

Abstract
Body fat distribution is a heritable trait and a well-established predictor of adverse metabolic outcomes, independent of overall adiposity. To increase our understanding of the genetic basis of body fat distribution and its molecular links to cardiometabolic traits, we conducted genome-wide association meta-analyses of waist and hip circumference-related traits in up to 224,459 individuals. We identified 49 loci (33 new) associated with waist-to-hip ratio adjusted for body mass index (WHRadjBMI) and an additional 19 loci newly associated with related waist and hip circumference measures (P<5×10−8). Twenty of the 49 WHRadjBMI loci showed significant sexual dimorphism, 19 of which displayed a stronger effect in women. The identified loci were enriched for genes expressed in adipose tissue and for putative regulatory elements in adipocytes. Pathway analyses implicated adipogenesis, angiogenesis, transcriptional regulation, and insulin resistance as processes affecting fat distribution, providing insight into potential pathophysiological mechanisms.
Depot-specific accumulation of fat, particularly in the central abdomen, confers an elevated risk of metabolic and cardiovascular diseases and mortality1. An easily accessible measure of body fat distribution is waist-to-hip ratio (WHR), a comparison of waist and hip circumferences. A larger WHR indicates more intra-abdominal fat deposition and is associated with higher risk for type 2 diabetes (T2D) and cardiovascular disease2,3. Conversely, a smaller WHR indicates greater gluteal fat accumulation and is associated with lower risk for T2D, hypertension, dyslipidemia, and mortality4-6. Our previous genome-wide association study (GWAS) meta-analyses have identified loci for WHR after adjusting for body mass index (WHRadjBMI)7,8. These loci are enriched for association with other metabolic traits7,8 and show that different fat distribution patterns can have distinct genetic components9,10.
To further elucidate the genetic architecture of fat distribution and to increase our understanding of molecular connections with cardiometabolic traits, we performed a meta-analysis of WHRadjBMI associations in 142,762 individuals with GWAS data and 81,697 individuals genotyped with the Metabochip11, all from the Genetic Investigation of ANthropometric Traits (GIANT) Consortium. Given the marked sexual dimorphism previously observed among established WHRadjBMI loci7,8, we performed analyses in men and women separately, the results of which were subsequently combined. To more fully characterize the genetic determinants of specific aspects of body fat distribution, we performed secondary GWAS meta-analyses for five additional traits: unadjusted WHR, BMI-adjusted and unadjusted waist (WCadjBMI and WC) and hip circumferences (HIPadjBMI and HIP). We evaluated the associated loci to understand their contributions to variation in fat distribution and adipose tissue biology, and their molecular links to cardiometabolic traits.
RESULTS
New loci associated with WHRadjBMI
We performed meta-analyses of GWAS of WHRadjBMI in up to 142,762 individuals of European ancestry from 57 new or previously described GWAS7, and separately in up to an additional 67,326 European ancestry individuals from 44 Metabochip studies (Extended Data Fig. 1; Supplementary Tables 1-3). The combination of these two meta-analyses included up to 2,542,447 autosomal SNPs in up to 210,088 European ancestry individuals. We defined new loci based on genome-wide significant association (P<5 × 10−8 after genomic control correction at both the study-specific and meta-analytic levels) and distance (>500 kb) from previously established loci7,8.
We identified 49 loci for WHRadjBMI, 33 of which were new and 16 previously described7,8. Of these, a European ancestry sex-combined analysis identified 39 loci, 24 of which were new (Table 1, Supplementary Table 4, and Supplementary Figs. 1-3)7,8. European ancestry sex-specific analyses identified nine additional loci, eight of which were new and significant in women but not in men (all Pmen>0.05; Table 1, Supplementary Fig. 4). The addition of 14,371 individuals of non-European ancestry genotyped on Metabochip identified one additional locus in women (rs1534696, near SNX10, Pwomen=2.1×10−8, Pmen=0.26, Table 1, Supplementary Tables 1-3), with no evidence of heterogeneity across ancestries (Phet=0.86, Supplementary Note).
Table 1. WHRadjBMI loci achieving genome-wide significance (P<5×10−8) in sex-combined and/or sex-specific meta-analyses.
|
|
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sex-combined | Women | Men | Sex diff.Pb | |||||||||||
|
| ||||||||||||||
| SNP | Chr | Locus | EAa | EAF | β | P | N | β | P | N | β | P | N | |
| Novel loci achieving genome-wide significance in European-ancestry meta-analyses | ||||||||||||||
|
| ||||||||||||||
| rs905938 | 1 | DCST2 | T | 0.74 | 0.025 | 7.3E-10 | 207,867 | 0.034 | 4.9E-10 | 115,536 | 0.015 | 1.10E-02 | 92,461 | 1.6E-02 |
