

Abstract
Purpose of Review
Recent genetic findings have identified new targets of investigation in the field of interstitial lung diseases and have the potential to change clinical care.
Recent Findings
These findings implicate abnormalities in (1) host defense, (2) cell-cell adhesion, and (3) aging and senescence in the pathophysiology of pulmonary fibrosis. At least one common genetic variant strongly associated with pulmonary fibrosis appears to have prognostic implications for patients.
Summary
The inherited risk for pulmonary fibrosis is substantial, and recent data suggests that genetic risk for familial and sporadic forms of the disease are similar. Further characterization of the genetic risk will influence clinical practice in terms of categorization, diagnosis, and screening of individuals for this disease.
Keywords: IPF, pulmonary fibrosis, MUC5B, telomere, desmosome
Introduction
Despite decades of research, the etiology of fibrosing idiopathic interstitial pneumonias (IIPs), also known as fibrosing interstitial lung diseases (ILDs), has remained elusive. In the past few years, genetic studies of IIPs have led to new insights into disease susceptibility and identified new targets for further investigation.
Idiopathic Pulmonary Fibrosis (IPF) is the most common of the fibrosing IIPs. IPF is characterized by scarring of the lung parenchyma, leading to characteristic peripheral and basilar-predominant reticular opacities, volume loss with traction bronchiectasis, and honeycombing on high-resolution computed tomography and the pattern of Usual Interstitial Pneumonia (UIP) on histopathology [1]. The prognosis is poor, with no effective therapies and a median survival time from diagnosis of three years [2].
The etiology of IPF remains unknown, though it has long been known that specific environmental exposures are associated with disease [3-6]. Numerous studies have indicated that there is a genetic basis for pulmonary fibrosis – twin studies and familial aggregation of cases provided the first signs that inherited factors played a role in disease development [7-10]. For instance, abnormalities in surfactant proteins in particular are strongly implicated in familial pulmonary fibrosis [11-13]. In addition, pleiotropic genetic disorders such as dyskeratosis congenita [14] and Hermansky-Pudlak syndrome [15] are associated with pulmonary fibrosis. However, these mutations account for a small proportion of the population risk of disease making further research into genetic predisposition necessary.
Recent population studies have pointed to numerous additional specific genetic variants that confer significant risk to development of IPF and other fibrosing IIPs [16-18*]. Notably, pre-clinical evidence of interstitial lung abnormalities appears in asymptomatic patients carrying these genetic variants [19**]. This finding could have implications for the clinical care of those found to have genotypes conferring disease risk, particularly in light of the recent finding that genotype may help predict prognosis [20**]. As the body of evidence illustrating the genetic predisposition to development of fibrosing lung disease grows, the distinction between “familial” and “idiopathic” forms of the disease is becoming less clear—indeed, recent findings indicate that both sporadic and familial presentations of IPF have similar genetic risk factors [17**, 21].
In this review, we summarize current knowledge about genetic risk for the development of ILDs with a focus on the most recent findings in the field. Genes implicated in the development of ILDs can be categorized into three main categories, which will create a framework for the discussion: (1) genes implicated in host defense; (2) genes involved in aging and senescence; (3) and genes involved in cell-cell adhesion. We conclude by addressing the implications of recent findings—specifically, that there may be no genetic difference between sporadic and familial cases of IPF.
1. Host Defense
Alterations in an individual's host defense mechanisms have been the target of previous research examining genetic predisposition to disease. Genetic variation in cytokine genes has been implicated in fibrosis in prior studies [22-24]. More recently, genome-wide association studies have further expanded investigators' focus from the alveolar epithelia and the fibroblast to the role of host defense in pathogenesis of disease.
Mucins
In 2011, Seibold and colleagues determined through genome-wide linkage analysis and subsequent fine-mapping/sequencing that a single nucleotide polymorphism (SNP) rs35705950 on the p-terminus of chromosome 11 is strongly associated with IPF and with Familial Interstitial Pneumonia (FIP), as defined by the presence of two or more cases of definite or probable idiopathic interstitial pneumonia within three generations of a family [16]. This SNP resides in the promoter region of the MUC5B gene, which codes for an airway mucin that is highly conserved across primate species. Analysis of other loci in the region showed that rs35705950 remained the most significant SNP. The odds ratios for disease among those who are heterozygous (GT) and homozygous (TT) for the minor allele at this site were 6.8 and 20.8 for FIP and 9.0 and 21.8, respectively, for IPF [16], indicating a strong influence of the SNP on disease development. Not only was IPF diagnosis associated with greater than 14-fold increase in expression of MUC5B in lung tissue regardless of genotype, but presence of the minor allele (T) at rs35705950 was associated with a 37.4-fold increase in gene expression among unaffected individuals [16]. In the healthy human lung, MUC5B is found in the cytoplasm of secretory columnar cells in the bronchi as well as in bronchioles. In the setting of IPF, MUC5B appears in these locations as well as the characteristic honeycomb cysts [25*].
