

Abstract
Understanding how ligands bind to G-protein-coupled receptors and how binding changes receptor structure to affect signaling is critical for developing a complete picture of the signal transduction process. The adenosine A2A receptor (A2AR) is a particularly interesting example, as it has an exceptionally long intracellular carboxyl terminus, which is predicted to be mainly disordered. Experimental data on the structure of the A2AR C-terminus is lacking, because published structures of A2AR do not include the C-terminus. Calmodulin has been reported to bind to the A2AR C-terminus, with a possible binding site on helix 8, next to the membrane. The biological meaning of the interaction as well as its calcium dependence, thermodynamic parameters, and organization of the proteins in the complex are unclear. Here, we characterized the structure of the A2AR C-terminus and the A2AR C-terminus-calmodulin complex using different biophysical methods, including native gel and analytical gel filtration, isothermal titration calorimetry, NMR spectroscopy, and small-angle X-ray scattering. We found that the C-terminus is disordered and flexible, and it binds with high affinity (Kd = 98 nM) to calmodulin without major conformational changes in the domain. Calmodulin binds to helix 8 of the A2AR in a calcium-dependent manner that can displace binding of A2AR to lipid vesicles. We also predicted and classified putative calmodulin-binding sites in a larger group of G-protein-coupled receptors.
Introduction
G-protein-coupled receptors (GPCRs), also known as seven transmembrane (TM) receptors, are the largest family of membrane proteins in the human genome, with ∼800 members. GPCRs are of great interest to the pharmaceutical industry, as they regulate many physiological processes and their dysfunction is related to different diseases. They all share a common organization of seven TM α-helices, an extracellular N-terminus, and an intracellular C-terminus. Although the TM regions of GPCRs are fairly homologous, both N- and C-termini are highly variable, ranging in size from 1 to 2348 amino acids for the N-terminus, and from 2 to 397 amino acids for the C-terminus. GPCR ligands also vary considerably, ranging from photons and ions to small organic molecules, peptides, and proteins, depending on the receptor (1).
During GPCR signaling, extracellular ligand binding induces a conformational change that activates the receptor. The activated receptor catalyzes a GDP-to-GTP exchange in an intracellular heterotrimeric G-protein, resulting in the dissociation of the GαGTP subunit from the Gβγ subunit. The GαGTP and Gβγ subunits then either activate or inhibit different effector molecules and pathways, depending on the type of ligand. In recent years, it has become clear that GPCRs can also signal through a G-protein-independent manner by binding to different TM or intracellular GPCR-interacting proteins. These interactions are known to regulate the receptor ligand specificity, cell surface expression, endocytosis, recycling, and other cellular processes (2). In vivo, the functional form of the GPCR is oligomeric rather than monomeric, through either homo- or heteromers (3,4).
The human adenosine A2A receptor
The human adenosine A2A receptor (A2AR) is a 44.7 kDa GPCR with an intracellular C-terminus that bears two interesting differences to the C-termini of the closely related adenosine A1, A2B, and A3 receptors. One difference is in length—the C-terminus of A2AR (A2A-ct) is 122 amino acids long, whereas the other receptors are 38, 40, and 34 amino acids, respectively. A second key difference is that A2A-ct lacks a canonical cysteine residue at the end of helix 8 that is perpendicular to the membrane, adjacent to the TM helix 7. A majority of the other members of the rhodopsin class of GPCRs have one or two cysteines at this position, which are putative palmitoylation sites (5). Because palmitoylation is known to anchor peripheral membrane proteins or protein segments to the membrane (6), it has been speculated that the lack of palmitoylation and the exceptional length of A2A-ct make it more flexible and accessible for interacting with other proteins.
A2A-ct has a number of putative and demonstrated phosphorylation sites that may impact function. Threonine 298 phosphorylation was reported to mediate the short-term, but not long-term, desensitization of the A2AR after agonist stimulation (7). Serine 374 phosphorylation has been shown to be required in the A2AR-mediated inhibition of the dopamine D2 receptor agonist binding and signaling (8,9).
The A2A-ct is predicted to be mostly disordered, but experimental data concerning its structure are not available. So far, 12 A2AR structures have been published, but all of these structures are based on engineered A2AR proteins that lack the C-terminus (10–16). The most comprehensive A2AR structure ends at residue 317 (12).
A2A-ct interacts with a number of proteins, including G-protein-coupled receptor kinases (GRKs), β-arrestins, α-actinins, calmodulin (CaM), neuronal calcium-binding protein 2 (NECAB2), translin-associated protein X (TRAX), Arf nucleotide-binding site opener (ARNO/cytohesin-2), ubiquitin-specific protease 4 (USP4), and neuronal calcium sensor protein 1 (NCS-1) (17–24). These interactions have been studied using yeast two-hybrid screens, colocalization, coimmunoprecipitation, pulldown, immunoelectron microscopy, mass spectrometry, and bioluminescence and fluorescence resonance energy transfer approaches using either crude brain membrane extracts containing A2AR, A2AR-transfected cell cultures, or small synthetic A2AR peptides. As far as we know, interactions of A2AR with other proteins have not been studied with the purified A2AR protein or its individual domains. The knowledge about binding thermodynamics and kinetics is very limited, and the meaning of these interactions is poorly understood.
Calmodulin and A2AR
CaM is a 16.7 kDa highly conserved, soluble, acidic, intracellular calcium-binding protein that is a key player in calcium-mediated signaling in mammalian cells. CaM has a dumbbell-like structure, in which two globular domains are connected by a helical linker. The globular domains contain two helix-loop-helix (EF-hand) motifs each and are capable of binding two calcium ions per domain (25). Typically, the helical linker region of CaM is cut into two shorter helices during complex formation, allowing the globular domains to wrap around the binding partner (26–28). CaM target domains are contiguous sequences of ∼20 amino acids, which have a tendency to form basic amphipathic helices. There is little sequence similarity between CaM targets, yet many of them have very similar nanomolar binding affinities (29,30). Of the GPCRs, CaM has been reported to bind to adenosine A2A, dopamine D2, μ- and δ-opioid, metabotrobic glutamate 5 and 7 (mGlu5/mGlu7), muscarinic, serotonin 5-hydroxytryptamine 1A, 2A, and 2C (5HT1A, 5HT2A, and 5HT2C), parathyroid hormone 1 and 2 (PTH1/PTH2), vasoactive intestinal peptide, pituitary adenylate cyclase activating peptide, corticotrophin releasing hormone, calcitonin, glucagon-like peptide 1 and 2, vasopressin 2 (V2), calcium-sensing (CaS), and melanocortin 1 (MC1) receptors (19,31–41).
Woods et al. (19) were the first ones to report CaM interaction with the human A2AR. They used mass spectrometry to study CaM binding to dopamine D2 and adenosine A2A synthetic peptides. Thereafter, interactions between these receptors and CaM have been studied using different resonance energy transfer -based techniques, immunoprecipitation and transinfected cell cultures, for example. These studies have confirmed the interaction and provided some information about the role of CaM as a calcium-dependent modulator of A2A and D2 receptors both alone and as a heteromer (42,43). Calmodulin has very acidic epitopes 80DSEEEI85, 117TDEEVDEM124, and 137NYEEFV142 that have been suggested to interact with positively charged epitopes on A2A and D2 receptors (19). Such an epitope on the A2AR is the arginine-rich sequence 291RIREFRQTFR300 on the proximal part of the C-terminus. It has been reported to bind CaM independently of calcium (19,42). Despite all these studies, very little is known about the strength and stoichiometry of the A2AR-CaM interaction, or the complex structure, at the protein level.
In this work, we show CaM binding to the A2A-ct and present the thermodynamic parameters of the interaction. In addition, we shed light on the A2A-ct-CaM complex structure in solution. In these biophysical experiments, we have used recombinant proteins instead of short synthetic peptides or cell-based approaches. In addition to in vitro experiments, we present CaM-binding site predictions for a group of proteins mainly belonging to class A GPCRs. Based on the location of the binding sites, we divide the proteins into different classes and speculate on the physiological relevance of this classification.
Materials and Methods
Constructs and protein production
Human A2AR C-terminal amino acids 293–412 and 321–412 were cloned into the pQE-T7-1 and pQE-T7-2 vectors (Qiagen, Hilden, Germany), using NdeI/XhoI cloning sites. This generated A2A-ct constructs with a His10-tag in either the N-terminal or C-terminal end. Human CaM in the pET-14b vector (Merck KGaA, Darmstadt, Germany) was kindly provided by Dr. Nobuhiro Hayashi. All constructs were verified by sequencing and then transformed into Escherichia coli BL21(DE3) cells.
Expression of A2A-ct constructs was tested in small scale, and based on the higher expression level, A2A-ct 293–412 (A2A-ctL) with a C-terminal and A2A-ct 321-412 (A2A-ctS) with an N-terminal 10× His-tag were selected for further studies. For protein production, BL21(DE3) cells with either the A2A-ctL or the A2A-ctS plasmid were cultured in Luria-Bertani medium (LB-medium) containing 50 μg ml−1 kanamycin at 37°C, shaking the culture at 220 rpm until the OD at 600 nm was 0.6. Expression was then induced by adding 0.4 mM isopropyl β-D-1-thiogalactopyranoside (IPTG), and culturing was continued at 22°C, shaking the culture at 220 rpm. After 2 h, the cells were harvested.
10 ml of LB-medium containing 100 μg ml−1 ampicillin were inoculated with BL21(DE3) cells containing the CaM plasmid, and the cells were cultured at 37°C with shaking (220 rpm). After 7 h, the culture was transferred to a shake flask containing 1 l LB-medium with 100 μg ml−1 ampicillin. Culturing was continued for 5 h, and then, protein production was induced by adding 0.8 mM IPTG. After overnight (∼11 h) expression, the cells were harvested.
