

Abstract
Aims/hypothesis
The regenerative process in the pancreas is of particular interest, since insulin-producing beta cells are lost in diabetes. Differentiation of new beta cells from pancreatic non-endocrine cells has been reported in vivo and in vitro, a finding that implies the existence of pancreatic stem/progenitor cells. However, while tissue-specific stem cells are well documented in skin, intestine and testis, pancreatic stem cells have been elusive. We hypothesised that pancreatic stem/progenitor cells within the non-endocrine fraction could be a source of new islets in vitro.
Methods
To test if there were such cells within the pancreas, we generated pancreatic cell aggregates from tissue remaining after islet isolation from mouse insulin promoter 1–green fluorescent protein (MIP-GFP) mice. To eliminate any contamination of insulin-positive cells, we deleted all GFP-positive aggregates using COPAS Select and cultured with Matrigel. Immunohistochemistry, quantitative real-time PCR and single-cell nested RT-PCR were performed to confirm formation of insulin-producing cells.
Results
The GFP-negative cells were expanded as monolayers and then differentiated into three-dimensional cystic structures. After 1 week of culture, GFP-positive cells were found as clusters or single cells. By quantitative real-time PCR, no insulin mRNA was detected immediately after COPAS sorting, but after differentiation insulin mRNA of the whole preparation was 1.91±0.31% that of purified MIP-GFP beta cells. All GFP-positive cells expressed insulin 1; most expressed insulin 2, pancreas duodenum homeobox-1 and cytokeratin 19 by single cell nested RT-PCR.
Conclusions/interpretation
Our data support the concept that within the exocrine (acinar and ductal) pancreas of the adult mouse there are cells that can give rise to insulin-positive cells in vitro.
Keywords: Beta cell, Duct, Neogenesis, Pancreas
Introduction
Beta cell replacement therapy has been shown to be effective as a cure for type 1 diabetes. However, a major obstacle to successful islet transplantation has been an inadequate supply of insulin-producing tissue [1]. This need for transplantable tissue has stimulated efforts to expand existing pancreatic islets and/or grow new ones. The regenerative process in the pancreas is of particular interest. While tissue-specific stem cells are well documented in skin, intestine and testis, pancreatic stem cells have been elusive at best [1-3]. It is clear in the pancreas that replication is an important mechanism for renewal and growth, but compelling data suggest that progenitors[4], or even possibly stem cells, have a role in renewal and growth after birth. Generation of insulin-containing islet buds from cultures enriched with murine or human duct epithelial cell in vitro has led to the suggestion that pancreas stem or progenitor cells exist within the pancreatic ducts [5-9]. There is also strong in vivo evidence of islet differentiation from pancreatic duct cells [10-14]. Additionally, purified (CA19-9 positive) duct cells from adult human pancreas can differentiate to insulin-producing cells under kidney capsule of NOD/SCID mouse [15, 16]. Thus, there has been renewed interest in the identification, expansion and differentiation of adult progenitors as well as in enhancing the expansion of pre-existing beta cells.
One problem of experiments performed with pancreatic cells is the possible presence of pre-existing islet cells at the onset of culture. In many of the above experiments, ductal cells were obtained by gradient centrifugation, manual isolation or cultivation of crude pancreatic fractions, so contamination of pre-existing beta cells cannot be definitively excluded. Therefore, to delete pre-existing beta cells completely, we used pancreas from transgenic (mouse insulin promoter 1–green fluorescent protein [MIP-GFP]) mice, in which all insulin-positive beta cells also GFP–positive [17], together with the large orifice flow cytometer COPAS Select (Union Biometrica, Holliston, MA, USA) to eliminate all GFP-positive cells for our starting culture material. With traditional flow cytometers, which use a large electrostatic charge sorting method, it is necessary to use single cell suspensions, which often results in very low cell viability. However, the COPAS Select is an air actuated large orifice flow sorter [18] that differs from traditional flow cytometers in two important design areas, thereby permitting larger objects to be analysed and dispensed. First, the large-bore fluidics and flow cell can accommodate objects as large as the 500 μm clusters in this study. Second, the pneumatic sorting mechanism (located right after the flow cell) sorts objects by temporarily switching an air diverter off and on again to generate a slice of liquid containing the object to be sorted. This sorting method is gentle enough to permit the collection of live biological materials and has been found to have little effect on post analysis viability. These tools enabled us to culture GFP-negative cell clusters in order to test their ability to differentiate to insulin-positive cells in vitro. We report here that insulin-positive cells can be differentiated from islet-depleted pancreatic cells after COPAS sorting, suggesting that pancreatic stem/progenitor cells do exist in the adult pancreas.
Methods
Cell isolation
MIP-GFP transgenic mice were a gift from M. Hara (Department of Medicine, University of Chicago, IL, USA) and were backcrossed ten times with C57BL/6J mice. Using a modification of murine islet isolation [19], pancreases from adult MIP-GFP mice were digested by collagenase (Liberase RI; Roche, Indianapolis, IN, USA), separated on Ficoll gradient and the pellet containing granulated acinar tissue and small intercalated ducts was collected. Less than 1% of the pellet was GFP-positive. This islet-deleted tissue was then suspended in RPMI (11 mmol/l glucose) medium with 10% (by volume) fetal bovine serum and antibiotics, and cultured on non-sticky dishes at 37°C. After 3 days of suspension, aggregated tissue was separated by gravity (5 min) three times and the supernatant aspirated to remove low-density components, including dead cells, each time. About 80 to 90% of the initial cells were dead at this stage. All animal procedures were approved by the Joslin Institutional Animal Care and Use Committees.
