

ABSTRACT
Our previous studies have established that the p53 populations that accumulate in normal human cells exposed to etoposide or infected by an E1B 55-kDa protein-null mutant of human adenovirus type 5 carry a large number of posttranslational modifications at numerous residues (C. J. DeHart, J. S. Chahal, S. J. Flint, and D. H. Perlman, Mol Cell Proteomics 13:1–17, 2014, http://dx.doi.org/10.1074/mcp.M113.030254). In the absence of this E1B protein, the p53 transcriptional program is not induced, and it has been reported that the viral E4 Orf3 protein inactivates p53 (C. Soria, F. E. Estermann, K. C. Espantman, and C. C. O'Shea, Nature 466:1076–1081, 2010, http://dx.doi.org/10.1038/nature09307). As the latter protein disrupts nuclear Pml bodies, sites at which p53 is modified, we used mass spectrometry to catalogue the posttranscriptional modifications of the p53 population that accumulates when neither the E1B 55-kDa nor the E4 Orf3 protein is made in infected cells. Eighty-five residues carrying 163 modifications were identified. The overall patterns of posttranslational modification of this population and p53 present in cells infected by an E1B 55-kDa-null mutant were similar. The efficiencies with which the two forms of p53 bound to a consensus DNA recognition sequence could not be distinguished and were lower than that of transcriptionally active p53. The absence of the E4 Orf3 protein increased expression of several p53-responsive genes when the E1B protein was also absent from infected cells. However, expression of these genes did not attain the levels observed when p53 was activated in response to etoposide treatment and remained lower than those measured in mock-infected cells.
IMPORTANCE The tumor suppressor p53, a master regulator of cellular responses to stress, is inactivated and destroyed in cells infected by species C human adenoviruses, such as type 5. It is targeted for proteasomal degradation by the action of a virus-specific E3 ubiquitin ligase that contains the viral E1B 55-kDa and E4 Orf6 proteins, while the E4 Orf3 protein has been reported to block its ability to stimulate expression of p53-dependent genes. The comparisons reported here of the posttranslational modifications and activities of p53 populations that accumulate in infected normal human cells in the absence of both mechanisms of inactivation or of only the E3 ligase revealed little impact of the E4 Orf3 protein. These observations indicate that E4 Orf3-dependent disruption of Pml bodies does not have a major effect on the pattern of p53 posttranslational modifications in adenovirus-infected cells. Furthermore, they suggest that one or more additional viral proteins contribute to blocking p53 activation and the consequences that are deleterious for viral reproduction, such as apoptosis or cell cycle arrest.
INTRODUCTION
The cellular p53 protein was discovered by virtue of its interaction with the major product of the simian virus 40 oncogene, large T antigen (1, 2). The p53 tumor suppressor is a master regulator of cellular responses to internal and external stresses, when it can induce inhibition of cell cycle progression, apoptosis, or other responses, such as changes in metabolism. Under normal conditions, the human p53 protein is maintained at low concentrations, for example, as a result of its targeting for proteasomal degradation by the E3 ubiquitin ligase Hdm2 (3–5). Once stabilized and activated in response to genotoxic and other forms of stress, p53 binds to specific promoter sequences to activate or repress the transcription of numerous target genes (6–10) and can also operate in the cytoplasm to induce apoptosis by transcription-independent mechanisms (reviewed in references 11 to 14).
One of the first interactions between human adenovirus type 5 (Ad5) and cellular proteins to be identified was the association of the viral E1B 55-kDa protein with p53 (15). In view of its crucial roles in regulating cell survival and other aspects of cellular physiology, considerable effort has since been devoted to elucidation of the impacts of adenoviral gene products on the activities and properties of p53. The viral immediate-early E1A proteins induce accumulation of p53 and p53-dependent apoptosis (16–19). Such stabilization of p53 depends on E1A sequences required for transformation of rodent cells in culture and induction of cell cycle progression (20, 21) and has been reported to be mediated by the Arf/p19 (22) and Mdm4 (23) proteins, which block targeting of p53 for proteasomal degradation by Hdm2. However, induction of cell cycle arrest or apoptosis by p53 is blocked in Ad5-infected cells by the actions of other viral gene products, notably those of the E1B 55-kDa protein.
Binding of this E1B protein to the N-terminal activation domain of p53 inhibits p53-dependent transcription in vitro and in transient assays (24–27). Such inhibition depends on a repression domain within the E1B 55-kDa protein (28) and correlates with the ability of the E1B protein to cooperate with E1A proteins in the transformation of rodent cells in culture (27, 29–33). In transformed cells, interaction of the E1B 55-kDa and p53 proteins also results in sequestration of p53 in juxtanuclear cytoplasmic structures (34–36).
The steady-state concentration of p53 increases during the early phase of Ad5 infection of permissive human cells (21, 37–39), consistent with the effects of E1A protein described above. However, the concentration of p53 then decreases rapidly, because the protein is targeted for proteasomal degradation by the E1B 55-kDa- and E4 Orf6 protein-containing E3 ubiquitin ligase that assembles in infected cells (21, 37, 40–46). Mutations that prevent synthesis of either the E1B or the E4 Orf6 protein, or that impair their interaction with one another, therefore, lead to accumulation of p53 as the infectious cycle progresses. Nevertheless, apoptosis is not induced (37, 41) and expression of several genes activated by p53 was not observed to increase in cells infected by E1B 55-kDa-null mutants (37, 47). Furthermore, no impact of the E1B 55-kDa protein on inhibition of the p53-dependent transcriptional program was detected in a genome-wide comparison of cellular gene expression in normal human cells infected by Ad5 and such a mutant (48). These observations indicate that, while the E1B 55-kDa protein and the E3 ubiquitin ligase in which it serves as the substrate recognition subunit (49–52) are necessary to prevent accumulation of p53, it is not responsible for inhibition of transcriptional activation (or repression) by p53. This function has been ascribed more recently to the E4 Orf3 protein: expression of p53-responsive genes was reported to be increased in cells infected by a mutant virus null for production of both the E1B 55-kDa and E4 Orf3 proteins compared to those infected by the wild-type virus or an E1B 55-kDa-null mutant (53). Furthermore, production of the E4 Orf3 protein in the absence of other viral proteins impaired activation of expression-responsive p53 genes (53).
The activity and function of p53 are governed by mechanisms that regulate the stability and localization of the protein, as well as such properties as tetramerization, DNA-binding activity, and the ability to alter the expression of particular sets of target genes. Prominent among the parameters that regulate p53 is posttranslational modification (PTM). The protein is subjected to multiple types of modification, including phosphorylation of Ser or Thr residues; acetylation, methylation, ubiquitinylation, sumoylation, and neddylation of Lys residues; methylation of Arg residues; ADP-ribosylation of Asp or Glu residues; and O-glycosylation of Ser residues (reviewed in references 54 to 58). Among these, the contributions of acetylation, phosphorylation, methylation, and ubiquitinylation to modulating the stability, localization, and transcriptional activity of p53 have been extensively documented (reviewed in references 54 and 59 to 61). One cellular site at which the p53 protein is modified in this way is in association with the nuclear foci termed promyelocytic leukemia (Pml) bodies. Mammalian cell nuclei contain a finite number of these structures (also called nuclear domains 10 [ND10]), which contain the Pml proteins, as well as numerous nonconstitutive components (reviewed in references 62 and 63). Both p53 and several kinases and acetyltransferases that catalyze the addition of activating modifications to p53 have been reported to be associated with Pml bodies (64–71).
The genomes of several families of DNA viruses are initially associated with Pml bodies, prior to the disruption of these foci by viral gene products (72–75). In adenovirus-infected cells, the E4 Orf3 protein is necessary and sufficient to induce dismantling of Pml bodies and relocalization of their components, including the Pml protein, to nuclear track-like structures formed by the viral protein (76–78). This function of the E4 Orf3 protein is important for protecting viral genome replication against inhibition induced by type I or type II interferon (79, 80). However, it could also contribute to the E4 Orf3-dependent inactivation of p53 (53) by inducing dissociation of p53 from the enzymes that add activating posttranslational modifications. We have reported recently that endogenous wild-type p53 synthesized in normal human cells treated with an inducer of DNA damage or infected with an E1B 55-kDa-null mutant of Ad5 is very extensively modified by phosphorylation; acetylation; mono-, di-, or trimethylation; and ubiquitinylation at many more sites than have been described previously (81). We have therefore applied the same approach, isolation of p53 for analysis by mass spectrometry, to catalogue the PTMs of p53 from infected cells that lack the E4 Orf3, as well as the E1B 55-kDa protein, for comparison to those of the two p53 populations characterized previously.
MATERIALS AND METHODS
Cells and viruses.
Human foreskin fibroblasts (HFFs) and 293 cells (82) were maintained in Dulbecco's modified Eagle's medium (DMEM) (GIBCO-BRL) supplemented with 7.5% bovine growth serum (Thermo Scientific HyClone) and 5% bovine growth serum plus 5% calf serum (GIBCO-BRL), respectively.
The E1B 55-kDa-null mutant AdEasyE1Δ2347 (ΔE1B) (83), the E4 Orf3-null mutant dl341 (84) (ΔOrf3), and the E1B 55-kDa-, E4 Orf3-null double mutant described below (ΔE1B/ΔOrf3) were propagated in 293 cells. The concentrations of infectious particles were determined by plaque assay on 293 cells (85) or by immunofluorescence for the E2 DNA-binding protein (DBP) after infection with serial dilutions of virus stocks for 18 h at 39°C or 24 h at 37°C.
Isolation of an E1B 55-kDa-, E4 Orf3-null mutant of Ad5.
