

ABSTRACT
Tetraspanins constitute a family of cellular proteins that organize various membrane-based processes. Several members of this family, including CD81, are actively recruited by HIV-1 Gag to viral assembly and release sites. Despite their enrichment at viral exit sites, the overall levels of tetraspanins are decreased in HIV-1-infected cells. Here, we identify Vpu as the main viral determinant for tetraspanin downregulation. We also show that reduction of CD81 levels by Vpu is not a by-product of CD4 or BST-2/tetherin elimination from the surfaces of infected cells and likely occurs through an interaction between Vpu and CD81. Finally, we document that Vpu-mediated downregulation of CD81 from the surfaces of infected T cells can contribute to preserving the infectiousness of viral particles, thus revealing a novel Vpu function that promotes virus propagation by modulating the host cell environment.
IMPORTANCE The HIV-1 accessory protein Vpu has previously been shown to downregulate various host cell factors, thus helping the virus to overcome restriction barriers, evade immune attack, and maintain the infectivity of viral particles. Our study identifies tetraspanins as an additional group of host factors whose expression at the surfaces of infected cells is lowered by Vpu. While the downregulation of these integral membrane proteins, including CD81 and CD82, likely affects more than one function of HIV-1-infected cells, we document that Vpu-mediated lowering of CD81 levels in viral particles can be critical to maintaining their infectiousness.
INTRODUCTION
Tetraspanins are integral membrane proteins that span the lipid bilayer four times. The 33 members (in humans) of this protein family, by homo- and hetero-oligomerizing and by laterally interacting with other proteins and with lipids, form a web that serves as the basis for their involvement in the organization of membranes. When triggered by intra- or extracellular cues, so-called tetraspanin-enriched microdomains (TEMs) can form, and these platforms then support or modulate various membrane-based processes, including cell adhesion, membrane fusion, signaling, and protein sorting. Consequently, tetraspanins play roles in a wide range of biological activities, such as fertilization, muscle formation and repair, generation of synaptic contacts at neuromuscular junctions, maintenance of skin integrity, and induction of immune responses (1–4). They are also implicated in pathologies, including cancer (e.g., metastasis [5]) and inherited disorders (6), as well as in the propagation and pathogenesis of numerous infectious agents (parasites, bacteria, and viruses) (7–11).
While one member of the tetraspanin family (CD63) was shown more than 2 decades ago to be specifically acquired by HIV-1 particles released from infected cells (12–14), only during the past decade has work by several groups documented that tetraspanins play roles during different stages of the viral replication cycle (for recent reviews, see references 9 and 15). The tetraspanins CD9, CD53, CD63, CD81, CD82, and tetraspanin 14 have been found to accumulate at the exit site and/or to be incorporated into newly formed viral particles (16–21). Indeed, HIV-1 Gag actively recruits tetraspanins to the release site (22, 23), possibly creating an environment that is favorable for HIV-1 assembly/release and also allowing tetraspanin incorporation into viral particles. How tetraspanins support assembly, however, remains unclear, and whether their presence at the viral exit site directly promotes release may depend on the physiological circumstances and on the cell type (24–28). Crucially, when incorporated into viral particles, tetraspanins render them less infectious by inhibiting fusion with and thus entry into target cells (20, 27). Why the virus would specifically incorporate a host factor that renders it less infectious is unclear; perhaps their acquisition is merely tolerated as a negative but acceptable by-product of a potentially positive function performed at the presynaptic side of the virological synapse (VS): because tetraspanins inhibit the fusion of producer and target cells (29, 30), they may preserve the integrity of the VS and thus foster particle transmission through this conduit, as well as the subsequent separation of producer and target cells (as discussed previously [31–33]). The dichotomy between beneficial (prevention of cell-cell fusion at the VS) and detrimental (inhibition of virus-cell fusion) tetraspanin functions in infected cells perhaps might explain an apparent paradox: while tetraspanins are actively enriched at the exit site, overall cellular levels of tetraspanins are lowered upon HIV-1 infection (27), as well as activation of chronically infected cells (20). By regulating cellular levels of tetraspanins, viral factors may (through yet unidentified mechanisms) help establish a balance between their beneficial and detrimental effects, ultimately promoting viral spread.
Here, we set out to identify the viral factor responsible for tetraspanin downregulation in HIV-1-infected cells. Because CD81 and CD82 are prominently expressed at the surfaces of many T cell lines and also because they have already been shown to play functional roles (e.g., control of signaling) at the immunological synapse (IS), the cell-cell interface that is thought to be closely related to the VS (as discussed recently [34]), we focused our investigations on these tetraspanins rather than CD9 or CD63, tetraspanins that either are barely expressed in some T cells (M. Lambelé and M. Thali, unpublished observation) or whose presence at the plasma membrane is of only a transient nature to start with (35), respectively.
MATERIALS AND METHODS
Cell culture and antibodies (Ab).
293T, HeLa, and TZM-bl cells were maintained in Dulbecco's modified Eagle's medium (CellGro) supplemented with 2 mM l-glutamine, 10% heat-inactivated fetal bovine serum (FBS) (Gibco-Invitrogen), and antibiotics (ATB) (100 U/ml penicillin and 100 μg/ml streptomycin; CellGro). Jurkat (clone E6-1) and CEMss cells were obtained from Arthur Weiss and from Peter L. Nara, respectively, through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH. The cells were maintained in RPMI 1640 medium supplemented with 10% heat-inactivated FBS and ATB. Primary CD4+ T cells were isolated from whole blood of healthy donors by Ficoll gradient centrifugation followed by negative selection (Miltenyi Biotec). They were subsequently activated in RPMI 1640 medium supplemented with 20% FBS, ATB, 5 μg/ml phytohemagglutinin (PHA) (Sigma), and 10 ng/ml interleukin 2 (Roche). After 3 days, the cells were washed and maintained in RPMI 1640 supplemented with FBS, ATB, and interleukin 2.
The following antibodies were used during this study: unconjugated (clone JS-81; BD Biosciences) or phycoerythrin (PE)-conjugated (clone JS-81; BD Biosciences) anti-CD81, unconjugated (clone BL-2; Abcam) or PE-conjugated (clone ASL-4; Biolegend) anti-CD82, PE-conjugated anti-CD4 (clone L120; BD Biosciences), PE-conjugated anti-CD3 (clone HIT3a; BD Biosciences), anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (clone 6C5; Abcam), anti-Vpu (polyclonal; Abcam), and fluorescein isothiocyanate (FITC)-conjugated anti-p24 (clone KC57; Beckman Coulter). The following reagents were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: HIV-1 NL4-3 Vpu antiserum (from Klaus Strebel), HIV-1 Nef antiserum (from Ronald Swanstrom), HIV-1 p24 hybridoma (183-H12-5C; from Bruce Chesebro), HIV-Ig (from Luiz Barbosa), and monoclonal antibody to HIV-1 p24 (AG3.0; from Jonathan Allan).