Protein purification
BL21(DE3) cells with either A2A-ctL or A2A-ctS were suspended in xTractor Buffer (Takara Bio Europe/Clontech, Saint-Germain-en-Laye, France), containing complete EDTA-free protease inhibitors (Roche, Basel, Switzerland). After a 10-min incubation on ice, the lysate was clarified by centrifugation. The soluble fraction was supplemented with 60 mM imidazole, and mixed with HisPur Ni-NTA Resin (Thermo Fisher Scientific, Waltham, MA), equilibrated beforehand with wash buffer (50 mM HEPES, 500 mM NaCl, 10% glycerol, 60 mM imidazole, 10 mM TCEP-HCl, 0.1 mM PMSF pH 7.4). After 1 h of binding, the mixture was transferred into an empty column, and unbound proteins were washed away using the wash buffer. Bound proteins were eluted with elution buffer (50 mM HEPES, 500 mM NaCl, 10% glycerol, 500 mM imidazole pH 7.4), and eluates were analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The buffer in the A2A-ct eluates was exchanged to cation exchange buffer (50 mM MES, 250 mM NaCl pH 5.8) using PD-10 columns (GE Healthcare, Little Chalfont, UK), and cation exchange was performed using a HiTrap CM FF column (GE Healthcare). The protein was eluted with a linear 250–1000 mM NaCl gradient in 50 mM MES, pH 5.8. The fractions were analyzed by SDS-PAGE, and A2A-ct-containing fractions were pooled. A2A-ct concentration was determined spectrophotometrically (1 mg ml−1 A280 = 0.490 or ε280 = 7090 M-1cm−1 for A2A-ctL, and 1 mg ml−1 A280 = 0.640 or ε280 = 7090 M-1cm−1 for A2A-ctS), and the protein was concentrated to 1-2 mg ml−1 by centrifugal ultrafiltration. All purification steps were done at 4°C.
The CaM purification protocol was modified from earlier studies (44,45). CaM-expressing BL21(DE3) cells were suspended in lysis buffer (10 mM HEPES, 1 mM EDTA, 10 mM DTT, 10 μg ml−1 DNase I pH 7.4) and lysed by sonication. After centrifugation, the soluble fraction was incubated for 10 min at 80°C and then centrifuged. 5 mM CaCl2 was added to the supernatant and the centrifugation was repeated. The supernatant containing CaM was applied onto a HiTrap Phenyl FF (High Sub) column (GE Healthcare) equilibrated with wash buffer (10 mM HEPES, 4 mM CaCl2 pH 7.4). The column was washed sequentially with wash buffer, wash buffer supplemented with 500 mM NaCl, and wash buffer again. CaM was then eluted with elution buffer (10 mM HEPES, 5 mM EGTA pH 7.4). The fractions were analyzed by SDS-PAGE, and CaM-containing fractions were pooled. The protein concentration was determined spectrophotometrically (1 mg ml−1 A280 = 0.178 or ε280 = 2980 M−1cm−1), and the protein was concentrated to 10 mg ml−1 by centrifugal ultrafiltration. All purification steps were done at 4°C unless stated otherwise.
Protein homogeneity and monodispersity were checked by SDS-PAGE, native PAGE, and analytical size exclusion chromatography. Based on electrospray ionization time-of-flight mass spectrometry, A2A-ctL, A2A-ctS, and CaM showed expected molecular masses of 14,467 Da (14,465 Da), 11,113 Da (11,113 Da), and 16,705 Da (16,706 Da), respectively. Theoretical molecular masses are indicated in parenthesis.
Synchrotron radiation far-UV circular dichroism spectropolarimetry
Synchrotron radiation circular dichroism (SRCD) spectra were measured on the CD1 beamline at the ASTRID storage ring, ISA, Århus, Denmark. A quartz cuvette with a path length of 100 μm was used, and the protein concentrations were in the range 0.5–1.0 mg ml−1. The protein was in a buffer containing 50 mM HEPES, 250 mM NaCl, and 1 mM TCEP-HCl pH 7.4, and the measurements were carried out at 25°C. In addition to the protein alone, spectra were collected in the presence of 30% TFE (trifluoroethanol), 0.1% DPC (n-dodecylphosphocholine), 0.1% DDM (n-dodecyl-β-D-maltopyranoside), 0.1% SDS, and 1 mg ml−1 DMPC/DMPG (1,2-dimyristoyl-sn-glycero-3-phosphocholine/1,2 dimyristoyl-sn-glycero-3-phospho-rac-(1-glycerol)) (1:1) vesicles (Larodan Fine Chemicals, Malmö, Sweden). Three scans were taken for each sample and the corresponding buffer, and after averaging, the buffer contribution was subtracted.
Fluorescence spectrometry
A2A-ctL fluorescence spectra were measured at 25°C in 50 mM HEPES, 250 mM NaCl, 1 mM TCEP-HCl pH 7.4 with the FluoroMax-4 spectrofluorometer (Horiba Scientific, Kyoto, Japan). 0.15 mg ml−1 protein in a 3 mm cuvette was used in the measurements. The excitation wavelength was 295 nm, and emission spectra were recorded from 300 to 450 nm. Measurements were repeated in the presence of 30% TFE, 0.1% DPC, 0.1% DDM, 0.1% SDS, and 1 mg ml−1 DMPC/DMPG (1:1) vesicles. Samples were measured in triplicate, buffer background was subtracted, and an average spectrum was calculated for each sample. Finally, the fluorescence intensities were normalized to allow comparison of peak maximum wavelengths.
NMR spectroscopy
NMR experiments required labeling of A2A-ctL with 15N and 13C. BL21(DE3) cells containing the A2A-ctL plasmid were cultured at 37°C in LB-medium to an OD of 0.6 at 600 nm. The cells were harvested and washed with unlabeled minimal medium. The cells were then suspended to the same density in minimal medium containing U-13C6 D-glucose, 15NH4Cl, (both from Cambridge Isotope Laboratories, Tewksbury, MA) and 0.1 mg ml−1 kanamycin, supplemented with 1× MEM Vitamin Solution (Sigma-Aldrich, St. Louis, MO). After 1 h growth recovery at 22°C, A2A-ctL expression was induced by adding 0.4 mM IPTG. Culturing was continued at 22°C for 2 h, and the cells were harvested by centrifugation. A2A-ctL was purified as described previously. All A2A-ctL NMR spectra were measured in 95%/5% 50 mM HEPES, 250 mM NaCl, 1 mM TCEP-HCl, 5 mM CaCl2 pH 7.2/D2O at 25°C at 800 MHz 1H frequency using a Varian UNITY INOVA 800 NMR spectrometer (Agilent, Santa Clara, California), equipped with a cryogenically cooled 1H, 13C, 15N probehead. The backbone assignment of A2A-ctL was carried out using a plethora of three-dimensional (3D) NH-detected experiments including experiments specially tailored for intrinsically disordered proteins (46,47) as well as the conventional HNCO, HNCACB, and CBCA(CO)NH (48). Proton chemical shifts were referenced with respect to residual solvent signal (4.79 ppm at 25°C and pH 7.2). The 13C and 15N chemical shifts were indirectly referenced. Secondary structure propensities were calculated with the secondary structure propensities (SSP) algorithm (49) that uses the relative difference of 13Cα and 13Cβ chemical shifts between α- and β-structures i.e., SSP is not sensitive toward potential referencing errors in 13C chemical shifts. The reference database of SSP is based on the random coil chemical shifts (50). To map the CaM binding epitope in A2A-ctL, unlabeled CaM was added in a stepwise manner to 100 μM 15N-labeled A2A-ctL (molar ratios 0.5:1, 1:1, 2:1 and 4:1 CaM/A2A-ctL).
Native polyacrylamide gel electrophoresis
A2A-ctL, A2A-ctS, and CaM were dialyzed against 50 mM HEPES, 250 mM NaCl, 1 mM TCEP-HCl pH 7.4. Samples containing 300 pmol of A2A-ctL or A2A-ctS alone or together with 300 pmol of CaM, and 5 mM CaCl2 or EDTA were analyzed on 16% native polyacrylamide gel electrophoresis (native PAGE) gel to test for CaM binding to the A2A-ct and the calcium dependence of the interaction. The synthetic peptides RIREFRQTFRKIIRSHVLRQQ and YAYAIAEFAQTFAKIIASHVLAQQ (JPT Peptide Technologies, Berlin, Germany), corresponding to the wild-type (WT) A2AR amino acids 291–311 and amino acids 288–311 with arginine to alanine mutations at positions 291, 293, 296, 300, 304, and 309, respectively, were used in the binding competition assay. 300 pmol A2A-ctL, 300 pmol CaM, and 3.7–24,300 pmol of peptide were mixed in the presence of 5 mM CaCl2 and analyzed on 16% native PAGE.
Isothermal titration calorimetry
A2A-ctL, A2A-ctS, and CaM were dialyzed against 50 mM HEPES, 250 mM NaCl, 1 mM TCEP-HCl pH 7.4 containing either 5 mM CaCl2 or EDTA. Isothermal titration calorimetry (ITC) was carried out using the VP-ITC instrument (MicroCal, Northampton, MA) at 25°C. All samples were degassed before the experiment. 200 μM CaM was placed in the syringe and 18 μM A2A-ctL or A2A-ctS in the cell. CaM was titrated into the A2A-ct solution at 4-min intervals so that the first injection was 2 μl, and the remaining 19 injections were 15 μl. Each CaM-A2A-ct titration was done twice. The heat of dilution and mixing was determined by titrating CaM into the buffer. Data were analyzed using Origin (MicroCal). For each titration, the first 2-μl injection was ignored, the heat of dilution and mixing was subtracted, and fitting was done with the single binding site model. This provided information about stoichiometry (n), association constant (Ka), entropy (ΔS), and enthalpy change (ΔH).
Analytical gel filtration
The ITC samples were recovered from the measurement cell, concentrated to 120 μl, and subjected to gel filtration using a Superdex 200 10/300 GL column (GE Healthcare) in 50 mM HEPES, 250 mM NaCl pH 7.4 with either 5 mM CaCl2 or EDTA. Gel filtration standard proteins (Sigma-Aldrich) were β-amylase (molecular mass 200 kDa, Stokes radius 5.30 nm), alcohol dehydrogenase (150 kDa, 4.55 nm), albumin (66 kDa, 3.60 nm), carbonic anhydrase (29 kDa, 2.14 nm), and cytochrome c (12 kDa, 1.75 nm). The standards were run in the same way as the samples in the presence of calcium or EDTA, and their Stokes radii (rs) were plotted against their elution volumes to obtain a standard curve. Stokes radii for the samples were calculated on the basis of their elution volumes using the standard curve.
