

Abstract
Introduction
The SIX homeodomain proteins and the EYA family of co-activators form a bipartite transcription factor complex that promotes the proliferation and survival of progenitor cells during organogenesis and is down-regulated in most adult tissues. Abnormal over-expression of SIX1 and EYA in adult tissue is associated with the initiation and progression of diverse tumor types. Importantly, SIX1 and EYA are often co-overexpressed in tumors, and the SIX1-EYA2 interaction has been shown to be critical for metastasis in a breast cancer model. The EYA proteins also contain protein tyrosine phosphatase activity, which plays an important role in breast cancer growth and metastasis as well as directing cells to the repair pathway upon DNA damage.
Areas covered
This review provides a summary of the SIX1/EYA complex as it relates to development and disease and the current efforts to therapeutically target this complex.
Expert opinion
Recently, there have been an increasing number of studies suggesting that targeting the SIX1/EYA transcriptional complex will potently inhibit tumor progression. Although current attempts to develop inhibitors targeting this complex are still in the early stages, continued efforts towards developing better compounds may ultimately result in effective anti-cancer therapies.
1. Introduction
The sine oculis homeodomain (SIX) proteins are a family of transcription factors evolutionarily conserved from lower invertebrates to humans1. The SIX proteins were originally identified in Drosophila as vital regulators of eye development, and have since been shown to participate in the expansion and differentiation of cell populations in numerous organs including the retina, craniofacial structures, auditory system, brain, lung, muscle, kidney, and gonads, amongst others2. SIX proteins promote cell proliferation through the regulation of multiple cell cycle genes known to affect the G1/S and G2/M transitions, as well as progression through S-phase3–7. Loss and gain-of-function studies have implicated SIX1 as a regulator of multiple tissue-specific differentiation factors that control cell fate8–10. Moreover, mutations within the SIX family, both naturally occurring and genetically engineered, have adverse consequences on the development of organs/tissues11–16, confirming its role as a critical regulator of organogenesis.
In humans, there are six family members (SIX1-6) that are homologous to the Drosophila sine oculis (so) gene17. All SIX proteins are composed of two conserved domains, the nucleic acid recognition homeodomain (HD) and the SIX domain (SD)18. DNA binding is primarily mediated through the HD, while protein-protein interactions are mediated through the SD. However, there is strong evidence suggesting that the SD also contributes to the SIX1-DNA interaction, though this has yet to be directly confirmed by structural analyses19. In addition to the SD and HD, SIX2, SIX4, and SIX5 also have intrinsic transcriptional activation domains (AD), and thus may not require co-activator proteins, such as the EYAs described below, to promote transcription under all circumstances20, 21.
The remaining SIX proteins, including SIX1, have no intrinsic activation domains and require a family of co-activators, EYA1-4, to mediate transcriptional activation. The human EYA proteins are homologs of the Drosophila eyes absent (eya) proteins that contain a highly conserved Eya domain (ED), responsible for interacting with the SD22. The SIX proteins actively translocate EYAs into the nucleus, where EYAs stimulate SIX-mediated transcription through their proline/serine/threonine (P/S/T) rich transactivation domains23, 24. However, in cases in which EYA co-activators are absent, SIX1 can bind to promoters in conjunction with the co-repressor DACH1 to block transcription of target genes4. Embedded within the P/S/T domain of the EYA proteins is a separate ED, known as Eya domain 2 (ED2). This domain possesses threonine phosphatase activity, which has been shown to play a role in innate immunity25, 26. Similar to SIX1, EYA proteins are also critical for the development of multiple organs in part by promoting proliferation and survival of progenitor cell populations27.
Interestingly, not only is the ED able to interact with SIX proteins to stimulate transcription, the ED also possesses Mg2+-dependent tyrosine phosphatase activity28, 29. The EYA proteins use a nucleophilic aspartic acid residue and a divalent metal ion to catalyze tyrosine dephosphorylation. Using SIX1-dependent reporter assays, the phosphatase activity of EYA3 has been shown to be required for the transcription of a subset of SIX1-induced genes4. However, a detailed understanding of the exact mechanism by which this occurs is lacking. To date, there are only two validated in vivo targets for EYA’s tyrosine phosphatase activity. The first target is the histone variant, H2AX30, 31. Upon DNA damage, EYAs dephosphorylate H2AX, directing cells to DNA repair rather than apoptotic pathways. More recently, EYA2 has been shown to inhibit the transcriptional activity of the estrogen receptor β (ERβ), through the dephosphorylation of the Y36 residue32. The phosphorylated state of Y36 promotes the interaction between ERβ and its co-activators, stimulating transcriptional activation and subsequent inhibition of cell proliferation and differentiation33.
Loss- and gain of-function analyses in fruit flies, fish, frogs, and mice have been used to confirm the importance of both SIX and EYA genes in development, which is underscored by the existence of naturally occurring mutations of SIX and EYA in various human genetic syndromes14, 16, 34–41. Proteins from both families are co-expressed in intricate spatio-temporal patterns in embryonic tissues to direct complex developmental processes2. There is much evidence that both EYA and SIX1 are required not only for cell proliferation but also for cell survival. For instance, the Eya1 knockout phenocopies the Six1 knockout in mice, and organ defects in both are attributed to a decrease in progenitor cell proliferation and an increase in cellular apoptosis, thus leading to reduced size or absence of organ development36, 42. In both Six1 and Eya1 knockout mice, embryonic lung epithelial cells show increased expression of differentiation markers and a loss of progenitor cell markers, supporting their role as critical regulators of epithelial progenitor cells8. Additionally, in kidney development, knockout studies have shown that Six1, Six2, Six4, and Eya1 each regulate critical genes required during development, including GDNF, which is important for the maintenance of the mesenchymal progenitor pool2, 9, 10, 35. The loss of any of these Six family members leads to premature differentiation and depletion of the progenitor cell population. The studies highlighted above reveal the numerous developmental roles of the SIX1/EYA transcriptional complex, such as maintaining progenitor pools, cellular proliferation, and directing differentiation programs, confirming that the complex is a critical regulator of tissue growth and patterning.
2. The role of SIX1 and EYA in genetic syndromes
Naturally occurring mutations in both SIX and EYA are implicated in multiple developmental disorders confirming their critical roles in organogenesis2. In particular, mutations in SIX1, SIX5, or EYA1 can lead to branchio-oto-renal (BOR) syndrome, a developmental disorder defined by hearing loss, bronchial fistulae, and renal anomalies43. EYA1 is the most commonly mutated gene in BOR syndrome with currently 14 known mutations located within the ED11, 44, 45, which contains both the SIX1 interaction site, as well as its tyrosine phosphatase activity46. Structural and biochemical analysis of EYA1/2 have provided insights into the molecular mechanisms of several BOR mutations19. Three of the BOR mutations (V496E, delV499, and L529P) are located in the SIX1 interface and are predicted to abrogate SIX1-EYA2 binding. Another mutant, N433P, resides in the middle of the catalytic domain and is shown to disrupt phosphatase activity; however it is still capable of binding SIX1 in cells47, 48. In addition to EYA1, mutations in SIX1 have also been reported in BOR patients. Biochemical analyses of 6 of these mutations reveal that five act by disrupting the SIX1-DNA interaction38, 46. The remaining SIX1 BOR mutation, V17E, is located at the SIX1-EYA interface and disrupts the SIX1-EYA interaction46. These natural mutations confirm that the SIX1/EYA transcriptional complex, including EYA’s phosphatase activity, is critical for normal organ development.
3. Oncogenic roles of the SIX1 transcription factor
The ability of the SIX transcriptional complex to direct multiple developmental programs necessary for the maintenance and expansion of progenitor populations, suggests that the abnormal overexpression of SIX transcriptional complex proteins in adult tissues could contribute to both tumorigenesis and tumor progression. Tumorigenic cells often resemble a more embryonic state, having been reprogrammed to maintain proliferation, survival, and additional cancer hallmark pathways (Figure 1), suggesting that correcting aberrant developmental transcriptional pathways in malignant cells may be a useful therapeutic approach49. To date, SIX1 is the most heavily implicated SIX family member in cancer. Therefore, we will focus the remainder of this article on SIX1 and its known oncogenic roles.
Figure 1.

A recreation of the Cancer Hallmarks115, illustrating the known hallmarks in which the SIX1 and EYA family members carry out their tumor-promoting properties. In total, the SIX1/EYA complex has been shown to play a role in four out of the six original hallmarks, one of the two enabling characteristics and one of the two emerging hallmarks, highlighting the importance of targeting this complex as a potential therapeutic.