Virus production and infections.
The proviral constructs used in this report include the following: pNL4-3 wild type (WT) (obtained from Malcolm Martin through the NIH AIDS Reagent Program); pNL4-3 with Nef deleted (ΔNef) (a kind gift from John Guatelli); pNL4-3 with Vpu deleted (ΔVpu), pNL4-3 52/56, and pNL4-3 Urd (kind gifts from Klaus Strebel); and pNL4-3 (with enhanced green fluorescent protein [eGFP] expressed from an internal ribosomal entry site [IRES]) WT, with Nef deleted (ΔNef), with Vpu deleted (ΔVpu), and with Vpu and Nef deleted (ΔVpu ΔNef) (which have been described previously [36]).
293T cells were transfected using calcium phosphate (Invitrogen) with 18 μg of proviral DNA and 2 μg of pVSV-G. At 48 h posttransfection, the virus-containing supernatants were precleared by centrifugation, passed through a 0.45-μm filter, assayed for p24 content using a homemade enzyme-linked immunosorbent assay (ELISA) capture assay (detailed in reference 37), and stored at −80°C.
T cells were infected by spinoculation with supernatants containing 50 ng (for Jurkat or CEMss cells) or 250 ng (for primary CD4+ T cells) of p24 per million cells for 2 h at 1,200 × g (unless otherwise indicated), washed with medium, and incubated at 37°C in 5% CO2.
Quantification of total protein levels by Western blotting.
Two or 3 days postinfection, 4 million CEMss or Jurkat T cells were pelleted, resuspended in TNE (50 mM Tris-HCl, pH 7.5, 200 mM NaCl, 2 mM EDTA) containing 1% Triton X-100 and protease inhibitor cocktail (Promega), and incubated on ice for 30 min. The lysates were centrifuged for 20 min at 4°C at 20,000 × g to remove cellular debris. The proteins were then resolved on SDS-polyacrylamide gels under nonreducing conditions. Following protein transfer onto a polyvinylidene difluoride (PVDF) membrane, the membrane was blocked by incubation with 5% milk powder in Tris-buffered saline (TBS) (10 mM Tris-HCl, pH 7.2, and 150 mM NaCl) with 0.1% Tween 20 (TBS-T), washed, and incubated with primary antibody for tetraspanins (diluted 1/1,000), GAPDH (diluted 1/50,000), or p24 (1/1,000) overnight at 4°C. The membranes were then washed, incubated with a horseradish peroxidase (HRP)-conjugated secondary antibody for 30 min at room temperature (RT), washed again, and developed with enhanced chemiluminescent substrate (Thermo Scientific). Quantifications of protein levels were performed using Fiji (NIH ImageJ software).
To assess the ability of Vpu to downregulate CD81 independently of CD4 or BST-2, 293T cells were transfected using X-tremeGene HP DNA transfection reagent (Roche), following the manufacturer's recommendations, with an empty vector or with a pVphu plasmid (a kind gift from Klaus Strebel). Two days posttransfection, the cells were lysed with lysis buffer, and Western blot assays were carried out as previously described.
Analysis of cell surface protein levels by flow cytometry.
One million cells infected with eGFP-expressing HIV-1 were precooled for 10 min on ice, washed once with cold phosphate-buffered saline containing 1% bovine serum albumin (PBS-BSA), and then stained on ice for 30 min with the indicated primary Ab diluted 1/50. Then, the cells were washed, fixed with cold 4% paraformaldehyde (PFA) for 10 min, washed, and resuspended in 350 μl PBS. When non-eGFP-expressing HIV-1 was used for infection, cells were surface stained and fixed as described above, and then the cells were permeabilized using PBS containing 0.2% Triton X-100 and 1% BSA for 20 min at 37°C, stained with an anti-p24 FITC-conjugated antibody diluted 1:500 for 30 min at 37°C, washed, and resuspended in 350 μl of PBS. Finally, samples were collected using a BD LSRII analytical cytometer, and the data were analyzed using FlowJo v 9.7 or v X (Treestar). Samples were gated for eGFP/p24 expression (Fig. 1B shows a representative dot plot), and the mean fluorescence intensity (MFI) of the protein of interest was quantified within this gate.
FIG 1.

Vpu is required for CD81 and CD82 downregulation in infected cells. (A) Western blot analysis of CD81 and CD82 in infected CEMss T cells. Two to 3 days postinfection, the cells were lysed, and CD81/GAPDH (left) or CD82/GAPDH (right) protein levels were analyzed. Representative blots are shown. The histograms represent the overall total cellular CD81 and CD82 levels normalized to the mock condition. (B to D) Analyses of surface levels of CD81, CD82, or CD4 in infected CEMss and primary CD4+ T cells by 2-color flow cytometry (the values are normalized to the mock condition). (B) Representative dot plots displaying the flow cytometry gating strategy used to obtain the MFI values used in panels C and D. The boxed area is the gate selected for analysis. (C) Quantification of the MFI of surface-expressed CD81, CD82, or CD4 in infected CEMss cells. (D) Analysis of cell surface levels of CD81, CD82, and CD4 in infected primary CD4+ T cells. (E) Quantification of the overall CD81 level in 293T cells by Western blotting. Two days posttransfection, 293T cells were lysed, and CD81, Vpu, and GAPDH levels were analyzed by Western blotting. A representative blot is shown. (F) Analyses of CD81 surface expression by 2-color flow cytometry. Jurkat T cells infected with WT NL4-3 or the ΔVpu, Urd, or 52/56 mutant were surface stained for CD81 and overall stained for p24 or lysed to quantify Vpu expression under the different conditions. Vpu was analyzed by Western blotting; a representative blot is shown. Three biological replicates, consisting of two technical replicates each, were performed in all experiments. n.s., nonsignificant; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001 (compared to the WT condition, except where indicated by a horizontal bar). The error bars indicate standard errors of the means.
Analysis of tetraspanin incorporation into viral particles.
CEMss T cells were infected as described above. Two days postinfection, virus-containing supernatants were collected, precleared by centrifugation, and filtered through 0.45-μm membrane. Viruses were then concentrated through a 20% sucrose cushion at 100,000 × g at 4°C for 2 h in a T865 Sorvall rotor. The viral pellets were resuspended in 200 μl PBS at pH 7.4.