The association of this MUC5B promoter variant with pulmonary fibrosis has been confirmed in multiple cohorts [17**, 18] [26-28*], and remains the most robust genetic finding association with IPF to date (Table 1). Intriguingly, the rs35705950 variant has also been shown in the general population to be associated with subclinical interstitial lung abnormalities, which may be precursor lesions to clinically evident pulmonary fibrosis [19]. The odds of having definite fibrosis on a CT scan were 6.3 times higher for each copy of the MUC5B variant [19]. Though this polymorphism is strongly associated with both subclinical interstitial lung abnormalities and with pulmonary fibrosis, it is also associated with improved survival in IPF patients, as shown by a retrospective analysis of two independent cohorts, suggesting that genotype may be a marker of prognosis [20**]. A remarkable aspect of the MUC5B variant finding is its high frequency, being found in approximately 20% of the European Centre d'Etude du Polymorphisme Humain (CEPH, individuals with Northern and Western European ancestry) population and 19% of the Framingham Heart Study population [19]. Yet the apparent effect size of this common variant is significant [16]. The frequency of the variant in the CEPH population and the relative infrequency of IPF in the general population suggest a significant role for gene by gene or gene by environment interactions in the development of disease. In addition, the MUC5B variant confers specific genetic risk for IPF, as studies of ILD secondary to sarcoidosis and scleroderma have failed to show an association [27-29*], though other groups have previously found MUC5B variants associated with diffuse panbronchiolitis in Asian populations [30].
Table 1. Summary of studies supporting association of rs35705950 minor allele with IPF.
This table summarizes the published literature confirming the association between the rs35705950 minor allele and IPF that was originally described in 2011 by Seibold and colleagues [16].
| Location | IPF (% SNP) | Controls (% SNP) | Odds Ratio (GT vs GG) | Odds Ratio (TT vs GG) | p-value | Reference |
|---|---|---|---|---|---|---|
| USA | 367 (60.8%) | 802 (20.7%) | 5.7 (4.3-7.5) | 9.6 (4.7-19.4) | 8.9×10-41 | Zhang et al. NEJM 2011 [26] |
| UK | 110 (67.0%) | 416 (19.6%) | 6.6 (4.1-10.7) | 11.8 (4.3-33.7) | 2.0×10-17 | Stock et al. Thorax 2013 [27*] |
| France | 142 (65.5%) | 1383 (20.2%) | 6.4 (4.5-9.0) | 19.1 (9.0-36.1) | 9.0×10-29 | Borie et al. PLoS One 2013 [28*] |
| USA | 2492 (63.1%) | 1890 (19.6%) | 4.5 (3.9-5.2) - overall | 7.2×10-95 | Fingerlin et al. Nat Genetics 2013 [17**] | |
| USA | 324 (57.2%) | 702 (21.3%) | 2.4 (2.1-2.8) - overall | 2.4×10-50 | Noth et al. Lancet Resp 2013 [18*] | |
Despite the strength and reproducibility of these findings, the mechanism by which the MUC5B variant leads to pulmonary fibrosis remains unknown. Recent, findings in murine models indicate that Muc5b is critical in airway response to pathogens [31**]. These findings suggest that dysregulation of MUC5B could impair host defense or contribute to poor clearance of inhaled particles and toxins via disordered mucociliary clearance. Alternatively, excess MUC5B may impair the alveolar repair response, leading to disordered signaling between alveolar epithelia and other matrix producing cells.
Inflammatory mediators
Toll like receptors (TLRs) are transmembrane receptors that recognize structurally conserved molecules derived from microbes. Another recent GWAS confirmed the MUC5B SNP association and identified three SNPs in Toll interacting protein (TOLLIP) significantly associated with pulmonary fibrosis [18]. These variants, all associated with differential expression of the gene, implicate the innate immune response in IPF pathogenesis. Intriguingly, one TOLLIP variant (rs5743890) was associated with mortality, providing another example of genotype affecting prognosis [18]. In a smaller study, a specific variant in Toll-like receptor 3 (TLR3) leading to a functional amino acid substitution (L412F) was found to be associated with decreased TLR3 activity in primary fibroblasts from IPF patients. This nonsynonymous mutation was also associated with early mortality and accelerated lung function decline in those carrying the variant [32]. In the case of asbestos-related lung disease, functional polymorphisms (rs35829419) in the NLRP3 gene, whose product is thought to play a role in inflammation and apoptosis, have also been linked to risk of pulmonary fibrosis [33*].
2. Aging & Senescence
The ability of the alveolar epithelium to respond to stress and injury has also long been a hypothesized etiology of pulmonary fibrosis, especially since so many presumably injurious exposures (e.g., asbestos, cigarette smoking) have been associated with the disease. Prior studies have indicated that the abnormalities in the ability of epithelial cells to divide and replace epithelia could be central to the pathophysiology of disease [34-36*], and correspondingly genetic variants in cell cycle genes have been associated with IPF and disease progression [37].