SIX1 overexpression in cancers was first observed in 1998, where high levels of SIX1 expression in primary breast cancers and metastatic lesions were argued to influence tumor progression, at least in part, through its ability to attenuate the G2 cell cycle checkpoint in response to DNA damage3. Subsequently, multiple studies have confirmed that overexpression of SIX1 is tied to the occurrence and progression of breast cancer as well as other cancer types, in part through its ability to influence cell cycle control50–55.
Over the past decade SIX1 has been implicated in multiple “hallmarks of cancer” including sustained proliferative signaling5, 56, 57, invasion and metastasis51, 57–61, evasion of growth suppressors62, induction of genomic instability63, and resistance of cell death53, 64–66 (Figure 1). SIX1 is able to maintain cellular proliferation through the transcriptional regulation of numerous genes involved in cell cycle progression. Cyclin D1 has been shown to be a direct transcriptional target of SIX1 in pancreatic cancer56 and rhabdomyosarcoma (RMS)57, while Cyclin A1 is transcriptionally activated by SIX1 in breast cancer5 and c-Myc is transcriptionally regulated by SIX1 in rhabdomyosarcoma (RMS)57.
The second cancer hallmark in which SIX1 has been implicated is activating invasion and metastasis. Indeed, SIX1 has been shown to be both pro-oncogenic and metastatic. Mammary specific Six1 overexpression in transgenic mice promotes highly aggressive tumors that undergo an epithelial to mesenchymal transition (EMT) and exhibit stem cell phenotypes59. Six1-induced EMT has been shown to be through its ability to stimulate TGF-β signaling, which is also critical for Six1-mediated metastasis in experimental metastasis mouse models of mammary cancer51, 58. In addition, SIX1 stimulates lymphangiogenesis in breast cancer through transcriptionally regulating VEGF-C, which then further contributes to its ability to mediate metastasis60. Finally, in RMS, SIX1 directly promotes the expression of Ezrin, a protein known to play an important role in cell surface adhesion, cellular migration and domain organization within the plasma membrane57. SIX1 has also been implicated in metastasis of other tumor types, including hepatocellular carcinoma, where it is associated with both proliferation and invasion61. Together, these studies demonstrate a critical role for SIX1 in tumor progression.
Another hallmark of cancer with which SIX1 is associated is the evasion of growth suppression. Indeed, we have shown in an experimental metastasis model that Six1 can induce a switch in TGF-β signaling from tumor suppressive to tumor promotional, overcoming the growth suppressive function of TGF-β to allow its pro-migratory and invasive functions to be dominant62. Not only is SIX1 involved in overcoming growth suppression, but it has also been implicated in resistance to cell death. Studies have shown that SIX1 overexpression induces resistance to apoptosis and more specifically to tumor necrosis factor-related apoptosis inducing ligand (TRAIL)-mediated apoptosis53, 64, 65. Furthermore, SIX1 overexpression can circumvent paclitaxel-induced apoptosis in breast cancer cells66. In addition to its documented effect on these original hallmarks of cancer, SIX1 has been shown to affect a cancer enabling characteristic, genome instability, where its overexpression has been shown to increase double strand breaks in breast cells63.
Importantly, the ability of SIX1 to mediate these hallmarks of cancer is not restricted to one tumor type. Instead, SIX1 overexpression has been correlated with poor prognosis in numerous cancers including breast51, 59, lung54, pancreatic56, cervical55, 67, colorectal68, 69, and ovarian cancers53, amongst others. Furthermore, knockdown of SIX1 in multiple tumor types, including breast60 and colorectal cancers70, hepatocellular carcinomas61, and rhabdomyosarcomas50, dramatically decreases tumor size and metastasis in animal models. Currently, few studies have examined the mechanisms leading to Six1 re-expression in cancer and thus this remains an important topic to be investigated. There is evidence that SIX1 can be amplified in a small percentage of breast cancers71, and that it can be regulated by microRNA-185, which is lost in cancers thereby allowing Six1 to be re-expressed64.
4. Oncogenic roles of the EYA family
EYAs are overexpressed in many cancers including Wilms’ tumor72, Ewing sarcoma73, breast74, ovarian75, and lung cancers76. In general, overexpression of EYAs has been shown to be pro-tumorigenic27, but in some cases the EYA proteins may have tumor suppressive functions. Thus, understanding the biology of EYAs in cancer may be more complex than that of SIX1, and this complexity may in part be attributable to its dual activities as both a transcriptional cofactor and phosphatase. For example, while EYA2 amplification and/or overexpression has been observed in numerous cancers19, 75, 77, the EYA2 gene is hypermethylated, resulting in decreased expression, in both colorectal78 and pancreatic cancers79, when compared to normal tissues. Similarly, EYA3, which has been shown to have a pro-oncogenic role in Ewing Sarcoma73, is deleted in pancreatic adenocarcimomas80. While EYA1 is increased downstream of the MLL-ENL fusion protein in acute leukemia and its expression in murine hematopoietic progenitor cells leads to their immortalization81 and transformation in cooperation with Six1, Eya overexpression has also been shown to activate an apoptotic program in a murine myeloid cell line82. Together, these data suggest that the EYA proteins may have multiple roles in cancer, and that they act in a highly context-dependent manner. Nonetheless, in the numerous cases where EYA overexpression is pro-tumorigenic, its overexpression has been implicated in a number of cancer hallmarks, including sustained proliferative signaling, resistance to cell death, angiogenesis, invasion and metastasis, and replicative immortality4, 19, 30, 32, 81, 83–85. Due to the multiple biochemical activities of the EYA proteins, it is likely that EYAs contribute to these hallmarks through various mechanisms of action. Below we address how the overexpression of Eya proteins in selected tissues may mediate these hallmarks, either through their role as transcriptional activators along with SIX1, or their role as tyrosine phosphatases. To date, the role of the threonine activity of EYA in cancer has not been explored, and remains an interesting avenue of investigation.
5. The role of the SIX1/EYA interaction in tumorigenesis and metastasis
Both SIX1 and EYA are overexpressed in multiple cancer types including ovarian53, 75, breast77, glioblastomas86, leukemia81, and Wilms’ tumor72. Moreover, we found that overexpression of either SIX1 or EYA commonly correlated with recurrence, metastasis, and a decreased overall survival in a variety of tumor types, using the Oncomine cancer microarray database19. Examination of SIX1 and EYA2 expression in the van de Vijver and Wang breast cancer gene expression datasets led to the observation that only when both proteins are overexpressed together does one observe a shortened time to metastasis and relapse, as well as shortened overall survival77. Furthermore, in breast cancer cell lines, EYA2 is required for many of the SIX1-induced pro-metastatic phenotypes, including enhanced TGF-β signaling and EMT19, 77. These data imply a coordinated action is required between the two proteins in breast and other cancer types and reinforce the hypothesis that EYA2 is required for the ability of SIX1 to mediate tumor progression.
Replicative immortality is a widely accepted hallmark of cancer. As outlined above, Eya1 has been shown to transform and immortalize hematopoietic progenitor cells (HPCs) in vitro81. Although Six1 alone was unable to immortalize HPCs, co-transduction of Six1 and Eya1 resulted in an increase in the number of immortalized cells over Eya1 alone, indicating that Six1 is able to enhance the transforming capacity of Eya181.
Our group recently demonstrated the importance of a direct protein interaction between SIX1 and EYA to mediate breast cancer associated metastasis, an additional hallmark of cancer, using the single amino acid BOR mutation, V17E, which disrupts the SIX1-EYA interaction19. In contrast to wildtype SIX1, SIX1 V17E is unable to activate TGF-β signaling or induce EMT-like characteristics in MCF7 breast cancer cells. In a mouse model of late stage metastasis, mice injected with MCF7 cells carrying the SIX1-V17E mutation did not display enhanced metastasis or shortened survival above controls cells, in contrast to MCF7 cells overexpressing wildtype SIX119. These data suggest that the SIX1-EYA interaction is critical for SIX1-mediated tumor progression and that inhibition of this protein complex could prevent tumor progression.