To obtain an Ad5 mutant that could direct synthesis of neither the E1B 55-kDa nor the E4 Orf3 protein, we first constructed a hybrid genome comprising a left segment from the genome of the E1B 55-kDa-null mutant AdEasyE1Δ2347 (83) and the right end of the E4 Orf3-null mutant dl341 (84). The sequence-verified plasmid containing the genome of the AdEasy (86) derivative AdEasyE1Δ2347 was amplified in Escherichia coli and purified by the Qiagen Maxiprep protocol. Following digestion with PacI (New England BioLabs), the released viral genome was isolated using the Qiaex II kit (Qiagen) following electrophoresis in 0.75% low-melting-point agarose (International Biotechnologies Inc.). To isolate dl341 DNA, 293 cells at 90% confluence were infected with 5 PFU/cell of the mutant for 44 h, and virus particles were isolated by sequential sedimentation in CsCl step and continuous gradients as described previously (87). Viral DNA was purified from the particles following dialysis into 0.01 M Tris-HCl, pH 7.5, containing 1 mM EDTA as described previously (88).
The purified viral genomes were digested with EcoRI (New England BioLabs), which cuts at the single EcoRI site (bp 27331) remaining in each genome. The long (27.3-kbp) left-end fragment from AdEasyE1Δ2347 was gel purified as described above. The short (8.67-kbp) fragment of dl341 was purified by electrophoresis in 1% agarose (Denville Scientific) and extracted using a gel extraction kit (Qiagen). Mixtures of various molar ratios (1:1, 1:2, and 1:4) of the two fragments were incubated with T4 DNA ligase (New England BioLabs) for 16 h at 16°C. The ligation products were then transfected into 293 cell monolayers in 6-well plates using Attractene (Qiagen) according to the manufacturer's protocol. The transfected cells were harvested 48 h later and lysed by five cycles of freezing and thawing in 0.01 M Tris-HCl, pH 7.5, containing 0.15 M NaCl, 5 mM KCl, 10 mM MgCl2, and 0.01% (wt/vol) dextrose. Viruses were isolated from plaques formed upon infection of 293 cells with the lysates and subjected to amplification by propagation in these cells and further plaque purification. The presence of the E1B 55-kDa mutation was validated by loss of an MwoI site following release of DNA and PCR amplification of the relevant segment of the E1B region as described previously (83). To assay for the E4 mutation, the region of the genome between bp 34353 and 34382 was amplified by PCR of viral DNAs containing the E1B mutation and sequenced (Genewiz). Once the presence of both mutations was confirmed, the mutant was subjected to an additional cycle of plaque purification and verification and was then propagated in 293 cells infected at 1 PFU/cell and maintained at 39°C for 44 h.
Purification of ΔE1B/ΔOrf3 p53.
Human foreskin fibroblasts were infected with 200 PFU/cell AdEasyE1Δ2347-dl341 for 44 h. The cells were harvested, washed twice with ice-cold phosphate-buffered saline (PBS) (GIBCO-BRL), and incubated for 20 min in lysis buffer, 20 mM Tris-HCl, pH 7.5, containing 0.25 M NaCl, 1% (vol/vol) N-lauroyl sarcosine, and a mixture of inhibitors of proteases and enzymes that remove specific PTMs (4 μM leupeptin, 1.7 μM antipain, 1.5 μM pepstatin, 1 mM phenylmethylsulfonyl fluoride [PMSF], 1 μM sodium orthovanadate, 1 mM sodium fluoride, 1.5 mM sodium butyrate, 1 mM β-glycerophosphate, and 5 μM microcystin). Genomic DNA and debris were removed by centrifugation at 13.2 krpm for 10 min at 4°C. The p53 protein was then purified by precipitation using an immunoaffinity resin comprising the anti-p53 monoclonal antibodies 421, 1620, and 1801 (89–91) covalently cross-linked to protein G-agarose beads (Thermo Scientific), as described previously (81).
Immunoblotting.
The viral E1B 55-kDa and E4 Orf3 proteins and the E2 DBP were detected by immunoblotting, as described previously (92), using the mouse monoclonal antibody (MAb) 2A6 (93), the rat MAb 6A11 (94), and the mouse MAb B6 (95), respectively, and horseradish peroxidase (HRP)-conjugated anti-mouse or anti-rat IgG (Chemicon). Human p53 and β-actin were examined using the HRP-conjugated mouse MAbs DO1 (Santa Cruz Biotechnologies) and AC-15 (Abcam), respectively.
Analysis of p53 posttranslational modifications by mass spectrometry.
Samples of ΔE1B/ΔOrf3 p53 recovered from immunoaffinity purification were subjected to gel electrophoresis, and the species of ∼50-kDa molecular mass was recovered, dehydrated, destained, reduced, alkylated, and digested with trypsin, chymotrypsin, or Lys C-endoproteinase, as described previously (81). The resulting peptides were subjected to reversed-phase nano-liquid chromatography-mass spectrometry (nano-LC-MS) and tandem MS (MS-MS) performed using a nanoflow ultra-high-performance liquid chromatography (UPLC) system (Nano Ultra 2D Plus; Eksigent) coupled to an LTQ-Orbitrap Velos hybrid mass spectrometer (Thermo) fitted with a Flex ion source (Thermo-Proxeon). Preprocessing of the collected tandem-MS data into .mgf files, searching against the Swiss-Prot human database using the Mascot search engine (v2.2.7; Matrix Science), and iterative PTM assignment were also performed as described previously (81). Collation of the results using Scaffold (Proteome Software) according to the PeptideProphet and Protein Prophet parsimony algorithms (ISB), calculation of Ascore-derived PTM site localization probabilities, and manual inspection and validation of PTM assignments were performed with the criteria described previously (81). The peptide spectral matches with ≥90% peptide identifier (ID) confidence and >95% PTM Ascore localization probabilities were used in spectral-counting-based quantification by residue using Scaffold PTM (v. 2.0; Proteome Software).
Analysis of DNA binding by p53.
Lysates were prepared as described above from HFFs infected with 100 PFU/cell AdEasyE1Δ2347-dl341, or its AdEasyE1Δ2347 parent, for 44 h or from uninfected cells treated with 125 μM etoposide for the same period. The p53 proteins were examined by immunoblotting as described above, and the relative concentrations were determined using Image J (96), with β-actin as the internal control. The DNA-binding activity of p53 was examined as a function of the p53 concentration using the p53 Transcription Factor Assay Kit (Cayman Chemical Company). In this assay, p53 present in cell lysates is incubated with a double-stranded DNA oligonucleotide containing a consensus p53 DNA-binding site immobilized on the wells of a 96-well plate. The plates are then washed extensively, and bound p53 is detected immunologically by an enzyme-linked immunosorbent assay (ELISA)-based method, with final absorbance measurement at 450-nm wavelength.
Measurement of RNA concentrations.
Human foreskin fibroblasts were mock infected; infected with 200 PFU/cell wild-type AdEasyE1, AdEasyE1Δ2347, or AdEasyE1Δ2347-dl341 for 30 h; or maintained in DMEM containing 125 μM etoposide for the same period. Whole-cell RNA was purified as described previously (97), and cDNA was prepared using reverse transcription by Superscript II (Life Technologies) with 2 μg RNA and 0.5 μg/μl random hexamer primers (Applied Biosystems) per reaction. Quantitative real-time (RT) PCR was performed as described previously (97), using the following primers for amplicons within human genes: BAX fwd, 5′-TTTCTGACGGCAACTTCAACTG3′, and rev, 5′GGTGCACAGGGCCTTGAG3′; HDM2 fwd, 5′TCCTCTCAAGCTCCGTGTTTG3′, and rev, 5′TCATGATGTGGTCAGGGTAGATG3′; p21 fwd, 5′TGTCACTGTCTTGTACCCTTG3′, and rev, 5′GGCGTTTGGAGTGGTAGAA3′; and PUMA fwd, 5′CGACCTCAACGCACAGTAC3′, and rev, 5′CCTAATTGGGCAGCCATCTCG3′. These primers detected spliced mRNAs. A glyceraldehyde-3-phosphate dehydrogenase (GAPDH) amplicon, described previously (97), was used as the internal control, and all measurements were made in triplicate. Relative RNA concentrations were determined by the ΔΔCT method (98) after normalization to the values obtained for GADPH mRNA.
RESULTS
Isolation of an Ad5 mutant null for production of the E1B 55-kDa and E4 Orf3 proteins.
To investigate the impact of the adenoviral E4 Orf3 protein on p53, we wished to compare the properties of the protein when it accumulates to high concentrations in the absence of the E1B 55-kDa protein and when made in the absence of both this E1B and the E4 Orf3 protein. A mutant virus carrying alterations that prevent accumulation of both these viral proteins was therefore isolated in the same background as the E1B 55-kDa-null mutant AdEasyE1Δ2347 described previously (83), as summarized in Fig. 1A and described in Materials and Methods. During all the steps in isolation and propagation of the double mutant, 293 cells were maintained at 39°C to offset, at least partially, the temperature-dependent defects in reproduction, export of viral late mRNAs, and synthesis of late proteins characteristic of E1B 55-kDa-null mutants (99, 100). To confirm the effects of the mutation on viral gene expression, HFFs were infected with 100 PFU/cell of the double mutant AdEasyE1Δ2347-dl341 (here designated ΔE1B/ΔOrf3), AdEasyE1Δ12347 (designated ΔE1B), dl341, or, as a positive control, the wild-type parent of dl341, dl309 (101), for 24 h, and the accumulation of viral proteins was examined by immunoblotting as described in Materials and Methods. Similar quantities of the E2 72-kDa single-stranded DBP were observed in cells infected by the wild-type and mutant viruses (Fig. 1B). Neither the E1B 55-kDa nor the E4 Orf3 protein could be detected in cells infected by the double mutant, while the single mutants failed to induce synthesis of the predicted proteins (Fig. 1B).
FIG 1.