COPAS sorting and MoFlo sorting
The aggregates were then sorted in RPMI medium with 10% fetal bovine serum on a COPAS Select (Union Biometrica) at a flow rate of 20 to 40 events/s. Clusters containing GFP-positive cells were detected by GFP excitation and deleted. By microscopic examination of sorted clusters, aggregates (40–250 μm diameter) were determined to efficiently include the viable epithelial cell clusters. To assess deletion of GFP-positive cells, 10,000 events were analysed for purity by fluorescence microscopy. Additionally, aliquots of GFP-negative fractions from COPAS sorts were dispersed in trypsin-DNase at 37°C for 15 min and then sorted for viability, forward scatter and GFP excitation on a MoFlo (Cytomation, Fort Collins, CO, USA) and analysed using Summit software (Cytomation).
Cell culture for differentiation
Sorted aggregates were collected, washed and plated on to collagen-coated plates (rat tail collagen type 1, at 5 μg/cm2; BD, Franklin Lakes, NJ, USA) at a density of 2000 clusters/cm2 in DMEM/F12 medium (17 mmol/l glucose) with 10% fetal bovine serum, 20 ng/ml epidermal growth factor (EGF) (BD) and antibiotics. After 24 h, media were changed to serum-free DMEM/F12 medium (17 mmol/l glucose) with 1 g/l insulin–transferrin–selenium supplement (Roche, Nutley, NJ, USA), 2 g/l BSA, 10 nmol/l nicotinamide (Sigma-Aldrich, St. Louis, MO, USA), 5 nmol/l exendin-4 (Sigma) and 20 ng/ml EGF (BD). Within 4 to 5 days adherent cells made monolayers that were maintained for 3 to 4 weeks with additional growth factors and Matrigel (BD).
Immunostaining
Cells/aggregates were cultured in culture slides (BD Falcon) and fixed in freshly made 4% (by weight) paraformaldehyde in PBS. For antigen retrieval, microwaving in 10 mmol/l citrate buffer pH 6.0 was done, followed by blocking in 3% (by volume) H2O2 and tyramide blocking buffer (PerkinElmer, Waltham, MA, USA). Slides were incubated overnight at 4°C with Dolichos biflorus (DBA) lectin (1:2000, biotinylated; VectorLabs, Burlingame, CA, USA), incubated for 15 min at room temperature with streptavidin horseradish peroxidase conjugate (1:100; PerkinElmer) and developed using Rhodamine-tyramide (1:50; PerkinElmer) for 5 min at room temperature. Subsequently, for insulin immunostaining, guinea pig anti-bovine insulin antibody (1:100; Linco Research, St. Charles, MO, USA) was applied for 1 h as primary antibody, then slides were washed and incubated with FITC-conjugated donkey anti-guinea pig immunoglobulin G (1:200; Jackson ImmunoResearch, West Grove, PA, USA) at room temperature for 1 h. Sections were examined in confocal mode on a Zeiss LSM 410 microscope using appropriate filters.
Quantitative real-time PCR
Quantitative real-time PCR was performed using the ABI 7300 real-time PCR system (Applied Biosystems, Forest City, CA, USA) according to the manufacturer’s instructions. The probe and primer sets of mouse insulin 1 (Assay ID Mm01259683_g1) and beta-actin (Assay ID Mm00607939_s1) were purchased from Applied Biosystems. Total RNA was isolated from total cultured population using a kit (RNeasy Mini kit; Qiagen, Valencia, MD, USA). To assess the integrity of total RNA, an aliquot of the RNA sample was run on a denaturing agarose gel. Reverse-transcription was performed using a kit (High-Capacity cDNA; Applied Biosystems). cDNA (10 ng) was applied to each well and levels of mRNA were determined as the average of triplet aliquots. Insulin mRNA expression was normalised to that of the internal control beta-actin and then compared using the delta delta Ct method with that of pure GFP-positive beta cells from MIP-GFP mice, which had been run in parallel.