6. The role of EYA’s phosphatase activity in tumorigenesis and metastasis
In addition to their role as co-activators of the SIX1 transcriptional complex, the EYA proteins are protein tyrosine phosphatases belonging to the haloacid dehalogenase (HAD) family of enzymes. The EYA phosphatase activity has been shown to play a role in chronic cellular proliferation, again, a hallmark of cancers. However, this role has been controversial. Although Hegde and colleagues detected no changes in cellular proliferation using phosphatase-deficient EYA2/384, Wu and colleagues, using a different breast cancer model, have demonstrated that EYA1 promotes cellular proliferation in a manner dependent on its phosphatase activity, through up-regulation of Cyclin D183. In support of the latter study, Rosenfeld and colleagues had earlier demonstrated that the Eya3 phosphatase activity plays an important role in cellular proliferation in mouse myoblasts4. More recently, work by Yuan and colleagues identified a second in vivo target for EYA2 tyrosine phosphatase activity that is an important mediator of cellular proliferation32. The dephosphorylation of residue Y36 on ERβ by EYA2 ablates ERβ transcriptional activation and thus reduces its ability to inhibit tumor cell growth32.
Overexpression experiments using either wildtype or phosphatase-dead EYA mutants in breast cancer cells demonstrate that the EYA tyrosine phosphatase activity is critical for EYA-dependent transformation, migration, invasion, and metastasis84. Of interest, the EYA3 tyrosine phosphatase activity has recently been shown to influence another cancer hallmark, angiogenesis, both in vitro and in vivo models using small molecule inhibitors and genetic approaches85.
The final cancer hallmark in which EYA’s phosphatase activity has been implicated is resisting cell death. EYA plays a role in the DNA damage response through its ability to dephosphorylate the minor histone protein, H2AX, upon DNA damage30, 31. EYA1, 2 and 3 dephosphorylate H2AX at the C-terminal tyrosine 142 residue in mouse and human embryonic cell lines30, 31. Dephosphorylation of H2AX by EYA leads to the recruitment of the MDC1/MRN repair complex and directs cells away from apoptotic pathways and towards DNA repair30. Moreover, knockdown of EYA increases the number of apoptotic cells in response to hypoxia or ionizing radiation30, and knockdown of EYA3 has been shown to sensitize Ewing sarcoma cells to DNA damaging chemotherapeutics73. While in the majority of circumstances EYA is shown to resist cell death, as outlined above, in some studies EYA has actually been shown to promote cell death82. Nonetheless, the bulk of studies suggest that therapeutically targeting the phosphatase activity of EYA may be beneficial in two ways, through the prevention of tumor progression (presumably in part through its role as a transcription factor with SIX1), as well as through sensitizing cells to DNA-damaging chemotherapeutics.
7. Opportunities for Targeting the SIX1/EYA Transcriptional Complex
As a global regulator of tumor progression, inhibition of the SIX1 transcriptional complex could be a powerful approach that is therapeutically beneficial for many different cancers. Although traditionally considered a difficult target, recently significant progress has been made in targeting various aspects of transcriptional regulation including enzymatic activity, protein-protein interactions, epigenetic alterations, and DNA binding87. A large body of work has been performed to validate the SIX1 transcriptional complex as a therapeutic target. The SIX1 transcriptional complex is an attractive target due to its decreased presence in normal adult tissues and its ability to influence a broad range of developmental programs critical for tumorigenesis/metastasis when aberrantly re-expressed88. Since it is traditionally difficult to target transcription factor-DNA interactions, we will focus on the importance and implications of targeting the SIX1-EYA protein-protein interaction and EYA phosphatase activity in SIX1-mediated tumor progression.
7.1. Disrupting the SIX1/EYA interaction
Since the SIX1-EYA developmental transcriptional complex is down-regulated in most adult tissues, inhibiting the SIX1-EYA interaction may be a unique and specific approach that can disrupt multiple stages of the tumorigenic process in many tumor types with limited side effects. Although developing therapeutics targeting protein interactions is considered a difficult endeavor, major strides have been made recently with more than 12 small-molecule protein-protein interaction inhibitors currently in clinical development89.
The co-crystal of the SIX1-EYA2 complex reveals that SIX1 predominately uses a single amphipathic α-helix, located in the SD, to bind into a groove on EYA2 ED through hydrophobic interactions, hydrogen bonds, and salt bridges (Figure 2)19. Sequence alignment of the EYA family members reveals at least 93% sequence identity among EYA1, 2, and 4 at the SIX1 interface, and a 67% sequence identity (93% similarity) to EYA3. In addition, the SIX1-EYA2 interface is only about 800 Å2, which is smaller than the average globular protein interface of 1,200–2,000 Å2 19, 89. Often, these helix-grooves can form smaller pockets more comparable to classic enzyme active sites than the flat planar surfaces of globular protein interfaces, due to their structured and well-defined clefts89. This suggests that a small molecule inhibitor could be designed to bind all EYA family members in this binding cleft to perturb SIX1 mediated transcription. Finally, the SD, specific to the SIX family, does not display structural homology to any protein currently deposited in the protein data bank, including other homeoproteins19. This suggests that the development of a small molecule capable of disrupting the SIX1/EYA2 interaction would display high selectivity.
Figure 2.
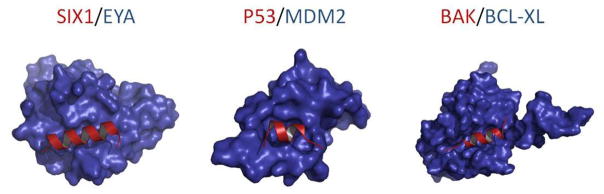
Surface representation illustrating the similarities between the protein interfaces of SIX1/EYA2 and two successfully targeted protein interactions, P53/MDM2 and BAK/BCL-XL. The red ribbon represents the single helix in SIX1, P53, and BAK that interacts with their corresponding protein partners in blue.
To date, the greatest successes in targeting protein-protein interactions with small molecules have been through targeting this type of protein-protein interaction. For example, the α-helix of the BAX BH3 domain interacting with BCL-XL is successfully disrupted by ABT-263 (Figure 2), which is currently in clinical trials for the treatment of multiple cancers90. ABT-263 is one of the first inhibitors to originate from fragment screening, which was ultimately crafted into a potent and selective inhibitor with a Ki of ≤0.5 nM91. Additionally, there are two mid-nanomolar inhibitors in clinical trials that inhibit the P53-MDM2 interaction (Figure 2), which are referred to as the Nutlins92, 93. The Nutlin family binds in the cleft on MDM2 in which a single helix of P53 binds, and was developed from compounds originally discovered through screening efforts by surface plasmon resonance94, 95.
The pioneering work of Wells and colleagues, using alanine scanning approaches, revealed the disparate contributions of residues at protein interfaces to the total binding energy96. This experiment confirmed that smaller patches at the protein interface largely contribute to the overall interaction such that, small molecules able to disrupt these “hot spots”, could effectively disrupt protein-protein interactions. This work, in combination with the recognized importance of the SIX1-EYA2 protein interaction (through the disruption of the interaction by mutating one residue in the SIX1 interface) in TGF-β signaling, EMT and metastasis19, suggest a small molecule inhibitor targeting this protein interaction may be an effective therapeutic approach.
One of the largest advantages to targeting the SIX1/EYA interface is that SIX1 is not expressed in most adult tissues with the exception of skeletal muscle, and thus side effects in response to such a therapy would likely be limited. In skeletal muscle, Six1 and Eya1 have been shown to reprogram adult slow-twitch oxidative fibers towards a fast twitch glycolytic97 and their expression is increased in response to muscle overload98. Of note, Six1 is specifically expressed in the satellite cells in skeletal muscle, which are the stem cells within muscle that are important for establishment and regeneration of the tissue99–101. In adult mice, a conditional knockout of Six1 in this population of cells demonstrated that Six1 was necessary for proper tissue regeneration and replenishment of the stem cell pool following muscle trauma99. However, loss of Six1 had no effect on muscle weight or histology in the adult mice, and importantly, did not affect the maintenance of the stem cell population in muscle102. Thus, although Six1 does not appear to be critical for normal adult muscle homeostasis, it is important in the muscle if trauma occurs. Because muscle wasting is often observed in cancer patients103, one would want to carefully monitor skeletal muscle enzymes in response to any inhibitors developed that target the Six1/Eya complex. Finally, it should be noted that Six1 is repressed by Ezh2 in the postnatal heart, and its inappropriate re-expression leads to cardiac hypertrophy102. Thus, the targeting and subsequent inactivation of Six1 is unlikely to have any adverse consequences in the heart. Overall, because we do not expect that inhibition of Six1/Eya will have an effect in most tissues (due to the paucity of Six1 expression in adult tissue), chronic use of inhibitors targeting the Six1 complex is expected to be well tolerated.