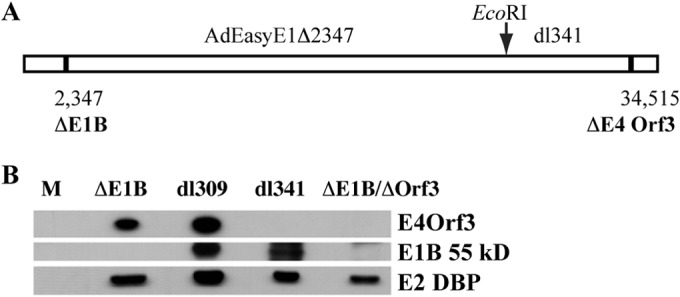
Construction and characterization of the E1B 55-kDa- and E4 Orf3-null double-mutant virus AdEasyE1Δ2347-dl341. (A) The origins of the segments of the double mutant virus genome are shown schematically, indicating the positions of the single-pair deletions that prevent production of the E1B 55-kDa or E4 Orf3 protein (ΔE1B and ΔE4 Orf3, respectively) and the EcoRI site exploited in the construction of the mutant. (B) HFFs were infected with 100 PFU/cell AdEasyE1Δ2347 (ΔE1B), dl309, dl341, or the double mutant (ΔE1B/ΔOrf3) for 24 h, and the viral proteins listed on the right were examined by immunoblotting.
As described in the introduction, when mutations prevent assembly of the virus-specific E3 ubiquitin ligase in Ad5-infected cells, high concentrations of p53 accumulate during the late phase of infection. To assess the effect, if any, of the E4 Orf3 protein on p53 stabilization and accumulation, HFFs were infected with the E1B 55-kDa-null mutant and the ΔE1B/ΔEOrf3 double mutant for increasing periods, and p53 was examined by immunoblotting, as described in Materials and Methods. By 24 h after infection, the initial period of the late phase in these cells (102), p53 was readily detected in cells infected by the mutant viruses, but not in cells infected by the phenotypically wild-type parent of ΔE1B, AdEasyE1 (Fig. 2A). This pattern was maintained at 44 h after infection. We wished to compare more directly the concentrations of p53 that accumulate in cells infected by the single and double mutants to one another and to the quantity of p53 that accumulates in response to DNA damage. HFFs were therefore infected with ΔE1B or the double mutant or exposed to the topoisomerase II inhibitor etoposide for 44 h. The p53 present in equal volumes of cell lysates was examined by immunoblotting (Fig. 2B), and the signals were quantified as described in Materials and Methods using β-actin as an internal control. Closely similar concentrations of p53 were present in cells infected by the E1B 55-kDa-null and the E1B- and E4Orf3-null double mutants (Fig. 2B), indicating that the E4 Orf3 protein has no significant impact on the stability and accumulation of p53 when the virus-specific E3 ubiquitin ligase is not present in infected cells. This conclusion is in agreement with previous observations in human small airway epithelial cells (53). The concentrations of both ΔE1B/ΔOrf3 and ΔE1B p53 at 44 h postinfection (p.i.) were more than 10-fold greater than those detected in HFFs treated in parallel with etoposide (Fig. 2B). The high concentrations to which p53 accumulated in cells infected by the ΔE1B/ΔOrf3 mutant facilitated characterization of the PTMs of this p53 population.
FIG 2.
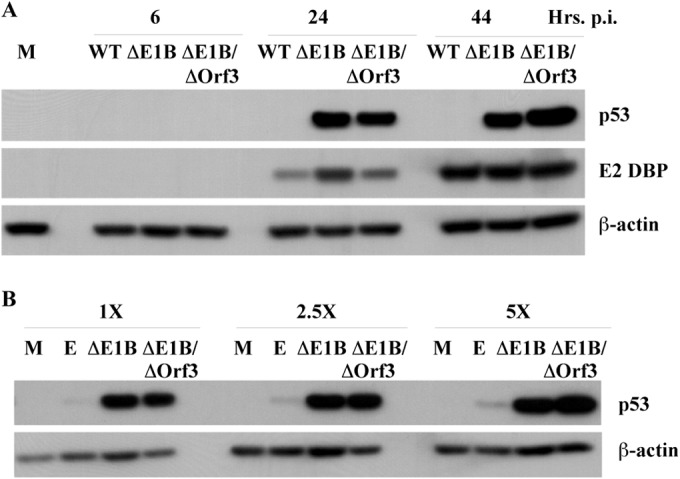
Accumulation of p53 in AdEasyE1Δ2347-dl341-infected cells. (A) HFFs were infected with 100 PFU/cell wild-type AdEasyE1 (WT), AdEasyE1Δ2347 (ΔE1B), or AdEasyE1Δ2347-dl341 (ΔE1B/ΔOrf3) for the periods indicated or mock infected (M). Whole-cell lysates were prepared, and the proteins listed on the right were examined by immunoblotting. (B) HFFs were infected for 44 h with the viruses indicated, mock infected, or incubated with medium containing 125 μM etoposide (E) for 44 h. Cell lysates were prepared, and p53 present in the increasing lysate volumes indicated at the top was visualized by immunoblotting.
Analysis of the posttranslational modifications of ΔE1B/ΔOrf3 p53 by mass spectrometry.
To characterize the PTMs of ΔE1B/ΔOrf3, HFFs were infected with 200 PFU/cell ΔE1B/ΔOrf3 for 44 h and lysed in the presence of N-lauroyl sarcosine, and p53 was purified by immunoaffinity as described in Materials and Methods. Following in-gel digestion of the eluted p53, as described in Materials and Methods, PTM-bearing peptides were identified by tandem mass spectrometry and a series of iterative searches using the Mascot search algorithm, with PTM site validation based on a series of increasingly stringent criteria, as described previously (81). This analysis was restricted to the class of PTMs well established to modulate the activity and properties of p53, namely, acetylation, methylation, phosphorylation, and ubiquitinylation.
Examples of high-confidence assignments of MS-MS spectra to PTM-bearing peptides of ΔE1B/ΔOrf3 p53 are shown in Fig. 3, and all such peptides identified are listed in Table S1 in the supplemental material. As catalogued in Table S1, the MS analyses identified 85 modified residues in ΔE1B/ΔOrf3 p53, 41 of which had not been described prior to our previous analysis (81). These modified residues were distributed across all functional domains of the p53 protein and carried a total of 163 PTMs. The location and identity of each of the modifications corresponded closely to those observed in populations of p53 isolated from etoposide-treated cells (E p53) or from cells infected by AdEasyE1Δ2347 (ΔE1B p53) (81). To facilitate comparison among the PTMs identified in these p53 populations, spectral counting was employed to provide estimates of the relative abundances of each type of PTM at each modified residue of ΔE1B/ΔOrf3 p53 or, for comparison, to those of ΔE1B p53, which we have described previously (81).
FIG 3.

MS-MS spectra of p53 isolated from AdEasyE1Δ2347-dl341-infected cells. The p53 protein was isolated from double-mutant-infected cells and subjected to protease digestion as described in Materials and Methods. The peptides were analyzed using reversed-phase nano-UPLC-MS and MS-MS on an LTQ Orbitrap Velos MS platform. Shown are representative examples of tandem mass spectra displaying the assignment of fragment ions, which are labeled with their empirically determined m/z values and b- and y-ion designations. The matched peptide sequences are shown above the spectra, with the PTMs detected color coded as follows; phosphorylation, red; acetylation, green; monomethylation, navy; trimethylation, turquoise; and carbamidomethylation, orange.
These analyses, which are summarized in Fig. 4, reinforced the conclusion that the overall patterns of PTMs of these two p53 populations are similar. However, clear differences in the relative degrees to which specific residues carried particular PTMs were observed in two regions of the protein. Serine and Thr residues in the N-terminal activation domain and the N-terminal segment of the DNA-binding domain displayed a lower relative degree of phosphorylation in ΔE1B/ΔOrf3 p53 than in ΔE1B p53 (Fig. 4A). Similarly, the relative degree of phosphorylation within the central portion of the DNA-binding domain, notably Ser183 and Ser185, was reduced compared to ΔE1B p53 and much lower than observed previously in active p53 isolated from etoposide-treated cells (Fig. 4B) (81). The majority of these sites of p53 phosphorylation have not been described previously, so the functional consequences of these differences cannot be inferred. While some differences in the relative degrees of methylation of arginine residues between the populations were observed, they were not obviously clustered in a particular domain or segment of the protein (data not shown). In contrast, a second region of difference in the relative degree of PTM in the ΔE1B/ΔOrf3 and ΔE1B p53 populations was revealed when lysine modifications were compared: Lys305, Lys319, Lys320, and Lys321, within the tetramerization domain, exhibited lower degrees of modifications of all types examined in ΔE1B/ΔOrf3 than in ΔE1B p53 (Fig. 4C). We therefore conclude that, while the ΔE1B/ΔOrf3 and ΔE1B p53 populations exhibit some differences in the relative degrees of modification of residues with the activation, DNA-binding, and tetramerization domains, the sites modified and the particular modifications carried by specific amino acids are generally similar.
FIG 4.

Comparison of the posttranslational modification profiles of ΔE1B p53 and ΔE1B/ΔOrf3 p53. Peptides from populations of ΔE1B/ΔOrf3 p53 were subjected to nano-UPLC-MS and MS-MS analyses on the LTQ Orbitrap Velos MS platform. Modification profiles were generated from triplicate LC-MS runs for each sample by spectral counting, as described previously (81). (A and C) Modification profiles for phosphorylation of Ser and Thr residues (A) and acetylation (A), ubiquitinylation (G), and mono-, di-, and trimethylation (M, D, and T, respectively) of Lys residues (C), which are compared to those of ΔE1B p53 (81). (B) Phosphorylation profiles of the two p53 populations compared to that of p53 isolated from etoposide-treated (E p53) cells reported previously in Molecular and Cellular Proteomics (81).
Comparison of DNA binding by ΔE1B/ΔOrf3 and ΔE1B p53.
In one approach to attempt to identify functional consequences of differences in the PTM patterns of the various p53 populations, we compared sequence-specific binding to DNA in vitro by the ΔE1B/ΔOrf3 and ΔE1B p53 populations. Transcriptionally active p53 that accumulates in etoposide-treated cells was included in these experiments to provide a positive control. Whole-cell lysates were prepared from HFFs infected with the ΔE1B or ΔE1B/ΔOrf3 mutant or treated with etoposide, as described in Materials and Methods. The relative quantities of p53 recovered in the extracts were determined by immunoblotting and densitometry, using β-actin as an internal control (see Materials and Methods). The binding of the p53 populations to a synthetic, consensus p53 recognition site was then examined as a function of the p53 concentration. At the higher concentrations tested, a somewhat greater quantity of E p53 than of the other two populations bound to DNA (Fig. 5A). At lower p53 concentrations, the efficiency of DNA binding by p53 from etoposide-treated cells was considerably greater than that exhibited by either ΔE1B or ΔE1B/ΔOrf3 p53 (Fig. 5B). Indeed, the DNA-binding activities of the last two p53 populations could not be distinguished.
