen, ventroposterior putamen, caudate nucleus, and accumbens). For each of the 6 regions, we investigated both common and differential connectivity between groups. Based on previous findings in idiopathic PD,5 we focused our group comparison on a single region of interest (ROI), i.e., the right inferior parietal cortex (12-mm sphere around MNI coordinates [+56, −20, +28]). We also performed a whole-brain search. The statistical threshold was set to p < 0.05 family-wise error (FWE)-corrected for the search volume. Finally, we correlated corticostriatal connectivity with age for both LRRK2 G2019S mutation carriers and noncarriers, using sex as a covariate of no interest.
Voxel-based morphometry.
Using the DARTEL toolbox in SPM8, we compared gray matter volume between groups. Age, sex, and total intracranial volume were added as covariates of no interest. We focused our group comparison on ROIs in the inferior parietal cortex and bilateral basal ganglia.
RESULTS
LRRK2 G2019S mutation carriers and noncarriers.
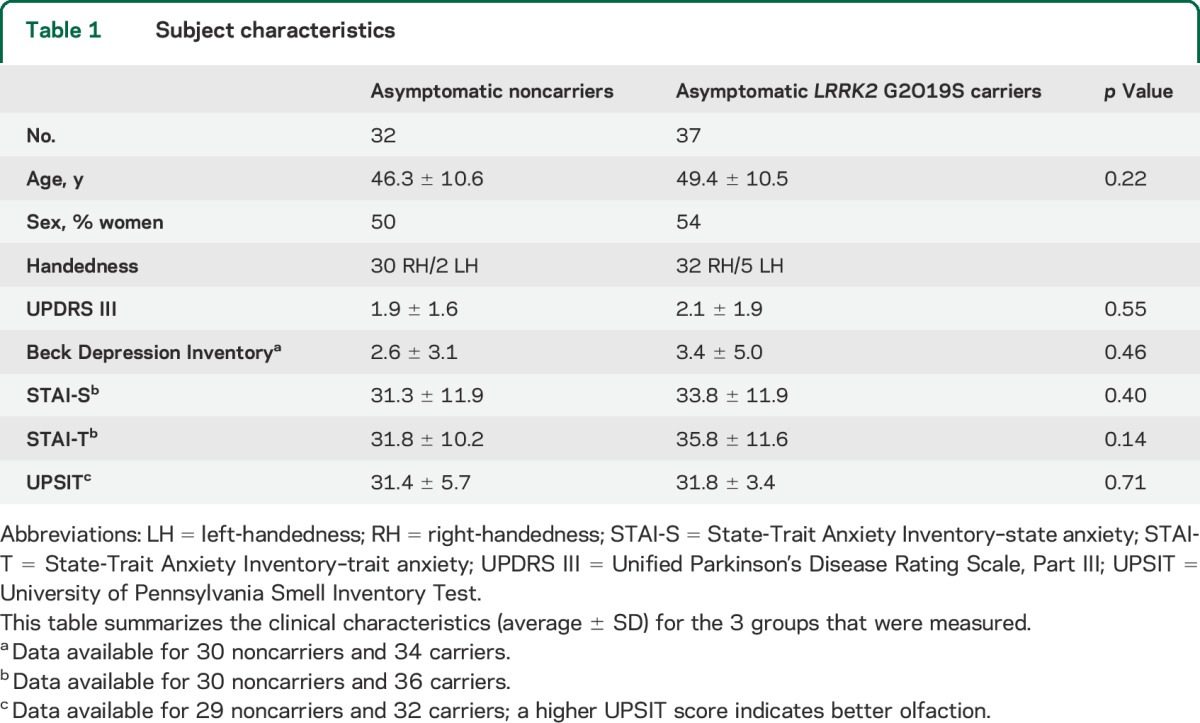
LRRK2 G2O19S mutation carriers (n = 37) and noncarriers (n = 32) did not differ in age, sex, or clinical scores (table 1). On average, carriers and noncarriers moved <0.5 mm in all 3 axes during scanning (no group difference; p > 0.5).
Table 1.
Subject characteristics
Similar corticostriatal connectivity between groups.