7.2. Inhibition of EYA phosphatase activity
A second approach to targeting the SIX1/EYA transcription complex is through targeting EYA’s phosphatase activity. Structurally, the EYA proteins have a well-defined and unique active site that should be capable of accommodating a small molecule inhibitor104. Traditionally, the identification of phosphatase inhibitors specific to their intended target is difficult, since many phosphatases have structurally similar active sites105. However, EYA is distinct in that it contains a unique catalytic core and helical cap104. EYA phosphatases utilize an aspartic acid as their catalytic residue, in contrast to the catalytic cysteine residue used in most cellular phosphatases. EYAs also distinguish themselves within the HAD family by targeting phosphorylated tyrosine residues, while substrates of most other HAD members are small molecules or phosphorylated serine/threonines29. The unique structure of EYA’s active site suggests that the identification of a selective EYA phosphatase inhibitor is possible. Indeed, both small molecule and virtual screening efforts have facilitated the discovery of specific EYA phosphatase inhibitors85, 106–109.
To date, benzbromarone (Figure 3A), and a few derivates and metabolites, are the most thoroughly investigated active site inhibitors of the EYA family85, 106. These inhibitors were originally identified through enzymatic screening of the NCI diversity library and were further validated in cell culture models85. These inhibitors display moderate IC50 values (> 8 μM) in enzymatic assays with either EYA2 or 3, and are effective in cellular assays, inhibiting cell motility, and angiogenic tubulogenesis as well as sprouting85. Additionally, the inhibitors were able to reduce cell proliferation after a 72 hour treatment, supporting the role of EYA phosphatase activity in proliferation106. Using docking studies and kinetic analysis, Hegde and colleagues proposed that benzbromarone binds immediately adjacent to the phosphotyrosine binding site and is an uncompetitive inhibitor of EYA2 and EYA385. In the clinic, these compounds hold promise, since benzbromarone has been used for many years in the treatment of gout, and its pharmacological profile is well known. However, due to reports of unwanted hepatotoxicity, this compound will require further chemical optimization before it can be re-purposed in the clinic as an EYA inhibitor.
Figure 3.
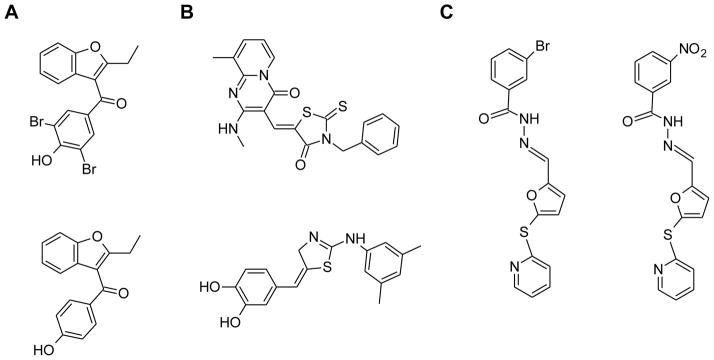
Chemical structures of the 3 series of compounds identified as inhibitors of EYA2 phosphatase activity. A. Benzbromarone and benzarone identified through the screening of a small NCI library. B. Two representative Mg2+ chelators identified through virtual screening. C. The top two allosteric N-arylidenebenzohydrazide compounds identified through a large scale HTS.
In addition, other classes of EYA active site inhibitors have been discovered through virtual screening efforts (Figure 3B) 107. These compounds are proposed to bind in the active site of EYA2 and chelate the essential Mg2+ ion. Park and colleagues further optimized the compounds through the creation of multiple hydrogen bonds and van der Waals contacts with nearby residues to achieve IC50 values ranging from 6–50 μM108. However, the selectivity and efficacy of these inhibitors in biochemical and cellular assays has not yet been validated, and thus require further study before their clinical potential can be determined.
Using quantitative high-throughput screening approaches our group has identified an additional class of EYA2 ED specific inhibitors, which appear to act through an allosteric mechanism109, 110. In this work, we used a fluorescent phosphatase assay to screen the NIH Molecular Libraries Probe Production Centers Network (MLPCN) library of over 330,000 compounds, which led to the identification of a series of N-arylidenebenzohydrazide-containing compounds that inhibit EYA2 (Figure 3C) 109. The top molecules within this series have low micromolar IC50 values and were validated in a secondary assay monitoring the dephosphorylation of the pH2AX peptide substrate by EYA2. These inhibitors display specificity towards EYA2 over other cellular phosphatases, including PTP1B (a prototypic tyrosine phosphatase), PPM1A (a Mg2+ dependent Ser/Thr phosphatase), and Scp1 (a HAD family serine phosphatase). Importantly, these compounds are able to reverse EYA2 phosphatase-dependent cell migration in MCF10A cells, suggesting that these compounds could ultimately inhibit metastasis110.
Extensive mutational and kinetic analyses indicate that these N-arylidenebenzohydrazide compounds are reversible inhibitors with a long occupation time and interact with EYA2 at an allosteric site opposite of the enzymatic active site110. The allosteric nature of these inhibitors provides additional selectivity for EYA2 over another EYA family member, EYA3110. Allosteric modulators can offer an advantage over orthosteric ones because of their ability to increase selectivity and modulatory control111. These molecules are promising leads that can potentially be developed into highly specific therapeutic agents targeting tumors overexpressing EYA2.
8. Expert Opinion
Many of the most successful cancer drugs developed to date have molecular targets that drive a known hallmark of cancer including evasion of growth suppressors, sustained proliferative signaling, enabling replicative immortality, resisting cell death, promoting invasion and metastasis, and inducing angiogenesis. Drugs with this directed mechanism of action, in theory, should have fewer off-target effects and subsequent nonspecific toxicity. Indeed, many anticancer therapeutics have been specifically designed to target pathways critical for tumor progression that are reduced or absent in normal adult tissues, thereby limiting harmful side effects. Unfortunately, therapies that target only one key hallmark pathway often lose potency over time as the cancer cells develop resistance, resulting in clinical relapse. Therefore, the development of therapies co-targeting multiple cancer hallmarks will result in a more effective and durable cancer treatment. Aberrant transcription factor expression can result in altered gene expression profiles, and is known to be fundamental to the acquisition of hallmark capabilities112. Indeed, many signaling pathways that are altered in cancer are believed to act through certain key transcription factors, and thus targeting transcriptional complexes may be a means of targeting a central node of the disease. Direct intervention of the transcriptional process is thus an exciting strategy to correct gene expression in cancer cells. In particular, the SIX1/EYA transcriptional complex is an ideal target for anti-cancer therapy, since it is largely a developmental complex that is abnormally overexpressed in many cancer types and regulates multiple pathways critical for tumorigenesis.
The combination of both structural and functional data strongly suggest that the perturbation of the SIX1 transcriptional complex using small molecules may be a feasible approach in developing novel anti-cancer agents. Two of the more attractive approaches towards achieving this goal are the inhibition of either the interaction between SIX1 and EYA or EYA’s enzymatic activity. Both approaches hold great promise, but are not without their own risks.
Protein-protein interactions are integral for most cellular functions, and they represent a substantial opportunity for drug development. Despite the evidence that protein interfaces have significant potential for pharmacological intervention; relatively few examples exist in the discovery and development of potent and selective protein-protein interaction inhibitors. Therefore, the disruption of protein interactions using small molecules is still considered a challenging task, owing to the often large planar surfaces of globular protein interactions and the relatively small footprints of a small molecule. Nevertheless, potent small molecule inhibitors of protein-protein interactions have been discovered and are currently in clinical trials. The SIX1-EYA interaction, in particular, has some characteristics that suggest it too is amenable to small molecule intervention. The high-resolution crystal structure of the SIX1/EYA2 complex and mutational analysis have provided key information to support this statement. SIX1 interacts with EYA through its unique SD, using predominantly a single α-helix. The interface is relatively small compared to a typical protein-protein interface and a single amino acid mutation on the SIX1 helix is able to disrupt the SIX1-EYA interaction. The molecular details of the topology of SIX1-EYA interface provided by the crystal structure will help facilitate any future high-throughput screening and virtual-based discovery efforts to identify and optimize small molecule inhibitors of the SIX1/EYA2 interaction.