| rs10919388 | 1 | GORAB | C | 0.72 | 0.024 | 3.2E-09 | 181,049 | 0.033 | 4.8E-10 | 102,446 | 0.013 | 2.98E-02 | 78,738 | 9.8E-03 |
| rs1385167 | 2 | MEIS1 | G | 0.15 | 0.029 | 1.9E-09 | 206,619 | 0.023 | 4.0E-04 | 114,668 | 0.036 | 2.32E-07 | 92,085 | 1.6E-01 |
| rs1569135 | 2 | CALCRL | A | 0.53 | 0.021 | 5.6E-10 | 209,906 | 0.023 | 6.9E-07 | 116,642 | 0.019 | 1.48E-04 | 93,398 | 5.8E-01 |
| rs10804591 | 3 | PLXND1 | A | 0.79 | 0.025 | 6.6E-09 | 209,921 | 0.040 | 6.1E-13 | 116,667 | 0.004 | 5.28E-01 | 93,387 | 5.7E-06 |
| rs17451107 | 3 | LEKR1 | T | 0.61 | 0.026 | 1.1E-12 | 207,795 | 0.023 | 1.0E-06 | 115,735 | 0.030 | 1.42E-08 | 92,194 | 3.5E-01 |
| rs3805389 | 4 | NMU | A | 0.28 | 0.012 | 1.5E-03 | 209,218 | 0.027 | 4.6E-08 | 116,226 | −0.007 | 2.09E-01 | 93,125 | 1.6E-06 |
| rs9991328 | 4 | FAM13A | T | 0.49 | 0.019 | 4.5E-08 | 209,925 | 0.028 | 3.4E-10 | 116,652 | 0.007 | 1.69E-01 | 93,407 | 8.5E-04 |
| rs303084 | 4 | SPATA5-FGF2 | A | 0.80 | 0.023 | 3.9E-08 | 209,941 | 0.029 | 3.4E-07 | 116,662 | 0.016 | 9.91E-03 | 93,412 | 1.1E-01 |
| rs9687846 | 5 | MAP3K1 | A | 0.19 | 0.024 | 7.1E-08 | 208,181 | 0.041 | 3.8E-12 | 115,897 | 0.000 | 9.69E-01 | 92,417 | 1.3E-06 |
| rs6556301 | 5 | FGFR4 | T | 0.36 | 0.022 | 2.6E-08 | 178,874 | 0.018 | 7.1E-04 | 101,638 | 0.029 | 1.00E-06 | 77,370 | 1.4E-01 |
| rs7759742 | 6 | BTNL2 | A | 0.51 | 0.023 | 4.4E-11 | 208,263 | 0.024 | 1.7E-07 | 115,648 | 0.023 | 5.49E-06 | 92,749 | 8.6E-01 |
| rs1776897 | 6 | HMGA1 | G | 0.08 | 0.030 | 1.1E-05 | 177,879 | 0.052 | 6.8E-09 | 100,516 | 0.003 | 7.42E-01 | 77,497 | 1.8E-04 |
| rs7801581 | 7 | HOXA11 | T | 0.24 | 0.027 | 3.7E-10 | 195,215 | 0.025 | 7.7E-06 | 108,866 | 0.029 | 2.39E-06 | 86,483 | 6.9E-01 |
| rs7830933 | 8 | NKX2-6 | A | 0.77 | 0.022 | 7.4E-08 | 209,766 | 0.037 | 1.2E-12 | 116,567 | 0.001 | 8.35E-01 | 93,333 | 1.4E-06 |
| rs12679556 | 8 | MSC | G | 0.25 | 0.027 | 2.1E-11 | 203,826 | 0.033 | 2.1E-10 | 114,369 | 0.017 | 4.15E-03 | 89,591 | 2.8E-02 |
| rs10991437 | 9 | ABCA1 | A | 0.11 | 0.031 | 1.0E-08 | 209,941 | 0.040 | 2.8E-08 | 116,644 | 0.022 | 6.13E-03 | 93,430 | 7.2E-02 |
| rs7917772 | 10 | SFXN2 | A | 0.62 | 0.014 | 5.6E-05 | 209,642 | 0.027 | 5.5E-09 | 116,514 | −0.001 | 8.57E-01 | 93,263 | 2.3E-05 |
| rs11231693 | 11 | MACROD1-VEGFB | A | 0.06 | 0.041 | 4.5E-08 | 198,072 | 0.068 | 2.7E-11 | 110,164 | 0.009 | 4.20E-01 | 88,043 | 2.5E-05 |
| rs4765219 | 12 | CCDC92 | C | 0.67 | 0.028 | 1.6E-15 | 209,807 | 0.037 | 1.0E-14 | 116,592 | 0.018 | 5.32E-04 | 93,350 | 5.7E-03 |
| rs8042543 | 15 | KLF13 | C | 0.78 | 0.026 | 1.2E-09 | 208,255 | 0.023 | 6.7E-05 | 115,760 | 0.030 | 1.01E-06 | 92,629 | 3.6E-01 |
| rs8030605 | 15 | RFX7 | A | 0.14 | 0.030 | 8.8E-09 | 208,374 | 0.031 | 1.0E-05 | 115,864 | 0.031 | 5.91E-05 | 92,644 | 9.9E-01 |
| rs1440372 | 15 | SMAD6 | C | 0.71 | 0.024 | 1.1E-10 | 207,447 | 0.022 | 1.1E-05 | 115,201 | 0.027 | 1.39E-06 | 92,380 | 5.2E-01 |
| rs2925979 | 16 | CMIP | T | 0.31 | 0.018 | 1.2E-06 | 207,828 | 0.032 | 3.4E-11 | 115,431 | −0.002 | 7.86E-01 | 92,531 | 1.2E-06 |
| rs4646404 | 17 | PEMT | G | 0.67 | 0.027 | 1.4E-11 | 198,196 | 0.034 | 5.3E-11 | 115,337 | 0.017 | 2.45E-03 | 87,857 | 2.6E-02 |
| rs8066985 | 17 | KCNJ2 | A | 0.50 | 0.018 | 1.4E-07 | 209,977 | 0.026 | 4.0E-09 | 116,683 | 0.007 | 1.89E-01 | 93,428 | 1.8E-03 |
| rs12454712 | 18 | BCL2 | T | 0.61 | 0.016 | 1.0E-04 | 169,793 | 0.035 | 1.1E-09 | 96,182 | −0.007 | 2.45E-01 | 73,576 | 1.6E-07 |
| rs12608504 | 19 | JUND | A | 0.36 | 0.022 | 8.8E-10 | 209,990 | 0.017 | 2.6E-04 | 116,689 | 0.028 | 1.05E-07 | 93,435 | 1.2E-01 |
| rs4081724 | 19 | CEBPA | G | 0.85 | 0.035 | 7.4E-12 | 207,418 | 0.033 | 9.2E-07 | 115,322 | 0.039 | 1.41E-07 | 92,230 | 5.0E-01 |
| rs979012 | 20 | BMP2 | T | 0.34 | 0.027 | 3.3E-14 | 209,941 | 0.026 | 1.0E-07 | 116,668 | 0.028 | 6.59E-08 | 93,407 | 6.7E-01 |
| rs224333 | 20 | GDF5 | G | 0.62 | 0.020 | 2.6E-08 | 208,025 | 0.009 | 7.4E-02 | 115,803 | 0.036 | 9.00E-12 | 92,356 | 6.4E-05 |
| rs6090583 | 20 | EYA2 | A | 0.48 | 0.022 | 6.2E-11 | 209,435 | 0.029 | 2.8E-10 | 116,382 | 0.015 | 2.37E-03 | 93,187 | 3.2E-02 |
|
| ||||||||||||||
| Novel loci achieving genome-wide significance in all-ancestry meta-analyses | ||||||||||||||
|
| ||||||||||||||
| rs1534696 | 7 | SNX10 | C | 0.43 | 0.011 | 1.3E-03 | 212,501 | 0.027 | 2.1E-08 | 118,187 | −0.006 | 2.64E-01 | 92,243 | 2.1E-06 |
|
| ||||||||||||||
| Previously reported loci achieving genome-wide significance in European-ancestry meta-analyses | ||||||||||||||
|
| ||||||||||||||
| rs2645294 | 1 | TBX15-WARS2 | T | 0.58 | 0.031 | 1.7E-19 | 209,808 | 0.035 | 1.5E-14 | 116,596 | 0.027 | 1.46E-07 | 93,346 | 2.0E-01 |
| rs714515 | 1 | DNM3-PIGC | G | 0.43 | 0.027 | 4.4E-15 | 203,401 | 0.029 | 1.8E-10 | 113,939 | 0.025 | 8.54E-07 | 89,596 | 5.1E-01 |
| rs2820443 | 1 | LYPLAL1 | T | 0.72 | 0.035 | 5.3E-21 | 209,975 | 0.062 | 5.7E-35 | 116,672 | 0.002 | 6.91E-01 | 93,437 | 2.6E-17 |
| rs10195252 | 2 | GRB14-COBLL1 | T | 0.59 | 0.027 | 5.9E-15 | 209,395 | 0.052 | 4.7E-30 | 116,329 | −0.003 | 5.33E-01 | 93,199 | 2.4E-17 |