Telomeres are repetitive nucleotide sequences at the ends of chromosomes that protect genes from being damaged through the normal DNA replication process, and so are critical to maintaining genomic stability and regulating a cell's replicative capacity [38*]. Once telomeres shorten past a threshold, they activate a DNA damage response leading to cell death or cell-cycle arrest. Telomerase maintains telomere length by adding nucleotide repeats to the ends of chromosomes during the replication process and is composed of a reverse transcriptase component (TERT) and an RNA component, which serves as a template for elongation (TERC). Not only is telomerase activity ubiquitous in cancerous cells, it is also found in cells undergoing injury and repair, including fibrogenesis [39].
Sequence variants in genes regulating telomere length have been associated with numerous age-related diseases, including pulmonary fibrosis [38*]. Familial pulmonary fibrosis has been linked to shortened telomeres [40], and more specifically to specifically TERT and TERC mutations [40-42] some of which are inherited in an autosomal dominant fashion and show evidence of genetic anticipation [40] [12]. Interestingly, IPF patients have shortened telomeres even without the presence of known telomerase mutations [43, 44], suggesting that there may be factors other than previously described mutations that affect telomere length and, by extension, risk of disease.
Subsequent studies have found similar associations of TERT mutations with sporadic IPF [45-47]. TERT mutations have been found in 10-15% of FIP families, but in the majority of FIP patients, the responsible genetic abnormality has not been found [48, 49*]. However, recently an X-linked mutation in a third telomerase-associated gene, dyskerin (DKC1), has been described in a family with FIP [49*]. DKC1 binds to TERC and stabilizes the telomerase complex; as such, this newly described mutation led to decreased telomerase activity in affected individuals [49*]. The genetic risk for pulmonary fibrosis conferred by variants in TERT and TERC was confirmed by a large GWAS of individuals with fibrotic IIPs that found common variants in the TERC and TERT loci, 3q26 and 5p15, respectively [17**]. Telomerase activity was further implicated in this GWAS by the finding that a common variant in OBFC1, a gene implicated in variation of leukocyte telomere length [50-52], was also associated with fibrotic IIP [17**].
There is mounting evidence that specific variants in genes affecting telomerase activity or telomere length confer risk of pulmonary fibrosis, but the mechanism(s) by which these genetic changes lead to fibrosis remains unclear. Better understanding of the gene's role in the pathophysiology of fibrosis has the potential to direct us to therapeutic targets.
3. Cell-Cell Adhesion
A third category of genes recently identified as targets for future investigation are those involved in cell-cell adhesion. Specifically, variants in desmoplakin (DSP) and dipeptidyl peptidase 9 (DPP9) are associated with fibrotic IIP [17**]. In addition, DSP expression in lung tissue varied with the number of copies of the most statistically significant common variant, rs2076295 [17**], and DSP and DPP9 have been shown in various organs (heart, skin, kidney) to be critical in epithelial integrity [53-55].
DSP is an important component of the desmosome, a transmembrane structure particularly abundant in cells undergoing constant stretch (heart, skin, airway). As such, DSP may be particularly important in the regions of the lung that experience constant mechanical stress, such as the peripheral and basilar segments, those that are preferentially affected in IPF patients [56]. Intriguingly, other investigations have illustrated that DSP acts as a tumor suppressor by suppressing the WNT/β -caten in pathway [57], a pathway that itself has been implicated in the pathogenesis of IPF [58-60]. Further investigations into desmosomes in IPF may suggest a conduit by which environmental exposures could induce intracellular signaling changes, since the intracellular DSP interacts closely with cytoskeleton. Non-small cell lung cancer and other cancers illustrate that the desmosome is not merely an adhesion structure, but instead is a dynamic part of the epithelial cell and can alter phenotype via intracelleular signaling [61].
Implications of recent findings
Recent genetic findings in the field of fibrotic lung disease have broadened the scope of inquiry into the pathogenesis of disease, prompting numerous questions requiring further investigation. In the case of the promoter polymorphism in MUC5B, the variant rs35705950 is not only associated with an increased risk of IPF, but also with a 2.8-fold increased risk of having interstitial lung abnormalities and a 6.3-fold increased risk of having definite radiographic evidence of pulmonary fibrosis [19**]. Yet, those IPF patients with the rs35705950 allele also had improved survival when compared to those without it [20**]. These findings have significant clinical implications, specifically in the use of clinical genetic testing and prospective screening for patients deemed at increased risk of disease. A more complete understanding of the genetic risk of IPF will allow early detection and more accurate prognostication for patients whose clinical course has been considered unpredictable.
Genes and the Environment
The frequency with which many of the variants strongly associated with pulmonary fibrosis (e.g., rs35705950, rs2076295) are found in the general population suggests that though these genetic variants confer risk for the development of fibrotic lung disease, there are likely environmental factors contributing to risk of disease development. As illustrated by the 2013 Framingham population study, 19% of the population carries the rs35705950 variant, but the incidence of IPF within that population itself is far less than 1% [19**].
Epidemiologists have long observed a link between environmental exposures and the development of both familial and sporadic pulmonary fibrosis, specifically: cigarettes smoke, farming, livestock exposure, wood dust, metal dust, and stone/sand [62-64]. Other occupational exposures such as asbestos, drug exposures such as bleomycin [65, 66], and therapeutic exposures such as radiation [67, 68] have been linked to various forms of pulmonary disease, including fibrosis [69]. Future investigation into the role of gene by environment interactions will be critical to understanding the role that genetic variation plays in disease pathogenesis.