Analysis of single cells by RT-PCR
Multiplex single-cell RT-PCR analysis was performed according to the method by Hu et al.[20] with minor modifications [21]. Briefly, propidium iodide-negative, GFP-positive differentiated cells were collected, after which a second sort for the same gate was performed. These double-sorted single cells were deposited into 96-well reaction plates containing lysis buffer (0.5% Triton X-100 by volume) and reverse transcription buffer. Cell lysates were reverse-transcribed using eight pairs of gene-specific reverse primers with final concentrations: 1X first strand buffer (Invitrogen, Carlsbad, CA, USA), 10 mmol/l dithiothreitol (Invitrogen), 1 mmol/l deoxycytidine triphosphates (New England BioLabs, Ipswich, MA, USA), 0.5% Triton X-100 (Sigma), 0.1% (by weight) BSA, 10 U/μl moloney murine leukaemia virus reverse transcriptase (Invitrogen), 0.1 U/μl RNase inhibitor (Invitrogen) and 0.4 pmol/l reverse primers. Cells were lysed by rapid up-and-down pipetting several times, after which cell lysates were transferred to 200 μl thin-wall PCR tubes. After incubation at 37°C for 90 min, the samples were incubated at 94°C for 30 s to inactivate the enzyme. The first-round PCR was carried out in the same tube by addition of premixed PCR buffer containing the gene-specific forward primers, i.e. 1X GeneAmp PCR Gold Buffer (Applied Biosystems), 2.5 mmol/l MgCl2, AmpliTaq Gold 0.1 U/μμl (Applied Biosystems) and 0.1 pmol/l forward primers. The total volume of the first PCR reactions was 30 μl; PCRs were performed as follows: 36 cycles of 30 s at 94°C, 90 s at 60°C and 90 s at 72°C. Two per cent of aliquots of the first-round PCR reactions were replica-plated into new PCR tubes for the second-round PCR, which was carried out separately for each gene with fully nested gene-specific primers (1X GeneAmp PCR Gold Buffer, 2.5 mmol/l MgCl2, AmpliTaq Gold 0.1 U/μl and 0.25 pmol/l forward primers). The second-round PCR was performed as follows: 35 cycles of 30 s at 94°C, 90 s at 60°C and 90 s at 72°C. Aliquots of second-round PCR products were then subjected to gel electrophoresis. Since the primers are designed to span at least one intron, genomic products can be excluded by their larger size. The sequences of external and internal oligonucleotide primers for each gene are in Electronic supplementary material (ESM) Table 1. Double-sorted GFP-positive cells from MIP-GFP mice and microdissected duct tissue were used as controls. Around 90% of single cell-deposited wells yielded successful hypoxanthine-guanine phosphoribosyl transferase (HPRT) amplification. Results from HPRT-positive wells were counted.
Results
Depletion of insulin-positive clusters by COPAS Select
After collagenase digestion, we started with material obtained from the pellet after Ficoll gradient, which was mainly composed of small clusters of acinar cells with some pieces of duct (Fig. 1a). After 48 h culture, most clusters were fragmented. By 72 h, 80 to 90% of the initial cells were dead and floating. Viable cells made dense, relatively larger clusters than the initial clumps of exocrine cells (Fig. 1b). Their size was similar to that of isolated islets with most from 50 to 200 μm in diameter. By gravity, these aggregates were collected in a tube and sorted with COPAS Select (Fig. 1c).
Fig. 1.
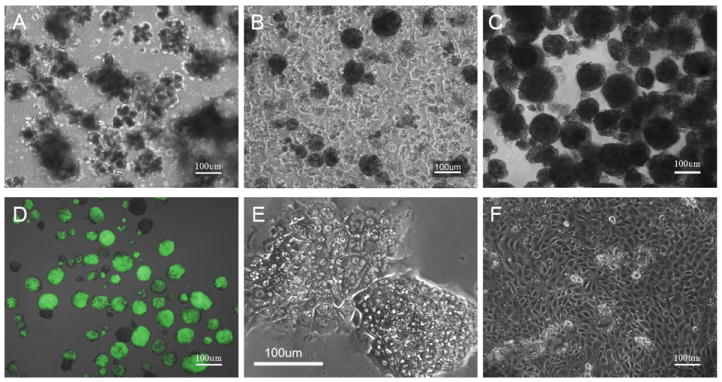
Cultured clusters from MIP-GFP mouse pancreas. a Immediately after digestion, cells are only loosely clustered. b, c Cells at 72 h after digestion before (b) and after (c) COPAS sort. d GFP-positive clusters including whole islets or only containing single GFP-positive cells were collected after COPAS sort. e After COPAS sort and 24 h culture, sorted GFP-negative aggregates attach and spread. f Epithelial monolayer 4 days after COPAS sort. Scale bars, 100 μm
Since the isolated pancreas was from MIP-GFP mice and GFP was intense in the beta cells, contaminated beta cells were easily detected even under a fluorescent microscope. Similarly, COPAS Select clearly separated the beta cells from other cells according to GFP emission, which is referred to as green peak height (Fig. 2). COPAS even detected single GFP-positive cells in a cluster. We kept the samples in medium at room temperature during the sort, since the GFP-negative clusters were relatively stable at room temperature in contrast to at 4°C.
Fig. 2.