Surface plasmon resonance (SPR)
SPR was carried out using the Biacore T100 system (GE Healthcare) at 25°C. DMPC/DMPG vesicles (1:1 ratio) were immobilized on a Sensor Chip L1 (GE Healthcare) according to the manufacturer’s instructions, and the surface was further blocked by injecting bovine serum albumin at 0.1 mg ml−1. The running buffer in the experiment contained 10 mM HEPES pH 7.4 and 150 mM NaCl. To follow possible detachment of A2A-ctL from the membrane by CaM, either 0.2 or 1.0 μg ml−1 A2A-ctL was injected over the surface for 120 s, followed by a dissociation phase of 360 s. Immediately after the dissociation phase, 20 μg ml−1 Ca2+-CaM was injected for 180 s in a similar manner. A flow rate of 30 μl min−1 was used throughout the run. For kinetic analysis of A2A-ctL binding to membranes, a titration experiment using 0.2–30 μg ml−1 A2A-ctL was carried out using the same settings, with regeneration using 100 mM NaOH between the injections. In another experiment to follow the effect of prior CaM complex formation on membrane binding, A2A-ctL (5 μg ml−1) and Ca2+-CaM (25 μg ml−1) were injected either separately or together, intervened by regeneration injections with 100 mM NaOH. All injections were 120 s in duration in the latter experiment.
Small-angle x-ray scattering (SAXS)
A2A-ctL and CaM were dialyzed against 50 mM HEPES, 250 mM NaCl, 5 mM CaCl2, 1 mM TCEP-HCl pH 7.4. SAXS measurements were carried out on the European Synchrotron Radiation Facility (ESRF) Bio-SAXS beamline ID14-3, Grenoble, France (51). Samples were measured at 4°C using an x-ray wavelength of 0.0931 nm and an exposure time of 10 s. The data were recorded using a PILATUS 1 M detector at a sample-detector distance of 2.5 m, covering a momentum transfer range, s (4πSinθ/λ) of 0.05–5.8 nm−1. 10 frames were collected for each sample in total. A2A-ctL and CaM were measured alone and in a 1:1 complex. Concentrations varied between 0.2 and 2.2 mg ml−1 for A2A-ctL, 0.75–6.0 mg ml−1 for CaM, and 1.55–3.1 mg ml−1 for the complex. No radiation damage was observed when comparing the 10 time frames with 10 s exposures. Solvent scattering was measured from the buffer before and after each sample, and the average background scattering was subtracted with PRIMUS (52). For data analysis and modeling, the data from different concentrations of the same sample were merged, such that the low-angle data were taken from the lowest concentration and the high-angle data from the highest concentration. This corrects possible minor concentration-dependent effects at low angles, while keeping high-angle data accurate. The maximum dimension Dmax and the distance distribution function p(r) were calculated using GNOM (53). Because A2A-ctL behaved like an intrinsically disordered protein, its radius of gyration was also determined using the Debye formalism (54). Furthermore, the expected dimensions based on a random conformation were calculated as described (55,56).
Molecular models were built using several approaches. Overall shapes of the samples were built using DAMMIF (57), and chain-like models of the individual proteins using GASBOR (58). The GASBOR models of A2A-ctL and CaM were further used to model the complex with rigid-body refinement in SASREF (59). In addition, two-phase models for the complex were made using MONSA (60). The ensemble optimization method (EOM) was used to detect possible populations of different conformations in A2A-ctL (61).
Calmodulin-binding site prediction
Ninety-eight proteins belonging to GPCR class A and eight proteins belonging to either class B or C and shown experimentally to bind CaM were selected for the prediction. Protein sequences were analyzed using the CaM target database (62) that scores the amino acids from 0–9 based on their predicted ability to interact with CaM. Continuous amino acid segments containing at least six amino acids with a score of 8 or 9 were considered as a putative binding site. Eleven sites located in nonsensical domains, like an extracellular loop, were omitted from the analysis. The predicted binding sites were classified as either C-terminus binders or other site binders. For each binding site, a binding score was calculated by dividing the sum of the CaM target database scores by 28, which was the number of amino acids in the longest continuous putative binding site. Finally, the sites were sorted in descending order based on the calculated score.
Results
The carboxyl terminal domain of the adenosine A2A receptor is intrinsically disordered
A2A-ct is predicted to contain large unstructured and/or flexible portions by several protein disorder servers, including DisEMBL, FoldIndex, DISOPRED2, and XtalPred-RF (63–66). Experimental data on the A2A-ct structure are missing, because this domain was almost completely truncated from the antagonist and agonist occupied crystal structures (Fig. 1, A and B) (10–16). We decided to experimentally characterize the disordered regions of this domain by various complementary techniques before conducting interaction studies with CaM. Because the CaM-binding motif is most likely in the proximal part of A2A-ct (19), we designed two different C-terminal constructs of the A2AR—the full-length domain (A2A-ctL) and the null-binding domain (A2A-ctS) (Fig. 1 C). Of these, only A2A-ctL has a predicted CaM-binding domain, and this construct was used in the secondary structure studies.
Figure 1.
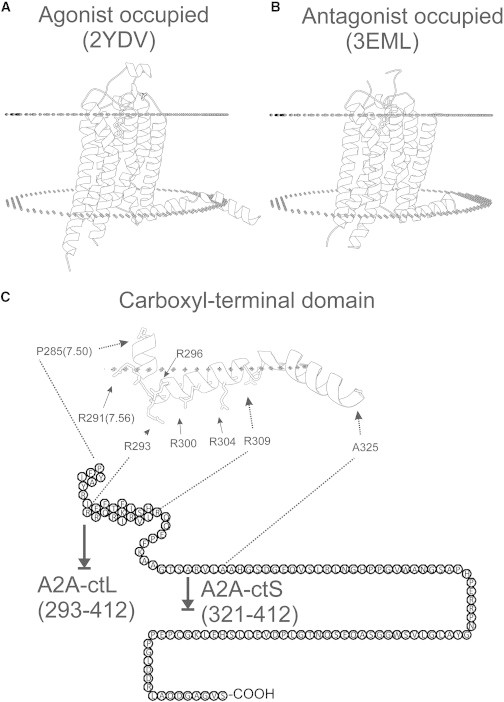
Helix 8 conformation of agonist versus antagonist bound A2AR and organization of the A2A-ct. (A) Agonist NECA bound A2AR (Protein Data Bank (PDB) ID 2YDV). (B) Antagonist ZM241385 bound to A2AR (PDB ID 3EML). Helix 8 is embedded in the membrane in both structures. (C) Helix 8 has many positively charged arginine residues that might be required for protein-protein interactions. R291 is fully embedded in the membrane and is probably unable to interact with other proteins. R293, R296, R300, R304, and R309 form a positively charged surface next to the membrane that might mediate interactions with negatively charged proteins or other molecules.
Synchrotron radiation far-UV CD spectropolarimetry and intrinsic tryptophan fluorescence spectrometry
The far-UV CD spectrum of A2A-ctL is typical for a protein without a globular conformation or secondary structure (Fig. 2 A) (67). It has a small negative shoulder around 222 nm, a strong negative minimum around 200 nm, and no positive signals in the far-UV range. Because deconvolution analysis methods are based on the usage of crystal structure-derived reference data sets, they are not able to predict the secondary structure reliably for such a flexible protein. Hence, helical content was simply estimated based on the CD signal at 222 nm (68). Interestingly, in the presence of the α-helix stabilizing TFE (16% helical), the negative minimum shifted from 200 to 208 nm. An addition of DDM or lipid vesicles did not have a significant effect on the spectra, suggesting that any interactions these compounds have with A2A-ctL do not cause conformational changes. On the other hand, DPC (7% helical) and SDS (14% helical) caused spectral changes that resembled the ones observed with TFE. This suggests the capability of A2A-ctL to adopt a more α-helical conformation in the presence of certain membrane-mimicking compounds. The head group of the detergent or lipid seems to be important, since SDS and DPC, unlike DDM, both have a negatively charged sulfate or phosphate group, respectively.
Figure 2.
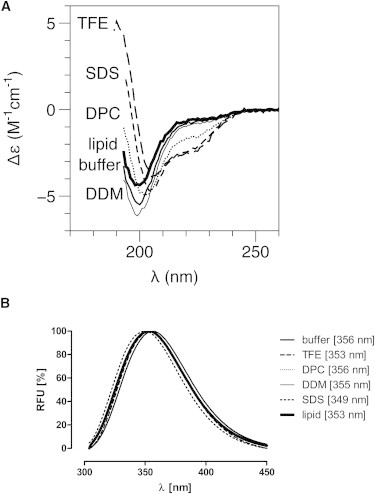
Characterization of secondary structure and folding of the A2A-ctL by SRCD spectropolarimetry and fluorescence spectrometry. (A) SRCD and (B) fluorescence spectra of A2A-ctL are shown without additives and with DDM, lipid vesicles, DPC, SDS, and TFE.
A2A-ct has a single tryptophan residue at position 346. According to fluorescence measurements, Trp-346 is exposed to an aqueous environment (Fig. 2 B). The addition of 0.1% DPC had no effect on the fluorescence maximum, and 0.1% DDM decreased the 356-nm peak maximum only to 355 nm. On the other hand, either of the 30% TFE and 0.1 mg ml−1 DMPC/DMPG vesicles shifted the fluorescence maximum from 356 to 353 nm, and 0.1% SDS even further to 349 nm, indicating a very slight decrease in the solvent accessibility of the tryptophan. Despite these small changes, Trp-346 remained mainly in an aqueous environment, and most probably, its surroundings do not take part in the helix formation detected by SRCD.