Both groups showed significant functional connectivity between each of the striatal subregions and other brain areas (tables e-1 to e-3 on the Neurology® Web site at Neurology.org) (figure 2). Specifically, the dorsoposterior putamen was connected to regions involved in motor execution (supplementary motor area [SMA], primary motor cortex); the ventroposterior putamen to the brainstem and cerebellum; the dorsoanterior putamen to regions involved in motor planning (pre-SMA and rostral dorsal premotor cortex; the ventroanterior putamen to temporoparietal regions; the caudate nucleus to regions involved in cognitive control (dorsolateral and dorsomedial prefrontal cortex); and the nucleus accumbens to regions involved in emotional processing (orbitofrontal cortex). The spatial distribution of these brain regions follows the known anatomy of corticostriatal loops,19 in line with a diffusion tensor imaging study in humans,20 previous resting-state fMRI studies,5,21 and a meta-analysis of cortical and striatal coactivation patterns.16
Figure 2. Shared corticostriatal connectivity between LRRK2 G2O19S mutation carriers and noncarriers.

The images represent SPM{t} maps of similar functional connectivity across groups, thresholded at p < 0.001 uncorrected (for graphical purposes), with a cluster extent threshold of 50 voxels, overlaid on anatomical images from a representative subject of the MNI (Montreal Neurological Institute) series.
Differential corticostriatal connectivity between groups.
ROI analyses.
There was a significant group (carriers > noncarriers) × striatal region (ventroanterior putamen > dorsoposterior putamen) interaction in the right rolandic operculum (MNI coordinates [+54, −10, +22], p = 0.023 FWE-corrected, t = 3.54) (figure 3). Anatomically, this region was assigned to OP4 (30% probability) in the right inferior parietal cortex. This finding indicates that corticostriatal connectivity of the right inferior parietal cortex was shifted from the dorsoposterior putamen to the ventroanterior putamen in LRRK2 G2O19S mutation carriers vs noncarriers. Using the equivalent ROI in the left hemisphere, we found the same interaction in the left inferior parietal cortex (MNI coordinates [−48, −12, +24], p = 0.011 FWE-corrected, t = 3.77). When we flipped the contrast images in the axial plane and statistically compared the strength of corticostriatal connectivity in a voxel-wise manner between both hemispheres, we observed no significant lateralization (p > 0.5). Post hoc separate analyses for the dorsoposterior and ventroanterior putamen did not reveal significant group differences. At a more liberal threshold of p < 0.001 uncorrected (cluster extent threshold of 10 voxels), we observed additional areas within the motor circuit showing a similar shift in corticostriatal connectivity (table 2).
Figure 3. Differential corticostriatal connectivity between LRRK2 G2O19S mutation carriers and noncarriers.
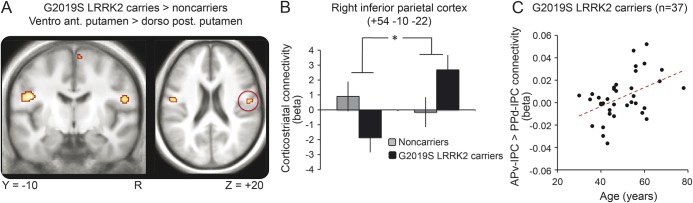
(A) SPM{t} of functional connectivity showing an interaction between group (LRRK2 G2O19S mutation carriers, noncarriers) and seed region (dorsoposterior putamen, ventroanterior putamen), overlaid onto the average MRI of all 69 subjects. For graphical purposes, the contrast is shown at a threshold of p < 0.001 uncorrected with a cluster extent threshold of 10 voxels. (B) Connectivity strength between the 2 seed regions of interest (dorsoposterior putamen, ventroanterior putamen) and the right IPC, separately for LRRK2 G2O19S mutation carriers (black bars) and matched noncarriers (gray bars). The y-axis indicates the β values (in SEM units) of a multiple regression analysis, averaged across subjects; that is, the unique contribution of each seed region's BOLD time series to the BOLD time series of the right IPC. Error bars indicate SEM (normalized to 1). (C) Correlation between age and the shift in corticostriatal connectivity in 37 LRRK2 G2O19S mutation carriers. On the y-axis, β values from the local maximum in the right IPC as shown in panel A (MNI coordinates: [+50, −18, +32] for the LRRK2 G2O19S mutation carriers). APv = ventroanterior putamen; BOLD = blood oxygen level dependent; IPC = inferior parietal cortex; MNI = Montreal Neurological Institute; PPd = dorsoposterior putamen.