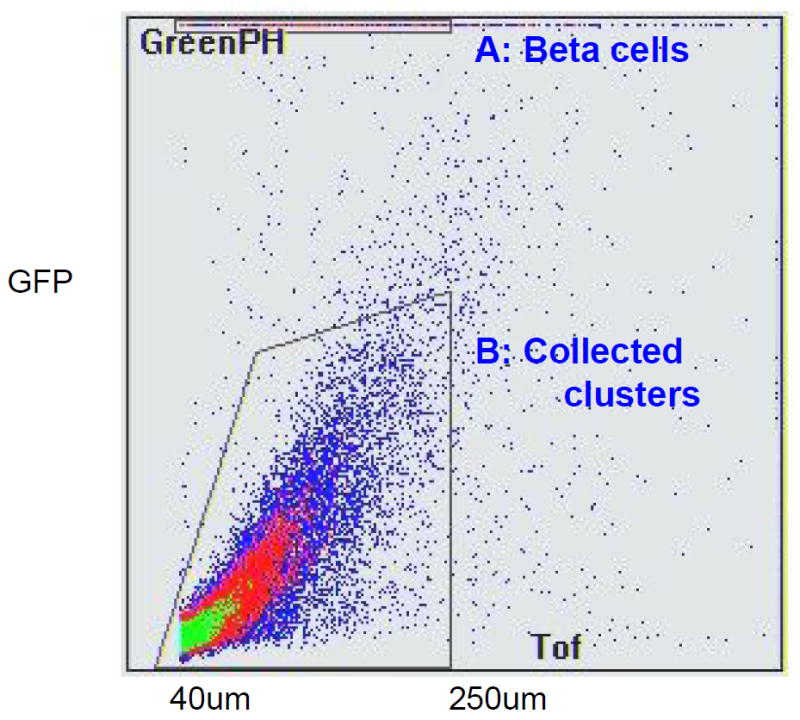
GFP-negative aggregates collected with COPAS sort. GFP-negative clusters (40 to 250 μm diameter) were collected for culturing (Gated area shown by black dashed lines). A:Beta cells, B: collected clusters. Time of flight represents axial length of the object (size); green-peak height (Greeen PH) shows GFP emission
We gated for the axial length of the object (size), which is referred to as time of flight, and collected clusters from 40 to 250 μm diameter. We chose to collect this population because smaller clusters had many dead cells and poor viability, while the bigger clusters had central necrosis. Approximately, 35 to 50% of all sorted clusters were collected. The sorting was gentle enough to continue the culture process after sorting. The lack of increased propidium iodide uptake and lack of change of size distribution of clusters before and after COPAS sorting suggested that the process did not affect viability. Sorted clusters in a hydrophobic dish could be maintained as healthy aggregates for about 1 week and were able to expand if plated on collagen-coated dishes. After COPAS sorting, we verified by fluorescence microscopy that there were no GFP-positive clusters. Additionally, to examine the accuracy of COPAS sorting, we dispersed aliquots of GFP-negative clusters into single cells by trypsin and sorted these on the MoFlo cytometer to examine whether any contamination of GFP-positive cells had occurred. As shown in Fig. 3, there was no contamination of GFP-positive cells after COPAS sorting.
Fig. 3.
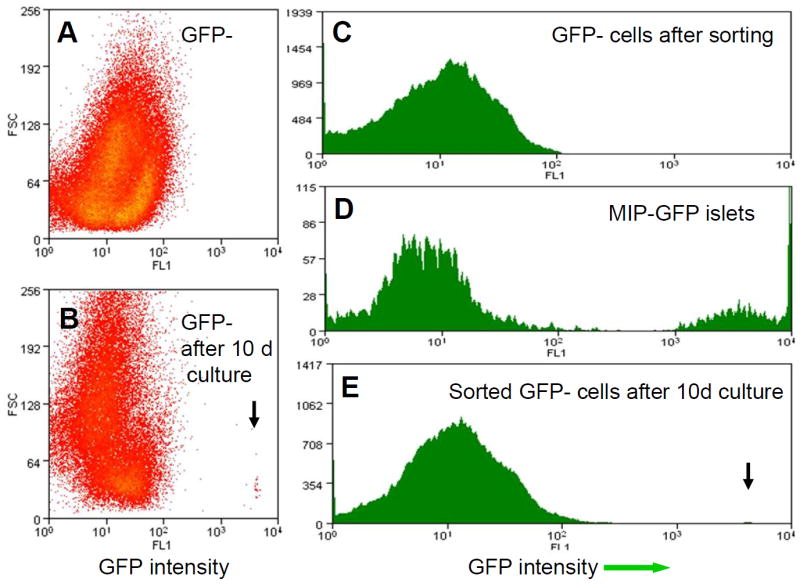
MoFlo sort. a, c MoFlo sort of dispersed GFP-negative samples after COPAS sort showed that there were no GFP cells in this population. b, e GFP-positive differentiated cells in the GFP-negative population 10 days after the COPAS sort. Arrow, new GFP-positive cells. d The MIP-GFP islet sample from COPAS serves as a positive control. FL1,conventionally used for green channel; FSC forward scatter which indicates cell volume
Growth and differentiation of epithelial monolayers
GFP-negative clusters started to attach soon after the sorting (Fig. 1e), particularly when plated on collagen- or fibronectin-coated dishes. The attachment rate at 24 h was 5 to 23% of total plated clusters (mean± SD 15± 5.9%, n= 3 experiments). At this point, the medium was changed to serum free medium. Serum-free medium was important for differentiation and to inhibit mesenchymal cell growth. Within a week, the aggregates made homogeneous monolayers (Fig. 1f) that resembled the cobblestone epithelium of cultured isolated duct tissue. Monolayers from GFP-negative aggregates and isolated pancreatic ducts had positive immunostaining for three duct cell markers: pan-cytokeratin (Dako,Glostrup, Denmark), DBA lectin and CD133 (eBioscience, San Diego, CA, USA) [22] (data not shown). Fibroblastic cells sometimes surrounded a monolayer plaque. At 1 week the percentage of vimentin-positive fibroblastic cells was usually less than 2%, unless the viability of sorted clusters was poor. The monolayer cells expanded well with EGF. Previously EGF receptor signalling has been reported to stimulate proliferation and branching of fetal pancreatic duct, with impaired proliferation and delayed islet cell differentiation when no EGF signal is present [23].
Once monolayers were formed (4–5 days), Matrigel was overlaid with additional growth factors. Within 3 days, Matrigel facilitated three-dimensional cystic structures, in which GFP-positive cells were often seen as clusters or single cells (Fig. 4). The percentage of GFP-positive cells was 1% to 3% of total cells (n= 3 experiments). The addition of hepatocyte growth factor, basic fibroblast growth factor, exendin 4, activin A and betacellulin separately or as a cocktail to facilitate differentiation did not change the proportion of GFP-positive cells (data not shown), even though these factors have been reported to induce differentiation of non-beta cells into insulin-producing cells.