Viral particles were further enriched by immunoprecipitation (see Results). Sepharose G beads or A/G agarose beads (Santa Cruz) were coated with 5 μl HIV-Ig for 3 h under gentle rocking at 4°C; then, 100 μl of concentrated viruses or PBS was added to the bead-antibody mixture and incubated overnight at 4°C under gentle rotation. After 3 washes in PBS-BSA, the beads were pelleted and resuspended in 50 μl Laemmli buffer (under nonreducing conditions). Boiled samples were then subjected to 10% SDS-acrylamide migration and transferred onto PVDF membranes. The membranes were then blocked for 30 min in TBS-T containing 5% milk (TBS-T–milk) and then incubated with anti-CD81 primary antibody diluted as described above in TBS-T–milk overnight at 4°C under gentle agitation. The membranes were then washed 3 times in TBS-T, incubated with secondary antibody conjugated to HRP (diluted 1:10,000) in TBS-T–milk for 30 min at RT, washed 3 times, and revealed using A Pierce ECL kit. Quantification of immunoblot bands was done using ImageJ (NIH).
Cell-free infectivity assay.
One day prior to infection, TZM-bl reporter cells, which express beta-galactosidase and luciferase under the control of the HIV-1 long terminal repeat (LTR), were plated in 96-well plates (10,000 cells/well). Supernatants from virus-producing CEMss or Jurkat T cells were precleared by centrifugation, filtered through a 0.45-μm membrane, and then assayed for p24 content using a homemade ELISA. One to 0.5 ng of p24 in 100 μl of medium containing 20 μg/ml DEAE-dextran was added to each well. Forty hours later, the medium was removed, the cells were washed, and 50 μl of All-in-One mammalian beta-galactosidase reagent (Thermo Scientific Pierce) was added to each well. The plate was incubated at 37°C for 30 min, and absorbance was read at 405 nm in a plate reader.
Immunofluorescence staining of infected cells.
CEMss cells were infected as described above. Two days postinfection, 250,000 infected cells were plated onto 35-mm glass-bottom dishes (MatTek Corporation) treated with poly-l-lysine (Sigma). After 1 h at 37°C, the cells were fixed with 4% PFA for 10 min at 37°C and then blocked and permeabilized using PBS containing 1% BSA and 0.2% Triton X-100 for 20 min at 37°C. The cells were then stained for CD81 and Vpu for 1 h at 37°C, washed 3 times using PBS, and incubated for 30 min at 37°C with secondary Alexa Fluor-conjugated antibodies. Images were acquired on a DeltaVision workstation (Applied Precision) with an Olympus IX-70 base equipped with a CoolSnap HQ charge-coupled-device (CCD) Camera (Photometrics) and an Olympus PlanApo 1.42-numerical-aperture (NA) 60× objective. Softworx 5 software was used to deconvolve and export the images. The images were then loaded into Fiji for analysis of the colocalization between Vpu and CD81 using the plug-in Coloc2. Colocalization between CD81 and Vpu was reported using Pearson's correlation R, where an R value of 0 corresponds to no colocalization and an R value of 1 is perfect colocalization.
Fluorescence-activated cell sorting (FACS)-based FRET assay.
peYFP- and peCFP-C1/N1 plasmids, the peCFP-eYFP Förster resonance energy transfer (FRET) positive control, the membrane-associated cyan fluorescent protein (MEM-CFP) control, and vectors encoding CD81 and HIV-1 NL4-3 Nef and Vpu fusion proteins have been described previously (38, 39). A plasmid expressing CD82 N-terminally labeled with yellow fluorescent protein (YFP) was generated by PCR amplifying CD82 from a HeLa cell-derived cDNA library, introducing 5′ XhoI and 3′ EcoRI restriction sites. The PCR product was subsequently inserted into the peYFP- and peCFP-C1 vectors by standard ligation procedures. All PCR-derived constructs were sequenced to confirm their identities.
FACS-FRET was essentially done as described previously (38, 39). In brief, 150,000 293T cells were seeded per 12-well plate and transfected with 2.5 μg total DNA (1.25 μg of each construct) using the calcium phosphate technique. Cells were harvested for FACS analysis 24 to 36 h later and resuspended in a total volume of 300 μl FACS buffer (PBS containing 1% FBS). Subsequently, samples were analyzed for FRET signal using a FACSCantoII flow cytometer (BD).
Statistical analysis.
All statistical analyses were performed using GraphPad Prism 5. Student's t test or one-way analysis of variance (ANOVA) was used with a Bonferroni post hoc test to correct for multiple comparisons. Statistical significance was indicated when the P value was ≤0.05.
RESULTS
Since both Vpu and Nef are known to downregulate various host cell proteins (reviewed in references 40, 41, and 42), we began our search for tetraspanin downregulators by analyzing whether the reduction of tetraspanin levels in HIV-1-infected T lymphocytes was dependent on the expression of these accessory proteins.
Vpu is the main viral determinant for tetraspanin downregulation in HIV-1-infected T cells.
We first analyzed the ability of NL4-3 viruses with Nef and/or Vpu deleted to downmodulate CD81 and CD82 in infected T cells. To minimize the effects of deleting Vpu and Nef on virus release and infectivity at the outset of the experiment, particles produced in 293T cells, pseudotyped with the envelope glycoprotein G of vesicular stomatitis virus (VSV-G), were used to infect CEMss and Jurkat cells. Forty eight to 72 h postinfection, total cellular levels (i.e., tetraspanin amounts at the plasma membrane and at internal membranes combined) of CD81 and CD82 were quantified by Western blotting, and the tetraspanin levels were normalized relative to the GAPDH content, which was used as a loading control. The results were further normalized to the uninfected (mock) condition.
CD81 levels were reduced by 80% and 85% when cells were infected with WT and ΔNef viruses, respectively, whereas viruses lacking Vpu downregulated CD81 less efficiently (by 32% for ΔVpu and 40% for ΔVpu ΔNef) (Fig. 1A, left). We observed a similar pattern of downregulation for the nonglycosylated form of CD82 with our panel of mutants (Fig. 1A, right). WT and ΔNef NL4-3-infected cells, which express Vpu, presented 62% and 55% decreased CD82 levels, while viruses lacking Vpu displayed a 1.5-fold increase in the level of CD82 (compared to the uninfected condition). Similar results were observed in Jurkat T cells (data not shown).