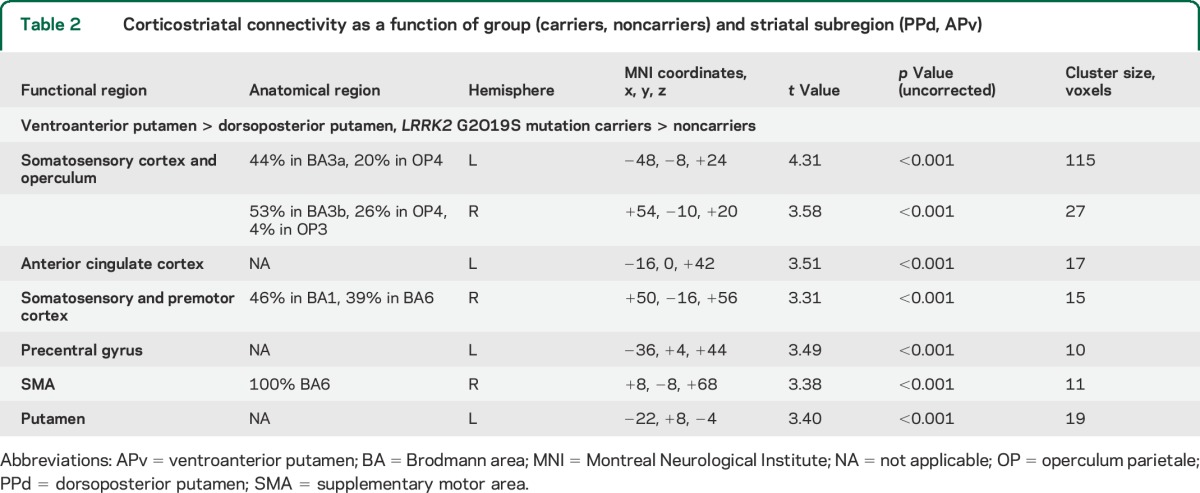
Table 2.
Corticostriatal connectivity as a function of group (carriers, noncarriers) and striatal subregion (PPd, APv)
Correlation of corticostriatal connectivity with age in LRRK2 G2O19S mutation carriers.
The shift in corticostriatal connectivity (from dorsoposterior to ventroanterior putamen) of the right inferior parietal cortex (MNI coordinates [+50, −18, +32], t = 3.63, p = 0.032 FWE-corrected) increased with age in the carriers (age range 30–78 years) but not in the noncarriers (age range 30–74 years) (figure 3). The interaction between group and age was not significant (p > 0.05). There was no significant relationship between age and functional connectivity of other striatal subregions.
Gray matter volume.
Gray matter volume in the bilateral basal ganglia or inferior parietal cortex did not differ between groups.
DISCUSSION
There are 2 main findings. First, corticostriatal connectivity of the right inferior parietal cortex was shifted from the dorsoposterior to the ventroanterior putamen in LRRK2 G2O19S mutation carriers, mirroring similar findings in idiopathic PD.5 We observed a similar shift in corticostriatal connectivity of the left inferior parietal cortex. The latter finding should be considered with more caution because our a priori hypothesis concerned the right hemisphere.5 Second, this shift in corticostriatal connectivity increased with age in LRRK2 G2O19S mutation carriers but not in noncarriers. These findings show that corticostriatal connectivity shifts from more affected to less affected striatal regions in healthy subjects at risk of developing PD.