Fig. 4.
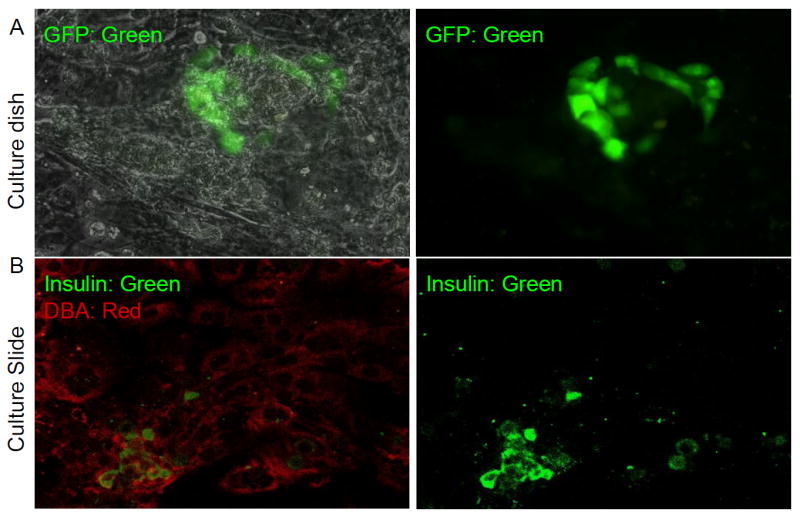
GFP/insulin protein is expressed in cystic structures of epithelial monolayer from GFP- population 10 days after COPAS sort. a A cluster of GFP-positive cells (green) seen within monolayer (overlaid on phase image on left and just green channel on right panel. b Co-staining for insulin (green) and the ductal marker DBA lectin (red) suggests that new insulin-positive cells are derived from duct cells in vitro (merged with both channela on left and just green for insulin on the right panel).
Ten days after COPAS sorting, we collected all cultured cells for RNA analysis and MoFlo determination of GFP-positive cells. Quantitative real-time PCR did not detect insulin mRNA immediately after COPAS sorting, but after differentiation using collagen-coated dishes, insulin mRNA of the whole preparation was induced to 1.91±0.31% of that seen in purified MIP-GFP sorted beta cells (Fig. 5). Compared with the GFP intensity of fresh isolated beta cells, the strength of GFP intensity of each differentiated GFP-positive cell was lower but still definitively GFP-positive as detected by MoFlo (Fig. 3b, e). These GFP-positive cells were sorted into 96-well plates for single-cell nested PCR to examine the gene expression of single cells. As shown in Fig. 6 in a representative experiment (n=3), all GFP-positive cells expressed insulin 1 and many expressed insulin 2, pancreas duodenum homeobox-1 (Pdx1) and cytokeratin 19 (CK19, also known as Krt19)). A few cells also expressed glucagon, pancreatic polypeptide and somatostatin.
Fig. 5.
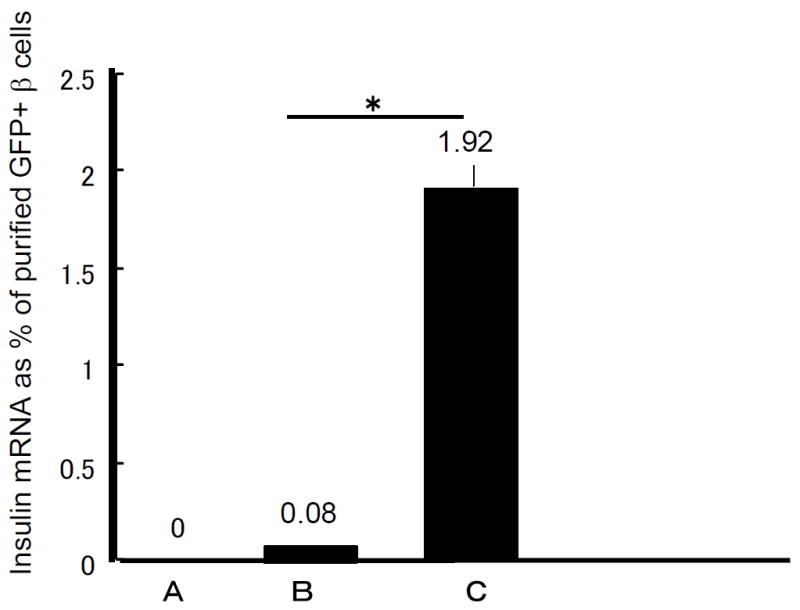
Quantitative real-time PCR of mouse insulin mRNA in the GFP-negative aggregates. Initially in the GFP-negative aggregates immediately after COPAS sort (bar 1) there was no detectable insulin mRNA. In samples cultured for 10 days in a hydrophobic dish (bar 2) or in a collagen-coated dish (bar 3) insulin mRNA levels, measured as per cent of purified GFP-positive beta cells, increased. n=3 for each stage (mean±SE); *p<0.05
Fig. 6.

Nested single cell PCR of differentiated GFP-positive cells. Representative gene expression of single new GFP-positive cells showed that these cells expressed insulin 1 (as would be expected with GFP-positive). Most also expressed insulin 2 (20/24) and the duct marker Ck19 (21/24). Only some cells had detectable levels of Pdx1 (10/24). Manually dissected duct tissue for CK19 and sorted GFP-positive beta cells from MIP-GFP mice served as positive control.