NMR spectroscopy
NMR spectroscopy can be employed to study structural features of disordered proteins at single-residue resolution (69). To confirm the disordered nature of A2A-ctL in solution, we measured a two-dimensional 1H, 15N-HSQC spectrum of A2A-ctL at 800 MHz 1H frequency (Fig. 3 A). The spectrum shows poorly dispersed crosspeaks, with the proton chemical shifts ranging from 7.5 to 8.5 1H ppm, indicating that A2A-ct undergoes fast conformational dynamics in solution, which is typical for highly disordered proteins (69). The conformational heterogeneity of intrinsically disordered proteins calls for special means for the NMR signal assignment. Indeed, to accomplish backbone assignment of A2A-ctL, we employed a panoply of novel NH-detected triple-resonance experiments that correlate 1HN, 13C’, and 15N frequencies and simultaneously link amino acid stretches flanking single proline residues (46,47). These data were supplemented with 13Cα and 13Cβ chemical shifts available from HNCACB/CBCA(CO)NH experiments (H. Tossavainen, M. Hellman, H. Piirainen, V.P. Jaakola, and P. Permi, unpublished). In this way a nearly complete backbone assignment of A2A-ctL was obtained.
Figure 3.
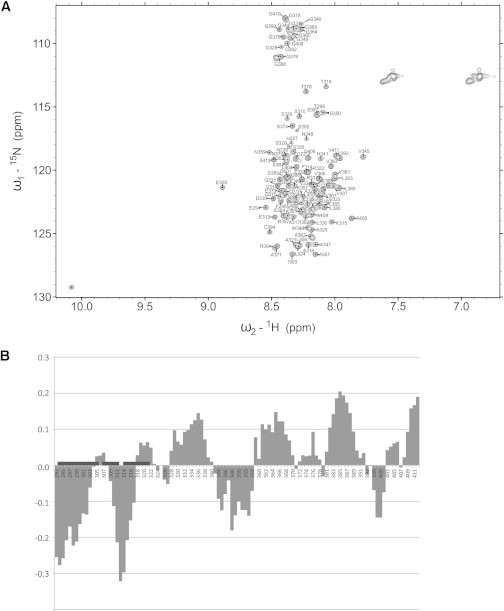
A2A-ctL characterization by NMR spectroscopy. (A) 1H, 15N-HSQC spectrum of A2A-ctL measured at 800 MHz 1H frequency. (B) Secondary structure propensities of A2A-ctL. Positive and negative numbers are indicative of α- and β-conformations, respectively. The N-terminal residues 293–321 that comprise the CaM binding epitope of A2A-ctL according to NMR titration studies are highlighted with boxes.
Next, to obtain a more quantitative description of conformational space sampling in A2A-ctL, we conducted an analysis of SSP present in A2A-ctL. Fig. 3 B displays the SSP score of A2A-ctL, as determined by comparing experimental 13Cα-13Cβ chemical shifts to random coil chemical shifts using the software package SSP (49). These data highlight a tendency of A2A-ctL toward extended conformation in the N-terminal CaM-binding site, where residues 293–303 and 310–317 populate βs/βp conformations 13.0% and 20.0%, respectively. NMR secondary chemical shift data reveal significant propensity of A2A-ctL to α-helical conformation in residues 329–340 (9.2%) and 382–389 (15.3%). Therefore, according to the NMR data, A2A-ctL transiently populates β-structures in the N-terminal part of the polypeptide chain, whereas the remainder of the A2A-ctL is partially in α-helical conformation.
We sought to understand the role of membrane mimicking detergents in conformational propensities of A2A-ctL by gradual addition of TFE to the NMR sample. Indeed, in accordance with far-UV CD spectral data, increasing TFE concentration induced significant chemical shift perturbations to the 1H, 15N-HSQC-spectrum of A2A-ctL (Fig. S1 in the Supporting Material). However, in most cases chemical shift perturbations were too large for crosspeaks to be identified reliably in the crowded spectrum and hence would have required crosspeak reassignment, which was not conducted in this study due to the limited lifetime of the NMR sample.
Biophysical characterization of the ACA-ct–CaM complex
After characterizing A2A-ct as an intrinsically disordered protein, we studied the binding of CaM to A2A-ct and the structural properties of the complex, using both A2A-ctL and the shorter A2A-ctS construct.
Native PAGE
CaM was mixed with either A2A-ctL or A2A-ctS in the presence of calcium or EDTA, and analyzed by native PAGE (Fig. 4 A). Both A2A-ct constructs behaved similarly in the presence and absence of calcium. The well-known calcium-induced conformational change of CaM (70) was clearly visible on the gel. When A2A-ctL and CaM were mixed in the presence of calcium, an extra band appeared, most likely corresponding to an A2A-ctL-CaM complex. This band was absent in the EDTA-containing sample, suggesting that the binding is calcium-dependent. Although A2A-ctL bound to CaM, no extra bands were detected in either of the samples containing A2A-ctS and CaM. This suggests that the CaM-binding site on A2A-ct locates between amino acids 293 and 320. This region on A2A-ct contains many positively charged arginine residues that are thought to be involved in CaM binding (19). The observed calcium dependence of the binding was surprising, because in previous studies, calcium was not thought to be involved in the interaction (42).
Figure 4.
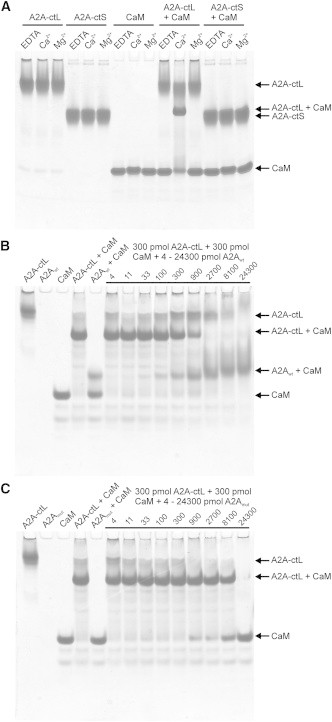
Binding of CaM to the C-terminal constructs of A2AR and competitive binding by A2A-ct-mimicking WT (A2Awt) and mutated (A2Amut) peptides on native PAGE. (A) A2A-ctL forms a complex with CaM in a calcium-dependent manner. The complex is not detected in the presence of EDTA or magnesium. A2A-ctS does not bind CaM on native PAGE. (B) A2Awt displaces A2A-ctL on CaM. (C) A2Amut does not form a complex with CaM and is unable to displace A2A-ctL bound to CaM.
To confirm the CaM-binding site on A2A-ctL and the importance of arginine residues for binding, we carried out binding competition experiments using a peptide corresponding to WT A2AR amino acids 291–311 and a peptide corresponding to A2AR amino acids 288–311, with six arginine to alanine mutations (Fig. 4, B and C, respectively). The WT peptide bound calmodulin as expected, because an extra protein band appeared on a native gel. This band was missing from the mutated peptide-CaM sample, demonstrating the importance of the mutated arginine residues for the interaction. When the amount of WT A2A peptide was increased from 3.7 pmol to 24.3 nmol, the peptide clearly displaced A2A-ctL from CaM. On the other hand, the mutated A2A peptide did not affect A2A-ctL-CaM binding, although some unbound CaM was present in samples containing the highest amount of the mutated peptide. We did not quantitatively analyze the results for a number of reasons, including the lack of quantitativeness of PAGE; the presence of a small amount of protein remaining in the wells, potentially due to aggregation; and the presence of multiple faint protein bands visible on the gels. For more quantitative analyses, other methods were used.
Isothermal titration calorimetry
As previous studies have not provided information about stoichiometry and affinity of the A2AR-calmodulin interaction, we performed ITC experiments for CaM binding to A2A-ctL in the presence of calcium and EDTA (Fig. 5, A and B). The A2A-ctL-CaM interaction is a calcium-dependent exothermic binding reaction with a favorable enthalpy (−12.93 ± 0.13 kcal mol−1), but an unfavorable entropy (3.4 kcal mol−1). The dissociation constant (Kd) for the complex is 97.9 ± 9.3 nM and the stoichiometry 0.92 ± 0.04, indicating 1:1 complex formation (Table 1). In the presence of EDTA, no binding was observed. To confirm the CaM-binding site on A2A-ctL, we also titrated A2A-ctS with CaM in the presence of calcium (Fig. 5 C). In this case, much smaller exothermic peaks were observed. Consequently, the removal of amino acids 293–320 from A2A-ct clearly abolished high-affinity binding to CaM. The remaining exothermic peaks were probably caused by unspecific or low affinity interactions between A2A-ctS and calmodulin.
Figure 5.
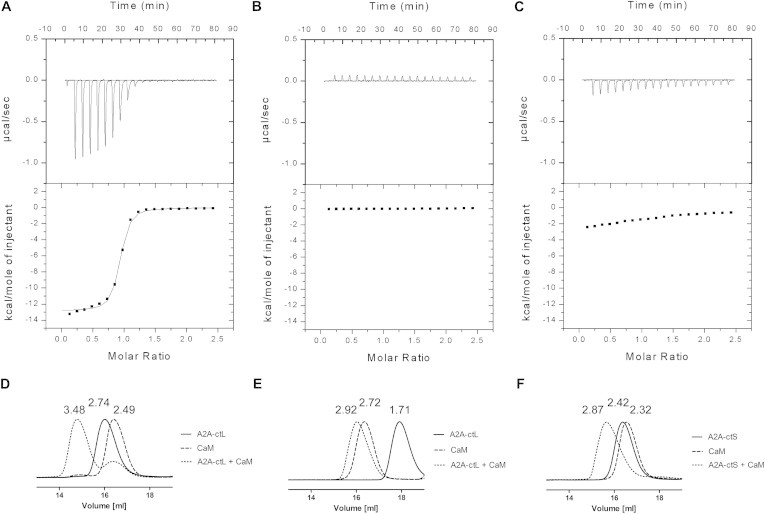
Binding of CaM to the A2A-ct constructs using ITC and size exclusion chromatography. (A) Titration of CaM to A2A-ctL in the presence of calcium and (B) in the presence of EDTA. (C) Titration of CaM to A2A-ctS in the presence of calcium. Size exclusion chromatography results for A2A-ctL, CaM, and A2A-ctL-CaM in the presence of (D) calcium or (E) EDTA, and (F) for A2A-ctS, CaM, and A2A-ctS-CaM in the presence of calcium. Hydrodynamic radii (nm) of the proteins are given above the peaks.