| rs17819328 | 3 | PPARG | G | 0.43 | 0.021 | 2.4E-09 | 208,809 | 0.035 | 4.6E-14 | 116,072 | 0.005 | 3.26E-01 | 92,871 | 5.1E-06 |
| rs2276824 | 3 | PBRM1c | C | 0.43 | 0.024 | 3.2E-11 | 208,901 | 0.028 | 3.7E-09 | 116,128 | 0.020 | 1.35E-04 | 92,907 | 2.0E-01 |
| rs2371767 | 3 | ADAMTS9 | G | 0.72 | 0.036 | 1.6E-20 | 194,506 | 0.056 | 1.2E-26 | 108,624 | 0.012 | 3.49E-02 | 86,016 | 3.6E-09 |
| rs1045241 | 5 | TNFAIP8-HSD17B4 | C | 0.71 | 0.019 | 4.4E-07 | 209,710 | 0.035 | 6.6E-12 | 116,560 | −0.001 | 9.29E-01 | 93,284 | 8.3E-07 |
| rs7705502 | 5 | CPEB4 | A | 0.33 | 0.027 | 4.7E-14 | 209,827 | 0.027 | 1.9E-08 | 116,609 | 0.027 | 2.30E-07 | 93,352 | 1.0E+00 |
| rs1294410 | 6 | LY86 | C | 0.63 | 0.031 | 2.0E-18 | 209,830 | 0.037 | 1.6E-15 | 116,624 | 0.025 | 1.37E-06 | 93,340 | 6.3E-02 |
| rs1358980 | 6 | VEGFA | T | 0.47 | 0.039 | 3.1E-27 | 206,862 | 0.060 | 3.7E-34 | 115,047 | 0.015 | 4.02E-03 | 91,949 | 3.7E-11 |
| rs1936805 | 6 | RSPO3 | T | 0.51 | 0.043 | 3.6E-35 | 209,859 | 0.052 | 3.7E-30 | 116,602 | 0.031 | 3.08E-10 | 93,392 | 1.0E-03 |
| rs10245353 | 7 | NFE2L3 | A | 0.20 | 0.035 | 8.4E-16 | 210,008 | 0.041 | 7.9E-13 | 116,704 | 0.027 | 1.43E-05 | 93,438 | 7.2E-02 |
| rs10842707 | 12 | ITPR2-SSPN | T | 0.23 | 0.032 | 4.4E-16 | 210,023 | 0.041 | 6.1E-15 | 116,704 | 0.022 | 1.44E-04 | 93,453 | 1.1E-02 |
| rs1443512 | 12 | HOXC13 | A | 0.24 | 0.028 | 6.9E-13 | 209,980 | 0.040 | 1.1E-14 | 116,688 | 0.013 | 2.77E-02 | 93,425 | 1.6E-04 |
| rs2294239 | 22 | ZNRF3 | A | 0.59 | 0.025 | 7.2E-13 | 209,454 | 0.028 | 6.9E-10 | 116,414 | 0.024 | 2.31E-06 | 93,173 | 5.0E-01 |
P values and β coefficients for the association with WHRadjBMI in the meta-analyses of combined GWAS and Metabochip studies. The smallest P value for each SNP is shown in bold.
The effect allele is the WHRadjBMI-increasing allele in the sex-combined analysis.
Test for sex difference; values significant at the table-wise Bonferroni threshold of 0.05/49=1.02× 10−3 are marked in bold.
Locus previously named NISCH-STAB1. Additional analyses that showed no significant evidence of heterogeneity between studies or due to ascertainment are provided in Supplementary Tables 27 and 28 (Supplementary Note). Chr, chromosome; EA, effect allele; EAF, effect allele frequency.
Genetic architecture of WHRadjBMI
To evaluate sexual dimorphism, we compared sex-specific effect size estimates of the 49 WHRadjBMI lead SNPs. The effect estimates were significantly different (Pdifference<0.05/49=0.001) at 20 SNPs, 19 of which showed larger effects in women (Table 1, Extended Data Fig. 2a), similar to previous findings7,8. The only SNP that showed a larger effect in men mapped near GDF5 (rs224333, βmen=0.036 and P=9.0×10−12, βwomen=0.009 and P=0.074, Pdifference= 6.4 × 10−5), a locus previously associated with height (rs6060369, r2=0.96 and rs143384, r2=0.96, 1000 Genomes Project CEU), though without significant differences between sexes12,13. Consistent with the larger number of loci identified in women, variance component analyses demonstrated a significantly larger heritability (h2) of WHRadjBMI in women than men in the Framingham Heart (h2women=0.46, h2men=0.19, Pdifference=0.0037) and TwinGene studies (h2women=0.56, h2men=0.32, Pdifference=0.001, Supplementary Table 5, Extended Data Fig. 2b).
To identify multiple association signals within observed loci, we performed approximate conditional analyses of the sex-combined and sex-specific summary statistics using GCTA14 (Supplementary Note). Multiple signals (P<5×10−8) were identified at nine loci (Extended Data Table 1). Fitting SNPs jointly identified different lead SNPs in the sex-specific and sex-combined analyses. For example, the MAP3K1-ANKRD55 locus showed near-independent (linkage disequilibrium (LD) r2<0.06) SNPs 54 kb apart that were significant only in women (rs3936510) or only in men (rs459193, Extended Data Table 1, Supplementary Table 4). Other signals are more complex. The TBX15-WARS2 locus showed different but correlated lead SNPs in men and women near WARS2 (r2=0.43), an independent signal near TBX15, and a distant independent signal near SPAG17 (Fig. 1). At the HOXC gene cluster, conditional analyses identified independent (r2<0.01) SNPs ~80 kb apart near HOXC12-HOXC13-HOTAIR and near HOXC4-HOXC6 (Fig. 1). These results suggest that association signals mapping to the same locus might act on different underlying genes and may not be relevant to the same sex.
Figure 1. Regional SNP association plots illustrating the complex genetic architecture at two WHRadjBMI loci.

Sex-combined meta-analysis SNP associations in European individuals were plotted with −log10 P values (left y-axis) and estimated local recombination rate in blue (right y-axis). Three index SNPs near HOXC6-HOXC13 (a–c) and four near TBX15-WARS2-SPAG17 (d–g) were identified through approximate conditional analyses of sex-combined or sex-specific associations (values shown as Pconditional <5×10−8, see Methods). The signals are distinguished by both color and shape, and linkage disequilibrium (r2) of nearby SNPs is shown by color intensity gradient.