There is a wide range of reported penetrance between 25% and 100% for the LRRK2 mutation overall.22 For Ashkenazy Jews carrying the G2019S LRRK2 mutation (i.e., the population tested here), lifetime penetrance until the age of 80 years has been estimated to be 26%.13 Thus, it is striking that we found altered corticostriatal connectivity in a group of LRRK2 G2O19S mutation carriers with an average age of ±50 years. Although the shift in corticostriatal connectivity was age-dependent, it was not driven by outliers: the cerebral effects were normally distributed over subjects (figure 3C), and removal of the 10% highest values (4 carriers) did not change the significance of the effect. Clinically healthy mutation carriers may not manifest motor symptoms because they do not develop nigrostriatal dopamine depletion, or, if they have dopamine deficiency, they may develop cerebral mechanisms that successfully compensate for nigrostriatal cell loss. Thus, the proportion of LRRK2 G2O19S mutation carriers developing symptomatic PD (26%) may not necessarily reflect the proportion of carriers developing cerebral changes. Accordingly, we previously showed that healthy LRRK2 G2O19S mutation carriers have task-related cerebral changes without behavioral impairments9,10 and subtle changes in gait and cognition23,24 without overt PD symptoms. These cerebral and behavioral markers may potentially serve to quantify cerebral dysfunction and compensatory reserve of LRRK2 G2O19S mutation carriers, in order to predict who will develop PD in the future.
Given the dorsoposterior to ventroanterior gradient of striatal dopamine depletion in PD, dopamine depletion in the premotor phase probably starts in the posterior putamen. Evidence for premotor nigrostriatal dopamine depletion has recently been shown in a large group of asymptomatic G2019S LRRK2 mutation carriers (aged 41–86 years), of whom 85% had abnormal substantia nigra hyperechogenicity (compared with 95% of patients with PD) and 44% had an abnormal dopamine transporter (DAT) scan. The carriers with an abnormal DAT scan were significantly older than the carriers with a normal DAT scan.25 Another study reported that 2 of 5 asymptomatic mutation carriers from a single R1441X LRRK2 family had abnormal DAT binding in the putamen, and one of those subjects showed a further reduction of DAT binding and subtle clinical parkinsonism a few years later.26 A third study in 2 families with Y1699C and R1441X mutations in the LRRK2 gene reported that 2 of 6 asymptomatic mutation carriers had abnormal DAT binding, with another 2 developing such abnormalities (but not clinical signs) over 4 years of follow-up.27 Finally, work in G2019S LRRK2 mice showed an age-dependent decrease in striatal dopamine content.28 These findings indicate that a large proportion of healthy LRRK2 G2O19S mutation carriers have ongoing nigrostriatal pathology, and that these pathologic changes increase with age. This fits with the age-dependent penetrance of the LRRK2 mutation.7,11,12 Based on these findings, we suggest that the age-dependent changes in corticostriatal connectivity in LRRK2 G2O19S mutation carriers reflect underlying striatal (dopaminergic) dysfunction.
In healthy LRRK2 G2O19S mutation carriers, corticostriatal connectivity of the inferior parietal cortex was shifted toward the ventroanterior putamen. This change may reflect compensatory adaptation, primary pathology, or a combination of both. A compensatory remapping of corticostriatal connectivity is constrained by existing anatomical connections and by the compensatory potential of the cortical region. In humans, the parietal cortex is anatomically connected to the dorsoposterior putamen but also to smaller portions of the ventroanterior putamen.20 The inferior parietal cortex provides strong visual and somatosensory input to the ventral premotor cortex,29 and in patients with PD, this ventral parieto-premotor circuit is hyperactivated during hand movements.30 This may serve to compensate for impaired activity in medial motor circuits (such as the SMA) that are functionally connected to the posterior putamen.31 The shift in corticostriatal connectivity of the inferior parietal cortex in LRRK2 G2O19S mutation carriers also occurred within the parietal area where these same subjects showed increased task-related activity during a Stroop task.9 The matched behavioral performance between LRRK2 G2O19S mutation carriers and noncarriers in that study indicates that the increased task-related parietal activity is compensatory in nature. In symptomatic PD, the shift in corticostriatal connectivity was larger in the least affected hemisphere than in the most affected hemisphere.5 Another study reported a shift in corticostriatal connectivity only in the right inferior parietal cortex, but most pronounced in patients with PD in whom the right hemisphere was less affected.32 This supports the idea that the change in corticostriatal connectivity reflects a compensatory mechanism. The lack of lateralization in our current sample of asymptomatic LRRK2 G2019S mutation carriers may be caused by the fact that we could not distinguish between most and least affected hemispheres, given the lack of clinical symptoms. Taken together, these findings suggest that LRRK2 G2O19S mutation carriers shift their computational resources to brain regions relatively unaffected by nigrostriatal dopamine depletion (i.e., the ventroanterior putamen), exploiting existing anatomical connections of this striatal region to cortical areas involved in motor control and cognition (i.e., the inferior parietal cortex).