Table 1.
ITC parameters for A2A-ctL-CaM interaction
| ΔH (kcal mol−1) | Kd (nM) | −TΔS (kcal mol−1) | N |
|---|---|---|---|
| −12.93 ± 0.13 | 97.9 ± 9.3 | 3.4 | 0.92 ± 0.04 |
Analytical gel filtration
After ITC measurements, we performed gel filtration for the samples recovered from the measurement cell. We determined Stokes radii for the peaks corresponding to A2A-ctL, A2A-ctS, CaM, and A2A-ctL/A2A-ctS-CaM samples, using proteins with known Stokes radii as standards. In the presence of calcium, the calculated Stokes radii for A2A-ctL, CaM, and the A2A-ctL-CaM complex were 2.74, 2.49, and 3.48 nm, respectively (Fig. 5 D). The relatively high Stokes radii values were understandable because of the elongated shape of calmodulin and the disordered nature of A2A-ct. According to the results, CaM clearly binds to A2A-ctL in the presence of calcium with relatively high affinity. Nearly all protein in the mixture elutes in the form of a complex (Fig. 5 D), indicating the complex is stable and monodisperse even in the separating conditions of a gel filtration column; the small amount of CaM in the chromatogram relates to its excess in the injected sample, and is unlikely to represent dissociation of the complex. When the samples were analyzed in the presence of EDTA, the corresponding Stokes radii for A2A-ctL, CaM, and A2A-ctL-CaM peaks were 1.71, 2.72, and 2.92 nm (Fig. 5 E). The radius of A2A-ctL almost doubled in the calcium-containing buffer. That might be either because of conformational change or dimer formation. Because the A2A-ct is not thought to be involved in dimerization, we are likely detecting a calcium-dependent conformational change of A2A-ct. In turn, the Stokes radii of peaks corresponding A2A-ctS, CaM, and the A2A-ctS-CaM samples in the presence of calcium were 2.42, 2.32, and 2.87 nm (Fig. 5 F). Although A2A-ctS did not bind CaM on native PAGE, and only very weak, if any, binding was visible on ITC, A2A-ctS seemed to somehow interact with CaM when analyzed by analytical gel filtration.
NMR spectroscopy
Protein-ligand interactions can be studied at single-residue resolution using NMR spectroscopy. Given that the calorimetric and gel filtration data suggested emergence of a relatively stable complex between A2A-ctL and CaM in the presence of calcium, we conducted NMR titration studies by adding unlabeled CaM to 15N, 13C-labeled A2A-ctL. Most significant chemical shift perturbations, in terms of disappearance of crosspeaks of the free A2A-ctL form upon addition of CaM, were observed in the proposed CaM binding region in the very N-terminal part of A2A-ctL (Fig. 3 B). This suggests that the CaM binding epitope of A2A-ctL comprises residues 293–321. Regrettably, because of the limited lifetime of the A2A-ctL sample and the low concentration of the A2A-ctL-CaM complex, we were not able to assign A2A-ctL crosspeaks in the CaM-bound form.
Surface plasmon resonance
Because A2A-ctL seemed to interact with negatively charged lipids, we studied the effect of CaM on the ability of A2A-ctL to bind lipid membranes. For this purpose, we immobilized a mixture of DMPC/DMPG on an L1 SPR sensor chip, and injected A2A-ctL over the lipid surface. A2A-ctL bound immediately to the lipids in a concentration-dependent manner, and no dissociation was observed (Fig. 6). CaM injection brought the response back to the starting level, corresponding to the response of the lipid-coated surface. A further experiment also demonstrated that a preformed complex of A2A-ctL and CaM resulted in much less binding to the membrane, compared to A2A-ctL alone (Fig. S2). Based on these results, we propose that Ca2+/CaM binding to the A2A-ctL disrupts the lipid-A2A-ctL interaction, releasing the complex from the surface. A titration of the same lipid vesicles with A2A-ctL in SPR (Fig. S3) allows to estimate a Kd ≈ 1 μM for the interaction, which is an order of magnitude weaker than that for the formation of the CaM complex. Hence, CaM is able to detach the overlapping membrane-binding site from the membrane surface by means of direct competition.
Figure 6.
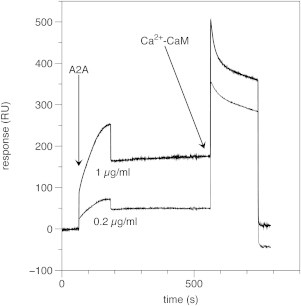
Binding of A2A-ctL to lipid vesicles and CaM-mediated release measured with SPR. Two different A2A-ctL concentrations (0.2 and 1.0 μg ml−1) are shown. Shown is one injection of A2A-ctL, followed by an equilibration period and an injection of CaM, which quantitatively releases bound A2A-ctL from the membrane.
SAXS
Obtaining crystals of the A2A-ctL-CaM complex seemed unlikely because of the flexible nature of A2A-ct, we decided to study the solution structures of A2A-ctL alone and in complex with CaM. Synchrotron SAXS experiments were used both to provide parameters describing the molecular shapes, as well as to build low-resolution 3D models representing the free and complexed proteins (Table 2, Fig. 7). Both A2A-ctL and CaM were monomeric in solution (Fig. S4), and the molecular mass calculated for their mixture indicated quantitative complex formation with 1:1 stoichiometry, which is in line with the affinity and stability of the complex described previously. A2A-ctL alone was highly elongated, whereas the conformation of CaM was consistent with its dumbbell-shaped crystal structure. CaM contains two folded lobes, separated by a flexible linker. For a fully random polymer chain, the Rg and Dmax values can be estimated from amino acid chain length; for A2A-ctL (134 residues), these values would be 3.78 nm and 10.5 nm, respectively. These results suggest A2A-ctL behaves more or less like a random chain in solution, and is even slightly more elongated than expected for a random polymer, suggesting intramolecular repulsion.
Table 2.
SAXS parameters for A2A-ctL, CaM, and the A2A-ctL-CaM complex
| Sample | Rg (nm) | Dmax (nm) | I(0)a (relative) | MW expected (kDa) | MWb (kDa) |
|---|---|---|---|---|---|
| A2A-ctL | 3.30 (53)a | 13c | 16.76 | 14.5 | 17.2 |
| 3.49c | |||||
| 3.78d | |||||
| 3.78e | 10.5e | ||||
| CaM | 2.15 (54)a | 7c | 16.13 | 16.7 | 16.5 |
| 2.19c | |||||
| A2A-ctL-CaM | 3.33 (32)a | 14c | 28.81 | 31.2 | 29.6 |
| 3.47c |
Analyzed using the Guinier plot tool in the Primus software. Number of data points used in the analysis is indicated in parentheses.
Molecular masses were estimated by the forward scattering comparison to a reference protein, bovine serum albumin (66 kDa).
Analyzed using the GNOM software.
Calculated from the Debye function (54).
Calculated based on random chain assumption (55).
Figure 7.
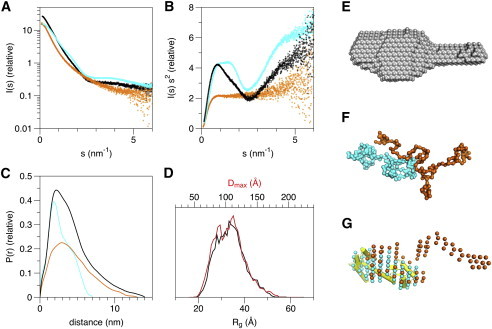
Solution structure determination of A2A-ctL, CaM, and the A2A-ctL-CaM complex by SAXS. (A) Raw scattering data, (B) Kratky plots, and (C) distance distribution functions of A2A-ctL (orange), CaM (cyan), and the A2A-ctL-CaM 1:1 molar mixture (black). (D) Rg (black), and Dmax (red) distributions for selected EOM ensembles of A2A-ctL suggest the presence of a slightly bimodal conformational distribution, which is anyway highly elongated. (E) Averaged ab initio dummy atom model of the complex generated by DAMMIF suggests binding of CaM to one end of A2A-ctL. (F) SASREF rigid body modeling using chain-like GASBOR models separately generated for both components. (G) The dummy atom multiphase model generated by MONSA also gives strong indications of the presence of a folded CaM molecule at one end of the complex, in which A2A-ctL remains largely disordered. The CaM crystal structure (yellow) (PDB ID 1CLL) has been superimposed on the CaM phase of the A2A-ctL-CaM complex MONSA model (G). In (F) and (G), CaM and A2A-ctL are colored cyan and orange, respectively.
The complex was even more elongated than A2A-ctL alone, suggesting A2A-ctL remains in a disordered conformation also in the complex. Shape restoration suggested CaM binding to one end of the extended A2A-ctL chain. The complex is similar to that observed for another intrinsically disordered protein, the myelin basic protein, in complex with CaM (71).
In addition to bead-based modeling, more detailed methods were used. In the first approach, separate chain-like models were first made for both proteins alone, and thereafter, these were used for rigid-body refinement. The obtained complex fit the measured data well, and also showed the binding of CaM to one end of A2A-ctL. As a complementary approach, two-phase ab initio modeling for the complex was carried out in MONSA. The results were again very similar, suggesting CaM binding to one end of A2A-ct, which remains in an extended conformation. Because A2A-ct is intrinsically disordered, methods providing single 3D models are descriptive at best, but they do give a realistic view into the corresponding molecular dimensions and the nature of protein-protein interactions in complexes. We also used the EOM method to characterize any conformational subpopulations of A2A-ct that would fit the measured SAXS data even better. The results indicate a slightly better fit to the raw data when a rather broad, somewhat bimodal, conformational ensemble is chosen. All SAXS fits are shown in Fig. S5 and fitting χ values in Table S1. Taken together, the A2A-ctL is very flexible and extended and has a high radius of gyration, which indicates a mostly disordered structure. CaM binds to one end of A2A-ctL, whereas the unbound end of A2A-ctL remains in the extended conformation.