Familial vs. Idiopathic Pulmonary Fibrosis
The framework under which clinicians and investigators have understood genetic risk and pulmonary fibrosis to date makes a clear distinction between “familial” forms of the disease (familial IIP) and “idiopathic” fibrosing lung diseases (sporadic IIP). Data from the Fingerlin 2013 study illustrates that odds ratios for development of pulmonary fibrosis for each of the significant genetic variants reported in the manuscript are equivalent in familial and sporadic forms of disease (Figure 1). While we have previously written of familial and sporadic disease as distinct entities, based on such data, from the perspective of genetic risk of developing pulmonary fibrosis, they appear equivalent.
Figure 1. The Genetic Basis of Familial and Sporadic Idiopathic Interstitial Pneumonia is Similar.
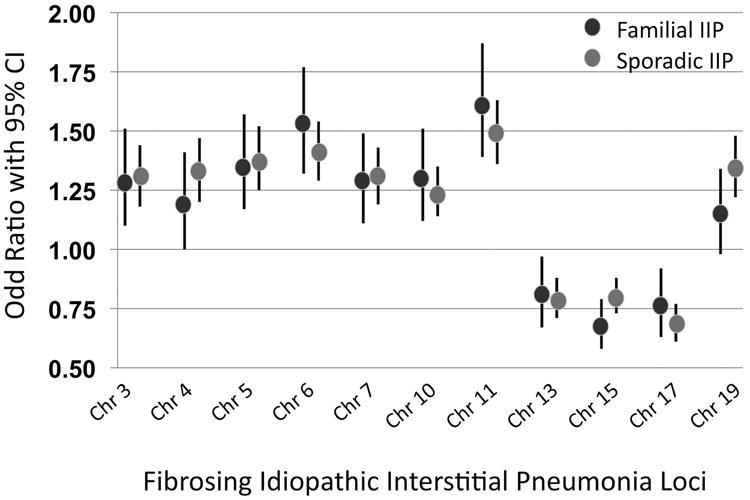
Data from Fingerlin et al. Nature Genetics 2013 depicted above illustrates that the genetic loci significantly associated with fibrotic IIP conferred the same increased risk of disease in both familial IIP and sporadic IIP cases [17**]. These data suggest that from the perspective of genetic risk, these diseases appear equivalent.
Conclusion
Many fibrotic IIP patients are diagnosed late in the course of their disease, limiting therapeutic options. Screening family members of affected individuals may prove a means of detecting and treating early-stage disease. As we understand better gene by environment interaction in the pathogenesis of fibrosis, we can target patients earlier in their disease course. Furthermore, given initial findings suggesting differential prognosis based on genotype and the overall genetic heterogeneity of fibrotic lung diseases, genetic variants may prove instrumental in redefining IIP subtypes. Finally, deeper understanding of genetic risk of disease points us to new investigational and therapeutic targets.
Key Points.
- Recently, genetic studies of IIPs have led to new insights into the susceptibility of individuals for IIPs.
- This progress provides investigators new targets to understand the pathophysiology of disease, specifically in the areas of (1) host defense, (2) cell-cell adhesion, and (3) aging and senescence.
- Genetic risk for development of pulmonary fibrosis appears to be similar in familial as well as sporadic forms of the disease.
- Further characterization of the genetic risk of developing pulmonary fibrosis will lead to novel approaches to prevent or delay the onset of this devastating disease.
- Better understanding of the genetic risk of developing pulmonary fibrosis also points investigators to new potential therapeutic targets.
Acknowledgments
SKM is supported by T32 HL007085 (NIH). DAS is supported by R01 HL097163 and P01 HL092870 (NIH).
Footnotes
There are no conflicts of interest to disclose.