Discussion
In this study, we have shown that differentiation of insulin-producing beta cells can be induced in vitro from murine non-endocrine pancreatic cells. One concern over recent studies [6-9] is that insulin-positive cells may have been derived from the residual beta cells in the starting material, with the situation further complicated by the capacity of these residual beta cells to replicate [24]. The problem is challenging because until now it has not been possible to efficiently separate and remove beta cells from a dispersed population of pancreatic cells. Indeed, recent studies have used manually microdissected tissue [8, 25], fractions from the gradients used for islet isolation [6, 7, 26, 27] or magnetic bead sorting [16]. All of these approaches risk contamination with small numbers of endocrine cells. On the presumption that human pancreatic tissue was like rodent, with neural cell adhesion molecule (NCAM) protein being selectively in islet endocrine cells, Gao et al [24] found that the depletion of NCAM-positive cells from human pancreatic tissue remaining after islet isolation resulted in the appearance of few insulin-positive cells, leading to the conclusion that non-islet cells were not precursors for new beta cells. However, we have observed that NCAM is expressed at the mRNA and protein levels in epithelium of small ducts as well as in the islets in the human pancreas (Bonner-Weir, unpublished data). Here, we took a different approach designed to completely eliminate insulin-positive cells from the starting material, namely the use of MIP-GFP mouse pancreas and large bore cytometric sorting. In fact, our COPAS-sorted GFP-negative fraction from digested MIP-GFP pancreas had no insulin mRNA by quantitative real-time PCR and not a single insulin-positive cell by flow cytometry (MoFLo) in three independent experiments.
In our preliminary experiments using microdissected ducts from MIP-GFP mice, we found that after culture about 3% of the cells were positive for GFP. However, the concern remained that whatever GFP-positive cells were found might have originated from pre-existing beta cells that may or may not, have proliferated. As a new approach to remove pre-existing beta cells more efficiently, we used the large bore COPAS cytometer, which enabled us to perform efficient deletion of beta cells and to culture a large number of residual pancreatic cells. However, the potential contribution of beta cell dedifferentiation and redifferentiation is an important issue for our interpretation. A recent lineage tracing study [28] showed that mouse pancreatic beta cells can dedifferentiate in vitro, with the loss of multiple beta cell markers, including PDX-1, insulin, GLUT2 and prohormone convertases 1/3, and a shift to mesenchymal morphology. However this dedifferentiation occurred over a 2 week culture of dispersed islet cells, even when using MIP-GFP mouse islets. Since we cultured tissue for only 72 h before elimination of beta cells by COPAS sorting, the possibility that some beta cells might start to dedifferentiate to GFP-negative phenotype only to then redifferentiate to GFP-positive phenotype later cannot be ruled out definitively. However, based on the steps we have taken, it is unlikely that our new beta cells are derived from dedifferentiated ‘false GFP-negative cells’ that avoided detection by both COPAS and MoFlo sorting. Thus, first, in preliminary experiments, we also observed new GFP-positive cells in GFP-negative samples after COPAS sorting immediately post digestion. However, since the low viability of the epithelial cells resulted in poor expansion, we decided to culture for 3 days so that the acinar cells would die. Second, we set the gate for cells that have very low green-peak height so as to have a safety margin from the intensely GFP-positive beta cells. There were always some intermediate-intensity GFP-positive cells when we ran COPAS and MoFlo on these cells or on cells directly after pancreatic digestion, but there was no difference in the proportion of these intermediate-intensity cells. If dedifferentiation of beta cells occurs over the 72 h, we should have seen more cells with intermediate intensity in the GFP-positive group; yet no such change in population distribution was seen, so these cells are not likely to be ‘fading’ beta cells. Additionally GFP protein has a fairly long half-life [29] and the intensity of GFP in beta cells is very strong. Furthermore, our COPAS sorted tissue had no detectable insulin mRNA by quantitative real-time PCR immediately after sorting (Fig. 5). Since insulin mRNA represents 2% of the RNA in beta cells and the half-life of insulin mRNA is 77 h at high glucose concentration (17 mmol/l) [30], the lack of detectable insulin mRNA in GFP-negative sorted aggregates cultured at 17 mmol/l glucose (Fig. 5) provides strong evidence for the absence of any beta cells in the sorted tissue.
Our data do not address which non-endocrine epithelial cell is the cell of origin. Single-cell nested PCR showed that most new GFP-positive cells expressed the duct marker, Ck19, and only 40% expressed Pdx1; some of them also expressed non-beta endocrine hormones. This expression does not indicate that there was initial non-beta endocrine contamination, since single-cell nested PCR found that 50% of single adult beta cells expressed other islet hormones in addition to insulin (Katsuta, unpublished data). Since early stages of insulin-positive cells produce only low amounts of PDX1 [31] and may have residual CK19 protein [16], we interpret the GFP-positive cells detected as immature, newly formed insulin-positive cells. Acinar cells have been shown to transdifferentiate into insulin-positive cells in suspension culture [32, 33], but our insulin-positive cells derived from epithelial monolayers that had no amylase staining (data not shown). We recently reported lineage-tracing experiments using carbonic anhydrase II promoter to selectively label the ductal cells and show that these give rise to new islets and acini during normal growth and after injury [4]. Using these latter mice in the current in vitro system could be a definitive way to identify the origin of our new beta cells.