The change in corticostriatal connectivity may also reflect pathologic changes occurring in the striatum. For instance, dopamine depletion in the basal ganglia is associated with a loss of segregation between parallel corticostriatal loops.33 This may lead to functional crosstalk between different neural systems, with detrimental consequences. For example, it may lead to impaired sensorimotor integration, which has been found in healthy Parkin mutation carriers.34 Furthermore, crosstalk between motor and cognitive corticostriatal loops may lead to impaired dual tasking, which is present both in patients with PD35 and in healthy LRRK2 G2O19S mutation carriers.23
Our voxel-based morphometry analysis suggests that the effects we report are unlikely caused by structural changes in LRRK2 G2O19S mutation carriers. Other studies in different cohorts have reported a subtle increase in gray matter volume of the caudate36 and parietal cuneus37 of asymptomatic LRRK2 mutation carriers, although the smaller sample size of these studies warrants caution. An important limitation of the current study is its cross-sectional nature. Future studies may longitudinally follow the cohort of LRRK2 G2O19S mutation carriers reported here, testing how the shift in corticostriatal connectivity changes after subjects develop PD. In a similar vein, primate models of PD may test corticostriatal connectivity at different time points during progressive MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) intoxication, in order to better outline the timing of compensatory mechanisms in a single cohort.38
We propose that the age-dependent shift in corticostriatal connectivity in healthy LRRK2 G2019S mutation carriers is compensatory in nature. This provides a window of opportunity for implementing compensation-enhancing therapies. Such therapies may involve stimulation of compensatory brain areas using repetitive transcranial magnetic stimulation,39 or behavioral training—for example, learning motor routines that involve the ventroanterior rather than the dorsoposterior putamen.40
Supplementary Material
ACKNOWLEDGMENT
The authors thank the participants of this project for their invaluable participation and the staff of the AJ consortium for their contribution: Beth Israel Medical Center, New York, NY: Brooke Johannes. Tel Aviv Sourasky Medical Center, Israel: Maayan Zalis, DMD, Yaacov Balash, MD, Shabtai Hertzel, MD, Ziv Gan Or, PhD, Hila Kobo, MSc, Noa Bregman, MD, Meir Kestenbaum, MD, Talma Hendler, MD, PhD, Hedva Lehrman, MD, Einat Even Sapir, MD, PhD, Shiran Levy, Anat Shkedy, MD, and Michal Shtaigman, RN.