FIG 5.
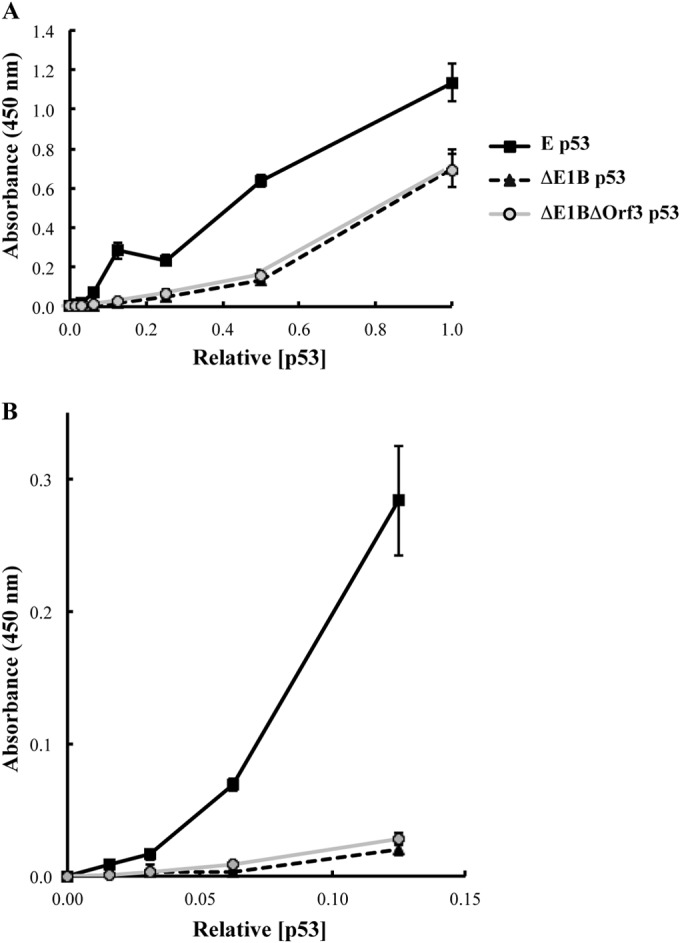
DNA-binding activities of p53 populations. The relative concentrations of p53 present in whole-cell extracts of HFFs infected with 100 PFU/cell AdEasyE1Δ2347 (ΔE1B) or AdEasyE1Δ2347-dl341 (ΔE1B/ΔOrf3) or treated with etoposide (E) were determined by immunoblotting and quantification of signals with β-actin as the internal control. The binding of equal concentrations of the p53 populations to a synthetic, consensus p53 binding site as a function of the p53 concentration was examined as described in Materials and Methods. All measurements were made in triplicate. The error bars indicate standard deviations.
Activation of expression of p53-responsive genes by three different p53 populations.
It has been previously reported that, when the virus-specific E3 ubiquitin ligase cannot form in the infected cells, mutations that prevent production of the E4 Orf3 protein result in activation of expression of p53-responsive genes (53). The closely similar DNA-binding properties of the p53 populations isolated from such infected cells and those in which the E4 Orf3 protein was present were therefore somewhat unexpected. To assess whether this property exhibited any correlation with expression of p53-responsive genes, HFFs were mock infected; infected with 200 PFU/cell AdEasyE1 (wild type) or the ΔE1B or ΔE1B/ΔOrf3 mutant, or, as a positive control, treated with etoposide for 30 h. Whole-cell RNA was then isolated, and the concentrations of specific mRNAs, relative to those of GAPDH mRNA, were compared by quantitative PCR after synthesis of cDNA from the same quantity of RNA, as described in Materials and Methods. These values, expressed relative to those measured for mock-infected cells, are shown in Fig. 6A.
FIG 6.
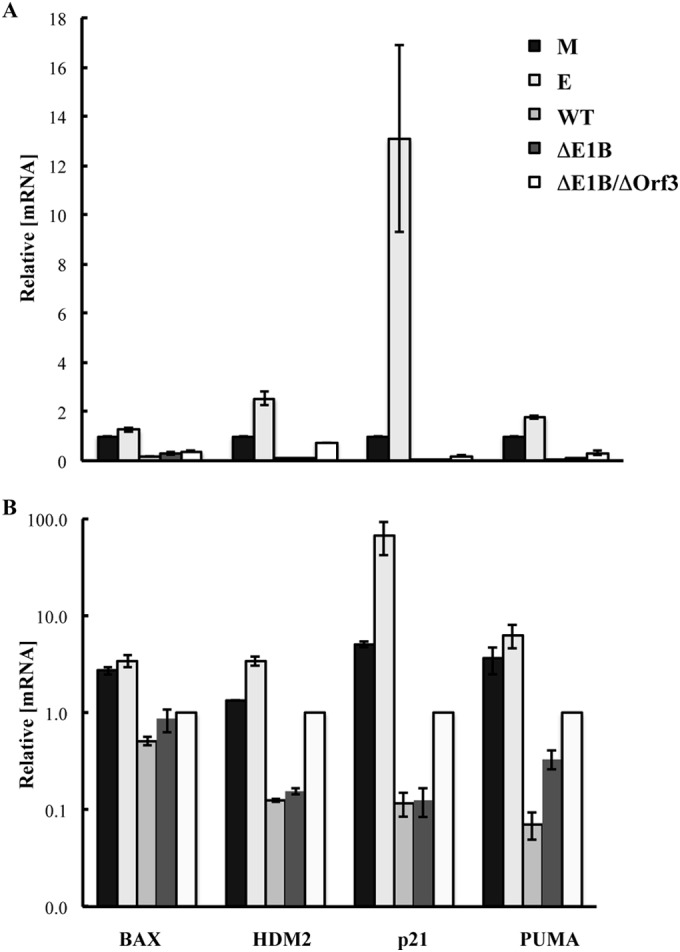
Effect of the E4 Orf3 protein on expression of p53-responsive genes. Total RNA was isolated 30 h after infection of HFFs with 200 PFU/cell AdEasy (WT), AdEasyE1Δ2347 (ΔE1B), or AdEasyE1Δ2347-dl341 (ΔE1B/ΔOrf3) or exposure of uninfected cells to medium containing 125 μM etoposide (E) or from mock-infected cells (M). The concentrations of the p53-responsive genes relative to those of GADPH mRNA were determined by quantitative RT-PCR as described in Materials and Methods. All measurements were made in triplicate, and the means and standard deviations of two independent experiments are shown relative to the values measured in mock-infected cells (A) or in cells infected by the E1B 55-kDa-, E4 Orf3-null double mutant (B).
As anticipated, exposure of uninfected HFFs to etoposide resulted in increased concentrations of the mRNAs for three of the four proteins encoded by p53-responsive genes examined in these experiments, BAX, HDM2, p21, and PUMA, although a large increase was observed only for p21 mRNA (Fig. 6A). In agreement with previous reports (see the introduction), expression of all the p53-responsive genes was reduced substantially in cells infected by wild-type Ad5 (AdEasyE1) or its E1B 55-kDa-null derivative (Fig. 6A). When the E4 Orf3 protein was also absent from infected cells, some increases in the concentrations of these mRNAs, relative to those in cells infected by the E1B 55-kDa-null mutant, were observed. However, these differences were modest, and BAX, HDM2, p21, and PUMA mRNAs did not attain even the concentrations observed in mock-infected cells, let alone those measured when p53 was activated in response to etoposide treatment (Fig. 6B).
DISCUSSION
As part of an effort to catalogue the PTMs of endogenous, wild-type human p53 (81), we have extended proteomic analyses of modifications to the p53 population that accumulates in normal human fibroblasts infected by an adenovirus mutant that can direct synthesis of neither the E1B 55-kDa nor the E4 Orf3 protein. The results reported here establish that this p53 population is also extensively modified by acetylation, methylation, phosphorylation, and ubiquitinylation: 85 modified residues bearing 163 modifications were detected, many of which had not been described prior to our analysis of p53 that accumulates in HFFs treated with etoposide or infected by an E1B 55-kDa-null mutant of Ad5 (81). These comparisons were undertaken in an attempt to assess the impacts of specific patterns of PTM on the properties and activities of p53, a crucial regulator of cell survival, proliferation, and death. However, we observed only a relatively small number of absolute differences in the modification states of particular residues, that is, the absence or presence of particular PTMs, in the three p53 populations isolated from normal human cells that we have now examined. This finding reinforces the hypothesis that the structure, activities, and functions of p53 are governed by combinational patterns of posttranslational modification rather than by the presence or absence of individual PTMs on specific residues (56, 61, 103–105). It also emphasizes the likelihood that this and the p53 populations examined previously comprise a substantial repertoire of proteoforms. These observations highlight the need for application of complementary methods to define the particular combinations of PTMs present on p53 molecules and to estimate their relative abundances in p53 populations exhibiting different properties, notably, analysis by “top-down” mass spectrometry (106–108).