Tyrosine phosphatases play critical roles in cell signaling and physiological processes. When mis-regulated, they can function as oncoproteins, modulating several key pathways contributing to the hallmarks of cancer113. The EYA family of atypical tyrosine phosphatases appears to be no exception, promoting proliferation, motility, and angiogenesis. In particular, EYA’s enzymatic activity is an attractive target for future anti-cancer therapies because of the uniqueness of its catalytic domain. EYA phosphatases utilize an aspartic acid, rather than the more common cysteine, as their catalytic residue and are unique within the HAD family, targeting phosphorylated tyrosine residues. Historically, small molecule inhibitors of tyrosine phosphatases have been easy to identify, including both natural products and synthetic compounds. Unfortunately, the close sequence identity between the different phosphatase family members has made the identification of specific inhibitors challenging. However, the unique structure and catalytic mechanism of EYA’s active site suggests that the identification of a selective EYA phosphatase inhibitor is possible.
To date, there are three classes of inhibitors that have been published targeting the enzymatic activity of EYA2, each class having their own advantages and limitations. Individually, these inhibitors all display moderate IC50 values, in the low micromolar range. Therefore, additional medicinal chemistry efforts to improve their potency could prove valuable. Benzbromarone is already well understood pharmacologically, however, due to unwanted drug-drug interactions with CYP2C9 substrates and reports of subsequent hepatotoxicity, benzbromarone is no longer marketed in the U.S. Additional chemical optimization of benzbromarone to remove this unwanted toxicity while retaining its activity may be necessary before it can be re-purposed in the clinic. The Mg2+-chelators, identified through in silico screening, are effective at inhibiting the enzymatic activity of EYA, but their selectivity and effectiveness in cell culture and in vivo have not yet been characterized. Moreover, metal chelators can present problems as potential therapeutics through the physiological depletion of metal ions and inhibition of other metalloenzymes. Finally, the allosteric inhibitors identified through HTS efforts may provide an additional level of selectivity, but have yet to be thoroughly characterized in vivo. Although certain sub-types of hydrazones (most notably hydroxyphenylhydrazones) have a reputation as pan assay interference compounds, appearing as hits in many high throughput screening assays, not all hydrazones are problematic and thus these compounds should not be ruled out as leads for further optimization and drug development114. Our hydrazone-containing compounds, for example, are clearly specific and not reactive to all proteins, as evidenced by their inactivity against the highly homologous EYA3 protein and other cellular phosphatases.
In conclusion, there is a growing body of evidence detailing the physiological and oncogenic roles of the SIX1/EYA transcriptional complex that emphasize the therapeutic potential of targeting this complex. The SIX1 transcriptional complex impacts multiple aspects of cancer biology, including properties related to both tumorigenesis and metastasis, through the regulation of several important cell cycle regulators, oncogenes, and other tumor promoting molecules. Although the progress made towards developing a clinically-sound small molecule inhibitor is still in its early stages, future efforts in this area may uncover potent and selective inhibitors that will ultimately lead to successful anti-cancer therapies.
References
- 1.Kumar JP. The sine oculis homeobox (SIX) family of transcription factors as regulators of development and disease. Cell Mol Life Sci. 2009;66(4):565–83. doi: 10.1007/s00018-008-8335-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu PX. The EYA-SO/SIX complex in development and disease. Pediatr Nephrol. 2012;28(6):843–54. doi: 10.1007/s00467-012-2246-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3*.Ford HL, Kabingu EN, Bump EA, et al. Abrogation of the G2 cell cycle checkpoint associated with overexpression of HSIX1: a possible mechanism of breast carcinogenesis. Proc Natl Acad Sci U S A. 1998;95(21):12608–13. doi: 10.1073/pnas.95.21.12608. This is the first study to demonstrate overexpression of SIX1 in any tumor type, and to implicate it in cell cycle control in cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4*.Li X, Oghi KA, Zhang J, et al. Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature. 2003;426(6964):247–54. doi: 10.1038/nature02083. One of the first studies to demonstrate that the Eya transcriptional cofactor contains intrinsic phosphatase activity, and that this may be critical for Six1-mediated transcription. [DOI] [PubMed] [Google Scholar]
- 5.Coletta RD, Christensen K, Reichenberger KJ, et al. The Six1 homeoprotein stimulates tumorigenesis by reactivation of cyclin A1. Proc Natl Acad Sci U S A. 2004;101(17):6478–83. doi: 10.1073/pnas.0401139101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li X, Perissi V, Liu F, et al. Tissue-specific regulation of retinal and pituitary precursor cell proliferation. Science. 2002;297(5584):1180–3. doi: 10.1126/science.1073263. [DOI] [PubMed] [Google Scholar]
- 7.Del Bene F, Tessmar-Raible K, Wittbrodt J. Direct interaction of geminin and Six3 in eye development. Nature. 2004;427(6976):745–9. doi: 10.1038/nature02292. [DOI] [PubMed] [Google Scholar]
- 8.El-Hashash AH, Al Alam D, Turcatel G, et al. Six1 transcription factor is critical for coordination of epithelial, mesenchymal and vascular morphogenesis in the mammalian lung. Dev Biol. 2011;353(2):242–58. doi: 10.1016/j.ydbio.2011.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Self M, Lagutin OV, Bowling B, et al. Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. EMBO J. 2006;25(21):5214–28. doi: 10.1038/sj.emboj.7601381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kobayashi H, Kawakami K, Asashima M, et al. Six1 and Six4 are essential for Gdnf expression in the metanephric mesenchyme and ureteric bud formation, while Six1 deficiency alone causes mesonephric-tubule defects. Mech Dev. 2007;124(4):290–303. doi: 10.1016/j.mod.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 11*.Abdelhak S, Kalatzis V, Heilig R, et al. A human homologue of the Drosophila eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family. Nat Genet. 1997;15(2):157–64. doi: 10.1038/ng0297-157. This is the first study to implicate EYA proteins in BOR syndrome. [DOI] [PubMed] [Google Scholar]
- 12.Hoskins BE, Cramer CH, Silvius D, et al. Transcription factor SIX5 is mutated in patients with branchio-oto-renal syndrome. Am J Hum Genet. 2007;80(4):800–4. doi: 10.1086/513322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13*.Laclef C, Hamard G, Demignon J, et al. Altered myogenesis in Six1-deficient mice. Development. 2003;130(10):2239–52. doi: 10.1242/dev.00440. The first of a series of papers describing developmental phenotypes of Six1 knockout mice. [DOI] [PubMed] [Google Scholar]
- 14.Laclef C, Souil E, Demignon J, et al. Thymus, kidney and craniofacial abnormalities in Six1 deficient mice. Mech Dev. 2003;120(6):669–79. doi: 10.1016/s0925-4773(03)00065-0. [DOI] [PubMed] [Google Scholar]
- 15.Xu PX, Zheng W, Huang L, et al. Six1 is required for the early organogenesis of mammalian kidney. Development. 2003;130(14):3085–94. doi: 10.1242/dev.00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng W, Huang L, Wei ZB, et al. The role of Six1 in mammalian auditory system development. Development. 2003;130(17):3989–4000. doi: 10.1242/dev.00628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Christensen KL, Patrick AN, McCoy EL, et al. The six family of homeobox genes in development and cancer. Adv Cancer Res. 2008;101:93–126. doi: 10.1016/S0065-230X(08)00405-3. [DOI] [PubMed] [Google Scholar]