GLOSSARY
- DAT
dopamine transporter
- FWE
family-wise error
- GBA
β-glucocerebrosidase
- LRRK2
leucine-rich repeat kinase 2
- MNI
Montreal Neurological Institute
- PD
Parkinson disease
- ROI
region of interest
- SMA
supplementary motor area
- SPM
Statistical Parametric Mapping
Footnotes
Supplemental data at Neurology.org
Contributor Information
Collaborators: Susan Bressman, Rachel Saunders-Pullman, Deborah Raymond, Laurie Ozelius, Vicki Shanker, Mark Groves, Christina Palmese, Akhila Iyer, Jeannie Soto-Valencia, Kaili Maloy Stanley, Marta San Luciano, Andres Deik, Matthew James Barrett, Jose Cabassa, Lawrence Severt, Ann Hunt, Naomi Lubarr, Rivka Sachdev, Joan Miravite, Sara Lewis, Lorraine N. Clark, Roy Alcalay, Stanley Fahn, Lucien Cote, Paul Greene, Cheryl Waters, Pietro Mazzoni, Blair Ford, Elan Louis, Oren Levy, Ming Xin Tang, Brian C. Rakitin, Helen Mejia, Ernest Roos, Martha Orbe Riley, Llency Rosado, Carol Moskowitz, Tsvyatko Dorovski, Lorraine Clark, Xinmin Liu, Sergey Kisselev, Itsik Peer, and Vladimir Vacic
AUTHOR CONTRIBUTIONS
Rick Helmich: analysis and interpretation of the data and drafting the manuscript. Avner Thaler: recruitment, clinical assessment, collection of the data, and revising the manuscript. Bart van Nuenen: interpretation of the data and revising the manuscript. Tanya Gurevich: clinical assessment and revision of the manuscript. Anat Mirelman: interpretation of the data and revising the manuscript. Karen Marder: design and conceptualization of the study, obtain funds, interpretation of data, revising the manuscript. Susan Bressman: design and conceptualization of the study, obtain funds, interpretation of data, revising the manuscript. Avi Orr-Urtreger: design and conceptualization of the study, obtain funds, genetic testing, revising the manuscript. Nir Giladi: design and conceptualization of the study, obtain funds, interpretation of data, revising the manuscript. Bas Bloem: design and conceptualization of the study, obtain funds, interpretation of data, revising the manuscript. Ivan Toni: design and conceptualization of the study, obtain funds, interpretation of data, revising the manuscript.
STUDY FUNDING
This work was supported by the Michael J. Fox Foundation for Parkinson Research and the Netherlands Organization for Scientific Research (VIDI grant 016.076.352 to B.R.B.).
DISCLOSURE
R. Helmich reports a grant from the Dutch Brain Foundation and the Van Leersum Foundation, outside the submitted work. A. Thaler and B. Van Nuenen report no disclosures relevant to the manuscript. T. Gurevich reports grants from Radboud University, grants from MJF Foundation, during the conduct of the study; grants from MJFF, grants from NPF, support from the pharmaceutical companies Teva, Novartis, AbbVie, and Medtronic, outside the submitted work. A. Mirelman reports no disclosures relevant to the manuscript. K. Marder reports grants from the Michael J. Fox Foundation, NIH grants NS036630, 1UL1RR024156, and Po412196, grants from U01NS052592, grants from Parkinson's Disease Foundation, grants from Huntington's Disease Foundation, and grants from CHDI, outside the submitted work. S. Bressman serves on the advisory boards of the Michael J. Fox Foundation, the Dystonia Medical Research Foundation, the Bachmann-Strauss Dystonia & Parkinson Foundation, and the board of We Move. She has consulted for Bristol-Myers Squibb. She has received research support from the Michael J. Fox Foundation, NIH, and Dystonia Medical Research Foundation. A. Orr-Urtreger reports no disclosures relevant to the manuscript. N. Giladi reports other from Teva Lundbeck, other from IntecPharma, other from NeuroDerm, other from Aramon Neuromedical Ltd., other from Pharma Two B, other from Novartis, other from UCB, other from MDS, grants from Michael J. Fox Foundation, grants from NPF, grants from EU 7th Framework, grants from ISF, and grants from Teva Innovative Ltd., outside the submitted work. B. Bloem reports grants from Netherlands Organization for Scientific Research, grants from Prinses Beatrix Foundation, grants from Stichting Internationaal Parkinson Fonds, grants