We assessed the aggregate effects of the primary association signals at the 49 WHRadjBMI loci by calculating sex-combined and sex-specific risk based on genotypes of the lead SNPs. In a linear regression model, the risk scores were associated with WHRadjBMI, with a stronger effect in women than in men (overall effect per allele β=0.001, P=6.7×10−4, women β=0.002, P=1.0×10−11, men β=7.0×10−4, P=0.02, Extended Data Fig. 3, Supplementary Note). The 49 SNPs explained 1.4% of the variance in WHRadjBMI overall, and more in women (2.4%) than in men (0.8%) (Supplementary Table 6). Compared to the 16 previously reported loci7,8, the new loci almost doubled the explained variance in women and tripled that in men. We further estimated that the sex-combined variance explained by all HapMap SNPs15 (h2G) is 12.1% (SE=2.9%).
At 17 loci with high-density coverage on the Metabochip11, we used association summary statistics to define credible sets of SNPs with a high probability of containing a likely functional variant. The 99% credible sets at seven loci spanned <20 kb, and at HOXC13 included only a single noncoding SNP (Supplementary Table 7, Supplementary Fig. 5). Imputation up to higher density reference panels will provide greater coverage and may have more potential to localize functional variants.
WHRadjBMI variants and other traits
Given the epidemiological correlations between central obesity and other anthropometric and cardiometabolic measures and diseases, we evaluated lead WHRadjBMI variants in association data from GWAS consortia for 22 traits. Seventeen of the 49 variants were associated (P<5×10−8) with at least one of the traits: high-density lipoprotein cholesterol (HDL-C; n=7 SNPs), triglycerides (TG; n=5), low-density lipoprotein cholesterol (LDL-C; n=2), adiponectin adjusted for BMI (n=3), fasting insulin adjusted for BMI (n=2), T2D (n=1), and height (n=7) (Supplementary Tables 8-9). WHRadjBMI SNPs also showed enrichment for directional consistency among nominally significant (P<0.05) associations with these traits and also with fasting and 2-hour glucose, diastolic and systolic blood pressure (DBP, SBP), BMI and coronary artery disease (CAD) (Pbinomial<0.05/23=0.0022, Extended Data Table 2); these results were generally supported by meta-regression analysis of the regression coefficient-estimates (Supplementary Table 10). Furthermore, our WHRadjBMI loci overlap with associations reported in the NHGRI GWAS Catalog (Table 2, Supplementary Table 11)16, the strongest of which is the locus near LEKR1, which is associated (P=2.0×10−35) with birthweight17. Unsupervised hierarchical clustering of the corresponding matrix of association Z-scores showed three major clusters characterized by patterns of anthropometric and metabolic traits (Extended Data Fig. 4). These data extend knowledge about genetic links between WHRadjBMI and insulin resistance-related traits; whether this reflects underlying causal relations between WHRadjBMI and these traits, or pleiotropic loci, cannot be inferred from our data.
Table 2. Candidate genes at new WHRadjBMI loci.
| SNP | Locus | Expression QTL (P<10−5)a | GRAIL (P<0.05)b | DEPICT (FDR<0.05)c | Literatured | Other GWAS signalse |
|---|---|---|---|---|---|---|
| rs905938 | DCST2 | ZBTB7B (PB, Blood) | - | - | - | - |
| rs10919388 | GORAB | - | - | - | - | - |
| rs1385167 | MEIS1 | - | - | - | MEIS1 | - |
| rs1569135 | CALCRL | - | TFPI | - | CALCRL | - |
| rs10804591 | PLXND1 | - | - | - | PLXND1 | - |
| rs17451107 | LEKR1 | TIPARP (S,O), LEKR1 (S) | - | - | - | Birthweight: CCNL1, LEKR1 |
| rs3805389 | NMU | - | - | - | NMU | - |
| rs9991328 | FAM13A | FAM13A (S) | - | FAM13A | - | FI: FAM13A |
| rs303084 | SPATA5-FGF2 | - | FGF2 | - | FGF2, NUDT6, SPRY1 | - |
| rs9687846 | MAP3K1 | - | MAP3K1 | - | MAP3K1 | FI, TG: ANKRD55, MAP3K1 |
| rs6556301 | FGFR4 | - | MXD3 | - | FGFR4 | Height |
| rs7759742 | BTNL2 | HLA-DRA (S), KLHL31 (S) | - | (not analyzed) | - | - |
| rs1776897 | HMGA1 | - | - | (not analyzed) | HMGA1 | Height: HMGA1, C6orf106, LBH |
| rs1534696 | SNX10 | SNX10 (S), CBX3 (S) | - | - | SNX10 | - |
| rs7801581 | HOXA11 | - | HOXA11 | HOXA11 | HOXA11 | - |
| rs7830933 | NKX2-6 | STC1 (S) | - | - | NKX2-6, STC1 | - |
| rs12679556 | MSC | - | EYA1 | RP11-1102P16.1 | MSC, EYA1 | - |
| rs10991437 | ABCA1 | - | - | - | ABCA1 | - |
| rs7917772 | SFXN2 | - | - | - | SFXN2 | Height |
| rs11231693 | MACROD1-VEGFB | - | VEGFB | MACROD1 | MACROD1, VEGFB | - |
| rs4765219 | CCDC92 | CCDC92 (S, O, L), ZNF664 (S, O) | FAM101A | - | - | Adiponectin, FI, HDL, TG: CCDC92, ZNF664 |
| rs8042543 | KLF13 | - | KLF13 | - | KLF13 | - |
| rs8030605 | RFX7 | - | - | - | - | |
| rs1440372 | SMAD6 | SMAD6 (Blood) | SMAD6 | SMAD6 | SMAD6 | Height |
| rs2925979 | CMIP | CMIP (S) | - | - | CMIP, PLCG2 | Adiponectin, FI, HDL: CMIP |
| rs4646404 | PEMT | - | - | PEMT | PEMT | - |
| rs8066985 | KCNJ2 | - | - | - | KCNJ2 | - |
| rs12454712 | BCL2 | - | - | - | BCL2 | - |
| rs12608504 | JUND | KIAA1683 (PB, O), JUND (LCL) | JUND | - | JUND | - |
| rs4081724 | CEBPA | - | CEBPA | - | CEBPA, CEBPG | - |
| rs979012 | BMP2 | - | BMP2 | BMP2 | BMP2 | Height: BMP2 |
| rs224333 | GDF5 | CEP250 (S, O), UQCC (Blood, S, O, L, LCL) | GDF5 | GDF5 | GDF5 | Height: GDF5, UQCC |
| rs6090583 | EYA2 | - | EYA2 | EYA2 | EYA2 | - |
Candidate genes based on secondary analyses or literature review. Details are provided in Supplementary Tables 8-9, 11-13, 15, 19, 21 and the Supplementary Note. The only nonsynonymous variant in high LD with an index SNP was GDF5 S276A. No copy number variants were identified.