Calmodulin binding predictions
Because a number of GPCRs have been shown to bind CaM at either the intracellular loop 3 or the C-terminus, we examined CaM binding to a group of GPCRs in silico using the CaM target database (62). The ones predicted to bind were further classified into C-terminal and other site binders (Table S1). To our knowledge, twenty-one GPCRs have been experimentally shown to bind CaM (19,31–35,37,38,40,72); some have been reported to have two separate binding sites. The CaM target database was able to correctly predict 18 different known binding sites from 17 different GPCRs. Here, if the residues taking part in CaM binding were known, the correspondence was checked based on them, but if not, the site was checked at the level of intracellular loop or C-terminus reported to possess the CaM binding region. The prediction of seven known binding sites failed. Almost all proteins in the prediction had one or more potential CaM-binding sites. Most often, the site was located in one of the three intracellular loops (20, 14, and 42 sites for the loops 1-3, respectively). A total of 26 binding sites were predicted in the C-terminus, including two previously known class A, five class B, and three class C GPCRs. According to the prediction, the CaM-binding site on the A2AR locates in the C-terminus, at residues 290–302 (YRIREFRQTFRKI). When the predicted binding sites were compared to the presence of one or more potentially palmitoylated cysteines in the proximal part of the receptor C-terminus, we noticed that a C-terminal binding site was almost three times more likely (37.3%) than in the case of palmitoylated receptors (14.3%) (Tables S2 and S3).
Discussion
In this study, our goal was biophysical characterization of the large cytoplasmic tail of human A2AR and the binding of CaM to it. Although experimental data related to the function of A2A-ct is very limited, it has been proposed to take part in different protein-protein interactions, including interactions with dopamine D2 receptor, ARNO/cytohesin-2, β-arrestins, and CaM (43,73,74).
Our SRCD, SAXS, and NMR results support the previously predicted disordered and extended conformation of the A2A-ct in solution. Interestingly, certain negatively charged membrane-mimicking compounds induce a conformational change, making the C-terminus more helical (Fig. 2). NMR revealed transient β-conformations in the proximal part of the C-terminus (Fig. 3 B). Although the A2AR lacks a putative palmitoylation site at the end of helix 8, which anchors to the lipid bilayer in many GPCRs (Table S2), this region adopts α-helical conformation in the solved A2A crystal structures and is partially buried in the membrane (10,12). Furthermore, the CXCR4 receptor lacks palmitoylation at the corresponding site, but it does not form helix 8 (75). More precise inspection of the A2AR crystal structures reveals that on this region the hydrophobic residues organize to the membrane side of the helix, whereas the polar and charged residues point toward the cytosol. Apart from Glu-294, charged residues in this helix are positive arginines. Thus, it is consistent that negative membrane-mimicking compounds affect the folding of the C-terminus, as the region corresponding to an amphipathic helix is induced to fold. A similar negative charge-assisted folding was observed in the case of peptides corresponding to the TM helices of A2AR (76). Consequently, the presence of transient β-conformations in aqueous environment is not surprising when considering that both hydrophobic as well as electrostatic interactions might play a role in folding of the C-terminus. In solution, these interactions are disturbed and the A2A-ct has more freedom to adopt different conformations.
A CaM-A2AR interaction has been reported in different cell-based and mass spectrometry studies (19,42,43), but nothing is known about its binding affinity or thermodynamics. In addition, the effect of calcium on binding is unclear. According to our native PAGE, ITC, and analytical gel filtration results, the A2A-ct binds CaM in a calcium-dependent manner (Figs. 4 and 5). When the C-terminal amino acids 293–320 are deleted or EDTA is added, high-affinity binding is abolished. The same effect is achieved by mutating arginines at positions 291, 293, 296, 300, 304, and 309 to alanines, demonstrating the importance of these positively charged amino acids for the interaction. Our SAXS and NMR data indicate that CaM binds to the proximal end of the A2A-ct tail with no major conformational changes—such as becoming more compact—occurring upon CaM binding (Fig. 7, Table 2).
In 2008, Woods, et al. (19), localized the CaM-binding site to the proximal portion of A2A-ct, and the A2AR C-terminal peptide always kept the first 291RIR293 residues when the A2A-ct-CaM complex was exposed to chymotryptic digestion. On the other hand, from the A2AR crystal structures, it is obvious that Arg-291 is buried in the membrane, and most probably unable to take part in the interaction in vivo. Our NMR results suggest CaM binding to residues 293–321 (Fig. 3 B). Thus, we think that Arg-293 and the subsequent arginines are involved in CaM binding. Based on ITC studies, the A2A-ct-CaM interaction has a 1:1 stoichiometry with a Kd of 97.9 ± 9.3 nM (Table 1). The affinity of binding is quite high for a protein-protein interaction, but CaM is known to bind many of its interaction partners with nanomolar affinity. For instance, Bofill-Cardona, et al., reported 80 nM affinity for the binding of CaM to dopamine D2 receptor peptide (72), and the affinity of CaM for example toward its protein kinase targets can be at least an order of magnitude higher (77,78).
A2A-ctL bound to lipid vesicles immobilized on a sensor surface, and CaM binding to A2A-ct released the whole A2A-ct-CaM complex from the vesicles. Binding of A2A-ct to vesicles was expected, as the vesicles contained negatively charged lipids, but the ability of CaM to release the whole C-terminus from the vesicles was an interesting finding. A similar competition between CaM and a lipid membrane surface was previously observed for the myelin basic protein (79). Most likely, CaM replaces the negative lipid head groups while binding to the arginine-rich epitope of the A2A-ct. It would be interesting to know to what extent the conformation of helix 8 has to change to bind CaM. Is CaM alone enough to disrupt the membrane-helix 8 interaction to an extent that enables A2A-ct-CaM binding, or is something else, like phosphorylation of Threonine 298, needed in vivo? One possible factor could also be a local change in the electrostatic environment during an action potential. It has been proposed that direct calcium binding to anionic membrane phospholipids changes the electrostatic environment surrounding the immunoreceptor tyrosine-based activation motif (ITAM) of a T-cell antigen receptor. This electrostatic change disrupts the ionic interaction between a positively charged ITAM and negatively charged phospholipids, exposing a tyrosine to phosphorylation and subsequently triggering an extensive signaling network (80). A similar mechanism could be possible in the case of A2AR.
We used the CaM Target Database (62) to examine potential CaM binding to 116 primarily class A GPCRs in silico. Seventy-six of the predicted binding sites localized to intracellular loops and 26 to the C-terminus. Of the three intracellular loops, binding was predicted to occur most frequently in loop 3. Presence of palmitoylable cysteine in the proximal part of the receptor C-terminus was observed to be related with almost three times lower probability of having a C-terminal CaM-binding site in the prediction compared to receptors lacking palmitoylable cysteine at this position. It seems that presence of palmitoylated cysteine in the proximal part of the GPRC C-terminus might restrain CaM binding into this domain. Is this a general regulatory mechanism of CaM binding among GPCRs? This remains to be answered in the further studies. There might be large differences in CaM binding among GPCRs belonging to the same GPCR family, let alone GPCRs belonging to the different families, and that ultimately, each GPCR-CaM interaction has to be verified separately. As discussed earlier, the CaM Target Database predicts CaM binding to the A2AR residues 290–302, but the first three amino acids T290, R291, and I292 of the predicted binding site are buried within the membrane in the crystal structures and possibly unavailable for direct CaM binding. Thus, when considering a potential CaM-binding site, it is important to also take into account such restrictions.
Overall, our study indicates that the carboxyl terminal domain of A2AR has significant structural flexibility and ability to adopt different conformations depending on its surrounding and interaction partners. This feature may play an important role in non-G-protein signaling. However, studying ligand-dependent changes and the effect of allosterism on these interactions requires full-length receptors. In the case of A2AR, it would be interesting to know how agonist/antagonist binding to the extracellular side or phosphorylation of threonine 298 affects CaM binding, and vice versa. Heteromerization of A2AR with D2, mGlu5, and CB1 receptors, for example, might also change the way these receptors bind CaM and which pathways they use for signaling. Comparison of affinities of CaM and other interaction partners would allow ranking their preference to bind to A2A-ct. Because most of the interaction partners of A2A-ct are calcium-binding proteins, finding out the possible role of calcium in each of these interactions would help in forming a general view about effects of varying intracellular calcium concentration on A2AR signaling, desensitization, and recycling, for example.
Acknowledgments
The authors thank the European Synchrotron Radiation Facility (Grenoble) and ISA, Center for Storage Ring Facilities (Århus) for the excellent experimental support and the use of beamline ID14-3 and CD1 data collection facilities, respectively. We are also grateful to Dr. Ulrich Bergmann for helpful discussions and technical guidance with mass spectrometric equipment. The authors acknowledge A. Pia Abola for assistance with manuscript preparation.
This work was supported by grants from Biocenter Oulu (University of Oulu, Finland), Academy of Finland (132138 to VPJ, 252066 to PK, and 259447 to PP), Sigrid Juselius Foundation, and FP7 Marie Curie European Reintegration Grant (IRG 249081). H.P. is a member of the Biocenter Oulu Doctoral Programme and University of Oulu Graduate School.
Supporting Material
Supporting Citations
References (81–87) appear in the Supporting Material.