References
- 1.Raghu G, Collard HR, Egan JJ, et al. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic Pulmonary Fibrosis: Evidence-based Guidelines for Diagnosis and Management. American Journal of Respiratory and Critical Care Medicine. 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.King TE, Tooze JA, Schwarz MI, et al. Predicting Survival in Idiopathic Pulmonary Fibrosis. American Journal of Respiratory and Critical Care Medicine. 2001;164:1171–1181. doi: 10.1164/ajrccm.164.7.2003140. [DOI] [PubMed] [Google Scholar]
- 3.Baumgartner KB, Samet JM, Stidley CA, et al. Cigarette smoking: A risk factor for idiopathic pulmonary fibrosis. American Journal of Respiratory and Critical Care Medicine. 1997;155:242–248. doi: 10.1164/ajrccm.155.1.9001319. [DOI] [PubMed] [Google Scholar]
- 4.Baumgartner KB, Samet JM, Coultas DB, et al. Occupational and environmental risk factors for idiopathic pulmonary fibrosis: A multicenter case-control study. Am J Epidemiol. 2000;152:307–315. doi: 10.1093/aje/152.4.307. [DOI] [PubMed] [Google Scholar]
- 5.Hubbard R, Lewis S, Richards K, et al. Occupational exposure to metal or wood dust and aetiology of cryptogenic fibrosing alveolitis. Lancet. 1996;347:284–289. doi: 10.1016/s0140-6736(96)90465-1. [DOI] [PubMed] [Google Scholar]
- 6.Hubbard R, Venn A, Smith C, et al. Exposure to commonly prescribed drugs and the etiology of cryptogenic fibrosing alveolitis - A case-control study. American Journal of Respiratory and Critical Care Medicine. 1998;157:743–747. doi: 10.1164/ajrccm.157.3.9701093. [DOI] [PubMed] [Google Scholar]
- 7.Bonanni PP, Frymoyer JW, Jacox RF. A family study of idiopathic pulmonary fibrosis - a possible dysproteinemic and genetically determined disease. American Journal of Medicine. 1965;39:411–21. doi: 10.1016/0002-9343(65)90208-1. [DOI] [PubMed] [Google Scholar]
- 8.Javaheri S, Lederer DH, Pella JA, et al. Idiopathic pulmonary fibrosis in monozygotic twins - the importance of genetic predisposition. Chest. 1980;78:591–594. doi: 10.1378/chest.78.4.591. [DOI] [PubMed] [Google Scholar]
- 9.Solliday NH, Williams JA, Gaensler EA, et al. Familial chronic interstitial pneumonia. The American review of respiratory disease. 1973;108:193–204. doi: 10.1164/arrd.1973.108.2.193. [DOI] [PubMed] [Google Scholar]
- 10.Bitterman PB, Rennard SI, Keogh BA, et al. Familial idiopathic pulmonary fibrosis - evidence of lung inflammation in unaffected family members. New England Journal of Medicine. 1986;314:1343–1347. doi: 10.1056/NEJM198605223142103. [DOI] [PubMed] [Google Scholar]
- 11.Nogee LM, Dunbar AE, Wert SE, et al. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. New England Journal of Medicine. 2001;344:573–579. doi: 10.1056/NEJM200102223440805. [DOI] [PubMed] [Google Scholar]
- 12.Fernandez BA, Fox G, Bhatia R, et al. A Newfoundland cohort of familial and sporadic idiopathic pulmonary fibrosis patients: clinical and genetic features. Respir Res. 2012;13 doi: 10.1186/1465-9921-13-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maitra M, Cano CA, Garcia CK. Mutant surfactant A2 proteins associated with familial pulmonary fibrosis and lung cancer induce TGF-β1 secretion. Proceedings of the National Academy of Sciences. 2012;109:21064–21069. doi: 10.1073/pnas.1217069110. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Armanios M. Telomerase and idiopathic pulmonary fibrosis. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2012;730:52–58. doi: 10.1016/j.mrfmmm.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gochuico BR, Huizing M, Golas GA, et al. Molecular medicine. Vol. 18. Cambridge, Mass: 2012. Interstitial lung disease and pulmonary fibrosis in Hermansky-Pudlak syndrome type 2, an adaptor protein-3 complex disease; pp. 56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seibold MA, Wise AL, Speer MC, et al. A Common MUC5B Promoter Polymorphism and Pulmonary Fibrosis. New England Journal of Medicine. 2011;364:1503–1512. doi: 10.1056/NEJMoa1013660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17**.Fingerlin TE, Murphy E, Zhang W, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nature Genet. 2013;45:613–620. doi: 10.1038/ng.2609. This large genome wide association study of non-Hispanic, white individuals with fibrotic idiopathic interstitial pneumonias confirmed associations between disease and TERT, MUC5B, TERC, and identified seven new loci associated with disease. Of particular interest are the novel variants found in DSP and DPP9, genes inolved in cell-cell adhesion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18*.Noth I, Zhang Y, Ma SF, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. The Lancet Respiratory Medicine. 2013;1:309–317. doi: 10.1016/S2213-2600(13)70045-6. This genome wide association study identified novel variants in TOLLIP and SPPL2C to be associated with IPF. One novel TOLLIP variant, rs5743890, was associated with mortality, and decreased expression of TOLLIP in IPF patients--these findings support the hypothesis that TOLLIP may be important in disease pathogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19**.Hunninghake GM, Hatabu H, Okajima Y, et al. MUC5B Promoter Polymorphism and Interstitial Lung Abnormalities. New England Journal of Medicine. 