Gene transcript levels associated with the SNP in the indicated tissue(s): PB, peripheral blood mononuclear cells; S, subcutaneous adipose; O, omental adipose; L, liver; lcl, lymphoblastoid cell line.
Genes in pathways identified as enriched by GRAIL analysis
Significant pathway genes derived by DEPICT using GWAS-only results.
Most plausible candidate genes based on literature review.
Traits associated at P<5 × 10−8 in GWAS or the GWAS catalog using the index SNP or a proxy, and the genes(s) named. FI, fasting insulin adjusted for BMI; HDL, high-density lipoprotein cholesterol; tg, triglycerides.
Potential functional WHRadjBMI variants
We next examined variants in LD with the WHRadjBMI lead SNPs (r2>0.7) for predicted effects on protein sequence, copy number, and cis-regulatory effects on expression (Table 2, Supplementary Tables 12-15, Supplementary Note). At 11 of the new loci, lead WHRadjBMI SNPs were in LD with cis-expression quantitative trait loci (eQTLs) for transcripts in subcutaneous adipose tissue, omental adipose tissue, liver, or blood cell types (Table 2, Supplementary Table 15). No additional sex-specific eQTLs were identified, perhaps reflecting limited power (Supplementary Table 16).
At the 11 WHRadjBMI loci harboring eQTLs, we compared the location of the candidate variants to regions of open chromatin (DNase I hypersensitivity and formaldehyde-assisted isolation of regulatory elements [FAIRE]) and histone modification enrichment (H3K4me1, H3K4me2, H3K4me3, H3K27ac, and H3K9ac) in adipose, liver, skeletal muscle, bone, brain, blood, and pancreatic islet tissues or cell lines (Supplementary Table 17). At seven of these 11 loci, at least one variant was located in a putative regulatory element in two or more datasets from the same tissue as the eQTL, suggesting that these elements may influence transcriptional activity (Supplementary Table 18). For example, at LEKR1, five variants in LD with the WHRadjBMI lead SNP are located in a 1.1 kb region with evidence of enhancer activity (H3K4me1 and H3K27ac) in adipose tissue (Extended Data Fig. 5a).
We also examined whether any variants overlapped with open chromatin or histone modifications from only one of the tested tissues, possibly reflecting tissue-specific regulatory elements (Supplementary Table 18). For example, five variants in a 2.2 kb region, located 77 kb upstream from a CALCRL transcription start site, overlapped with peaks in at least five datasets in endothelial cells (Extended Data Fig. 5b), suggesting that one or more of these variants may influence transcriptional activity. CALCRL, which is expressed in endothelial cells, is required for lipid absorption in the small intestine, and influences body weight in mice18. Other variants located in tissue-specific regulatory elements were detected at NMU for endothelial cells, at KLF13 and MEIS1 for liver, and at GORAB and MSC for bone (Supplementary Table 18).
Biological mechanisms
To identify potential functional connections between genes mapping to the 49 WHRadjBMI loci, we used three approaches (Supplementary Note). A survey of literature using GRAIL19 identified 15 genes with nominal significance (P<0.05) for potential functional connectivity (Table 2, Supplementary Table 19). The predefined gene set relationships across loci identified using MAGENTA20 highlighted signaling pathways involving vascular endothelial growth factor (VEGF), phosphatase and tensin (PTEN) homolog, the insulin receptor, and peroxisome proliferator-activated receptors (Supplementary Table 20). VEGF signaling plays a central, complex role in angiogenesis, insulin resistance, and obesity21, and PTEN signaling promotes insulin resistance22. Analyses using DEPICT23 facilitated prioritization of genes at associated loci, analyses of tissue specificity, and enrichment of reconstituted gene sets through integration of association results with expression data, protein-protein interactions, phenotypic data from gene knockout studies in mice, and predefined gene sets. DEPICT identified at least one prioritized gene (false discovery rate (FDR)<5%) at nine loci (Table 2, Supplementary Table 21) and identified 234 reconstituted gene sets (161 after pruning of overlapping gene sets) enriched for genes at WHRadjBMI loci. Among these we highlight biologically plausible gene sets suggesting roles in body fat regulation (including adiponectin signaling, insulin sensitivity, and regulation of glucose levels), skeletal growth, transcriptional regulation, and development (Fig. 2, Supplementary Table 22). We also note gene sets that are specific for abundance or development of metabolically active tissues including adipose, heart, liver, and muscle. Specific genes at the loci were significantly enriched (FDR<5%) for expression in adipocyte-related tissues, including abdominal subcutaneous fat (Fig. 2, Supplementary Table 23). Together, these analyses identified processes related to insulin and adipose biology and highlight mesenchymal tissues, especially adipose tissue, as important to WHRadjBMI.
Figure 2. Gene set enrichment and tissue expression of genes at WHRadjBMI-associated loci (GWAS-only P<10−5).

a, Reconstituted gene sets found to be significantly enriched by DEPICT (FDR<5%) are represented as nodes, with pairwise overlap denoted by the width of connecting lines and empirical enrichment P value indicated by color intensity (darker is more significant). b, The ‘Decreased Liver Weight’ meta-node, which consisted of 12 overlapping gene sets, including adiponectin signaling and insulin sensitivity. c, Based on expression patterns in 37,427 human microarray samples, annotations found to be significantly enriched by DEPICT are shown, grouped by type and significance.
We also tested variants at the 49 WHRadjBMI loci for overlap with elements from 60 selected regulatory datasets from the ENCODE24 and Epigenomic RoadMap25 data and found evidence of enrichment in 12 datasets (P<0.05/60=8.3×10−4, Extended Data Table 3). The strongest enrichments were detected for datasets typically attributed to enhancer activity (H3K4me1 and H3K27ac) in adipose, muscle, endothelial cells, and bone, suggesting that variants may regulate transcription in these tissues. These analyses point to mechanisms linking WHRadjBMI loci to regulation of gene expression in tissues highly relevant for adipocyte metabolism and insulin resistance.
We also reviewed functions of candidate genes located near new and previously established WHRadjBMI loci7,8, identifying genes involved in adipogenesis, angiogenesis, and transcriptional regulation (Table 2, literature review in the Supplementary Note). Adipogenesis candidate genes include CEBPA, PPARG, BMP2, HOXC/miR196, SPRY1, TBX15, and PEMT. Of these, CEBPA and PPARG are essential for white adipose tissue differentiation26, BMP2 induces differentiation of mesenchymal stem cells toward adipogenesis or osteogenesis27, and HOXC8 is a repressor of brown adipogenesis in mice that is regulated by miR-196a28, also located within the HOXC region (Fig. 1). Angiogenesis genes may influence expansion and loss of adipose tissue29; they include VEGFA, VEGFB, RSPO3, STAB1, WARS2, PLXND1, MEIS1, FGF2, SMAD6, and CALCRL. VEGFB is involved in endothelial targeting of lipids to peripheral tissues30, and PLXND1 limits blood vessel branching, antagonizes VEGF, and affects adipose inflammation31,32. Transcriptional regulators at WHRadjBMI loci include CEBPA, PPARG, MSC, SMAD6, HOXA, HOXC, ZBTB7B, JUND, KLF13, MEIS1, RFX7, NKX2-6, and HMGA1. Other candidate genes include NMU, FGFR4, and HMGA1, for which mice deficient for the corresponding genes exhibit obesity, glucose intolerance, and/or insulin resistance33-35.