References
- 1.Unal H., Karnik S.S. Domain coupling in GPCRs: the engine for induced conformational changes. Trends Pharmacol. Sci. 2012;33:79–88. doi: 10.1016/j.tips.2011.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Magalhaes A.C., Dunn H., Ferguson S.S.G. Regulation of G protein-coupled receptor activity, trafficking and localization by GPCR-interacting G proteins. Br. J. Pharmacol. 2011;165:1717–1736. doi: 10.1111/j.1476-5381.2011.01552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gurevich V.V., Gurevich E.V. GPCR monomers and oligomers: it takes all kinds. Trends Neurosci. 2008;31:74–81. doi: 10.1016/j.tins.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Milligan G. A day in the life of a G protein-coupled receptor: the contribution to function of G protein-coupled receptor dimerization. Br. J. Pharmacol. 2008;153(Suppl 1):S216–S229. doi: 10.1038/sj.bjp.0707490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chini B., Parenti M. G-protein-coupled receptors, cholesterol and palmitoylation: facts about fats. J. Mol. Endocrinol. 2009;42:371–379. doi: 10.1677/JME-08-0114. [DOI] [PubMed] [Google Scholar]
- 6.Dunphy J.T., Linder M.E. Signalling functions of protein palmitoylation. Biochim. Biophys. Acta. 1998;1436:245–261. doi: 10.1016/s0005-2760(98)00130-1. [DOI] [PubMed] [Google Scholar]
- 7.Palmer T.M., Stiles G.L. Identification of an A2a adenosine receptor domain specifically responsible for mediating short-term desensitization. Biochemistry. 1997;36:832–838. doi: 10.1021/bi962290v. [DOI] [PubMed] [Google Scholar]
- 8.Borroto-Escuela D.O., Marcellino D., Fuxe K. A serine point mutation in the adenosine A2AR C-terminal tail reduces receptor heteromerization and allosteric modulation of the dopamine D2R. Biochem. Biophys. Res. Commun. 2010;394:222–227. doi: 10.1016/j.bbrc.2010.02.168. [DOI] [PubMed] [Google Scholar]
- 9.Borroto-Escuela D.O., Romero-Fernandez W., Fuxe K. Characterization of the A2AR-D2R interface: focus on the role of the C-terminal tail and the transmembrane helices. Biochem. Biophys. Res. Commun. 2010;402:801–807. doi: 10.1016/j.bbrc.2010.10.122. [DOI] [PubMed] [Google Scholar]
- 10.Jaakola V.-P., Griffith M.T., Stevens R.C. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu F., Wu H., Stevens R.C. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lebon G., Warne T., Tate C.G. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature. 2011;474:521–525. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doré A.S., Robertson N., Marshall F.H. Structure of the adenosine A(2A) receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure. 2011;19:1283–1293. doi: 10.1016/j.str.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hino T., Arakawa T., Murata T. G-protein-coupled receptor inactivation by an allosteric inverse-agonist antibody. Nature. 2012;482:237–240. doi: 10.1038/nature10750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Congreve M., Andrews S.P., Marshall F.H. Discovery of 1,2,4-triazine derivatives as adenosine A(2A) antagonists using structure based drug design. J. Med. Chem. 2012;55:1898–1903. doi: 10.1021/jm201376w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu W., Chun E., Stevens R.C. Structural basis for allosteric regulation of GPCRs by sodium ions. Science. 2012;337:232–236. doi: 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klaasse E.C., Ijzerman A.P., Beukers M.W. Internalization and desensitization of adenosine receptors. Purinergic Signal. 2008;4:21–37. doi: 10.1007/s11302-007-9086-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burgueño J., Blake D.J., Ciruela F. The adenosine A2A receptor interacts with the actin-binding protein α-actinin. J. Biol. Chem. 2003;278:37545–37552. doi: 10.1074/jbc.M302809200. [DOI] [PubMed] [Google Scholar]
- 19.Woods A.S., Marcellino D., Fuxe K. How calmodulin interacts with the adenosine A(2A) and the dopamine D(2) receptors. J. Proteome Res. 2008;7:3428–3434. doi: 10.1021/pr8001782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Canela L., Luján R., Ciruela F. The neuronal Ca(2+) -binding protein 2 (NECAB2) interacts with the adenosine A(2A) receptor and modulates the cell surface expression and function of the receptor. Mol. Cell. Neurosci. 2007;36:1–12. doi: 10.1016/j.mcn.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 21.Sun C.-N., Cheng H.-C., Chern Y. Rescue of p53 blockage by the A(2A) adenosine receptor via a novel interacting protein, translin-associated protein X. Mol. Pharmacol. 2006;70:454–466. doi: 10.1124/mol.105.021261. [DOI] [PubMed] [Google Scholar]
- 22.Gsandtner I., Charalambous C., Zezula J. Heterotrimeric G protein-independent signaling of a G protein-coupled receptor. Direct binding of ARNO/cytohesin-2 to the carboxyl terminus of the A2A adenosine receptor is necessary for sustained activation of the ERK/MAP kinase pathway. J. Biol. Chem. 2005;280:31898–31905. doi: 10.1074/jbc.M506515200. [DOI] [PubMed] [Google Scholar]
- 23.Milojević T., Reiterer V., Nanoff C. The ubiquitin-specific protease Usp4 regulates the cell surface level of the A2A receptor. Mol. Pharmacol. 2006;69:1083–1094. doi: 10.1124/mol.105.015818. [DOI] [PubMed] [Google Scholar]
- 24.Navarro G., Hradsky J., Mikhaylova M. NCS-1 associates with adenosine A(2A) receptors and modulates receptor function. Front Mol. Neurosci. 2012;5:53. doi: 10.3389/fnmol.2012.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chin D., Means A.R. Calmodulin: a prototypical calcium sensor. Trends Cell Biol. 2000;10:322–328. doi: 10.1016/s0962-8924(00)01800-6. [DOI] [PubMed] [Google Scholar]
- 26.Ikura M., Clore G.M., Bax A. Solution structure of a calmodulin-target peptide complex by multidimensional NMR. Science. 1992;256:632–638. doi: 10.1126/science.1585175. [DOI] [PubMed] [Google Scholar]
- 27.Meador W.E., Means A.R., Quiocho F.A. Target enzyme recognition by calmodulin: 2.4 A structure of a calmodulin-peptide complex. Science. 1992;257:1251–1255. doi: 10.1126/science.1519061. [DOI] [PubMed] [Google Scholar]
- 28.Meador W.E., Means A.R., Quiocho F.A. Modulation of calmodulin plasticity in molecular recognition on the basis of x-ray structures. Science. 1993;262:1718–1721. doi: 10.1126/science.8259515. [DOI] [PubMed] [Google Scholar]
- 29.Radivojac P., Vucetic S., Dunker A.K. Calmodulin signaling: analysis and prediction of a disorder-dependent molecular recognition. Proteins. 2006;63:398–410. doi: 10.1002/prot.20873. [DOI] [PubMed] [Google Scholar]
- 30.Rhoads A.R., Friedberg F. Sequence motifs for calmodulin recognition. FASEB J. 1997;11:331–340. doi: 10.1096/fasebj.11.5.9141499. [DOI] [PubMed] [Google Scholar]
- 31.Wang D., Sadée W., Quillan J.M. Calmodulin binding to G protein-coupling domain of opioid receptors. J. Biol. Chem. 1999;274:22081–22088. doi: 10.1074/jbc.274.31.22081. [DOI] [PubMed] [Google Scholar]
- 32.Minakami R., Jinnai N., Sugiyama H. Phosphorylation and calmodulin binding of the metabotropic glutamate receptor subtype 5 (mGluR5) are antagonistic in vitro. J. Biol. Chem. 1997;272:20291–20298. doi: 10.1074/jbc.272.32.20291. [DOI] [PubMed] [Google Scholar]
- 33.Nakajima Y., Yamamoto T., Nakanishi S. A relationship between protein kinase C phosphorylation and calmodulin binding to the metabotropic glutamate receptor subtype 7. J. Biol. Chem. 1999;274:27573–27577. doi: 10.1074/jbc.274.39.27573. [DOI] [PubMed] [Google Scholar]
- 34.Lucas J.L., Wang D., Sadée W. Calmodulin binding to peptides derived from the i3 loop of muscarinic receptors. Pharm. Res. 2006;23:647–653. doi: 10.1007/s11095-006-9784-9. [DOI] [PubMed] [Google Scholar]
- 35.Turner J.H., Gelasco A.K., Raymond J.R. Calmodulin interacts with the third intracellular loop of the serotonin 5-hydroxytryptamine1A receptor at two distinct sites: putative role in receptor phosphorylation by protein kinase C. J. Biol. Chem. 2004;279:17027–17037. doi: 10.1074/jbc.M313919200. [DOI] [PubMed] [Google Scholar]
- 36.Turner J.H., Raymond J.R. Interaction of calmodulin with the serotonin 5-hydroxytryptamine2A receptor. A putative regulator of G protein coupling and receptor phosphorylation by protein kinase C. J. Biol. Chem. 2005;280:30741–30750. doi: 10.1074/jbc.M501696200. [DOI] [PubMed] [Google Scholar]
- 37.Labasque M., Reiter E., Marin P. Physical interaction of calmodulin with the 5-hydroxytryptamine2C receptor C-terminus is essential for G protein-independent, arrestin-dependent receptor signaling. Mol. Biol. Cell. 2008;19:4640–4650. doi: 10.1091/mbc.E08-04-0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mahon M.J., Shimada M. Calmodulin interacts with the cytoplasmic tails of the parathyroid hormone 1 receptor and a sub-set of class b G-protein coupled receptors. FEBS Lett. 2005;579:803–807. doi: 10.1016/j.febslet.2004.12.056. [DOI] [PubMed] [Google Scholar]
- 39.Nickols H.H., Shah V.N., Limbird L.E. Calmodulin interacts with the V2 vasopressin receptor: elimination of binding to the C-terminus also eliminates arginine vasopressin-stimulated elevation of intracellular calcium. J. Biol. Chem. 2004;279:46969–46980. doi: 10.1074/jbc.M407351200. [DOI] [PubMed] [Google Scholar]
- 40.Huang Y., Zhou Y., Yang J.J. Calmodulin regulates Ca2+-sensing receptor-mediated Ca2+ signaling and its cell surface expression. J. Biol. Chem. 2010;285:35919–35931. doi: 10.1074/jbc.M110.147918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y., Wang D., Sadée W. Calmodulin interaction with peptides from G-protein coupled receptors measured with S-Tag labeling. Biochem. Biophys. Res. Commun. 2005;333:390–395. doi: 10.1016/j.bbrc.2005.05.129. [DOI] [PubMed] [Google Scholar]