Supplementary Material
Acknowledgments
The authors thank J. Lock, J. LaVecchio and G. Buruzula for excellent technical assistance. We also especially thank J. Thompson (Union Biometrica) for expertise in setting the COPAS sort. This study was supported by grants from the National Institutes of Health DK44523 (to S. Bonner-Weir), DK66056 (to S. Bonner-Weir) and P30 DK36836 to the Joslin Diabetes and Endocrinology Research Center (Flow Cytometry and Advanced Microscopy Cores). Support also came from the Diabetes Research and Wellness Foundation and from private donors.
Abbreviations
- CK19
Cytokeratin 19
- DBA
Dolichos biflorus
- HPRT
Hypoxanthine-guanine phosphoribosyl transferase
- MIP-GFP mouse
Mouse insulin promoter 1–green fluorescent protein
- NCAM
Neural cell adhesion molecule
- PDX-1
Pancreas duodenum homeobox-1
- EGF
epidermal growth factor
Footnotes
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
References
- 1.Bonner-Weir S, Weir GC. New sources of pancreatic beta cells. Nature Biotechnology. 2005;23:857–861. doi: 10.1038/nbt1115. [DOI] [PubMed] [Google Scholar]
- 2.Suzuki A, Nakauchi H, Taniguchi H. Prospective isolation of multipotent pancreatic progenitors using flow-cytometric cell sorting. Diabetes. 2004;53:2143–2152. doi: 10.2337/diabetes.53.8.2143. [DOI] [PubMed] [Google Scholar]
- 3.Seaberg RM, Smukler SR, Kieffer TJ, et al. Clonal identification of multipotent precursors from adult mouse pancreas that generate neural and pancreatic lineages. Nat Biotechnology. 2004;22:1115–1124. doi: 10.1038/nbt1004. [DOI] [PubMed] [Google Scholar]
- 4.Inada A, Nienaber C, Katsuta H, et al. CAII positive pancreatic cells are progenitors for both endocrine and exocrine pancreas after birth revision. Proc Natl Acad Sci USA. 2008;105:19915–19919. doi: 10.1073/pnas.0805803105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramiya VK, Maraist M, Arfors KE, Schatz DA, Peck AB, Cornelius JG. Reversal of insulin-dependent diabetes using islets generated in vitro from pancreatic stem cells. Nat Medicine. 2000;6:278–282. doi: 10.1038/73128. [DOI] [PubMed] [Google Scholar]
- 6.Bonner Weir S, Taneja M, Weir GC, et al. In vitro cultivation of human islets from expanded ductal tissue. Proc Natl Acad Sci U S A. 2000;97:7999–8004. doi: 10.1073/pnas.97.14.7999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao R, Ustinov J, Pulkkinen MA, Lundin K, Korsgren O, Otonkoski T. Characterization of endocrine progenitor cells and critical factors for their differentiation in human adult pancreatic cell culture. Diabetes. 2003;52:2007–2015. doi: 10.2337/diabetes.52.8.2007. [DOI] [PubMed] [Google Scholar]
- 8.Katdare MR, Bhonde RR, Parab PB. Analysis of morphological and functional maturation of neoislets generated in vitro from pancreatic ductal cells and their suitability for islet banking and transplantation. J Endocrinology. 2004;182:105–112. doi: 10.1677/joe.0.1820105. [DOI] [PubMed] [Google Scholar]
- 9.Hao E, Tyrberg B, Itkin-Ansari P. Beta cell differentiation from non endocrine epithelial cells of the adult human pancreas. Nat Medicine. 2006;12:310–316. doi: 10.1038/nm1367. [DOI] [PubMed] [Google Scholar]
- 10.Sharma A, Zangen DH, Reitz P, et al. The homeodomain protein IDX-1 increases after an early burst of proliferation during pancreatic regeneration. Diabetes. 1999;48:507–513. doi: 10.2337/diabetes.48.3.507. [DOI] [PubMed] [Google Scholar]
- 11.Li L, Seno M, Yamada H, Kojima I. Betacellulin improves glucose metabolism by promoting conversion of intraislet precursor cells to beta cells in streptozotocin-treated mice. Am J Physiology Endocrinol Metab. 2003;285:E577–E583. doi: 10.1152/ajpendo.00120.2003. [DOI] [PubMed] [Google Scholar]
- 12.Gu D, Sarvetnick N. Epithelial cell proliferation and islet neogenesis in IFN-gamma transgenic mice. Development. 1993;118:33–46. doi: 10.1242/dev.118.1.33. [DOI] [PubMed] [Google Scholar]
- 13.Wang TC, Bonner-Weir S, Oates PS, et al. Pancreatic gastrin stimulates islet differentiation of transforming growth factor alpha-induced ductular precursor cells. J Clin Invest. 1993;92:1349–1356. doi: 10.1172/JCI116708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bonner-Weir S, Baxter LA, Schuppin GT, Smith FE. A second pathway for regeneration of the adult exocrine and endocrine pancreas: A possible recapitulation of embryonic development. Diabetes. 1993;42:1715–1720. doi: 10.2337/diab.42.12.1715. [DOI] [PubMed] [Google Scholar]