2013;368:2192–2200. doi: 10.1056/NEJMoa1216076. This study found that the MUC5B promoter polymorphism associated with IPF was found to be associated with interstitial lung abnormalities on CT scan in the general population (i.e., persons without diagnosed interstitial lung disease); this association was most apparent in older subjects, but was not influenced by smokin status. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20**.Peljto AL, Zhang YZ, Fingerlin TE, et al. Association Between the MUC5B Promoter Polymorphism and Survival in Patients With Idiopathic Pulmonary Fibrosis. JAMA-J Am Med Assoc. 2013;309:2232–2239. doi: 10.1001/jama.2013.5827. This study found that the rs35705950 MUC5B promoter polymorphism previously associated with IPF is associated with improved survival in IPF. This association of the variant with survival was independent of age, sex, forced vital capacity, diffusing capacity of carbon monoxide, MMP-7, and treatment status. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.García-Sancho C, Buendía-Roldán I, Fernández-Plata MR, et al. Familial pulmonary fibrosis is the strongest risk factor for idiopathic pulmonary fibrosis. Respiratory Medicine. 2011;105:1902–1907. doi: 10.1016/j.rmed.2011.08.022. [DOI] [PubMed] [Google Scholar]
- 22.Ahn MH, Park BL, Lee SH, et al. A promoter SNP rs4073T > A in the common allele of the interleukin 8 gene is associated with the development of idiopathic pulmonary fibrosis via the IL-8 protein enhancing mode. Respir Res. 2011;12:73. doi: 10.1186/1465-9921-12-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hutyrova B, Pantelidis P, Drabek J, et al. Interleukin-1 gene cluster polymorph isms in sarcoidosis and idiopathic pulmonary fibrosis. American Journal of Respiratory and Critical Care Medicine. 2002;165:148–151. doi: 10.1164/ajrccm.165.2.2106004. [DOI] [PubMed] [Google Scholar]
- 24.Whyte M, Hubbard R, Meliconi R, et al. Increased risk of fibrosing alveolitis associated with interleukin-1 receptor antagonist and tumor necrosis factor-alpha gene polymorphisms. American Journal of Respiratory and Critical Care Medicine. 2000;162:755–758. doi: 10.1164/ajrccm.162.2.9909053. [DOI] [PubMed] [Google Scholar]
- 25*.Seibold MA, Smith RW, Urbanek C, et al. The idiopathic pulmonary fibrosis honeycomb cyst contains a mucocilary pseudostratified epithelium. PLoS ONE. 2013;8:e58658. doi: 10.1371/journal.pone.0058658. This study describes the cells of the honeycomb cyst, the characteristic pathologic finding in IPF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y, Noth I, Garcia JGN, Kaminski N. A Variant in the Promoter of MUC5B and Idiopathic Pulmonary Fibrosis. New England Journal of Medicine. 2011;364:1576–1577. doi: 10.1056/NEJMc1013504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27*.Stock CJ, Sato H, Fonseca C, et al. Mucin 5B promoter polymorphism is associated with idiopathic pulmonary fibrosis but not with development of lung fibrosis in systemic sclerosis or sarcoidosis. Thorax. 2013;68:436–441. doi: 10.1136/thoraxjnl-2012-201786. This study confirmed the MUC5B promoter polymorphism's association with IPF. The findings also suggest that lung fibrosis in the context of other diseases such as systemic sclerosis and sarcoidosis are not asscoiated with this variant, highlighting what are likely to be differences in genetic susceptibility to these diseases. [DOI] [PubMed] [Google Scholar]
- 28*.Borie R, Crestani B, Dieude P, et al. The MUC5B Variant Is Associated with Idiopathic Pulmonary Fibrosis but Not with Systemic Sclerosis Interstitial Lung Disease in the European Caucasian Population. PLoS ONE. 2013;8(8):e70621. doi: 10.1371/journal.pone.0070621. This study confirms the association between the minor allele at the MUC5B promoter polymorphism and IPF and indicates an absence of association between the same polymorphism and systemic sclerosis-ILD, aruging that the two disease have different genetic risks and likely different pathophysiology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peljto AL, Steele MP, Fingerlin TE, et al. The Pulmonary Fibrosis-Associated MUC5B Promoter Polymorphism Does Not Influence the Development of Interstitial Pneumonia in Systemic Sclerosis. Chest. 2012;142:1584–1588. doi: 10.1378/chest.12-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kamio K, Matsushita I, Hijikata M, et al. Promoter analysis and aberrant expression of the MUC5B gene in diffuse panbronchiolitis. American Journal of Respiratory and Critical Care Medicine. 2005;171:949–957. doi: 10.1164/rccm.200409-1168OC. [DOI] [PubMed] [Google Scholar]