Even with the methods applied in these studies, some similarities and differences among the various p53 populations are evident. In particular, it is striking that the PTM profiles of ΔE1B/ΔOrf3 p53 were more similar to those of transcriptionally inert ΔE1B p53 than the profiles exhibited by p53 isolated from etoposide-treated cells (Fig. 4B; see also Fig. 8 in reference 81). The greatest differences in the degrees of relative modification among the three p53 populations point to possible roles for these regions in the modulation of p53 transcriptional activity. The N-terminal activation domain exhibited differences in both the character and extent of phosphorylation in the three p53 populations. This property is consistent with the model that phosphorylation of sites within this region by stress-specific effector kinases facilitates the activation of particular subsets of p53-dependent genes for a targeted stress response. However, the majority of these Ser and Thr residues were observed to be modified on both transcriptionally active and inert p53 (Fig. 4A and B), implying that patterns of phosphorylation, rather than the modification of individual residues, may be responsible for directing both promoter selectivity and transcriptional activity of p53, as well as its ability to bind to consensus DNA sequences and transcriptional coactivators, as proposed previously (109). The DNA-binding domains of transcriptionally active and inactive p53 populations also exhibited differences in Ser/Thr phosphorylation modifications (Fig. 4). For example, the relative degree of phosphorylation of Ser183 and Ser185 was very high in active E p53 and considerably lower in p53 from cells infected by both the E1B 55-kDa-null mutant and the E1B 55-kDa-null, E4 Orf3-null double mutant (Fig. 4B). Phosphorylation of residues within this domain has been demonstrated to alter its conformation, thereby impairing both DNA binding and the interaction of p53 with transcriptional coactivators (110, 111). The tetramerization and C-terminal basic domains exhibited many differences in the extent and character of modifications at specific Ser residues, Arg residues, and Lys residues for all classes of PTM among the p53 populations. These differences emphasize the presence of a daunting number of possible PTM combinations within these regions. This degree of difference, in addition to those observed within the domains described above, strongly reinforces the likelihood that multiple subpopulations of p53, with highly substoichiometric modifications, exist within ΔE1B/ΔOrf3 p53, ΔE1B p53, and E p53 populations. The complicated array of potential modifications for each residue of the p53 protein provides considerable support for the existence of hierarchical networks of combinatorial PTM.
It is well established that in adenovirus-infected cells, the viral E4 Orf3 protein induces dismantling of Pml bodies, which serve as intranuclear sites at which p53 is posttranslationally modified (see the introduction). Consequently, the E4 Orf3 protein might be predicted to alter both the localization of p53 and p53 posttranslational modifications made by Pml-associated enzymes. The p53 protein that accumulates to very high concentrations when the E1B 55-kDa protein is not produced in infected cells remains largely within the nucleus (41, 53). However, its association with structures resembling the peripheral zones of viral replication centers is lost, and p53 becomes dispersed throughout the nucleus (except for nucleoli) (41). This property indicates that, in the absence of the E1B protein, p53 is unlikely to associate specifically with Pml proteins, which become associated with viral replication centers following E4 Orf3-dependent disruption of Pml bodies (77, 112). Consistent with this view, the nuclear patterns of p53 were reported to be indistinguishable in normal human cells infected by E1B 55-kDa-null and E1B 55-kDa, E4Orf3 mutants, nor does p53 colocalize with the E4 protein (and, by inference, Pml) under the former condition (53).
Posttranscriptional modifications of p53 associated with Pml bodies include phosphorylation of Ser46 by homeodomain-interacting protein kinase 2 (65, 66) and acetylation of Lys120 and Lys382 by the monocycle leukemia zinc finger protein (71). Association with Pml has also been reported to enhance phosphorylation of Thr18 by casein kinase 1 (65, 66, 71, 113). Some differences in the relative degrees of phosphorylation of Ser and Thr residues in the N-terminal 100 or so amino acids were observed in the ΔE1B and ΔE1B/ΔOrf3 p53 populations (Fig. 4A). However, these populations did not yield significant quantities of peptides carrying Pml-associated modifications (Thr18 and Ser46), or only few differences were detected (K120 and K382) (Fig. 4). These modifications have been implicated in stabilization of p53 or its induction of particular responses, that is, activation of p53 (65, 66, 71, 113). Their absence might, therefore, be related to the relative inactivity of the ΔE1B and ΔE1B/ΔOrf3 populations. Be that as it may, the results presented here, in conjunction with the previous observations summarized above, indicate that E4 Orf3 protein-dependent disruption of the Pml nuclear bodies leads to relatively minor alterations in the posttranslational modification of p53.
As noted in the introduction, p53 from Ad5-infected cells in which neither the E1B 55-kDa nor the E4 Orf3 protein is synthesized was of interest as an alternative population of transcriptionally active p53, based on the report that this E4 Orf3 protein inactivates p53, even when p53 accumulates to high concentrations in the absence of the E1B 55-kDa protein (53). The closer similarity of the PTM profiles of the ΔE1B/ΔOrf3 and ΔE1B p53 populations to one another than to that of p53 from etoposide-treated cells, therefore, was somewhat unexpected. However, this pattern of similarity and difference was also evident when we examined the efficiency of DNA binding by the various forms of p53. Although the degree of DNA binding by the three p53 populations was not very different at higher concentrations of the protein, the efficiencies of DNA binding by ΔE1B/ΔOrf3 p53 and ΔE1B p53 were closely similar at lower p53 concentrations and, in both cases, lower than that of active p53 (Fig. 5). In the assay used in these experiments, the quantity of p53 bound to an immobilized consensus DNA-binding site is measured as a function of the protein concentration. As p53 binds to DNA as a tetramer (114, 115), the degree of binding is determined by both how efficiently the various p53 populations form tetramers and the affinity of tetramers for the DNA-binding site. In the absence of additional experiments, we cannot determine the relative contributions of these two parameters to the greater DNA binding by active p53 from etoposide-treated cells. It remains possible that the affinities of these forms of p53 for p53-responsive promoters, for example, that of the HDM2 or p21 gene (Fig. 6), differ. Nevertheless, the data indicate that the binding of ΔE1B/ΔOrf3 p53 to a consensus p53 recognition sequence closely resembles that of ΔE1B p53 and are consistent with the previous report that the DNA-binding domains of both of these p53 proteins adopt conformations competent for DNA binding (53).
Unexpectedly, we observed only modest effects of the E4 Orf3 protein on expression of p53-responsive genes in infected cells. As anticipated (see the introduction), infection with wild-type adenovirus was accompanied by large (up to 40-fold) reductions in the expression of the four p53-responsive genes examined compared to their expression in mock-infected cells. Also in agreement with previous reports (37, 47, 48), no increases in the concentrations of these mRNAs were detected when the E1B 55-kDa protein was not synthesized in the infected cells (Fig. 6A). When the E4 Orf3 protein was also absent, some increases in the concentrations of the mRNAs were observed in cells infected by AdEasyE1Δ2347-dl341 compared to the E1B 55-kDa-null mutant (Fig. 6): 6.5-, 8-, and 3-fold-higher concentrations of HDM2, p21, and PUMA mRNAs, respectively. A qualitatively and quantitatively similar pattern was observed in small airway epithelial cells infected by an independent pair of E1B 55-kDa-null and E1B 55-kDa-, E4 Orf3-null mutants and correlated with increased binding of p53 to p53-dependent promoters in the absence of the E4 Orf3 protein, leading to the proposal that the E4 Orf3 protein inactivates p53 (53). However, in the experiments reported here, the mutation that prevented synthesis of the viral E4 Orf3 protein in the E1B 55-kDa-null background did not lead to increases in the concentrations of p53-responsive mRNAs in infected cells above the values measured in mock-infected cells or, in most cases, return the p53 mRNA levels to these values (Fig. 6). In contrast, increases above mock-infected-cell values were reported by Soria et al. (53). This difference may result from infection of proliferating versus quiescent cells: it seems likely that the expression of p53-regulated genes is even lower in the latter state, such as the quiescent small airway epithelial cells employed previously (53), than in proliferating cells, like the HFFs used in these experiments. More importantly, the direct comparison of the efficiency of expression of p53-responsive genes in infected cells and in cells in which p53 was activated in response to treatment with etoposide (Fig. 6) demonstrated that the absence of the E4 Orf3 protein was not sufficient to restore p53 transcriptional activity: all of the p53-responsive mRNAs examined accumulated to higher concentrations in etoposide-treated cells than in those infected by the E1B 55-kDa-, E4 Orf3-null double mutant, and in the case of p21 (which increased the most in response to etoposide), a difference of some 60-fold was observed (Fig. 6B). We therefore conclude that, while the E4 Orf3 protein contributes to inactivation of p53 during the adenovirus infectious cycle, it is unlikely to be sufficient to block the transcriptional activity of this cellular regulator.
This conclusion implies that one or more additional viral proteins fuel this function. Obvious candidates are the E1A proteins, which block p53-depdendent stimulation of transcription from various promoters via N-terminal sequences required for interaction with the transcriptional coactivators and acetyltransferases p300/Cbp (116–119). Indeed, an E1A segment containing a p300/Cbp binding site is a very effective competitor for association of the p53 transcriptional activation domain with its binding site in the coactivator (120). It is therefore possible that binding of the E1A proteins to this domain of p53 overrides any effects of activating posttranslational modifications acquired as p53 accumulates in adenovirus-infected cells that lack the E1B 55-kDa protein. Indeed, mutations in the p300/Cbp-binding region of E1A have been reported to partially restore expression of the p53-dependent p21 gene in infected cells that do not synthesize the E1B 55-kDa protein (121).
Supplementary Material
ACKNOWLEDGMENTS
We thank Arnold Levine and Thomas Dobner for the gifts of anti-p53 and anti-E4 Orf3 antibodies, respectively; Henry Shwe and Saw Kyin for expert technical assistance; and Ellen Brindle-Clark for help with preparation of the manuscript.
This work was funded by a grant from the National Institute of Allergy and Infectious Diseases (R56A111091785) to S.J.F. C.J.D was supported in part by a fellowship from the New Jersey Commission on Cancer Research (NJCCR 10-249-CCR-E0).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.03072-14.