- 15.Gmyr V, Belaich S, Muharram G, et al. Rapid purification of human ductal cells from human pancreatic fractions with surface antibody CA19-9. Biochem Biophys Res Commun. 2004;320:27–33. doi: 10.1016/j.bbrc.2004.05.125. [DOI] [PubMed] [Google Scholar]
- 16.Yatoh S, Dodge R, Akashi T, et al. Differentiation of affinity-purified human pancreatic duct cells to beta cells. Diabetes. 2007;56:1802–1809. doi: 10.2337/db06-1670. [DOI] [PubMed] [Google Scholar]
- 17.Hara M, Dizon RF, Glick BS, et al. Imaging pancreatic beta cells in the intact pancreas. Am J Physiol Endocrinol Metab. 2006;290:E1041–E1047. doi: 10.1152/ajpendo.00365.2005. [DOI] [PubMed] [Google Scholar]
- 18.Fernandez LA, Hatch EW, Armann B, et al. Validation of large particle flow cytometry for the analysis and sorting of intact pancreatic islets. Transplantation. 2005;80:729–737. doi: 10.1097/01.tp.0000179105.95770.cd. [DOI] [PubMed] [Google Scholar]
- 19.Gotoh M, Maki T, Kiyoizumi T, Satomi S, Monaco AP. An improved method for isolation of mouse pancreatic islets. Transplantation. 1985;40:437–438. doi: 10.1097/00007890-198510000-00018. [DOI] [PubMed] [Google Scholar]
- 20.Hu M, Krause D, Greaves M, et al. Multilineage gene expression precedes commitment in the hematopoietic system. Genes Dev. 1997;11:774–785. doi: 10.1101/gad.11.6.774. [DOI] [PubMed] [Google Scholar]
- 21.Miyamoto T, Iwasaki H, Reizis B, et al. Myeloid or lymphoid promiscuity as a critical step in hematopoietic lineage commitment. Dev Cell. 2002;3:137–141. doi: 10.1016/s1534-5807(02)00201-0. [DOI] [PubMed] [Google Scholar]
- 22.Oshima Y, Suzuki A, Kawashimo K, Ishikawa M, Ohkohchi N, Taniguchi H. Isolation of mouse pancreatic ductal progenitor cells expressing CD133 and c-Met by flow cytometric cell sorting. Gastroenterology. 2007;132:720–732. doi: 10.1053/j.gastro.2006.11.027. [DOI] [PubMed] [Google Scholar]
- 23.Miettinen PJ, Huotari M, Koivisto T, et al. Impaired migration and delayed differentiation of pancreatic islet cells in mice lacking EGF-receptors. Development. 2000;127:2617–2627. doi: 10.1242/dev.127.12.2617. [DOI] [PubMed] [Google Scholar]
- 24.Gao R, Ustinov J, Korsgren O, Otonkoski T. In vitro neogenesis of human islets reflects the plasticity of differentiated human pancreatic cells. Diabetologia. 2005;48:2296–2304. doi: 10.1007/s00125-005-1935-8. [DOI] [PubMed] [Google Scholar]
- 25.Leng SH, Lu FE. Induction of pancreatic duct cells of neonatal rats into insulin-producing cells with fetal bovine serum: A natural protocol and its use for patch clamp experiments. World J Gastroenterol. 2005;11:6968–6974. doi: 10.3748/wjg.v11.i44.6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bogdani M, Lefebvre V, Buelens N, et al. Formation of insulin-positive cells in implants of human pancreatic duct cell preparations from young donors. Diabetologia. 2003;46:830–838. doi: 10.1007/s00125-003-1118-4. [DOI] [PubMed] [Google Scholar]
- 27.Ogata T, Park KY, Seno M, Kojima I. Reversal of streptozotocin-induced hyperglycemia by transplantation of pseudoislets consisting of beta cells derived from duct cells. Endocrine Journal. 2004;51:381–386. doi: 10.1507/endocrj.51.381. [DOI] [PubMed] [Google Scholar]
- 28.Weinberg N, Ouziel-Yahalom L, Knoller S, Efrat S, Dor Y. Lineage tracing evidence for in vitro dedifferentiation but rare proliferation of mouse pancreatic beta cells. Diabetes. 2007;56:1299–1304. doi: 10.2337/db06-1654. [DOI] [PubMed] [Google Scholar]
- 29.Corish P, Tyler-Smith C. Attenuation of green fluorescent protein half-life in mammalian cells. Protein Eng. 1999;12:1035–40. doi: 10.1093/protein/12.12.1035. [DOI] [PubMed] [Google Scholar]
- 30.Welsh M, Welsh M, Casadaban MJ, Steiner DF. Control of insulin gene expression in pancreatic beta cells and in an insulin producing cell line, RIN-5F cells. Regulation of insulin mRNA stability. J Biol Chem. 1985;260:13590–13594. [PubMed] [Google Scholar]
- 31.Nishimura W, Kondo T, Salameh T, et al. A switch from MafB to MafA expression accompanies differentiation to pancreatic beta cells. Developmental Biology. 2006;293:526–539. doi: 10.1016/j.ydbio.2006.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Minami K, Okuno M, Miyawaki K. Lineage tracing and characterization of insulin-secreting cells generated from adult pancreatic acinar cells. Proc Natl Acad Sci USA. 2005;102:15116–15121. doi: 10.1073/pnas.0507567102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baeyens L, de Breuck S, Lardon J, Mfopou JK, Rooman I, Bouwens L. In vitro generation of insulin-producing beta cells from adult exocrine pancreatic cells. Diabetologia. 2005;48:49–57. doi: 10.1007/s00125-004-1606-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.