Five additional central obesity traits
To determine whether the WHRadjBMI variants exert their effects primarily through WC or HIP and to identify loci that are not reported for WHRadjBMI, BMI, or height36,37, we performed association analyses for five additional traits: WCadjBMI, HIPadjBMI, WHR, WC, and HIP. Based on phenotypic data alone, WC and HIP are highly correlated with BMI (r=0.59-0.92), and WHR is highly correlated with WHRadjBMI (r=0.82-0.95), while WCadjBMI and HIPadjBMI are moderately correlated with height (r=0.24-0.63, Supplementary Table 24). In contrast to WHRadjBMI, which has almost no genetic correlation (see Methods) with height (rG<0.04, Extended Data Fig. 2c), WCadjBMI (rG=0.42) and HIPadjBMI (rG=0.82) have moderate genetic correlations with height. These data suggest that some, but not all, WCadjBMI and HIPadjBMI loci would be associated with height.
Across all meta-analyses, we identified an additional 19 loci associated with one of the five traits (P<5×10−8), nine of which showed significantly larger effects (Pdifference<0.05/19=0.003) in one sex than in the other (Table 3, Supplementary Figs. 1-4, Supplementary Table 25). Three of four new loci with larger effects in women were associated with HIPadjBMI and three of five new loci with larger effects in men were associated with WCadjBMI. Most of the 19 loci showed some evidence of association with WHRadjBMI in sex-combined or sex-specific analyses, but four loci showed no association (P>0.01) with WHRadjBMI, BMI, or height (Supplementary Tables 8, 26).
Table 3. New loci achieving genome-wide evidence of association (P<5×10−8) with additional waist and hip circumference traits.
|
|
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sex-combined | Women | Men | Sex diff. | ||||||||||||
|
| |||||||||||||||
| SNP | Trait | Chr | Locus | EAa | EAF | β | P | N | β | P | N | β | P | N | P b |
| Loci achieving genome-wide significance in European-ancestry meta-analyses | |||||||||||||||
|
| |||||||||||||||
| rs10925060 | WCadjBMI | 1 | OR2W5-NLRP3 | T | 0.03 | 0.017 | 2.2E-05 | 140,515 | 0.002 | 6.8E-01 | 85,186 | 0.045 | 9.1E-13 | 55,522 | 1.7E-08 |
| rs10929925 | HIP | 2 | SOX11 | C | 0.55 | 0.020 | 4.5E-08 | 207,648 | 0.021 | 9.0E-06 | 115,428 | 0.018 | 3.2E-04 | 92,499 | 6.1E-01 |
| rs2124969 | WCadjBMI | 2 | ITGB6 | C | 0.42 | 0.020 | 7.1E-09 | 231,284 | 0.016 | 3.5E-04 | 127,437 | 0.025 | 2.3E-07 | 104,039 | 1.4E-01 |
| rs17472426 | WCadjBMI | 5 | CCNJL | T | 0.92 | 0.014 | 3.1E-02 | 217,564 | −0.014 | 1.0E-01 | 119,804 | 0.052 | 4.3E-08 | 97,954 | 3.9E-08 |
| rs7739232 | HIPadjBMI | 6 | KLHL31 | A | 0.07 | 0.037 | 5.4E-05 | 131,877 | 0.063 | 1.0E-08 | 80,475 | −0.004 | 7.5E-01 | 51,589 | 2.9E-05 |
| rs13241538 | HIPadjBMI | 7 | KLF14 | C | 0.48 | 0.017 | 1.6E-06 | 210,935 | 0.033 | 9.9E-14 | 117,210 | −0.003 | 5.0E-01 | 93,911 | 2.0E-09 |
| rs7044106 | HIPadjBMI | 9 | C5 | C | 0.24 | 0.023 | 4.1E-05 | 143,412 | 0.039 | 5.7E-09 | 86,733 | −0.003 | 6.9E-01 | 56,865 | 1.3E-05 |
| rs11607976 | HIP | 11 | MYEOV | C | 0.70 | 0.022 | 4.2E-08 | 212,815 | 0.019 | 1.9E-04 | 118,391 | 0.024 | 7.7E-06 | 94,701 | 4.4E-01 |
| rs1784203 | WCadjBMI | 11 | KIAA1731 | A | 0.01 | 0.031 | 1.3E-08 | 63,892 | 0.000 | 9.9E-01 | 35,539 | 0.075 | 1.0E-19 | 28,353 | 1.2E-01 |
| rs1394461 | WHR | 11 | CNTN5 | C | 0.25 | 0.017 | 4.7E-04 | 144,349 | 0.035 | 3.6E-08 | 87,441 | −0.011 | 1.6E-01 | 57,094 | 1.1E-06 |
| rs319564 | WHR | 13 | GPC6 | C | 0.45 | 0.014 | 3.4E-05 | 212,137 | 0.003 | 5.3E-01 | 117,970 | 0.027 | 1.6E-08 | 94,350 | 6.0E-05 |
| rs2047937 | WCadjBMI | 16 | ZNF423 | C | 0.50 | 0.019 | 4.7E-08 | 231,009 | 0.022 | 5.5E-07 | 127,288 | 0.014 | 3.6E-03 | 103,914 | 2.0E-01 |
| rs2034088 | HIPadjBMI | 17 | VPS53 | T | 0.53 | 0.021 | 4.8E-09 | 210,737 | 0.028 | 9.6E-10 | 117,142 | 0.014 | 6.5E-03 | 93,781 | 2.5E-02 |
| rs1053593 | HIPadjBMI | 22 | HMGXB4 | T | 0.65 | 0.021 | 3.9E-08 | 202,070 | 0.029 | 1.8E-09 | 114,347 | 0.011 | 5.1E-02 | 87,908 | 6.2E-03 |
|
| |||||||||||||||
| Loci achieving genome-wide significance in all-ancestry meta-analyses | |||||||||||||||
|
| |||||||||||||||
| rs1664789 | WCadjBMI | 5 | ARL15 | C | 0.41 | 0.014 | 2.6E-05 | 244,110 | 0.005 | 2.8E-01 | 133,052 | 0.026 | 3.6E-08 | 109,025 | 4.4E-04 |
| rs722585 | HIPadjBMI | 6 | GMDS | G | 0.68 | 0.015 | 2.1E-04 | 205,815 | −0.001 | 8.8E-01 | 113,965 | 0.032 | 9.2E-09 | 89,831 | 4.3E-06 |
| rs1144 | WCadjBMI | 7 | SRPK2 | C | 0.34 | 0.019 | 3.1E-08 | 239,342 | 0.020 | 1.2E-05 | 131,398 | 0.018 | 4.1E-04 | 105,911 | 7.8E-01 |
| rs2398893 | WHR | 9 | PTPDC1 | A | 0.71 | 0.020 | 4.0E-08 | 226,572 | 0.019 | 5.1E-05 | 124,577 | 0.019 | 2.7E-04 | 99,968 | 9.5E-01 |
| rs4985155c | HIP | 16 | PDXDC1 | A | 0.66 | 0.018 | 4.5E-07 | 227,296 | 0.011 | 1.6E-02 | 125,048 | 0.029 | 9.7E-09 | 100,313 | 6.3E-03 |
P values and β coefficients for the association with the trait indicated in the meta-analysis of combined GWAS and Metabochip studies. The smallest P value for each SNP is shown in bold.