REFERENCES
- 1.Linzer DI, Levine AJ. 1979. Characterization of a 54K Dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 17:43–52. doi: 10.1016/0092-8674(79)90293-9. [DOI] [PubMed] [Google Scholar]
- 2.Lane DP, Crawford LV. 1979. T antigen is bound to a host protein in SV40-transformed cells. Nature 278:261–263. doi: 10.1038/278261a0. [DOI] [PubMed] [Google Scholar]
- 3.Deffie A, Wu H, Reinke V, Lozano G. 1993. The tumor suppressor p53 regulates its own transcription. Mol Cell Biol 13:3415–3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haupt S, Louria-Hayon I, Haupt Y. 2003. P53 licensed to kill? Operating the assassin. J Cell Biochem 88:76–82. doi: 10.1002/jcb.10311. [DOI] [PubMed] [Google Scholar]
- 5.Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM. 2000. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem 275:8945–8951. doi: 10.1074/jbc.275.12.8945. [DOI] [PubMed] [Google Scholar]
- 6.Kannan K, Amariglio N, Rechavi G, Jakob-Hirsch J, Kela I, Kaminski N, Getz G, Domany E, Givol D. 2001. DNA microarrays identification of primary and secondary target genes regulated by p53. Oncogene 20:2225–2234. doi: 10.1038/sj.onc.1204319. [DOI] [PubMed] [Google Scholar]
- 7.Menendez D, Inga A, Resnick MA. 2009. The expanding universe of p53 targets. Nat Rev Cancer 9:724–737. doi: 10.1038/nrc2730. [DOI] [PubMed] [Google Scholar]
- 8.Riley T, Sontag E, Chen P, Levine A. 2008. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 9:402–412. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- 9.Gottlieb E, Vousden KH. 2010. p53 regulation of metabolic pathways. Cold Spring Harb Perspect Biol 2:a001040. doi: 10.1101/cshperspect.a001040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. 1992. Definition of a consensus binding site for p53. Nat Genet 1:45–49. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- 11.Chipuk JE, Green DR. 2006. Dissecting p53-dependent apoptosis. Cell Death Differ 13:994–1002. doi: 10.1038/sj.cdd.4401908. [DOI] [PubMed] [Google Scholar]
- 12.Chipuk JE, Bouchier-Hayes L, Green DR. 2006. Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario. Cell Death Differ 13:1396–1402. doi: 10.1038/sj.cdd.4401963. [DOI] [PubMed] [Google Scholar]
- 13.Moll UM, Wolff S, Speidel D, Deppert W. 2005. Transcription-independent pro-apoptotic functions of p53. Curr Opin Cell Biol 17:631–636. doi: 10.1016/j.ceb.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 14.Green DR, Kroemer G. 2009. Cytoplasmic functions of the tumour suppressor p53. Nature 458:1127–1130. doi: 10.1038/nature07986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarnow P, Ho YS, Williams J, Levine AJ. 1982. Adenovirus E1b-58kd tumor antigen and SV40 large tumor antigen are physically associated with the same 54 kd cellular protein in transformed cells. Cell 28:387–394. doi: 10.1016/0092-8674(82)90356-7. [DOI] [PubMed] [Google Scholar]
- 16.Debbas M, White E. 1993. Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes Dev 7:546–554. doi: 10.1101/gad.7.4.546. [DOI] [PubMed] [Google Scholar]
- 17.Lowe SW, Ruley HE. 1993. Stabilization of the p53 tumor suppressor is induced by adenovirus 5 E1A and accompanies apoptosis. Genes Dev 7:535–545. doi: 10.1101/gad.7.4.535. [DOI] [PubMed] [Google Scholar]
- 18.Rao L, Debbas M, Sabbatini P, Hockenbery D, Korsmeyer S, White E. 1992. The adenovirus E1A proteins induce apoptosis, which is inhibited by the E1B 19-kDa and Bcl-2 proteins. Proc Natl Acad Sci U S A 89:7742–7746. doi: 10.1073/pnas.89.16.7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Braithwaite A, Nelson C, Skulimowski A, McGovern J, Pigott D, Jenkins J. 1990. Transactivation of the p53 oncogene by E1a gene products. Virology 177:595–605. doi: 10.1016/0042-6822(90)90525-V. [DOI] [PubMed] [Google Scholar]
- 20.Chiou SK, White E. 1997. p300 binding by E1A cosegregates with p53 induction but is dispensable for apoptosis. J Virol 71:3515–3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Querido E, Marcellus RC, Lai A, Charbonneau R, Teodoro JG, Ketner G, Branton PE. 1997. Regulation of p53 levels by the E1B 55-kilodalton protein and E4orf6 in adenovirus-infected cells. J Virol 71:3788–3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Stanchina E, McCurrach ME, Zindy F, Shieh SY, Ferbeyre G, Samuelson AV, Prives C, Roussel MF, Sherr CJ, Lowe SW. 1998. E1A signaling to p53 involves the p19(ARF) tumor suppressor. Genes Dev 12:2434–2442. doi: 10.1101/gad.12.15.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Z, Day CP, Yang JY, Tsai WB, Lozano G, Shih HM, Hung MC. 2004. Adenoviral E1A targets Mdm4 to stabilize tumor suppressor p53. Cancer Res 64:9080–9085. doi: 10.1158/0008-5472.CAN-04-2419. [DOI] [PubMed] [Google Scholar]
- 24.Kao CC, Yew PR, Berk AJ. 1990. Domains required for in vitro association between the cellular p53 and the adenovirus 2 E1B 55K proteins. Virology 179:806–814. doi: 10.1016/0042-6822(90)90148-K. [DOI] [PubMed] [Google Scholar]
- 25.Martin ME, Berk AJ. 1998. Adenovirus E1B 55K represses p53 activation in vitro. J Virol 72:3146–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yew PR, Berk AJ. 1992. Inhibition of p53 transactivation required for transformation by adenovirus early 1B protein. Nature 357:82–85. doi: 10.1038/357082a0. [DOI] [PubMed] [Google Scholar]
- 27.Teodoro JG, Branton PE. 1997. Regulation of p53-dependent apoptosis, transcriptional repression, and cell transformation by phosphorylation of the 55-kilodalton E1B protein of human adenovirus type 5. J Virol 71:3620–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yew PR, Liu X, Berk AJ. 1994. Adenovirus E1B oncoprotein tethers a transcriptional repression domain to p53. Genes Dev 8:190–202. doi: 10.1101/gad.8.2.190. [DOI] [PubMed] [Google Scholar]
- 29.Yew PR, Kao CC, Berk AJ. 1990. Dissection of functional domains in the adenovirus 2 early 1B 55k polypeptide by suppressor-linker-insertional mutagenesis. Virology 179:795–805. doi: 10.1016/0042-6822(90)90147-J. [DOI] [PubMed] [Google Scholar]
- 30.Martin KJ, Lillie JW, Green MR. 1990. Evidence for interaction of different eukaryotic transcriptional activators with distinct cellular targets. Nature 346:147–152. doi: 10.1038/346147a0. [DOI] [PubMed] [Google Scholar]
- 31.Teodoro JG, Halliday T, Whalen SG, Takayesu D, Graham FL, Branton PE. 1994. Phosphorylation at the carboxy terminus of the 55-kilodalton adenovirus type 5 E1B protein regulates transforming activity. J Virol 68:776–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Endter C, Hartl B, Spruss T, Hauber J, Dobner T. 2005. Blockage of CRM1-dependent nuclear export of the adenovirus type 5 early region 1B 55-kDa protein augments oncogenic transformation of primary rat cells. Oncogene 24:55–64. doi: 10.1038/sj.onc.1208170. [DOI] [PubMed] [Google Scholar]
- 33.Endter C, Kzhyshkowska J, Stauber R, Dobner T. 2001. SUMO-1 modification required for transformation by adenovirus type 5 early region 1B 55-kDa oncoprotein. Proc Natl Acad Sci U S A 98:11312–11317. doi: 10.1073/pnas.191361798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hutton FG, Turnell AS, Gallimore PH, Grand RJ. 2000. Consequences of disruption of the interaction between p53 and the larger adenovirus early region 1B protein in adenovirus E1 transformed human cells. Oncogene 19:452–462. doi: 10.1038/sj.onc.1203316. [DOI] [PubMed] [Google Scholar]
- 35.Blair Zajdel ME, Blair GE. 1988. The intracellular distribution of the transformation-associated protein p53 in adenovirus-transformed rodent cells. Oncogene 2:579–584. [PubMed] [Google Scholar]
- 36.Zantema A, Fransen JA, Davis-Olivier A, Ramaekers FC, Vooijs GP, DeLeys B, Van der Eb AJ. 1985. Localization of the E1B proteins of adenovirus 5 in transformed cells, as revealed by interaction with monoclonal antibodies. Virology 142:44–58. doi: 10.1016/0042-6822(85)90421-0. [DOI] [PubMed] [Google Scholar]
- 37.O'Shea C, Johnson L, Bagus B, Choi S, Nicholas C, Shen A, Boyle L, Pandey K, Soria C, Kunich J, Shen Y, Habets G, Ginzinger D, McCormick F. 2004. Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity. Cancer Cell 6:611–623. doi: 10.1016/j.ccr.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 38.Grand RJ, Grant ML, Gallimore PH. 1994. Enhanced expression of p53 in human cells infected with mutant adenoviruses. Virology 203:229–240. doi: 10.1006/viro.1994.1480. [DOI] [PubMed] [Google Scholar]
- 39.Chahal JS, Flint SJ. 2013. The p53 protein does not facilitate adenovirus type 5 replication in normal human cells. J Virol 87:6044–6046. doi: 10.1128/JVI.00129-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boyer JL, Ketner G. 2000. Genetic analysis of a potential zinc-binding domain of the adenovirus E4 34k protein. J Biol Chem 275:14969–14978. doi: 10.1074/jbc.M000566200. [DOI] [PubMed] [Google Scholar]
- 41.Cardoso FM, Kato SE, Huang W, Flint SJ, Gonzalez RA. 2008. An early function of the adenoviral E1B 55 kDa protein is required for the nuclear relocalization of the cellular p53 protein in adenovirus-infected normal human cells. Virology 378:339–346. doi: 10.1016/j.virol.2008.06.016. [DOI] [PubMed] [Google Scholar]