- 31**.Roy MG, Livraghi-Butrico A, Fletcher AA, et al. Muc5b is required for airway defence. Nature. 2014;505:412–416. doi: 10.1038/nature12807. This study describes the importance of Muc5b in mucociliary clearance in the murine airway, suggesting that in the human airways, it may also be critical in host defence and in maintaining airway homeostasis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O'Dwyer DN, Armstrong ME, Trujillo G, et al. The Toll-like Receptor 3 L412F Polymorphism and Disease Progression in Idiopathic Pulmonary Fibrosis. American Journal of Respiratory and Critical Care Medicine. 2013;188:1442–1450. doi: 10.1164/rccm.201304-0760OC. [DOI] [PubMed] [Google Scholar]
- 33*.Kukkonen MK, Vehmas T, Piirila P, Hirvonen A. Genes involved in innate immunity associated with asbestos-related fibrotic changes. Occup Environ Med. 2014;71:48–54. doi: 10.1136/oemed-2013-101555. This study finds functional polymorphisms in genes involved in innate immunity to be associated with asbestos-related pulmonary fibrosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Minagawa S, Araya J, Numata T, et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-beta-induced senescence of human bronchial epithelial cells. Am J Physiol-Lung Cell Mol Physiol. 2011;300:L391–L401. doi: 10.1152/ajplung.00097.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chilosi M, Poletti V, Murer B, et al. Abnormal re-epithelialization and lung remodeling in idiopathic pulmonary fibrosis: The role of Delta N-p63. Lab Invest. 2002;82:1335–1345. doi: 10.1097/01.lab.0000032380.82232.67. [DOI] [PubMed] [Google Scholar]
- 36*.Chilosi M, Carloni A, Rossi A, Poletti V. Premature lung aging and cellular senescence in the pathogenesis of idiopathic pulmonary fibrosis and COPD/emphysema. Transl Res. 2013;162:156–173. doi: 10.1016/j.trsl.2013.06.004. This review synthesizes a large amount of data supporting the role of premature aging and cellular senescence in the pathogenesis of IPF and COPD. [DOI] [PubMed] [Google Scholar]
- 37.Korthagen NM, van Moorsel CHM, Barlo NP, et al. Association between Variations in Cell Cycle Genes and Idiopathic Pulmonary Fibrosis. PLoS ONE. 2012;7(1):e30442. doi: 10.1371/journal.pone.0030442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38*.Codd V, Nelson CP, Albrecht E, et al. Identification of seven loci affecting mean telomere length and their association with disease. Nature Genet. 2013;45:422–427. doi: 10.1038/ng.2528. This study describes seven loci associated with leukocyte telomere length and argues that telomere-length variation may be causal in age-related disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu TJ, Ullenbruch M, Choi YY, et al. Telomerase and Telomere Length in Pulmonary Fibrosis. American Journal of Respiratory Cell and Molecular Biology. 2013;49:260–268. doi: 10.1165/rcmb.2012-0514OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Armanios MY, Chen JJL, Cogan JD, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. New England Journal of Medicine. 2007;356:1317–1326. doi: 10.1056/NEJMoa066157. [DOI] [PubMed] [Google Scholar]
- 41.Alder JK, Cogan JD, Brown AF, et al. Ancestral Mutation in Telomerase Causes Defects in Repeat Addition Processivity and Manifests As Familial Pulmonary Fibrosis. PLoS Genet. 2011;7(3):e1001352. doi: 10.1371/journal.pgen.1001352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nunes H, Monnet I, Kannengiesser C, et al. Is telomeropathy the explanation for combined pulmonary fibrosis and emphysema syndrome?: report of a family with TERT mutation. Am J Respir Crit Care Med. 2014;189:753–754. doi: 10.1164/rccm.201309-1724LE. [DOI] [PubMed] [Google Scholar]
- 43.Cronkhite JT, Xing C, Raghu G, et al. Telomere shortening in familial and sporadic pulmonary fibrosis. American Journal of Respiratory and Critical Care Medicine. 2008;178:729–737. doi: 10.1164/rccm.200804-550OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alder JK, Chen JJL, Lancaster L, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:13051–13056. doi: 10.1073/pnas.0804280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsakiri KD, Cronkhite JT, Kuan PJ, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:7552–7557. doi: 10.1073/pnas.0701009104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mushiroda T, Wattanapokayakit S, Takahashi A, et al. A genome-wide association study identifies an association of a common variant in TERT with susceptibility to idiopathic pulmonary fibrosis. J Med Genet. 2008;45:654–656. doi: 10.1136/jmg.2008.057356. [DOI] [PubMed] [Google Scholar]
- 47.Tsang AR, Wyatt HDM, Ting NSY, Beattie TL. hTERT mutations associated with idiopathic pulmonary fibrosis affect telomerase activity, telomere length, and cell growth by distinct mechanisms. Aging Cell. 2012;11:482–490. doi: 10.1111/j.1474-9726.2012.00810.x. [DOI] [PubMed] [Google Scholar]