The effect allele is the trait-increasing allele in the sex-combined analysis.
Test for sex difference; values significant at the table-wise Bonferroni threshold of 0.05/19=2.63×10−3 are marked in bold.
P=7.3×10−6 with height in Okada et al.43 (index SNP rs1136001; r2=0.79, distance=2,515 bp). Chr, chromosome; EA, effect allele; EAF, effect allele frequency.
We next asked whether the genes and pathways influencing these five traits are shared with WHRadjBMI or are distinct. Candidate genes were identified based on association with other traits, eQTLs, GRAIL, and literature review (Extended Data Table 4, Supplementary Tables 8, 11-13, 15-16, 19). Candidate variants identified based on LD (r2>0.7) included coding variants in NTAN1 and HMGXB4, and six loci showed significant eQTLs in subcutaneous adipose tissue. Based on the literature, several candidate genes are involved in adipogenesis and insulin resistance. For example, delayed induction of preadipocyte transcription factor ZNF423 in fibroblasts results in delayed adipogenesis38, and NLRP3 is part of inflammasome and pro-inflammatory T-cell populations in adipose tissue that contribute to inflammation and insulin resistance39. GRAIL analyses identified connections that partially overlap with those identified for WHRadjBMI (Supplementary Table 19). Taken together, the additional loci appear to function in processes similar to the WHRadjBMI loci. The identification of loci that are more strongly associated with WCadjBMI or HIPadjBMI than the other anthropometric traits suggests that the additional traits characterize aspects of central obesity and fat distribution that are not captured by WHRadjBMI or BMI alone.
DISCUSSION
These meta-analyses of GWAS and Metabochip data in up to 224,459 individuals identified additional loci associated with waist and hip circumference measures and help elucidate the role of common genetic variation in body fat distribution that is distinct from BMI and height. Our results emphasize the strong sexual dimorphism in the genetic regulation of fat distribution traits, a characteristic not observed for overall obesity as assessed by BMI36. Differences in body fat distribution between the sexes emerge in childhood, become more apparent during puberty40, and change with menopause, generally attributed to the influence of sex hormones41,42. At loci with stronger effects in one sex than the other, these hormones may interact with transcription factors to regulate gene activity.
Annotation of the loci emphasized the role for mesenchymally-derived tissues, especially adipose tissue, in fat distribution and central obesity. The development and regulation of adipose tissue deposition is closely associated with angiogenesis29, a process highlighted by candidate genes at several WHRadjBMI loci. These tissues are implicated in insulin resistance, consistent with the enrichment of shared GWAS signals with lipids, T2D, and glycemic traits. The identification of skeletal growth processes suggests that the underlying genes affect early development and/or differentiation of adipocytes from mesenchymal stem cells. In contrast, BMI has a significant neuronal component, involving processes such as appetite regulation36. Our results provide a foundation for future biological research in the regulation of body fat distribution and its connections with cardiometabolic traits, and offer potential target mechanisms for interventions in the risks associated with abdominal fat accumulation.
METHODS
Study overview
Our study included 224,459 individuals of European, East Asian, South Asian, and African American ancestry. The European ancestry arm included 142,762 individuals from 57 cohorts genotyped with genome-wide SNP arrays and 67,326 individuals from 44 cohorts genotyped with the Metabochip11 (Extended Data Fig. 1, Supplementary Table 1). The non-European ancestry arm comprised ~1,700 individuals from one cohort of East Asian ancestry, ~3,400 individuals from one cohort of South Asian ancestry, and ~9,200 individuals from six cohorts of African American ancestry, all genotyped with the Metabochip. There was no overlap between individuals genotyped with genome-wide SNP arrays and Metabochip. For each study, local institutional committees approved study protocols and confirmed that informed consent was obtained.
Traits
Our primary trait was WHRadjBMI, the ratio of waist and hip circumferences adjusted for age, age2, study-specific covariates if necessary, and BMI. For each cohort, residuals were calculated for men and women separately and then transformed by the inverse standard normal function. Cohorts with related men and women provided inverse standard normal transformed sex-combined residuals. For each cohort, the same transformations were applied to other traits: (i) WHR without adjustment for BMI (WHR); (ii) waist circumference with (WCadjBMI) and without (WC) adjustment for BMI; and (iii) hip circumference with (HIPadjBMI) and without (HIP) adjustment for BMI.
European ancestry meta-analysis for genome-wide SNP array data
Sample and SNP quality control (QC) were undertaken within each cohort (Supplementary Table 3)44. The GWAS scaffold in each cohort was imputed up to CEU haplotypes from HapMap resulting in ~2.5 million SNPs. Each directly typed and imputed SNP passing QC was tested for association with each trait under an additive model in a linear regression framework (Supplementary Table 3). SNP positions are reported based on NCBI Build 36. For each cohort, sex-specific association summary statistics were corrected for residual population structure using the genomic control inflation factor45 (median λGC=1.01, range=0.99 – 1.08). SNPs were removed prior to meta-analysis if they had a minor allele count ≤ 3, deviation from Hardy-Weinberg equilibrium exact P<10−6, directly genotyped SNP call rate<95%, or low imputation quality (below 0.3 for MACH, 0.4 for IMPUTE, and 0.8 for PLINK). Association summary statistics for each trait were combined via inverse-variance weighted fixed-effects meta-analysis and corrected for a second round of genomic control to account for structure between cohorts (Extended Data Fig. 1, Supplementary Fig. 1).