- 42.Harada JN, Shevchenko A, Pallas DC, Berk AJ. 2002. Analysis of the adenovirus E1B-55K-anchored proteome reveals its link to ubiquitination machinery. J Virol 76:9194–9206. doi: 10.1128/JVI.76.18.9194-9206.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nevels M, Rubenwolf S, Spruss T, Wolf H, Dobner T. 2000. Two distinct activities contribute to the oncogenic potential of the adenovirus type 5 E4orf6 protein. J Virol 74:5168–5181. doi: 10.1128/JVI.74.11.5168-5181.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Querido E, Morisson MR, Chu-Pham-Dang H, Thirlwell SW, Boivin D, Branton PE. 2001. Identification of three functions of the adenovirus e4orf6 protein that mediate p53 degradation by the E4orf6-E1B55K complex. J Virol 75:699–709. doi: 10.1128/JVI.75.2.699-709.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shen Y, Kitzes G, Nye JA, Fattaey A, Hermiston T. 2001. Analyses of single-amino-acid substitution mutants of adenovirus type 5 E1B-55K protein. J Virol 75:4297–4307. doi: 10.1128/JVI.75.9.4297-4307.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Steegenga WT, Riteco N, Jochemsen AG, Fallaux FJ, Bos JL. 1998. The large E1B protein together with the E4orf6 protein target p53 for active degradation in adenovirus infected cells. Oncogene 16:349–357. doi: 10.1038/sj.onc.1201540. [DOI] [PubMed] [Google Scholar]
- 47.Hobom U, Dobbelstein M. 2004. E1B-55-kilodalton protein is not required to block p53-induced transcription during adenovirus infection. J Virol 78:7685–7697. doi: 10.1128/JVI.78.14.7685-7697.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miller DL, Rickards B, Mashiba M, Huang W, Flint SJ. 2009. The adenoviral E1B 55-kilodalton protein controls expression of immune response genes but not p53-dependent transcription. J Virol 83:3591–3603. doi: 10.1128/JVI.02269-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheng CY, Blanchette P, Branton PE. 2007. The adenovirus E4orf6 E3 ubiquitin ligase complex assembles in a novel fashion. Virology 364:36–44. doi: 10.1016/j.virol.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 50.Luo K, Ehrlich E, Xiao Z, Zhang W, Ketner G, Yu XF. 2007. Adenovirus E4orf6 assembles with Cullin5-ElonginB-ElonginC E3 ubiquitin ligase through an HIV/SIV Vif-like BC-box to regulate p53. FASEB J 21:1742–1750. doi: 10.1096/fj.06-7241com. [DOI] [PubMed] [Google Scholar]
- 51.Blanchette P, Cheng CY, Yan Q, Ketner G, Ornelles DA, Dobner T, Conaway RC, Conaway JW, Branton PE. 2004. Both BC-box motifs of adenovirus protein E4orf6 are required to efficiently assemble an E3 ligase complex that degrades p53. Mol Cell Biol 24:9619–9629. doi: 10.1128/MCB.24.21.9619-9629.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schwartz RA, Lakdawala SS, Eshleman HD, Russell MR, Carson CT, Weitzman MD. 2008. Distinct requirements of adenovirus E1b55K protein for degradation of cellular substrates. J Virol 82:9043–9055. doi: 10.1128/JVI.00925-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Soria C, Estermann FE, Espantman KC, O'Shea CC. 2010. Heterochromatin silencing of p53 target genes by a small viral protein. Nature 466:1076–1081. doi: 10.1038/nature09307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kruse JP, Gu W. 2009. Modes of p53 regulation. Cell 137:609–622. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gu B, Zhu WG. 2012. Surf the post-translational modification network of p53 regulation. Int J Biol Sci 8:672–684. doi: 10.7150/ijbs.4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meek DW, Anderson CW. 2009. Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb Perspect Biol 1:a000950. doi: 10.1101/cshperspect.a000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Murray-Zmijewski F, Slee EA, Lu X. 2008. A complex barcode underlies the heterogeneous response of p53 to stress. Nat Rev Mol Cell Biol 9:702–712. doi: 10.1038/nrm2451. [DOI] [PubMed] [Google Scholar]
- 58.Brooks CL, Gu W. 2011. p53 regulation by ubiquitin. FEBS Lett 585:2803–2809. doi: 10.1016/j.febslet.2011.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brooks CL, Gu W. 2011. The impact of acetylation and deacetylation on the p53 pathway. Protein Cell 2:456–462. doi: 10.1007/s13238-011-1063-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dai C, Gu W. 2010. p53 post-translational modification: deregulated in tumorigenesis. Trends Mol Med 16:528–536. doi: 10.1016/j.molmed.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vousden KH, Prives C. 2009. Blinded by the light: the growing complexity of p53. Cell 137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 62.Bernardi R, Pandolfi PP. 2007. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol 8:1006–1016. doi: 10.1038/nrm2277. [DOI] [PubMed] [Google Scholar]
- 63.Lallemand-Breitenbach V, de The H. 2010. PML nuclear bodies. Cold Spring Harb Perspect Biol 2:a000661. doi: 10.1101/cshperspect.a000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.de Stanchina E, Querido E, Narita M, Davuluri RV, Pandolfi PP, Ferbeyre G, Lowe SW. 2004. PML is a direct p53 target that modulates p53 effector functions. Mol Cell 13:523–535. doi: 10.1016/S1097-2765(04)00062-0. [DOI] [PubMed] [Google Scholar]
- 65.D'Orazi G, Cecchinelli B, Bruno T, Manni I, Higashimoto Y, Saito S, Gostissa M, Coen S, Marchetti A, Del Sal G, Piaggio G, Fanciulli M, Appella E, Soddu S. 2002. Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat Cell Biol 4:11–19. doi: 10.1038/ncb714. [DOI] [PubMed] [Google Scholar]
- 66.Möller A, Sirma H, Hofmann TG, Rueffer S, Klimczak E, Dröge W, Will H, Schmitz ML. 2003. PML is required for homeodomain-interacting protein kinase 2 (HIPK2)-mediated p53 phosphorylation and cell cycle arrest but is dispensable for the formation of HIPK domains. Cancer Res 63:4310–4314. [PubMed] [Google Scholar]
- 67.Louria-Hayon I, Grossman T, Sionov RV, Alsheich O, Pandolfi PP, Haupt Y. 2003. The promyelocytic leukemia protein protects p53 from Mdm2-mediated inhibition and degradation. J Biol Chem 278:33134–33141. doi: 10.1074/jbc.M301264200. [DOI] [PubMed] [Google Scholar]
- 68.Fogal VGM, Sandy P, Zacchi P, Sternsdorf T, Jensen K, Pandolfi PP, Will H, Schneider C, Del Sal G. 2000. Regulation of p53 activity in nuclear bodies by a specific PML isoform. EMBO J 19:6185–6195. doi: 10.1093/emboj/19.22.6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Torok D, Ching RW, Bazett-Jones DP. 2009. PML nuclear bodies as sites of epigenetic regulation. Front Biosci 14:1325–1336. doi: 10.2741/3311. [DOI] [PubMed] [Google Scholar]
- 70.Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S, Higashimoto Y, Appella E, Minucci S, Pandolfi PP, Pelicci PG. 2000. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature 406:207–210. doi: 10.1038/35018127. [DOI] [PubMed] [Google Scholar]
- 71.Rokudai S, Laptenko O, Arnal SM, Taya Y, Kitabayashi I, Prives C. 2013. MOZ increases p53 acetylation and premature senescence through its complex formation with PML. Proc Natl Acad Sci U S A 110:3895–3900. doi: 10.1073/pnas.1300490110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Everett RD. 2006. Interactions between DNA viruses, ND10 and the DNA damage response. Cell Microbiol 8:365–374. doi: 10.1111/j.1462-5822.2005.00677.x. [DOI] [PubMed] [Google Scholar]
- 73.Everett RD, Chelbi-Alix MK. 2007. PML and PML nuclear bodies: implications in antiviral defence. Biochimie 89:819–830. doi: 10.1016/j.biochi.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 74.Maul GG. 1998. Nuclear domain 10, the site of DNA virus transcription and replication. Bioessays 20:660–667. doi:. [DOI] [PubMed] [Google Scholar]
- 75.Rivera-Molina YA, Martinez FP, Tang Q. 2013. Nuclear domain 10 of the viral aspect. World J Virol 2:110–122. doi: 10.5501/wjv.v2.i3.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Carvalho T, Seeler JS, Ohman K, Jordan P, Pettersson U, Akusjärvi G, Carmo-Fonseca M, Dejean A. 1995. Targeting of adenovirus E1A and E4-ORF3 proteins to nuclear matrix-associated PML bodies. J Cell Biol 131:45–56. doi: 10.1083/jcb.131.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Doucas V, Ishov AM, Romo A, Juguilon H, Weitzman MD, Evans RM, Maul GG. 1996. Adenovirus replication is coupled with the dynamic properties of the PML nuclear structure. Genes Dev 10:196–207. doi: 10.1101/gad.10.2.196. [DOI] [PubMed] [Google Scholar]
- 78.Leppard KN, Everett RD. 1999. The adenovirus type 5 E1b 55K and E4 Orf3 proteins associate in infected cells and affect ND10 components. J Gen Virol 80:997–1008. [DOI] [PubMed] [Google Scholar]
- 79.Ullman AJ, Hearing P. 2008. Cellular proteins PML and Daxx mediate an innate antiviral defense antagonized by the adenovirus E4 ORF3 protein. J Virol 82:7325–7335. doi: 10.1128/JVI.00723-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ullman AJ, Reich NC, Hearing P. 2007. Adenovirus E4 ORF3 protein inhibits the interferon-mediated antiviral response. J Virol 81:4744–4752. doi: 10.1128/JVI.02385-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.DeHart CJ, Chahal JS, Flint SJ, Perlman DH. 2014. Extensive post-translational modification of active and inactivated forms of endogenous p53. Mol Cell Proteomics 13:1–17. doi: 10.1074/mcp.M113.030254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Graham FL, Smiley J, Russell WC, Nairn R. 1977. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol 36:59–72. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- 83.Kato SE, Huang W, Flint SJ. 2011. Role of the RNA recognition motif of the E1B 55 kDa protein in the adenovirus type 5 infectious cycle. Virology 417:9–17. doi: 10.1016/j.virol.2011.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sarnow P, Hearing P, Anderson CW, Reich N, Levine AJ. 1982. Identification and characterization of an immunologically conserved adenovirus early region 11,000 Mr protein and its association with the nuclear matrix. J Mol Biol 162:565–583. doi: 10.1016/0022-2836(82)90389-8. [DOI] [PubMed] [Google Scholar]