Since CD81 and CD82 are both situated primarily at the plasma membrane, we next measured the surface levels of these proteins in cells infected with the same panel of viruses. CD4 and CD3 levels were analyzed in parallel as positive and negative controls, respectively. Figure 1B depicts a representative dot plot with the gating strategy used to select the infected cell population within which the MFI of the corresponding surface protein was determined. While the surface levels of CD3 remained unaltered or were even slightly increased (not shown), CD4 surface levels were strongly reduced, even in the absence of either Vpu or Nef expression; this was due to a known function of viral Env that also causes rapid downregulation of the viral receptor (43, 44) (Fig. 1C, right). Surface levels of CD81 and CD82 were reduced by 50% and 30%, respectively, when cells were infected with WT virus, and this effect was abrogated in the absence of Vpu expression (Fig. 1C, left and middle, respectively). Interestingly, though Nef deficiency also led to a slight loss of the downregulation ability of the virus, cells infected with ΔVpu ΔNef virus had significantly higher CD81 and CD82 surface levels than all the other viruses (Fig. 1C). Next, we assessed CD81 and CD82 downregulation in HIV-1-infected primary CD4+ T cells (Fig. 1D). Surface levels of both tetraspanins were reduced by about 50% when cells were infected with WT viruses. Deletion of Nef and/or Vpu abolished this effect, and, similarly to T cell lines, primary CD4+ T cells infected with the double-mutant virus showed increased levels of CD81 and CD82 at the plasma membrane. Together, these results suggest that while Vpu is the main determinant for both the overall downregulation of tetraspanins (Fig. 1A) and the reduction of their surface levels (Fig. 1C and D), Nef also contributes to the phenomenon, specifically by downregulating the levels of tetraspanins at the plasma membrane. This finding parallels the results shown in a report by Haller and colleagues (45), which reveals a role in tetraspanin downregulation for Nef, as well as for Vpu.
Given that there were no qualitative differences between HIV-1-induced downregulation of CD81 and CD82, and because the effects on CD81 were consistently stronger, we focused on this tetraspanin for the remainder of the study. We next tested whether Vpu-induced downregulation of CD81 was a by-product of CD4 or BST-2/tetherin downregulation. First, we analyzed whether Vpu expression in 293T cells, which express neither CD4 nor BST-2/tetherin, affects the levels of endogenous CD81. Figure 1E documents that Vpu expression led to an approximately 2-fold reduction of (total) CD81 levels. This result confirms that the reduction of CD81 levels was not merely a by-product of CD4 and/or BST-2/tetherin downregulation, and it also shows that Vpu expression alone is sufficient to induce downregulation of endogenous CD81.
Next, we tested whether Vpu-mutant viruses that can no longer downregulate either CD4 (52/56) (38, 46) or BST-2/tetherin (Urd) (38, 47) can still lower CD81 levels in infected Jurkat T cells. While the Urd mutant fails to decrease BST-2 cell surface expression because it no longer binds to this restriction/host cellular factor (38, 47), the 52/56 mutant (38, 46) still associates with CD4 but fails to downregulate CD4 because of the lack of phosphorylation of serines 52 and 56, which are essential for the recruitment of β-TrCP that bridges CD4-Vpu complexes to an E3-Ub ligase, thus targeting it to the proteasomal degradation pathway. Figure 1F shows that these mutants were still able to downregulate CD81, indicating that Vpu-mediated CD81 downregulation is independent of the 52/56 serine motif or the transmembrane domain of Vpu. This further confirms that this effect is not a by-product of either CD4 or BST-2/tetherin downregulation but, rather, constitutes a previously unrecognized function of Vpu, as was also revealed in the report by Haller and colleagues (45).
Vpu-mediated decreased CD81 incorporation correlates with enhanced virion infectivity.
As documented previously, incorporation of tetraspanins into HIV-1 particles decreases their infectivity (20, 27). Therefore, we reasoned that by decreasing CD81 at the cell surface, cells infected with WT viruses should release particles that contain less CD81 than particles released from cells infected with mutant (ΔVpu or ΔNef) viruses and, thus, that WT particles should be more infectious than ΔVpu or ΔNef viral particles. To test this hypothesis, viruses produced in T cells were used in parallel assays to quantify CD81 incorporation into virions by Western blotting and to test their infectivity using a β-galactosidase reporter cell line. Due to background CD81 signal (likely from exosomes that copurify with virus), we added an immunoprecipitation step to enrich viral particles. As documented in Fig. 2A and B, compared to WT virus, ΔVpu viruses incorporated substantially more CD81. Interestingly, and in line with the data shown in Fig. 1C and D, which show that Nef contributes to reduced surface levels of CD81, we found that the CD81/p24 ratio was significantly increased, not only for ΔVpu particles, but also for particles released from cells infected with ΔNef or ΔVpu ΔNef virus.
FIG 2.
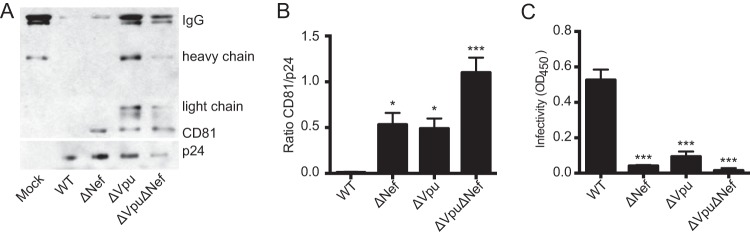
Increased CD81 incorporation into ΔVpu virions correlates with decreased infectivity. (A) Western blot analysis of CD81 incorporation into viral particles. Viruses produced in CEMss cells were purified and enriched as described in Materials and Methods. A representative blot from three independent experiments is shown. (B) Protein levels were analyzed by densitometry of the Western blots shown in panel A; the histogram represents the ratio of the CD81 level to the p24 level. (C) The viruses shown in panel A were assayed for infectivity in TZM-bl reporter cells. Equal levels of p24 were used. *, P ≤ 0.05; ***, P ≤ 0.001 (compared to the WT condition). The error bars indicate standard errors of the means.
We next assayed the infectivity of the same viruses and found that loss of either or both Vpu and Nef resulted in greatly decreased infectivity of the mutant viruses (Fig. 2C), which directly correlated with increased CD81 incorporation (Fig. 2A and B). Because previous studies documented that Vpu deletion does not affect Env levels in virions (48) and because tetraspanin overincorporation results in reduced infectivity (20, 27), the decreased infectivity of ΔVpu particles likely is a direct consequence of increased CD81 incorporation. Decreased infectivity of ΔNef and ΔVpu ΔNef virus, however, while partially the result of increased tetraspanin acquisition, most likely was due mainly to the lack here (in these mutants) of Nef's well-documented infectivity-enhancing functions, which include functional interactions with host cell proteins, such as CD4, dynamin-2, ERM proteins, and EHD4 (49–51). In summary, we found that Vpu may modulate the infectivity of viral particles by manipulating the levels of CD81.
Vpu colocalizes and interacts with CD81, thus inducing its degradation.
When downregulating CD4 and BST-2/tetherin, Vpu acts as an adaptor by marking these host cell proteins for degradation through the proteasomal and lysosomal degradation pathways (52, 53). Based on that and on the findings shown in Fig. 1, we hypothesized that Vpu and CD81 colocalize and interact in HIV-1-infected cells.