- 18.Kawakami K, Sato S, Ozaki H, et al. Six family genes--structure and function as transcription factors and their roles in development. Bioessays. 2000;22(7):616–26. doi: 10.1002/1521-1878(200007)22:7<616::AID-BIES4>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 19*.Patrick AN, Cabrera JH, Smith AL, et al. Structure-function analyses of the human SIX1-EYA2 complex reveal insights into metastasis and BOR syndrome. Nat Struct Mol Biol. 2013;20(4):447–53. doi: 10.1038/nsmb.2505. The first study to elucidate molecular details of the SIX1/EYA2 binding interface and conclusively demonstrate that a direct interaction between SIX1 and EYA is required for SIX1-induced metastasis in breast cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawakami K, Ohto H, Ikeda K, et al. Structure, function and expression of a murine homeobox protein AREC3, a homologue of Drosophila sine oculis gene product, and implication in development. Nucleic Acids Res. 1996;24(2):303–10. doi: 10.1093/nar/24.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brodbeck S, Besenbeck B, Englert C. The transcription factor Six2 activates expression of the Gdnf gene as well as its own promoter. Mech Dev. 2004;121(10):1211–22. doi: 10.1016/j.mod.2004.05.019. [DOI] [PubMed] [Google Scholar]
- 22.Rebay I, Silver SJ, Tootle TL. New vision from Eyes absent: transcription factors as enzymes. Trends Genet. 2005;21(3):163–71. doi: 10.1016/j.tig.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 23.Ohto H, Kamada S, Tago K, et al. Cooperation of six and eya in activation of their target genes through nuclear translocation of Eya. Mol Cell Biol. 1999;19(10):6815–24. doi: 10.1128/mcb.19.10.6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu PX, Cheng J, Epstein JA, et al. Mouse Eya genes are expressed during limb tendon development and encode a transcriptional activation function. Proc Natl Acad Sci U S A. 1997;94(22):11974–9. doi: 10.1073/pnas.94.22.11974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25*.Okabe Y, Sano T, Nagata S. Regulation of the innate immune response by threonine-phosphatase of Eyes absent. Nature. 2009;460(7254):520–4. doi: 10.1038/nature08138. First study to demonstrate that Eya’s N-terminal phosphatase activity is involved in immune responses. [DOI] [PubMed] [Google Scholar]
- 26.Sano T, Nagata S. Characterization of the threonine-phosphatase of mouse eyes absent 3. FEBS Lett. 2011;585(17):2714–9. doi: 10.1016/j.febslet.2011.07.029. [DOI] [PubMed] [Google Scholar]
- 27.Tadjuidje E, Hegde RS. The Eyes Absent proteins in development and disease. Cell Mol Life Sci. 2013;70(11):1897–913. doi: 10.1007/s00018-012-1144-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28*.Tootle TL, Silver SJ, Davies EL, et al. The transcription factor Eyes absent is a protein tyrosine phosphatase. Nature. 2003;426(6964):299–302. doi: 10.1038/nature02097. Amongst the first studies to demonstrate that the Eya co-activator has intrinsic phosphatase activity. [DOI] [PubMed] [Google Scholar]
- 29*.Rayapureddi JP, Kattamuri C, Steinmetz BD, et al. Eyes absent represents a class of protein tyrosine phosphatases. Nature. 2003;426(6964):295–8. doi: 10.1038/nature02093. Amongst the first studies to demonstrate that the Eya co-activator has intrinsic phosphatase activity. [DOI] [PubMed] [Google Scholar]
- 30*.Cook PJ, Ju BG, Telese F, et al. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature. 2009;458(7238):591–6. doi: 10.1038/nature07849. First study to identify an in vivo target for Eya phoshatase activity, and to demonstrate that Eya regulates the response to DNA damage. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krishnan N, Jeong DG, Jung SK, et al. Dephosphorylation of the C-terminal tyrosyl residue of the DNA damage-related histone H2A.X is mediated by the protein phosphatase eyes absent. J Biol Chem. 2009 doi: 10.1074/jbc.C900032200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32*.Yuan B, Cheng L, Chiang HC, et al. A phosphotyrosine switch determines the antitumor activity of ERbeta. J Clin Invest. 2014 doi: 10.1172/JCI74085. This study identifies a second target for the EYA tyrosine phosphatase, estrogen receptor(ER)-beta, and identifies yet another mechanism by which EYA may promote proliferation through dephosphorylation and inactivation of ER-beta. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thomas C, Gustafsson JA. The different roles of ER subtypes in cancer biology and therapy. Nat Rev Cancer. 2011;11(8):597–608. doi: 10.1038/nrc3093. [DOI] [PubMed] [Google Scholar]
- 34.Nica G, Herzog W, Sonntag C, et al. Eya1 is required for lineage-specific differentiation, but not for cell survival in the zebrafish adenohypophysis. Dev Biol. 2006;292(1):189–204. doi: 10.1016/j.ydbio.2005.12.036. [DOI] [PubMed] [Google Scholar]
- 35*.Xu PX, Adams J, Peters H, et al. Eya1-deficient mice lack ears and kidneys and show abnormal apoptosis of organ primordia. Nat Genet. 1999;23(1):113–7. doi: 10.1038/12722. First Eya knockout mouse demonstrating a critical role for the protein in organogenesis. [DOI] [PubMed] [Google Scholar]
- 36.Zou D, Silvius D, Fritzsch B, et al. Eya1 and Six1 are essential for early steps of sensory neurogenesis in mammalian cranial placodes. Development. 2004;131(22):5561–72. doi: 10.1242/dev.01437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brugmann SA, Pandur PD, Kenyon KL, et al. Six1 promotes a placodal fate within the lateral neurogenic ectoderm by functioning as both a transcriptional activator and repressor. Development. 2004;131(23):5871–81. doi: 10.1242/dev.01516. [DOI] [PubMed] [Google Scholar]
- 38*.Ruf RG, Xu PX, Silvius D, et al. SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes. Proc Natl Acad Sci U S A. 2004;101(21):8090–5. doi: 10.1073/pnas.0308475101. The first study to implicate SIX1 in BOR syndrome. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kozlowski DJ, Whitfield TT, Hukriede NA, et al. The zebrafish dog-eared mutation disrupts eya1, a gene required for cell survival and differentiation in the inner ear and lateral line. Dev Biol. 2005;277(1):27–41. doi: 10.1016/j.ydbio.2004.08.033. [DOI] [PubMed] [Google Scholar]
- 40.Bricaud O, Collazo A. The transcription factor six1 inhibits neuronal and promotes hair cell fate in the developing zebrafish (Danio rerio) inner ear. J Neurosci. 2006;26(41):10438–51. doi: 10.1523/JNEUROSCI.1025-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Y, Manaligod JM, Weeks DL. EYA1 mutations associated with the branchio-oto-renal syndrome result in defective otic development in Xenopus laevis. Biol Cell. 2010;102(5):277–92. doi: 10.1042/BC20090098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zou D, Silvius D, Rodrigo-Blomqvist S, et al. Eya1 regulates the growth of otic epithelium and interacts with Pax2 during the development of all sensory areas in the inner ear. Dev Biol. 2006;298(2):430–41. doi: 10.1016/j.ydbio.2006.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Song MH, Kwon TJ, Kim HR, et al. Mutational analysis of EYA1, SIX1 and SIX5 genes and strategies for management of hearing loss in patients with BOR/BO syndrome. PLoS One. 2013;8(6):e67236. doi: 10.1371/journal.pone.0067236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Azuma N, Hirakiyama A, Inoue T, et al. Mutations of a human homologue of the Drosophila eyes absent gene (EYA1) detected in patients with congenital cataracts and ocular anterior segment anomalies. Hum Mol Genet. 2000;9(3):363–6. doi: 10.1093/hmg/9.3.363. [DOI] [PubMed] [Google Scholar]
- 45.Orten DJ, Fischer SM, Sorensen JL, et al. Branchio-oto-renal syndrome (BOR): novel mutations in the EYA1 gene, and a review of the mutational genetics of BOR. Hum Mutat. 2008;29(4):537–44. doi: 10.1002/humu.20691. [DOI] [PubMed] [Google Scholar]
- 46*.Patrick AN, Schiemann BJ, Yang K, et al. Biochemical and functional characterization of six SIX1 Branchio-oto-renal syndrome mutations. J Biol Chem. 2009;284(31):20781–90. doi: 10.1074/jbc.M109.016832. This paper describes the characterization of numerous SIX1 mutations found in BOR syndrome, and identifies a naturally occurring point mutation in SIX1 which disrupts the SIX1/EYA interaction. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buller C, Xu X, Marquis V, et al. Molecular effects of Eya1 domain mutations causing organ defects in BOR syndrome. Hum Mol Genet. 2001;10(24):2775–81. doi: 10.1093/hmg/10.24.2775. [DOI] [PubMed] [Google Scholar]