- 85.Williams JF. 1970. Enhancement of adenovirus plaque formation on HeLa cells by magnesium chloride. J Gen Virol 9:251–253. doi: 10.1099/0022-1317-9-3-251. [DOI] [PubMed] [Google Scholar]
- 86.He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. 1998. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A 95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Finnen RL, Biddle JF, Flint J. 2001. Truncation of the human adenovirus type 5 L4 33-kDa protein: evidence for an essential role of the carboxy-terminus in the viral infectious cycle. Virology 289:388–399. doi: 10.1006/viro.2001.1130. [DOI] [PubMed] [Google Scholar]
- 88.Chahal JS, Flint SJ. 2012. Timely synthesis of the adenovirus type 5 E1B 55-kilodalton protein is required for efficient genome replication in normal human cells. J Virol 86:3064–3072. doi: 10.1128/JVI.06764-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Harlow E, Crawford LV, Pim DC, Williamson NM. 1981. Monoclonal antibodies specific for simian virus 40 tumor antigens. J Virol 39:861–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ball RK, Siegl B, Quellhorst S, Brandner G, Braun DG. 1984. Monoclonal antibodies against simian virus 40 nuclear large T tumour antigen: epitope mapping, papova virus cross-reaction and cell surface staining. EMBO J 3:1485–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Banks L, Matlashewski G, Crawford L. 1986. Isolation of human-p53-specific monoclonal antibodies and their use in the studies of human p53 expression. Eur J Biochem 159:529–534. doi: 10.1111/j.1432-1033.1986.tb09919.x. [DOI] [PubMed] [Google Scholar]
- 92.Gonzalez RA, Flint SJ. 2002. Effects of mutations in the adenoviral E1B 55 kDa protein coding sequence on viral late mRNA metabolism. J Virol 76:4507–4519. doi: 10.1128/JVI.76.9.4507-4519.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sarnow P, Sullivan CA, Levine AJ. 1982. A monoclonal antibody detecting the adenovirus type 5-E1b-58Kd tumor antigen: characterization of the E1b-58Kd tumor antigen in adenovirus-infected and -transformed cells. Virology 120:510–517. doi: 10.1016/0042-6822(82)90054-X. [DOI] [PubMed] [Google Scholar]
- 94.Nevels M, Spruss T, Wolf H, Dobner T. 1999. The adenovirus E4orf6 protein contributes to malignant transformation by antagonizing E1A-induced accumulation of the tumor suppressor protein p53. Oncogene 18:9–17. doi: 10.1038/sj.onc.1202284. [DOI] [PubMed] [Google Scholar]
- 95.Reich NC, Sarnow P, Duprey E, Levine AJ. 1983. Monoclonal antibodies which recognise native and denatured forms of the adenovirus DNA-binding protein. Virology 128:480–484. doi: 10.1016/0042-6822(83)90274-X. [DOI] [PubMed] [Google Scholar]
- 96.Rasband WS. 2009. ImageJ National Institutes of Health, Bethesda, MD. [Google Scholar]
- 97.Chahal JS, Qi J, Flint SJ. 2012. The human adenovirus type 5 E1B 55 kDa protein obstructs inhibition of viral replication by type I interferon in normal human cells. PLoS Pathog 8:e1002853. doi: 10.1371/journal.ppat.1002853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 99.Williams J, Karger BD, Ho YS, Castiglia CL, Mann T, Flint SJ. 1986. The adenovirus E1B 495R protein plays a role in regulating the transport and stability of the viral late messages. Cancer Cells 4:275–284. [Google Scholar]
- 100.Goodrum FD, Ornelles DA. 1999. Roles for the E4 orf6, orf3, and E1B 55-kilodalton proteins in cell cycle-independent adenovirus replication. J Virol 73:7474–7488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jones N, Shenk T. 1979. Isolation of adenovirus type 5 host range deletion mutants defective for transformation of rat embryo cells. Cell 17:683–689. doi: 10.1016/0092-8674(79)90275-7. [DOI] [PubMed] [Google Scholar]
- 102.Gonzalez R, Huang W, Finnen R, Bragg C, Flint SJ. 2006. Adenovirus E1B 55-kilodalton protein is required for both regulation of mRNA export and efficient entry into the late phase of infection in normal human fibroblasts. J Virol 80:964–974. doi: 10.1128/JVI.80.2.964-974.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ivanov GS, Ivanova T, Kurash J, Ivanov A, Chuikov S, Gizatullin F, Herrera-Medina EM, Rauscher F III, Reinberg D, Barlev NA. 2007. Methylation-acetylation interplay activates p53 in response to DNA damage. Mol Cell Biol 27:6756–6769. doi: 10.1128/MCB.00460-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sims R III, Reinberg D. 2008. Is there a code embedded in proteins that is based on post-translational modifications? Nat Rev Mol Cell Biol 9:815–820. doi: 10.1038/nrm2502. [DOI] [PubMed] [Google Scholar]
- 105.West LE, Gozani O. 2011. Regulation of p53 function by lysine methylation. Epigenomics 3:361–369. doi: 10.2217/epi.11.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ahlf DR, Compton PD, Tran JC, Early BP, Thomas PM, Kelleher NL. 2012. Evaluation of the compact high-field orbitrap for top-down proteomics of human cells. J Proteome Res 11:4308–4314. doi: 10.1021/pr3004216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tipton JD, Tran JC, Catherman AD, Ahlf DR, Durbin KR, Lee JE, Kellie JF, Kelleher NL, Hendrickson CL, Marshall AG. 2012. Nano-LC FTICR tandem mass spectrometry for top-down proteomics: routine baseline unit mass resolution of whole cell lysate proteins up to 72 kDa. Anal Chem 84:2111–2117. doi: 10.1021/ac202651v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Michalski A, Damoc E, Lange O, Denisov E, Nolting D, Muller M, Viner R, Schwartz J, Remes P, Belford M, Dunyach JJ, Cox J, Horning S, Mann M, Makarov A. 2012. Ultra high resolution linear ion trap Orbitrap mass spectrometer (Orbitrap Elite) facilitates top down LC MS/MS and versatile peptide fragmentation modes. Mol Cell Proteomics 11:O111.013698. doi: 10.1074/mcp.O111.013698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lee CW, Ferreon JC, Ferreon AC, Arai M, Wright PE. 2010. Graded enhancement of p53 binding to CREB-binding protein (CBP) by multisite phosphorylation. Proc Natl Acad Sci U S A 107:19290–19295. doi: 10.1073/pnas.1013078107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fraser JA, Madhumalar A, Blackburn E, Bramham J, Walkinshaw MD, Verma C, Hupp TR. 2010. A novel p53 phosphorylation site within the MDM2 ubiquitination signal: II. A model in which phosphorylation at SER269 induces a mutant conformation to p53. J Biol Chem 285:37773–37786. doi: 10.1074/jbc.M110.143107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fraser JA, Vojtesek B, Hupp TR. 2010. A novel p53 phosphorylation site within the MDM2 ubiquitination signal: I. Phosphorylation at SER269 in vivo is linked to inactivation of p53 function. J Biol Chem 285:37762–37772. doi: 10.1074/jbc.M110.143099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Berscheminski J, Wimmer P, Brun J, Ip WH, Groitl P, Horlacher T, Jaffray E, Hay RT, Dobner T, Schreiner S. 2014. Sp100 isoform-specific regulation of human adenovirus 5 gene expression. J Virol 88:6076–6092. doi: 10.1128/JVI.00469-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Alsheich-Bartok O, Haupt S, Alkalay-Snir I, Saito S, Appella E, Haupt Y. 2008. PML enhances the regulation of p53 by CK1 in response to DNA damage. Oncogene 27:3653–3661. doi: 10.1038/sj.onc.1211036. [DOI] [PubMed] [Google Scholar]
- 114.Wang Y, Prives C. 1995. Increased and altered DNA binding of human p53 by S and G2/M but not G1 cyclin-dependent kinases. Nature 376:88–91. doi: 10.1038/376088a0. [DOI] [PubMed] [Google Scholar]
- 115.McLure KG, Lee PW. 1998. How p53 binds DNA as a tetramer. EMBO J 17:3342–3350. doi: 10.1093/emboj/17.12.3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Steegenga WT, van Laar T, Riteco N, Mandarino A, Shvarts A, van der Eb AJ, Jochemsen AG. 1996. Adenovirus E1A proteins inhibit activation of transcription by p53. Mol Cell Biol 16:2101–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lill NL, Grossman SR, Ginsberg D, DeCaprio J, Livingston DM. 1997. Binding and modulation of p53 by p300/CBP coactivators. Nature 387:823–827. doi: 10.1038/42981. [DOI] [PubMed] [Google Scholar]
- 118.Somasundaram K, El-Deiry WS. 1997. Inhibition of p53-mediated transactivation and cell cycle arrest by E1A through its p300/CBP-interacting region. Oncogene 14:1047–1057. doi: 10.1038/sj.onc.1201002. [DOI] [PubMed] [Google Scholar]
- 119.Sang N, Avantaggiati ML, Giordano A. 1997. Roles of p300, pocket proteins, and hTBP in E1A-mediated transcriptional regulation and inhibition of p53 transactivation activity. J Cell Biochem 66:277–285. doi:. [DOI] [PubMed] [Google Scholar]
- 120.Ferreon JC, Lee CW, Arai M, Martinez-Yamout MA, Dyson HJ, Wright PE. 2009. Cooperative regulation of p53 by modulation of ternary complex formation with CBP/p300 and HDM2. Proc Natl Acad Sci U S A 106:6591–6596. doi: 10.1073/pnas.0811023106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Savelyeva I, Dobbelstein M. 2011. Infection with E1B-mutant adenovirus stabilizes p53 but blocks p53 acetylation and activity through E1A. Oncogene 30:865–875. doi: 10.1038/onc.2010.461. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.