To assess CD81 and Vpu colocalization in infected cells, we performed immunofluorescence analyses of permeabilized CEMss T cells infected with HIV-expressing eGFP via an IRES (i.e., not Gag-GFP). As depicted in Fig. 3A, we observed a striking difference in CD81 localization between WT-virus-infected cells and cells infected with ΔVpu virus. In the absence of Vpu, CD81 primarily localized at the plasma membrane (Fig. 3A, middle row), as observed in uninfected CEMss T cells (data not shown). In contrast, when Vpu was present (Fig. 3A, top and bottom rows), CD81 was localized to an intracellular compartment, consistent with the surface downmodulation observed by flow cytometry in Fig. 1C and D. In addition to the change in localization, CD81 also colocalized extensively with Vpu upon both WT and ΔNef virus infection (the Pearson correlation R values are shown in Fig. 3A, far right), suggesting that Vpu and CD81 may associate with each other.
FIG 3.

Vpu and CD81 association leads to CD81 downregulation via both lysosomal and proteasomal pathways. (A) Analysis of CD81 and Vpu localization by fluorescence microscopy. HIV-1-infected CEMss T cells were fixed and stained for Vpu (red) or CD81 (cyan). Representative cells are shown for the different conditions (WT, ΔVpu, and ΔNef). The viruses used here result in expression of free eGFP (shown in the left column) with the use of an IRES. The images correspond to a single z-section. The scale bar represents 6 μm. Pearson's correlation values were calculated using the Coloc 2 plug-in in Fiji (ImageJ; NIH) (17 cells per condition from 2 independent experiments were analyzed). (B) FACS-FRET analysis of interactions between CD81 or CD82 and Vpu or Nef, conducted as described in Materials and Methods. Shown are the mean values for total percentages of cells scoring FRET positive from a minimum of 3 independent experiments. Statistical comparisons were made to the membrane-associated MEM-CFP control; only significant comparisons are indicated (***, P ≤ 0.001; one-way ANOVA with a Bonferroni post hoc test). (C and D) HIV-1-infected Jurkat T cells were either left untreated or treated with 100 nM bafilomycin A1 (BFL A1) (C) or 25 μM MG132 (D) for 12 h. At least three independent experiments consisting of two technical replicates each were performed for each inhibitor. (C) Analysis of CD81 total levels, as quantified by Western blotting densitometry. A representative blot is depicted. *, P ≤ 0.05. (D) Quantification of surface levels of CD81 by flow cytometry. The mean fluorescence intensities for CD81 within infected cells are shown, normalized to the mock condition. The error bars indicate standard errors of the means.
To test whether Vpu and Nef interact with CD81 and CD82, we used a FACS-based FRET assay (38) in 293T cells, in which FRET signal is produced only when the FRET-capable YFP and CFP tags are in very close proximity. The expression levels of the CD81-YFP and CD82-YFP fusion proteins were evaluated by Western blotting and found to be (roughly) comparable (data not shown). Coexpression of Vpu-CFP and CD81-YFP in 293T cells resulted in significant FRET signal, but this was not observed when Nef-CFP was expressed instead (Fig. 3B). This result suggested that Vpu and CD81 most likely interact directly with each other. Notably, using the FRET assay, CD81 and CD82 were shown to also interact and hence form hetero-oligomers. In contrast, neither expression of Vpu-CFP nor that of Nef-CFP together with CD82-YFP resulted in detectable FRET signals (Fig. 3B). Therefore, although Vpu binds only to CD81, this interaction might indirectly lead to downregulation of CD82 by mediating internalization of CD81-CD82 hetero-oligomers.
Though we are interested primarily in elucidating the consequences for virus spread of the observed tetraspanin downregulation, we nevertheless sought to identify the cellular pathway(s) involved in the degradation of CD81. We thus used inhibitors of either vacuolar-type H+-ATPase and thus protein degradation in lysosomes (bafilomycin A1 [54]) or multiple peptidases in the proteasome (the peptide aldehyde MG132 [55]). As documented in Fig. 3C and D, we found that, comparable to the situation with BST-2/tetherin (42, 52), both pathways contribute to CD81 degradation in HIV-1-infected cells.
DISCUSSION
The data presented here reveal a previously unknown function of Vpu: we document that this accessory protein is the main viral determinant responsible for the downregulation of tetraspanins in HIV-1-infected cells. In line with an independent study by Haller and colleagues (45), we also show that Nef can contribute to lowering the levels of CD81, the tetraspanin focused on in this study. Notably, we show that this novel Vpu function can limit CD81 incorporation into virions, thus likely helping to preserve virion infectivity.
Early on during our studies aimed at investigating potential functions of tetraspanins at the HIV-1 assembly/release sites, we were intrigued by the observation that these integral membrane proteins, while accumulating at the exit sites, overall appear to be downregulated in infected cells (see Fig. 6 in reference 27). Koyanagi and colleagues reported a likely related phenomenon: they showed that tetraspanin levels in MOLT-4/IIIB cells (chronically infected T lymphocytes) were lowered upon activation with PHA/phorbol myristate acetate (PMA) (20). Given that both Nef and Vpu are known modulators of host cell factors, we were not surprised to find that one of these viral factors, Vpu, was required for CD81 and CD82 downregulation in T lymphocytes (regarding both total and surface levels) (Fig. 1A to D). In the case of Nef, we found a contribution to downregulation only of tetraspanin surface levels (Fig. 1C and D), which was also in line with our observation (shown in Fig. 3A) that CEMss cells infected with ΔNef virus showed considerably increased surface CD81 levels, but unchanged internal localization, compared to WT virus. This intriguing finding falls in line with a major role of Nef in the retrieval of proteins directly from the surfaces of infected cells, as opposed to Vpu, which intercepts surface proteins during their trafficking to the plasma membrane (56). Notably, work by the Fackler group (presented in the report by Haller et al. [45]), who performed a comprehensive screen of host cell receptors downregulated by Nef and Vpu, yielded the same information regarding these two accessory proteins.
Corroborating previous findings from our laboratory and the Koyanagi laboratory (20, 27), we subsequently found that the presence of Vpu (and consequently CD81 downregulation) appears to help maintain viral infectivity in CEMss cells (Fig. 2). We should note, however, that we did not observe the same phenotype in Jurkat T cells (Lambelé and Thali, unpublished). The latter result is also in line with our previous finding that short hairpin RNA (shRNA)-mediated CD81 ablation in HeLa cells did not increase the infectivity of particles released from such cells (27). Furthermore, Fackler and colleagues (45) did not observe decreased infectivity of ΔVpu particles produced in (HeLa-derived) TZM-bl cells. In contrast to these results, and more in line with the CEMss data presented here (Fig. 2), two previous reports found that ablation of CD81 (by small interfering RNA [siRNA] in MOLT-4 cells and in 293T cells [26, 50]) can increase viral infectivity. It thus appears likely that this novel Vpu function, just like previously reported ones, including, e.g., cell-to-cell transmission (57, 58), depends on the cell type and physiological circumstances.