- 48.Rayapureddi JP, Hegde RS. Branchio-oto-renal syndrome associated mutations in Eyes Absent 1 result in loss of phosphatase activity. FEBS Lett. 2006;580(16):3853–9. doi: 10.1016/j.febslet.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 49.Abate-Shen C. Deregulated homeobox gene expression in cancer: cause or consequence? Nat Rev Cancer. 2002;2(10):777–85. doi: 10.1038/nrc907. [DOI] [PubMed] [Google Scholar]
- 50*.Yu Y, Khan J, Khanna C, et al. Expression profiling identifies the cytoskeletal organizer ezrin and the developmental homeoprotein Six-1 as key metastatic regulators. Nat Med. 2004;10(2):175–81. doi: 10.1038/nm966. The first paper to demonstrate a critical role for Six1 in mediating metastasis of any tumor type. [DOI] [PubMed] [Google Scholar]
- 51*.Micalizzi DS, Christensen KL, Jedlicka P, et al. The Six1 homeoprotein induces human mammary carcinoma cells to undergo epithelial-mesenchymal transition and metastasis in mice through increasing TGF-beta signaling. J Clin Invest. 2009;119(9):2678–90. doi: 10.1172/JCI37815. This paper demonstrates that Six1 enhances metastasis through regulation of TGF-beta signaling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ng KT, Man K, Sun CK, et al. Clinicopathological significance of homeoprotein Six1 in hepatocellular carcinoma. Br J Cancer. 2006;95(8):1050–5. doi: 10.1038/sj.bjc.6603399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Behbakht K, Qamar L, Aldridge CS, et al. Six1 overexpression in ovarian carcinoma causes resistance to TRAIL-mediated apoptosis and is associated with poor survival. Cancer Res. 2007;67(7):3036–42. doi: 10.1158/0008-5472.CAN-06-3755. [DOI] [PubMed] [Google Scholar]
- 54.Mimae T, Okada M, Hagiyama M, et al. Upregulation of notch2 and six1 is associated with progression of early-stage lung adenocarcinoma and a more aggressive phenotype at advanced stages. Clin Cancer Res. 2012;18(4):945–55. doi: 10.1158/1078-0432.CCR-11-1946. [DOI] [PubMed] [Google Scholar]
- 55.Zheng XH, Liang PH, Guo JX, et al. Expression and clinical implications of homeobox gene Six1 in cervical cancer cell lines and cervical epithelial tissues. Int J Gynecol Cancer. 2010;20(9):1587–92. [PubMed] [Google Scholar]
- 56.Li Z, Tian T, Lv F, et al. Six1 promotes proliferation of pancreatic cancer cells via upregulation of cyclin D1 expression. PLoS One. 2013;8(3):e59203. doi: 10.1371/journal.pone.0059203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu Y, Davicioni E, Triche TJ, et al. The homeoprotein six1 transcriptionally activates multiple protumorigenic genes but requires ezrin to promote metastasis. Cancer Res. 2006;66(4):1982–9. doi: 10.1158/0008-5472.CAN-05-2360. [DOI] [PubMed] [Google Scholar]
- 58.Iwanaga R, Wang CA, Micalizzi DS, et al. Expression of Six1 in luminal breast cancers predicts poor prognosis and promotes increases in tumor initiating cells by activation of extracellular signal-regulated kinase and transforming growth factor-beta signaling pathways. Breast Cancer Res. 2012;14(4):R100. doi: 10.1186/bcr3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59*.McCoy EL, Iwanaga R, Jedlicka P, et al. Six1 expands the mouse mammary epithelial stem/progenitor cell pool and induces mammary tumors that undergo epithelial-mesenchymal transition. J Clin Invest. 2009;119(9):2663–77. doi: 10.1172/JCI37691. This is the first paper to demonstrate that aberrant expression of Six1 can induce tumors in a transgenic mouse model of mammary cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60*.Wang CA, Jedlicka P, Patrick AN, et al. SIX1 induces lymphangiogenesis and metastasis via upregulation of VEGF-C in mouse models of breast cancer. J Clin Invest. 2012;122(5):1895–906. doi: 10.1172/JCI59858. This paper demonstrates that Six1 influences metastasis not only through cell autonomous effects, but also through engaging the microenvironment by stimulating lymphangiogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ng KT, Lee TK, Cheng Q, et al. Suppression of tumorigenesis and metastasis of hepatocellular carcinoma by shRNA interference targeting on homeoprotein Six1. Int J Cancer. 2009 doi: 10.1002/ijc.25105. [DOI] [PubMed] [Google Scholar]
- 62.Micalizzi DS, Wang CA, Farabaugh SM, et al. Homeoprotein Six1 increases TGF-beta type I receptor and converts TGF-beta signaling from suppressive to supportive for tumor growth. Cancer Res. 2010;70(24):10371–80. doi: 10.1158/0008-5472.CAN-10-1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Coletta RD, Christensen KL, Micalizzi DS, et al. Six1 overexpression in mammary cells induces genomic instability and is sufficient for malignant transformation. Cancer Res. 2008;68(7):2204–13. doi: 10.1158/0008-5472.CAN-07-3141. [DOI] [PubMed] [Google Scholar]
- 64.Imam JS, Buddavarapu K, Lee-Chang JS, et al. MicroRNA-185 suppresses tumor growth and progression by targeting the Six1 oncogene in human cancers. Oncogene. 2010;29(35):4971–9. doi: 10.1038/onc.2010.233. [DOI] [PubMed] [Google Scholar]
- 65.Hua L, Fan L, Aichun W, et al. Inhibition of Six1 promotes apoptosis, suppresses proliferation, and migration of osteosarcoma cells. Tumour Biol. 2014;35(3):1925–31. doi: 10.1007/s13277-013-1258-1. [DOI] [PubMed] [Google Scholar]
- 66.Li Z, Tian T, Hu X, et al. Six1 mediates resistance to paclitaxel in breast cancer cells. Biochem Biophys Res Commun. 2013;441(3):538–43. doi: 10.1016/j.bbrc.2013.10.131. [DOI] [PubMed] [Google Scholar]
- 67.Tan J, Zhang C, Qian J. Expression and significance of Six1 and Ezrin in cervical cancer tissue. Tumour Biol. 2011;32(6):1241–7. doi: 10.1007/s13277-011-0228-8. [DOI] [PubMed] [Google Scholar]
- 68.Ono H, Imoto I, Kozaki K, et al. SIX1 promotes epithelial-mesenchymal transition in colorectal cancer through ZEB1 activation. Oncogene. 2012;31(47):4923–34. doi: 10.1038/onc.2011.646. [DOI] [PubMed] [Google Scholar]
- 69.Zhao H, Xu Z, Qin H, et al. miR-30b regulates migration and invasion of human colorectal cancer via SIX1. Biochem J. 2014;460(1):117–25. doi: 10.1042/BJ20131535. [DOI] [PubMed] [Google Scholar]
- 70.Li Z, Tian T, Hu X, et al. Targeting Six1 by lentivirus-mediated RNA interference inhibits colorectal cancer cell growth and invasion. Int J Clin Exp Pathol. 2014;7(2):631–9. [PMC free article] [PubMed] [Google Scholar]
- 71.Reichenberger KJ, Coletta RD, Schulte AP, et al. Gene amplification is a mechanism of Six1 overexpression in breast cancer. Cancer Res. 2005;65(7):2668–75. doi: 10.1158/0008-5472.CAN-04-4286. [DOI] [PubMed] [Google Scholar]
- 72.Li CM, Guo M, Borczuk A, et al. Gene expression in Wilms’ tumor mimics the earliest committed stage in the metanephric mesenchymal-epithelial transition. Am J Pathol. 2002;160(6):2181–90. doi: 10.1016/S0002-9440(10)61166-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Robin TP, Smith A, McKinsey E, et al. EWS/FLI1 regulates EYA3 in Ewing sarcoma via modulation of miRNA-708, resulting in increased cell survival and chemoresistance. Mol Cancer Res. 2012;10(8):1098–108. doi: 10.1158/1541-7786.MCR-12-0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang Y, Klijn JG, Zhang Y, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365(9460):671–9. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- 75*.Zhang L, Yang N, Huang J, et al. Transcriptional coactivator Drosophila eyes absent homologue 2 is up-regulated in epithelial ovarian cancer and promotes tumor growth. Cancer Res. 2005;65(3):925–32. First paper to demonstrate a role for Eya proteins in promoting tumor growth. [PubMed] [Google Scholar]
- 76.Guo JT, Ding LH, Liang CY, et al. Expression of EYA2 in non-small cell lang cancer. Zhonghua Zhong Liu Za Zhi. 2009;31(7):528–31. [PubMed] [Google Scholar]
- 77.Farabaugh SM, Micalizzi DS, Jedlicka P, et al. Eya2 is required to mediate the pro-metastatic functions of Six1 via the induction of TGF-beta signaling, epithelial-mesenchymal transition, and cancer stem cell properties. Oncogene. 2012;31(5):552–62. doi: 10.1038/onc.2011.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zou H, Harrington JJ, Shire AM, et al. Highly methylated genes in colorectal neoplasia: implications for screening. Cancer Epidemiol Biomarkers Prev. 2007;16(12):2686–96. doi: 10.1158/1055-9965.EPI-07-0518. [DOI] [PubMed] [Google Scholar]