from Alkemade-Keuls fonds, grants from Michael J. Fox Foundation, personal fees from Danone, personal fees from GlaxoSmithKline, personal fees from UCB, and personal fees from AbbVie, outside the submitted work. I. Toni reports no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Kordower JH, Olanow CW, Dodiya HB, et al. Disease duration and the integrity of the nigrostriatal system in Parkinson's disease. Brain 2013;136:2419–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palop JJ, Chin J, Mucke L. A network dysfunction perspective on neurodegenerative diseases. Nature 2006;443:768–773. [DOI] [PubMed] [Google Scholar]
- 3.Brück A, Aalto S, Nurmi E, Vahlberg T, Bergman J, Rinne JO. Striatal subregional 6-[18F]fluoro-l-dopa uptake in early Parkinson's disease: a two-year follow-up study. Mov Disord 2006;21:958–963. [DOI] [PubMed] [Google Scholar]
- 4.Mounayar S, Boulet S, Tande D, et al. A new model to study compensatory mechanisms in MPTP-treated monkeys exhibiting recovery. Brain 2007;130:2898–2914. [DOI] [PubMed] [Google Scholar]
- 5.Helmich RC, Derikx LC, Bakker M, Scheeringa R, Bloem BR, Toni I. Spatial remapping of cortico-striatal connectivity in Parkinson's disease. Cereb Cortex 2010;20:1175–1186. [DOI] [PubMed] [Google Scholar]
- 6.Poulopoulos M, Levy OA, Alcalay RN. The neuropathology of genetic Parkinson's disease. Mov Disord 2012;27:831–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Healy DG, Falchi M, O'Sullivan SS, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson's disease: a case-control study. Lancet Neurol 2008;7:583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mirelman A, Heman T, Yasinovsky K, et al. Fall risk and gait in Parkinson's disease: the role of the LRRK2 G2019S mutation. Mov Disord 2013;28:1683–1690. [DOI] [PubMed] [Google Scholar]
- 9.Thaler A, Mirelman A, Helmich RC, et al. Neural correlates of executive functions in healthy G2019S LRRK2 mutation carriers. Cortex 2013;49:2501–2511. [DOI] [PubMed] [Google Scholar]
- 10.van Nuenen BFL, Helmich RC, Ferraye M, et al. Cerebral pathological and compensatory mechanisms in the premotor phase of leucine-rich repeat kinase 2 parkinsonism. Brain 2012;135:3687–3698. [DOI] [PubMed] [Google Scholar]
- 11.Hulihan MM, Ishihara-Paul L, Kachergus J, et al. LRRK2 Gly2019Ser penetrance in Arab-Berber patients from Tunisia: a case-control genetic study. Lancet Neurol 2008;7:591–594. [DOI] [PubMed] [Google Scholar]
- 12.Kachergus J, Mata IF, Hulihan M, et al. Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations. Am J Hum Genet 2005;76:672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marder K, Tang M, Alcalay R, et al. Age specific penetrance of the LRRK2 G2019S mutation in the Michael J. Fox Ashkenazi Jewish (AJ) LRRK2 Consortium. Neurology 2014;82(suppl):S17.002 Abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gan-Or Z, Giladi N, Rozovski U, et al. Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology 2008;70:2277–2283. [DOI] [PubMed] [Google Scholar]
- 15.Fox MD, Raichle ME. Spontaneous fluctuations in brain activity observed with functional magnetic resonance imaging. Nat Rev Neurosci 2007;8:700–711. [DOI] [PubMed] [Google Scholar]
- 16.Postuma RB, Dagher A. Basal ganglia functional connectivity based on a meta-analysis of 126 positron emission tomography and functional magnetic resonance imaging publications. Cereb Cortex 2005;16:1508–1521. [DOI] [PubMed] [Google Scholar]
- 17.Lund TE, Nørgaard MD, Rostrup E, Rowe JB, Paulson OB. Motion or activity: their role in intra- and inter-subject variation in fMRI. NeuroImage 2005;26:960–964. [DOI] [PubMed] [Google Scholar]
- 18.Friston KJ, Ashburner JT, Kiebel SJ, Nichols TE, Penny WD. Statistical Parametric Mapping: The Analysis of Functional Brain Images. Waltham, MA: Academic Press; 2011. [Google Scholar]
- 19.Alexander GE, DeLong MR, Strick PL. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu Rev Neurosci 1986;9:357–381. [DOI] [PubMed] [Google Scholar]