As a first step toward elucidating when and how Vpu functions to lower the levels of CD81, we then set out to perform quantitative imaging and coimmunoprecipitation experiments. While technical reasons have so far prevented us from unequivocally showing an association between Vpu and CD81 by coimmunoprecipitation, the results of our imaging studies provide strong evidence for a very close interaction. As shown in Fig. 3A, there was a striking redistribution of CD81 from the plasma membrane to a perinuclear area if T cells were infected with WT virus, but not if they were infected with ΔVpu virus. Further, not only did Vpu expression lead to CD81 redistribution, this tetraspanin then also extensively colocalized with Vpu, strongly suggesting that the two proteins interact. The results of the FRET analysis shown in Fig. 3B confirm this finding and suggest that Vpu interacts directly with CD81, and quite possibly (given the data shown in Fig. 1E and F) independently of CD4 or BST-2/tetherin. CD81 (and likely the other tetraspanins that are downregulated in HIV-1-infected cells) may, however, share the fate of CD4 and BST-2/tetherin: as shown in Fig. 3C and D, both the lysosomal and the proteasomal pathways appear to contribute to the demise of these surface antigens. Additionally, we note that the effect of Vpu on CD81 localization is reminiscent of patterns observed in the cases of CD155 (59), BST-2/tetherin (60, 61), and CCR7 (62).
In conclusion, this report provides further support for the idea that Vpu, rather than merely helping the virus to overcome a restriction factor barrier, facilitates virus transmission and immune evasion by interacting specifically with various host cell factors (63, 64). Also, the fact that tetraspanins are downregulated in all cell types upon HIV-1 infection/production, irrespective of whether this preserves particle infectivity, strongly suggests that this downmodulation serves additional purposes. For example, CD81 is known to regulate signaling at the IS (65), and CD82 (and to a lesser extent CD81) is involved in modulating cell migration (66, 67), both of which are functions likely to be consequential for HIV-1 spread in vivo.
ACKNOWLEDGMENTS
We thank Oliver Fackler and his group for sharing their findings with us before submission of both manuscripts (reference 45 and the present report). We also thank Christopher Huston for allowing us to use his plate reader and Roxana del Rio-Guerra and the UVM flow cytometry and cell-sorting facility. Finally, we thank members of the Thali laboratory, as well as Jason Botten and his team, for discussions.
Support for this work was provided by NIH grant R01 AI080302 to M.T. and training grant T32 AI055402-06A1 to N.H.R. M. Schindler received funding from the Deutsche Forschungsgemeinschaft (DFG) and the Else Kröner-Fresenius Stiftung (EKFS).
REFERENCES
- 1.Hemler ME. 2005. Tetraspanin functions and associated microdomains. Nat Rev Mol Cell Biol 6:801–811. doi: 10.1038/nrm1736. [DOI] [PubMed] [Google Scholar]
- 2.Yanez-Mo M, Barreiro O, Gordon-Alonso M, Sala-Valdes M, Sanchez-Madrid F. 2009. Tetraspanin-enriched microdomains: a functional unit in cell plasma membranes. Trends Cell Biol 19:434–446. doi: 10.1016/j.tcb.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 3.Charrin S, le Naour F, Silvie O, Milhiet PE, Boucheix C, Rubinstein E. 2009. Lateral organization of membrane proteins: tetraspanins spin their web. Biochem J 420:133–154. doi: 10.1042/BJ20082422. [DOI] [PubMed] [Google Scholar]
- 4.Wright M, Levy S. 2013. Tetraspanins and immunity, p 233–256 InBerditchevski F, Rubinstein E (ed), Tetraspanins. Springer Netherland, Dordrecht, Netherlands. [Google Scholar]
- 5.Hemler ME. 2014. Tetraspanin proteins promote multiple cancer stages. Nat Rev Cancer 14:49–60. doi: 10.1038/nrc3640. [DOI] [PubMed] [Google Scholar]
- 6.Hemler ME. 2013. Genetic evidence for tetraspanin functions, p 169–186 InBerditchevski F, Rubinstein E (ed), Tetraspanins. Springer Netherland, Dordrecht, Netherlands. [Google Scholar]
- 7.Martin F, Roth DM, Jans DA, Pouton CW, Partridge LJ, Monk PN, Moseley GW. 2005. Tetraspanins in viral infections: a fundamental role in viral biology? J Virol 79:10839–10851. doi: 10.1128/JVI.79.17.10839-10851.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Spriel AB, Figdor CG. 2010. The role of tetraspanins in the pathogenesis of infectious diseases. Microbes Infect 12:106–112. doi: 10.1016/j.micinf.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 9.Monk PN, Partridge LJ. 2012. Tetraspanins: gateways for infection. Infect Disord Drug Targets 12:4–17. doi: 10.2174/187152612798994957. [DOI] [PubMed] [Google Scholar]
- 10.Feneant L, Levy S, Cocquerel L. 2014. CD81 and hepatitis C virus (HCV) infection. Viruses 6:535–572. doi: 10.3390/v6020535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scheffer KD, Berditchevski F, Florin L. 2014. The tetraspanin CD151 in papillomavirus infection. Viruses 6:893–908. doi: 10.3390/v6020893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meerloo T, Parmentier HK, Osterhaus AD, Goudsmit J, Schuurman HJ. 1992. Modulation of cell surface molecules during HIV-1 infection of H9 cells. An immunoelectron microscopic study. AIDS 6:1105–1116. [DOI] [PubMed] [Google Scholar]
- 13.Meerloo T, Sheikh MA, Bloem AC, de Ronde A, Schutten M, van Els CA, Roholl PJ, Joling P, Goudsmit J, Schuurman HJ. 1993. Host cell membrane proteins on human immunodeficiency virus type 1 after in vitro infection of H9 cells and blood mononuclear cells. An immuno-electron microscopic study. J Gen Virol 74:129–135. [DOI] [PubMed] [Google Scholar]
- 14.Orentas RJ, Hildreth JE. 1993. Association of host cell surface adhesion receptors and other membrane proteins with HIV and SIV. AIDS Res Hum Retroviruses 9:1157–1165. doi: 10.1089/aid.1993.9.1157. [DOI] [PubMed] [Google Scholar]