- 79.Vincent A, Hong SM, Hu C, et al. Epigenetic silencing of EYA2 in pancreatic adenocarcinomas promotes tumor growth. Oncotarget. 2014;5(9):2575–87. doi: 10.18632/oncotarget.1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gutierrez ML, Munoz-Bellvis L, del Abad MM, et al. Association between genetic subgroups of pancreatic ductal adenocarcinoma defined by high density 500 K SNP-arrays and tumor histopathology. PLoS One. 2011;6(7):e22315. doi: 10.1371/journal.pone.0022315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang QF, Wu G, Mi S, et al. MLL fusion proteins preferentially regulate a subset of wild-type MLL target genes in the leukemic genome. Blood. 2011;117(25):6895–905. doi: 10.1182/blood-2010-12-324699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Clark SW, Fee BE, Cleveland JL. Misexpression of the eyes absent family triggers the apoptotic program. J Biol Chem. 2002;277(5):3560–7. doi: 10.1074/jbc.M108410200. [DOI] [PubMed] [Google Scholar]
- 83.Wu K, Li Z, Cai S, et al. EYA1 phosphatase function is essential to drive breast cancer cell proliferation through cyclin D1. Cancer Res. 2013;73(14):4488–99. doi: 10.1158/0008-5472.CAN-12-4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84*.Pandey RN, Rani R, Yeo EJ, et al. The Eyes Absent phosphatase-transactivator proteins promote proliferation, transformation, migration, and invasion of tumor cells. Oncogene. 2010 doi: 10.1038/onc.2010.122. First paper to demonstrate that the Eya tyrosine phosphatase activity is required for breast cancer metastasis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85*.Tadjuidje E, Wang TS, Pandey RN, et al. The EYA tyrosine phosphatase activity is pro-angiogenic and is inhibited by benzbromarone. PLoS One. 2012;7(4):e34806. doi: 10.1371/journal.pone.0034806. Demonstration that a novel compound targeting the Eya tyrosine phosphatase can inhibit angiogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Auvergne RM, Sim FJ, Wang S, et al. Transcriptional differences between normal and glioma-derived glial progenitor cells identify a core set of dysregulated genes. Cell Rep. 2013;3(6):2127–41. doi: 10.1016/j.celrep.2013.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yan C, Higgins PJ. Drugging the undruggable: transcription therapy for cancer. Biochim Biophys Acta. 2013;1835(1):76–85. doi: 10.1016/j.bbcan.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wu W, Ren Z, Li P, et al. Six1: A critical transcription factor in tumorigenesis. Int J Cancer. 2014 doi: 10.1002/ijc.28755. [DOI] [PubMed] [Google Scholar]
- 89.Nero TL, Morton CJ, Holien JK, et al. Oncogenic protein interfaces: small molecules, big challenges. Nat Rev Cancer. 2014;14(4):248–62. doi: 10.1038/nrc3690. [DOI] [PubMed] [Google Scholar]
- 90.Rudin CM, Hann CL, Garon EB, et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin Cancer Res. 2012;18(11):3163–9. doi: 10.1158/1078-0432.CCR-11-3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tse C, Shoemaker AR, Adickes J, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68(9):3421–8. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 92.Ding Q, Zhang Z, Liu JJ, et al. Discovery of RG7388; a potent and selective p53-MDM2 inhibitor in clinical development. J Med Chem. 2013;56(14):5979–83. doi: 10.1021/jm400487c. [DOI] [PubMed] [Google Scholar]
- 93.Ray-Coquard I, Blay JY, Italiano A, et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol. 2012;13(11):1133–40. doi: 10.1016/S1470-2045(12)70474-6. [DOI] [PubMed] [Google Scholar]
- 94.Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303(5659):844–8. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 95.Carry JC, Garcia-Echeverria C. Inhibitors of the p53/hdm2 protein-protein interaction-path to the clinic. Bioorg Med Chem Lett. 2013;23(9):2480–5. doi: 10.1016/j.bmcl.2013.03.034. [DOI] [PubMed] [Google Scholar]
- 96.Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science. 1995;267(5196):383–6. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 97.Grifone R, Laclef C, Spitz F, et al. Six1 and Eya1 expression can reprogram adult muscle from the slow-twitch phenotype into the fast-twitch phenotype. Mol Cell Biol. 2004;24(14):6253–67. doi: 10.1128/MCB.24.14.6253-6267.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gordon BS, Delgado Diaz DC, White JP, et al. Six1 and Six1 cofactor expression is altered during early skeletal muscle overload in mice. J Physiol Sci. 2012;62(5):393–401. doi: 10.1007/s12576-012-0214-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99*.Le Grand F, Grifone R, Mourikis P, et al. Six1 regulates stem cell repair potential and self-renewal during skeletal muscle regeneration. J Cell Biol. 2012;198(5):815–32. doi: 10.1083/jcb.201201050. This paper demonstrates an important role for Six1 in the regeneration of skeletal muscle in adult mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nord H, Nygard Skalman L, von Hofsten J. Six1 regulates proliferation of Pax7-positive muscle progenitors in zebrafish. J Cell Sci. 2013;126(Pt 8):1868–80. doi: 10.1242/jcs.119917. [DOI] [PubMed] [Google Scholar]
- 101.Liu Y, Chakroun I, Yang D, et al. Six1 regulates MyoD expression in adult muscle progenitor cells. PLoS One. 2013;8(6):e67762. doi: 10.1371/journal.pone.0067762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Delgado-Olguin P, Huang Y, Li X, et al. Epigenetic repression of cardiac progenitor gene expression by Ezh2 is required for postnatal cardiac homeostasis. Nat Genet. 2012;44(3):343–7. doi: 10.1038/ng.1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fearon K, Strasser F, Anker SD, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol. 2011;12(5):489–95. doi: 10.1016/S1470-2045(10)70218-7. [DOI] [PubMed] [Google Scholar]
- 104.Jung SK, Jeong DG, Chung SJ, et al. Crystal structure of ED-Eya2: insight into dual roles as a protein tyrosine phosphatase and a transcription factor. FASEB J. 2010;24(2):560–9. doi: 10.1096/fj.09-143891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.He R, Zeng LF, He Y, et al. Small molecule tools for functional interrogation of protein tyrosine phosphatases. FEBS J. 2013;280(2):731–50. doi: 10.1111/j.1742-4658.2012.08718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pandey RN, Wang TS, Tadjuidje E, et al. Structure-activity relationships of benzbromarone metabolites and derivatives as EYA inhibitory anti-angiogenic agents. PLoS One. 2013;8(12):e84582. doi: 10.1371/journal.pone.0084582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107*.Park H, Jung SK, Yu KR, et al. Structure-based virtual screening approach to the discovery of novel inhibitors of eyes absent 2 phosphatase with various metal chelating moieties. Chem Biol Drug Des. 2011;78(4):642–50. doi: 10.1111/j.1747-0285.2011.01192.x. Discovery of novel EYA2 phosphatase inhibitors using virtual approaches. [DOI] [PubMed] [Google Scholar]
- 108.Park H, Ryu SE, Kim SJ. Structure-based de novo design of Eya2 phosphatase inhibitors. J Mol Graph Model. 2012;38:382–8. doi: 10.1016/j.jmgm.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 109*.Krueger AB, Dehdashti SJ, Southall N, et al. Identification of a selective small-molecule inhibitor series targeting the eyes absent 2 (Eya2) phosphatase activity. J Biomol Screen. 2013;18(1):85–96. doi: 10.1177/1087057112453936. Discovery of a small molecule inhibitor of the EYA2 phosphatase using high throughput screening. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110*.Krueger AB, Drasin DJ, Lea WA, et al. Allosteric Inhibitors of the Eya2 Phosphatase Are Selective and Inhibit Eya2-mediated Migration. J Biol Chem. 2014 doi: 10.1074/jbc.M114.566729. Demonstration that identified EYA2 phosphatase inhibitors act allosterically, and do not inhibit several other cellular phosphatases or the related EYA3 phosphatase. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.van Westen GJ, Gaulton A, Overington JP. Chemical, target, and bioactive properties of allosteric modulation. PLoS Comput Biol. 2014;10(4):e1003559. doi: 10.1371/journal.pcbi.1003559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Darnell JE., Jr Transcription factors as targets for cancer therapy. Nat Rev Cancer. 2002;2(10):740–9. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- 113.Ostman A, Hellberg C, Bohmer FD. Protein-tyrosine phosphatases and cancer. Nat Rev Cancer. 2006;6(4):307–20. doi: 10.1038/nrc1837. [DOI] [PubMed] [Google Scholar]
- 114.Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53(7):2719–40. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 115.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]