- 42.Navarro G., Aymerich M.S., Franco R. Interactions between calmodulin, adenosine A2A, and dopamine D2 receptors. J. Biol. Chem. 2009;284:28058–28068. doi: 10.1074/jbc.M109.034231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferré S., Woods A.S., Franco R. Calcium-mediated modulation of the quaternary structure and function of adenosine A2A-dopamine D2 receptor heteromers. Curr. Opin. Pharmacol. 2010;10:67–72. doi: 10.1016/j.coph.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hayashi N., Matsubara M., Taniguchi H. An expression system of rat calmodulin using T7 phage promoter in Escherichia coli. Protein Expr. Purif. 1998;12:25–28. doi: 10.1006/prep.1997.0807. [DOI] [PubMed] [Google Scholar]
- 45.Kursula P., Majava V. A structural insight into lead neurotoxicity and calmodulin activation by heavy metals. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2007;63:653–656. doi: 10.1107/S1744309107034525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hellman M., Piirainen H., Permi P. Bridge over troubled proline: assignment of intrinsically disordered proteins using (HCA)CON(CAN)H and (HCA)N(CA)CO(N)H experiments concomitantly with HNCO and i(HCA)CO(CA)NH. J. Biomol. NMR. 2014;58:49–60. doi: 10.1007/s10858-013-9804-0. [DOI] [PubMed] [Google Scholar]
- 47.Mäntylahti S., Tossavainen H., Permi P. An intraresidual i(HCA)CO(CA)NH experiment for the assignment of main-chain resonances in 15N, 13C labeled proteins. J. Biomol. NMR. 2009;45:301–310. doi: 10.1007/s10858-009-9373-4. [DOI] [PubMed] [Google Scholar]
- 48.Permi P., Annila A. Coherence transfer in proteins. Prog. Nucl. Magn. Reson. Spectrosc. 2004;44:97–137. [Google Scholar]
- 49.Marsh J.A., Singh V.K., Forman-Kay J.D. Sensitivity of secondary structure propensities to sequence differences between α- and γ-synuclein: implications for fibrillation. Protein Sci. 2006;15:2795–2804. doi: 10.1110/ps.062465306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang H., Neal S., Wishart D.S. RefDB: a database of uniformly referenced protein chemical shifts. J. Biomol. NMR. 2003;25:173–195. doi: 10.1023/a:1022836027055. [DOI] [PubMed] [Google Scholar]
- 51.Pernot P., Theveneau P., Cipriani F. New beamline dedicated to solution scattering from biological macromolecules at the ESRF. J. Phys. Conf. Ser. 2010;247:012009. [Google Scholar]
- 52.Konarev P.V., Volkov V.V., Svergun D.I. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Cryst. 2003;36:1277–1282. [Google Scholar]
- 53.Svergun D. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Cryst. 1992;25:495–503. [Google Scholar]
- 54.Calmettes P., Durand D., Smith J.C. How random is a highly denatured protein? Biophys. Chem. 1994;53:105–113. doi: 10.1016/0301-4622(94)00081-6. [DOI] [PubMed] [Google Scholar]
- 55.Fitzkee N.C., Rose G.D. Reassessing random-coil statistics in unfolded proteins. Proc. Natl. Acad. Sci. USA. 2004;101:12497–12502. doi: 10.1073/pnas.0404236101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ruskamo S., Chukhlieb M., Kursula P. Juxtanodin is an intrinsically disordered F-actin-binding protein. Sci Rep. 2012;2:899. doi: 10.1038/srep00899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Franke D., Svergun D.I. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Cryst. 2009;42:342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Svergun D.I., Petoukhov M.V., Koch M.H.J. Determination of domain structure of proteins from X-ray solution scattering. Biophys. J. 2001;80:2946–2953. doi: 10.1016/S0006-3495(01)76260-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Petoukhov M.V., Svergun D.I. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 2005;89:1237–1250. doi: 10.1529/biophysj.105.064154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Svergun D.I. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 1999;76:2879–2886. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bernadó P., Mylonas E., Svergun D.I. Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 2007;129:5656–5664. doi: 10.1021/ja069124n. [DOI] [PubMed] [Google Scholar]
- 62.Yap K.L., Kim J., Ikura M. Calmodulin target database. J. Struct. Funct. Genomics. 2000;1:8–14. doi: 10.1023/a:1011320027914. [DOI] [PubMed] [Google Scholar]
- 63.Linding R., Jensen L.J., Russell R.B. Protein disorder prediction: implications for structural proteomics. Structure. 2003;11:1453–1459. doi: 10.1016/j.str.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 64.Prilusky J., Felder C.E., Sussman J.L. FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics. 2005;21:3435–3438. doi: 10.1093/bioinformatics/bti537. [DOI] [PubMed] [Google Scholar]
- 65.Ward J.J., McGuffin L.J., Jones D.T. The DISOPRED server for the prediction of protein disorder. Bioinformatics. 2004;20:2138–2139. doi: 10.1093/bioinformatics/bth195. [DOI] [PubMed] [Google Scholar]
- 66.Slabinski L., Jaroszewski L., Godzik A. XtalPred: a web server for prediction of protein crystallizability. Bioinformatics. 2007;23:3403–3405. doi: 10.1093/bioinformatics/btm477. [DOI] [PubMed] [Google Scholar]
- 67.Receveur-Bréchot V., Bourhis J.-M., Longhi S. Assessing protein disorder and induced folding. Proteins. 2006;62:24–45. doi: 10.1002/prot.20750. [DOI] [PubMed] [Google Scholar]
- 68.Morrisett J.D., David J.S., Gotto A.M., Jr. Interaction of an apolipoprotein (apoLP-alanine) with phosphatidylcholine. Biochemistry. 1973;12:1290–1299. doi: 10.1021/bi00731a008. [DOI] [PubMed] [Google Scholar]
- 69.Hellman M., Tossavainen H., Permi P. Characterization of intrinsically disordered prostate associated gene (PAGE5) at single residue resolution by NMR spectroscopy. PLoS ONE. 2011;6:e26633. doi: 10.1371/journal.pone.0026633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ikura M. Calcium binding and conformational response in EF-hand proteins. Trends Biochem. Sci. 1996;21:14–17. [PubMed] [Google Scholar]
- 71.Majava V., Wang C., Kursula P. Structural analysis of the complex between calmodulin and full-length myelin basic protein, an intrinsically disordered molecule. Amino Acids. 2010;39:59–71. doi: 10.1007/s00726-009-0364-2. [DOI] [PubMed] [Google Scholar]
- 72.Bofill-Cardona E., Kudlacek O., Nanoff C. Binding of calmodulin to the D2-dopamine receptor reduces receptor signaling by arresting the G protein activation switch. J. Biol. Chem. 2000;275:32672–32680. doi: 10.1074/jbc.M002780200. [DOI] [PubMed] [Google Scholar]
- 73.Zezula J., Freissmuth M. The A(2A)-adenosine receptor: a GPCR with unique features? Br. J. Pharmacol. 2008;153(Suppl 1):S184–S190. doi: 10.1038/sj.bjp.0707674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Keuerleber S., Gsandtner I., Freissmuth M. From cradle to twilight: the carboxyl terminus directs the fate of the A(2A)-adenosine receptor. Biochim. Biophys. Acta. 2011;1808:1350–1357. doi: 10.1016/j.bbamem.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 75.Wu B., Chien E.Y.T., Stevens R.C. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lazarova T., Brewin K.A., Robinson C.R. Characterization of peptides corresponding to the seven transmembrane domains of human adenosine A2a receptor. Biochemistry. 2004;43:12945–12954. doi: 10.1021/bi0492051. [DOI] [PubMed] [Google Scholar]
- 77.de Diego I., Kuper J., Wilmanns M. Molecular basis of the death-associated protein kinase–calcium/calmodulin regulator complex. Sci. Signal. 2010;3:ra6. doi: 10.1126/scisignal.2000552. [DOI] [PubMed] [Google Scholar]
- 78.Brokx R.D., Lopez M.M., Makhatadze G.I. Energetics of target peptide binding by calmodulin reveals different modes of binding. J. Biol. Chem. 2001;276:14083–14091. doi: 10.1074/jbc.M011026200. [DOI] [PubMed] [Google Scholar]
- 79.Wang C., Neugebauer U., Kursula P. Charge isomers of myelin basic protein: structure and interactions with membranes, nucleotide analogues, and calmodulin. PLoS ONE. 2011;6:e19915. doi: 10.1371/journal.pone.0019915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shi X., Bi Y., Xu C. Ca2+ regulates T-cell receptor activation by modulating the charge property of lipids. Nature. 2013;493:111–115. doi: 10.1038/nature11699. [DOI] [PubMed] [Google Scholar]
- 81.Sadeghi H.M., Innamorati G., Birnbaumer M. Palmitoylation of the V2 vasopressin receptor. Mol. Pharmacol. 1997;52:21–29. doi: 10.1124/mol.52.1.21. [DOI] [PubMed] [Google Scholar]
- 82.O’Dowd B.F., Hnatowich M., Bouvier M. Palmitoylation of the human beta 2-adrenergic receptor. Mutation of Cys-341 in the carboxyl tail leads to an uncoupled nonpalmitoylated form of the receptor. J. Biol. Chem. 1989;264:7564–7569. [PubMed] [Google Scholar]
- 83.Cherezov V., Rosenbaum D.M., Stevens R.C. High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hanson M.A., Cherezov V., Stevens R.C. A specific cholesterol binding site is established by the 2.8 A structure of the human β2-adrenergic receptor. Structure. 2008;16:897–905. doi: 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Okamoto Y., Ninomiya H., Masaki T. Palmitoylation of human endothelinB. Its critical role in G protein coupling and a differential requirement for the cytoplasmic tail by G protein subtypes. J. Biol. Chem. 1997;272:21589–21596. doi: 10.1074/jbc.272.34.21589. [DOI] [PubMed] [Google Scholar]
- 86.Jin H., Xie Z., O’Dowd B.F. Palmitoylation occurs at cysteine 347 and cysteine 351 of the dopamine D(1) receptor. Eur. J. Pharmacol. 1999;386:305–312. doi: 10.1016/s0014-2999(99)00727-x. [DOI] [PubMed] [Google Scholar]
- 87.Heakal Y., Woll M.P., Kester M. Neurotensin receptor-1 inducible palmitoylation is required for efficient receptor-mediated mitogenic-signaling within structured membrane microdomains. Cancer Biol. Ther. 2011;12:427–435. doi: 10.4161/cbt.12.5.15984. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.