- 48.Kropski JA, Lawson WE, Young LR, Blackwell TS. Genetic studies provide clues on the pathogenesis of idiopathic pulmonary fibrosis. Dis Model Mech. 2013;6:9–17. doi: 10.1242/dmm.010736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49*.Kropski JA, Mitchell DB, Markin C, et al. A novel dyskerin (DKC1) mutation is associated with Familial Interstitial Pneumonia. Chest. 2014 Feb 6; doi: 10.1378/chest.13-2224. Epub ahead of print. This study describes a variant in DKC1, the third telomere-related gene identified in familial interstitial pneumonia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Levy D, Neuhausen SL, Hunt SC, et al. Genome-wide association identifies OBFC1 as a locus involved in human leukocyte telomere biology. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:9293–9298. doi: 10.1073/pnas.0911494107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maubaret CG, Salpea KD, Romanoski CE, et al. Association of TERC and OBFC1 Haplotypes with Mean Leukocyte Telomere Length and Risk for Coronary Heart Disease. PLoS ONE. 2013;8 doi: 10.1371/journal.pone.0083122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee JH, Cheng R, Honig LS, et al. Genome wide association and linkage analyses identified three loci-4q25, 17q23.2, and 10q11.21-associated with variation in leukocyte telomere length: the Long Life Family Study. Frontiers in genetics. 2013;4:310. doi: 10.3389/fgene.2013.00310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rasmussen TB, Hansen J, Nissen PH, et al. Protein expression studies of desmoplakin mutations in cardiomyopathy patients reveal different molecular disease mechanisms. Clin Genet. 2013;84:20–30. doi: 10.1111/cge.12056. [DOI] [PubMed] [Google Scholar]
- 54.den Haan AD, Tan BY, Zikusoka MN, et al. Comprehensive Desmosome Mutation Analysis in North Americans With Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Circ-Cardiovasc Genet. 2009;2:428–U436. doi: 10.1161/CIRCGENETICS.109.858217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu DMT, Wang XM, Ajami K, et al. DP8 and DP9 have extra-enzymatic roles in cell adhesion, migration and apoptosis. In: Lendeckel U, Reinhold D, Bank U, editors. Dipeptidyl Aminopeptidases: Basic Science and Clinical Applications. New York: Springer; 2006. pp. 63–72. [DOI] [PubMed] [Google Scholar]
- 56.Carloni A, Poletti V, Fermo L, et al. Heterogeneous distribution of mechanical stress in human lung: A mathematical approach to evaluate abnormal remodeling in IPF. J Theor Biol. 2013;332:136–140. doi: 10.1016/j.jtbi.2013.04.038. [DOI] [PubMed] [Google Scholar]
- 57.Yang IV. Epigenomics of idiopathic pulmonary fibrosis. Epigenomics. 2012;4:195–203. doi: 10.2217/epi.12.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chilosi M, Poletti V, Zamo A, et al. Aberrant Wnt/beta-catenin pathway activation in idiopathic pulmonary fibrosis. Am J Pathol. 2003;162:1495–1502. doi: 10.1016/s0002-9440(10)64282-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Konigshoff M, Balsara N, Pfaff EM, et al. Functional Wnt Signaling Is Increased in Idiopathic Pulmonary Fibrosis. PLoS ONE. 2008;3 doi: 10.1371/journal.pone.0002142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Konigshoff M, Kramer M, Balsara N, et al. WNT1-inducible signaling protein-1 mediates pulmonary fibrosis in mice and is upregulated in humans with idiopathic pulmonary fibrosis. J Clin Invest. 2009;119:772–787. doi: 10.1172/JCI33950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen YJ, Lee LY, Chao YK, et al. DSG3 facilitates cancer cell growth and invasion through the DSG3-plakoglobin-TCF/LEF-Myc/cyclin D1/MMP signaling pathway. PLoS ONE. 2013;8:e64088. doi: 10.1371/journal.pone.0064088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Steele MP, Speer MC, Loyd JE, et al. Clinical and pathologic features of familial interstitial pneumonia. American Journal of Respiratory and Critical Care Medicine. 2005;172:1146–1152. doi: 10.1164/rccm.200408-1104OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Taskar VS, Coultas DB. Is idiopathic pulmonary fibrosis an environmental disease? Proceedings of the American Thoracic Society. 2006;3:293–298. doi: 10.1513/pats.200512-131TK. [DOI] [PubMed] [Google Scholar]
- 64.Taskar V, Coultas D. Exposures and Idiopathic Lung Disease. Semin Respir Crit Care Med. 2008;29:670–679. doi: 10.1055/s-0028-1101277. [DOI] [PubMed] [Google Scholar]
- 65.Rabinowits M, Souhami L, Gil RA, et al. Increased pulmonary toxicity with bleomycin and cisplatin chemotherapy combinations. American journal of clinical oncology. 1990;13:132–138. doi: 10.1097/00000421-199004000-00009. [DOI] [PubMed] [Google Scholar]
- 66.Brusamolino E, Lunghi F, Orlandi E, et al. Treatment of early-stage Hodgkin's disease with four cycles of ABVD followed by adjuvant radio-therapy: analysis of efficacy and long-term toxicity. Haematologica. 2000;85:1032–1039. [PubMed] [Google Scholar]
- 67.Abratt RP, Morgan GW, Silvestri G, Willcox P. Pulmonary complications of radiation therapy. Clinics in chest medicine. 2004;25:167–177. doi: 10.1016/S0272-5231(03)00126-6. [DOI] [PubMed] [Google Scholar]
- 68.Mehta V. Radiation pneumonitis and pulmonary fibrosis in non-small-cell lung cancer: pulmonary function, prediction, and prevention. International journal of radiation oncology, biology, physics. 2005;63:5–24. doi: 10.1016/j.ijrobp.2005.03.047. [DOI] [PubMed] [Google Scholar]
- 69.Prazakova S, Thomas PS, Sandrini A, Yates DH. Asbestos and the lung in the 21st century: an update. The clinical respiratory journal. 2014;8:1–10. doi: 10.1111/crj.12028. [DOI] [PubMed] [Google Scholar]