- 15.Thali M. 2011. Tetraspanin functions during HIV-1 and influenza virus replication. Biochem Soc Trans 39:529–531. doi: 10.1042/BST0390529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nydegger S, Khurana S, Krementsov DN, Foti M, Thali M. 2006. Mapping of tetraspanin-enriched microdomains that can function as gateways for HIV-1. J Cell Biol 173:795–807. doi: 10.1083/jcb.200508165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chertova E, Chertov O, Coren LV, Roser JD, Trubey CM, Bess JW Jr, Sowder RC II, Barsov E, Hood BL, Fisher RJ, Nagashima K, Conrads TP, Veenstra TD, Lifson JD, Ott DE. 2006. Proteomic and biochemical analysis of purified human immunodeficiency virus type 1 produced from infected monocyte-derived macrophages. J Virol 80:9039–9052. doi: 10.1128/JVI.01013-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jolly C, Sattentau QJ. 2007. Human immunodeficiency virus type 1 assembly, budding, and cell-cell spread in T cells take place in tetraspanin-enriched plasma membrane domains. J Virol 81:7873–7884. doi: 10.1128/JVI.01845-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ott DE. 2008. Cellular proteins detected in HIV-1. Rev Med Virol 18:159–175. doi: 10.1002/rmv.570. [DOI] [PubMed] [Google Scholar]
- 20.Sato K, Aoki J, Misawa N, Daikoku E, Sano K, Tanaka Y, Koyanagi Y. 2008. Modulation of human immunodeficiency virus type 1 infectivity through incorporation of tetraspanin proteins. J Virol 82:1021–1033. doi: 10.1128/JVI.01044-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ono A. 2010. Relationships between plasma membrane microdomains and HIV-1 assembly. Biol Cell 102:335–350. doi: 10.1042/BC20090165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krementsov DN, Rassam P, Margeat E, Roy NH, Schneider-Schaulies J, Milhiet PE, Thali M. 2010. HIV-1 assembly differentially alters dynamics and partitioning of tetraspanins and raft components. Traffic 11:1401–1414. doi: 10.1111/j.1600-0854.2010.01111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hogue IB, Grover JR, Soheilian F, Nagashima K, Ono A. 2011. Gag induces the coalescence of clustered lipid rafts and tetraspanin-enriched microdomains at HIV-1 assembly sites on the plasma membrane. J Virol 85:9749–9766. doi: 10.1128/JVI.00743-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ruiz-Mateos E, Pelchen-Matthews A, Deneka M, Marsh M. 2008. CD63 is not required for production of infectious human immunodeficiency virus type 1 in human macrophages. J Virol 82:4751–4761. doi: 10.1128/JVI.02320-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen H, Dziuba N, Friedrich B, von Lindern J, Murray JL, Rojo DR, Hodge TW, O'Brien WA, Ferguson MR. 2008. A critical role for CD63 in HIV replication and infection of macrophages and cell lines. Virology 379:191–196. doi: 10.1016/j.virol.2008.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grigorov B, Attuil-Audenis V, Perugi F, Nedelec M, Watson S, Pique C, Darlix JL, Conjeaud H, Muriaux D. 2009. A role for CD81 on the late steps of HIV-1 replication in a chronically infected T cell line. Retrovirology 6:28. doi: 10.1186/1742-4690-6-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krementsov DN, Weng J, Lambele M, Roy NH, Thali M. 2009. Tetraspanins regulate cell-to-cell transmission of HIV-1. Retrovirology 6:64. doi: 10.1186/1742-4690-6-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li G, Endsley MA, Somasunderam A, Gbota SL, Mbaka MI, Murray JL, Ferguson MR. 2014. The dual role of tetraspanin CD63 in HIV-1 replication. Virol J 11:23. doi: 10.1186/1743-422X-11-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weng J, Krementsov DN, Khurana S, Roy NH, Thali M. 2009. Formation of syncytia is repressed by tetraspanins in human immunodeficiency virus type 1-producing cells. J Virol 83:7467–7474. doi: 10.1128/JVI.00163-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Symeonides M, Lambele M, Roy NH, Thali M. 2014. Evidence showing that tetraspanins inhibit HIV-1-induced cell-cell fusion at a post-hemifusion stage. Viruses 6:1078–1090. doi: 10.3390/v6031078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feldmann J, Schwartz O. 2010. HIV-1 Virological synapse: live imaging of transmission. Viruses 2:1666–1680. doi: 10.3390/v2081666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen BK. 2012. T cell virological synapses and HIV-1 pathogenesis. Immunol Res 54:133–139. doi: 10.1007/s12026-012-8320-8. [DOI] [PubMed] [Google Scholar]
- 33.Roy NH, Lambele M, Chan J, Symeonides M, Thali M. 2014. Ezrin is a component of the HIV-1 virological presynapse and contributes to the inhibition of cell-cell fusion. J Virol 88:7645–7658. doi: 10.1128/JVI.00550-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vasiliver-Shamis G, Dustin ML, Hioe CE. 2010. HIV-1 virological synapse is not simply a copycat of the immunological synapse. Viruses 2:1239–1260. doi: 10.3390/v2051239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Janvier K, Bonifacino JS. 2005. Role of the endocytic machinery in the sorting of lysosome-associated membrane proteins. Mol Biol Cell 16:4231–4242. doi: 10.1091/mbc.E05-03-0213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schindler M, Munch J, Kirchhoff F. 2005. Human immunodeficiency virus type 1 inhibits DNA damage-triggered apoptosis by a Nef-independent mechanism. J Virol 79:5489–5498. doi: 10.1128/JVI.79.9.5489-5498.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wehrly K, Chesebro B. 1997. p24 antigen capture assay for quantification of human immunodeficiency virus using readily available inexpensive reagents. Methods 12:288–293. doi: 10.1006/meth.1997.0481. [DOI] [PubMed] [Google Scholar]
- 38.Banning C, Votteler J, Hoffmann D, Koppensteiner H, Warmer M, Reimer R, Kirchhoff F, Schubert U, Hauber J, Schindler M. 2010. A flow cytometry-based FRET assay to identify and analyse protein-protein interactions in living cells. PLoS One 5:e9344. doi: 10.1371/journal.pone.0009344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koppensteiner H, Banning C, Schneider C, Hohenberg H, Schindler M. 2012. Macrophage internal HIV-1 is protected from neutralizing antibodies. J Virol 86:2826–2836. doi: 10.1128/JVI.05915-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lindwasser OW, Chaudhuri R, Bonifacino JS. 2007. Mechanisms of CD4 downregulation by the Nef and Vpu proteins of primate immunodeficiency viruses. Curr Mol Med 7:171–184. doi: 10.2174/156652407780059177. [DOI] [PubMed] [Google Scholar]