- 20.Draganski B, Kherif F, Kloppel S, et al. Evidence for segregated and integrative connectivity patterns in the human basal ganglia. J Neurosci 2008;28:7143–7152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Di Martino A, Scheres A, Margulies D, et al. Functional connectivity of human striatum: a resting state fMRI study. Cereb Cortex 2008;18:2735–2747. [DOI] [PubMed] [Google Scholar]
- 22.Goldwurm S, Tunesi S, Tesei S, et al. Kin-cohort analysis of LRRK2-G2019S penetrance in Parkinson's disease. Mov Disord 2011;26:2144–2145. [DOI] [PubMed] [Google Scholar]
- 23.Mirelman A, Gurevich T, Giladi N, Bar-Shira A, Orr-Urtreger A, Hausdorff JM. Gait alterations in healthy carriers of the LRRK2 G2019S mutation. Ann Neurol 2011;69:193–197. [DOI] [PubMed] [Google Scholar]
- 24.Thaler A, Mirelman A, Gurevich T, et al. Lower cognitive performance in healthy G2019S LRRK2 mutation carriers. Neurology 2012;79:1027–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sierra M, Sánchez-Juan P, Martínez-Rodríguez MI, et al. Olfaction and imaging biomarkers in premotor LRRK2 G2019S-associated Parkinson disease. Neurology 2013;80:621–626. [DOI] [PubMed] [Google Scholar]
- 26.Nandhagopal R, Mak E, Schulzer M, et al. Progression of dopaminergic dysfunction in a LRRK2 kindred: a multitracer PET study. Neurology 2008;71:1790–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams JR, Van Netten H, Schulzer M, et al. PET in LRRK2 mutations: comparison to sporadic Parkinson's disease and evidence for presymptomatic compensation. Brain 2005;128:2777–2785. [DOI] [PubMed] [Google Scholar]
- 28.Li X, Patel JC, Wang J, et al. Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson's disease mutation G2019S. J Neurosci 2010;30:1788–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gharbawie OA, Stepniewska I, Qi H, Kaas JH. Multiple parietal-frontal pathways mediate grasping in macaque monkeys. J Neurosci 2011;31:11660–11677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Samuel M, Ceballos-Baumann AO, Blin J, et al. Evidence for lateral premotor and parietal overactivity in Parkinson's disease during sequential and bimanual movements: a PET study. Brain 1997;120:963–976. [DOI] [PubMed] [Google Scholar]
- 31.Lehericy S, Ducros M, Krainik M, et al. 3-D diffusion tensor axonal tracking shows distinct SMA and pre-SMA projections to the human striatum. Cereb Cortex 2004;14:1302–1309. [DOI] [PubMed] [Google Scholar]
- 32.Hacker CD, Perlmutter JS, Criswell SR, Ances BM, Snyder AZ. Resting state functional connectivity of the striatum in Parkinson's disease. Brain 2012;135:3699–3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bergman H, Feingold A, Nini A, et al. Physiological aspects of information processing in the basal ganglia of normal and parkinsonian primates. Trends Neurosci 1998;21:32–38. [DOI] [PubMed] [Google Scholar]
- 34.Baumer T, Pramstaller PP, Siebner HR, et al. Sensorimotor integration is abnormal in asymptomatic Parkin mutation carriers: a TMS study. Neurology 2007;69:1976–1981. [DOI] [PubMed] [Google Scholar]
- 35.Pessiglione M, Czernecki V, Pillon B, et al. An effect of dopamine depletion on decision-making: the temporal coupling of deliberation and execution. J Cogn Neurosci 2005;17:1886–1896. [DOI] [PubMed] [Google Scholar]
- 36.Reetz K, Tadic V, Kasten M, et al. Structural imaging in the presymptomatic stage of genetically determined parkinsonism. Neurobiol Dis 2010;39:402–408. [DOI] [PubMed] [Google Scholar]
- 37.Brockmann K, Gröger A, Di Santo A, et al. Clinical and brain imaging characteristics in leucine-rich repeat kinase 2-associated PD and asymptomatic mutation carriers. Mov Disord 2011;26:2335–2342. [DOI] [PubMed] [Google Scholar]
- 38.Bezard E. Presymptomatic compensation in Parkinson's disease is not dopamine-mediated. Trends Neurosci 2003;26:215–221. [DOI] [PubMed] [Google Scholar]
- 39.van Nuenen BFL, Helmich RC, Buenen N, Van De Warrenburg BPC, Bloem BR, Toni I. Compensatory activity in the extrastriate body area of Parkinson's disease patients. J Neurosci 2012;32:9546–9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cramer SC, Sur M, Dobkin BH, et al. Harnessing neuroplasticity for clinical applications. Brain 2011;134:1591–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.