- 41.Malim MH, Emerman M. 2008. HIV-1 accessory proteins—ensuring viral survival in a hostile environment. Cell Host Microbe 3:388–398. doi: 10.1016/j.chom.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 42.Tokarev A, Guatelli J. 2011. Misdirection of membrane trafficking by HIV-1 Vpu and Nef: keys to viral virulence and persistence. Cell Logist 1:90–102. doi: 10.4161/cl.1.3.16708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wildum S, Schindler M, Munch J, Kirchhoff F. 2006. Contribution of Vpu, Env, and Nef to CD4 down-modulation and resistance of human immunodeficiency virus type 1-infected T cells to superinfection. J Virol 80:8047–8059. doi: 10.1128/JVI.00252-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen BK, Gandhi RT, Baltimore D. 1996. CD4 down-modulation during infection of human T cells with human immunodeficiency virus type 1 involves independent activities of vpu, env, and nef. J Virol 70:6044–6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haller C, Muller B, Fritz JV, Lamas-Murua M, Stolp B, Pujol FM, Keppler OT, Fackler OT. 2014. HIV-1 Nef and Vpu are functionally redundant broad-spectrum modulators of cell surface receptors, including tetraspanins. J Virol 88:14241–14257. doi: 10.1128/JVI.02333-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schubert U, Strebel K. 1994. Differential activities of the human immunodeficiency virus type 1-encoded Vpu protein are regulated by phosphorylation and occur in different cellular compartments. J Virol 68:2260–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Van Damme N, Goff D, Katsura C, Jorgenson RL, Mitchell R, Johnson MC, Stephens EB, Guatelli J. 2008. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 3:245–252. doi: 10.1016/j.chom.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Levesque K, Zhao YS, Cohen EA. 2003. Vpu exerts a positive effect on HIV-1 infectivity by down-modulating CD4 receptor molecules at the surface of HIV-1-producing cells. J Biol Chem 278:28346–28353. doi: 10.1074/jbc.M300327200. [DOI] [PubMed] [Google Scholar]
- 49.Pizzato M, Helander A, Popova E, Calistri A, Zamborlini A, Palu G, Gottlinger HG. 2007. Dynamin 2 is required for the enhancement of HIV-1 infectivity by Nef. Proc Natl Acad Sci U S A 104:6812–6817. doi: 10.1073/pnas.0607622104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bregnard C, Zamborlini A, Leduc M, Chafey P, Camoin L, Saib A, Benichou S, Danos O, Basmaciogullari S. 2013. Comparative proteomic analysis of HIV-1 particles reveals a role for Ezrin and EHD4 in the Nef-dependent increase of virus infectivity. J Virol 87:3729–3740. doi: 10.1128/JVI.02477-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Basmaciogullari S, Pizzato M. 2014. The activity of Nef on HIV-1 infectivity. Front Microbiol 5:232. doi: 10.3389/fmicb.2014.00232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dube M, Bego MG, Paquay C, Cohen EA. 2010. Modulation of HIV-1-host interaction: role of the Vpu accessory protein. Retrovirology 7:114. doi: 10.1186/1742-4690-7-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Andrew A, Strebel K. 2010. HIV-1 Vpu targets cell surface markers CD4 and BST-2 through distinct mechanisms. Mol Aspects Med 31:407–417. doi: 10.1016/j.mam.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y. 1991. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem 266:17707–17712. [PubMed] [Google Scholar]
- 55.Tsubuki S, Saito Y, Tomioka M, Ito H, Kawashima S. 1996. Differential inhibition of calpain and proteasome activities by peptidyl aldehydes of di-leucine and tri-leucine. J Biochem 119:572–576. doi: 10.1093/oxfordjournals.jbchem.a021280. [DOI] [PubMed] [Google Scholar]
- 56.Landi A, Iannucci V, Nuffel AV, Meuwissen P, Verhasselt B. 2011. One protein to rule them all: modulation of cell surface receptors and molecules by HIV Nef. Curr HIV Res 9:496–504. doi: 10.2174/157016211798842116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Casartelli N, Sourisseau M, Feldmann J, Guivel-Benhassine F, Mallet A, Marcelin AG, Guatelli J, Schwartz O. 2010. Tetherin restricts productive HIV-1 cell-to-cell transmission. PLoS Pathog 6:e1000955. doi: 10.1371/journal.ppat.1000955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jolly C, Booth NJ, Neil SJ. 2010. Cell-cell spread of human immunodeficiency virus type 1 overcomes tetherin/BST-2-mediated restriction in T cells. J Virol 84:12185–12199. doi: 10.1128/JVI.01447-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bolduan S, Reif T, Schindler M, Schubert U. 2014. HIV-1 Vpu mediated downregulation of CD155 requires alanine residues 10, 14 and 18 of the transmembrane domain. Virology 464-465:375–384. doi: 10.1016/j.virol.2014.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dube M, Roy BB, Guiot-Guillain P, Mercier J, Binette J, Leung G, Cohen EA. 2009. Suppression of Tetherin-restricting activity upon human immunodeficiency virus type 1 particle release correlates with localization of Vpu in the trans-Golgi network. J Virol 83:4574–4590. doi: 10.1128/JVI.01800-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hauser H, Lopez LA, Yang SJ, Oldenburg JE, Exline CM, Guatelli JC, Cannon PM. 2010. HIV-1 Vpu and HIV-2 Env counteract BST-2/tetherin by sequestration in a perinuclear compartment. Retrovirology 7:51. doi: 10.1186/1742-4690-7-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ramirez PW, Famiglietti M, Sowrirajan B, DePaula-Silva AB, Rodesch C, Barker E, Bosque A, Planelles V. 2014. Downmodulation of CCR7 by HIV-1 Vpu results in impaired migration and chemotactic signaling within CD4(+) T cells. Cell Rep 7:2019–2030. doi: 10.1016/j.celrep.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Strebel K. 2013. HIV accessory proteins versus host restriction factors. Curr Opin Virol 3:692–699. doi: 10.1016/j.coviro.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kahrstrom CT. 2014. Viral pathogenesis: Vpu puts the brakes on ADCC. Nat Rev Microbiol 12:397. doi: 10.1038/nrmicro3282. [DOI] [PubMed] [Google Scholar]
- 65.Rocha-Perugini V, Zamai M, Gonzalez-Granado JM, Barreiro O, Tejera E, Yanez-Mo M, Caiolfa VR, Sanchez-Madrid F. 2013. CD81 controls sustained T cell activation signaling and defines the maturation stages of cognate immunological synapses. Mol Cell Biol 33:3644–3658. doi: 10.1128/MCB.00302-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hong IK, Byun HJ, Lee J, Jin YJ, Wang SJ, Jeoung DI, Kim YM, Lee H. 2014. The Tetraspanin CD81 protein increases melanoma cell motility by up-regulating metalloproteinase MT1-MMP expression through the pro-oncogenic Akt-dependent Sp1 activation signaling pathways. J Biol Chem 289:15691–15704. doi: 10.1074/jbc.M113.534206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tsai YC, Weissman AM. 2011. Dissecting the diverse functions of the metastasis suppressor CD82/KAI1. FEBS Lett 585:3166–3173. doi: 10.1016/j.febslet.2011.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]