European ancestry meta-analysis for Metabochip data
Sample and SNP QC analyses were undertaken in each cohort (Supplementary Table 3). Each SNP passing QC was tested for association with each trait under an additive model using linear regression. The Metabochip array11 is enriched, by design, for loci associated with anthropometric and cardiometabolic traits, thus, we based our correction on 4,425 SNPs selected for inclusion based on associations with QT-interval that were not expected to be associated with anthropometric traits (>500 kb from variants on Metabochip46 for these traits). These study-specific inflation factors had a median λGC=1.01(range 0.93–1.11), with only one study exceeding 1.10. After removing SNPs for QC as described in the previous section, association summary statistics were combined via inverse-variance weighted fixed-effects meta-analysis and corrected for a second round of genomic control on the basis of QT-interval SNPs to account for structure between cohorts.
European ancestry meta-analyses
Association summary statistics from the two parts of the European ancestry arm were combined via inverse-variance weighted fixed-effects meta-analysis using METAL47 with no further genomic control correction. Results were reported for SNPs with a sex-combined sample size≥50,000. The meta-analyses were repeated for men and women separately for each trait. Analyses were corrected for population structure within each sex. The meta-analysis of WHRadjBMI in men included up to 93,480 individuals, and in women up to 116,742 individuals.
Meta-analyses of studies of all ancestries
Sample and SNP QC, tests of association, genomic control correction (median λGC=1.01, range=0.90–1.17, with only one study exceeding 1.10), and meta-analyses were performed as described above. Association summary statistics from the European and non-European ancestry meta-analyses were combined via inverse-variance weighted fixed-effects meta-analysis without further genomic control correction.
Heterogeneity
For each lead SNP, we tested for sex differences based on the sex-specific beta estimates and standard errors, while accounting for potential correlation between estimates as previously used in Randall et al10. Similarly, we tested for potential differences in effects between European and non-European samples, comparing the effects from GWAS+Metabochip data for Europeans and Metabochip data for non-Europeans, and we tested for differences between population-based studies and samples ascertained on diabetes status, and cardiovascular disease, or both. In assessing effects of ascertainment overall, we compared effects in seven subsets of our study sample using population-based studies (i.e., those not ascertained on any phenotype) as the referent population: 1) all studies ascertained on any phenotype, 2) T2D cases, 3) T2D controls, 4) T2D cases+controls, 5) CAD cases, 6) CAD controls, and 7) CAD cases+controls. We evaluated significance for heterogeneity tests within each comparison using a Bonferroni-corrected p-value of 0.05/49=0.05/49=1.02×10−3 as well as an FDR threshold48 of <5% (Supplementary Table 28). Between-study heterogeneity in all meta-analyses was assessed using I2 statistics49.
Heritability and genetic and phenotypic correlations of waist traits
We calculated the heritability and genetic correlations of several central obesity traits using variance component models50,51 in the Framingham Heart Study (FHS) and TWINGENE study. In this approach, the phenotypic variance is decomposed into additive genetic, non-additive genetic, and environmental sources of variation (including model error), and for sets of traits, the covariances between traits. We report narrow sense heritability (h2), the ratio of the additive genetic variance to the total phenotypic variance. Sex-specific inverse normal trait residuals, adjusted for age (and cohort in FHS), were used to estimate heritability separately in men and women, using variance components analysis in SOLARv.4.2.752 (FHS) or M×1.70353 (TWINGENE). Additionally, the sex-specific residuals were used to conduct bivariate quantitative variance component genetic analyses that calculate genetic and environmental correlations between traits. The genetic correlations obtained are estimates of the additive effects of shared genes, and a genetic correlation significantly different from zero suggests a direct influence of the same genes on more than one trait. Similarly, significant environmental correlations suggest shared environmental effects.
We estimated sex-stratified correlations between all waist traits, as well as BMI, height, and weight in TWINGENE, FHS, KORA, and EGCUT. In TWINGENE and FHS, age-adjusted Pearson correlations were used; in EGCUT and KORA, correlations were adjusted for age and age2.
European ancestry approximate conditional analyses
To evaluate the evidence for multiple association signals within identified loci, we performed approximate conditional analyses of sex-combined, women-specific, and men-specific data as implemented in the GCTA software14,54. This approach makes use of association summary statistics from the combined European ancestry meta-analysis and a reference dataset of individual-level genotype data to estimate LD between variants and hence also the approximate correlation between allelic effect estimates in a joint association model.
To evaluate robustness of the GCTA results, we performed analyses using two reference datasets: Prospective Investigation of the Vasculature in Uppsala Seniors (PIVUS) consisting of 949 individuals from Uppsala County, Sweden with both GWAS and Metabochip genotype data; and Atherosclerosis Risk in Communities (ARIC) consisting of 6,654 individuals of European descent from four communities in the USA with GWAS data. Both GWAS datasets were imputed using data from Phase II of the International HapMap Project55. Results shown use the PIVUS reference dataset because Metabochip genotypes are available (see a comparison in the Supplementary Note). Assuming that the LD correlations between SNPs more than 10 Mb away are zero, and using each reference dataset in turn, we performed a genome-wide stepwise selection procedure to select associated SNPs one-by-one at a P value<5×10−8. For each locus at which multiple association signals were observed in the sex-combined, women-, and/or men-specific data, the SNPs selected by GCTA as independently associated with WHRadjBMI in any of the three meta-analyses are reported, with the SNP identified in the sex-combined analysis taken by default when proxies are identified in the women- and/or men-specific analyses. For SNPs not selected by a particular joint conditional analysis, but identified by either of the other two analyses, summary statistics were calculated for association analysis of the SNP conditioned on the GCTA-selected SNP(s).
Genetic risk score
We calculated a genetic risk score for each individual in the population-based KORA study (1,670 men and 1,750 women) using the 49 identified variants, weighted by the allelic effect from the European ancestry meta-analyses of WHRadjBMI. Sex-combined scores were computed on the basis of the sex-combined meta-analysis. Sex-stratified scores were calculated on the basis of men- and women-specific meta-analyses, where SNPs not achieving nominal significance in the respective sex (P≥0.05) were excluded. For each individual, the sex-combined and sex-stratified risk scores were rounded to the nearest integer for plotting. Risk scores were then tested for association with WHRadjBMI using linear regression.
Explained variance
We calculated the variance explained by a single SNP as:
where MAF is the minor allele frequency, β is the SNP effect estimate computed by meta-analysis, and Var(Y) is the variance of the phenotype Y as it went into the study-specific association testing. To derive the total variance explained by a set of independent SNPs, we computed the sum of single-SNP explained variances under the assumption of independent contributions.
To estimate the polygenic variance explained by all HapMap SNPs, we used the all-SNP estimation approach implemented in GCTA and analysed individuals in the ARIC and TwinGene cohorts, including the first 20 principal components as fixed covariates. After removing one of each pair of individuals with estimated genetic relatedness>0.025, 11,898 unrelated individuals with WHRadjBMI were available.
Fine-mapping analyses
We considered each identified locus, defined as 500 kb upstream and downstream of the lead SNP, and computed 95% credible intervals using a Bayesian approach. On the basis of association summary statistics from the European ancestry, non-European ancestry, or all ancestries sex-combined meta-analyses, we calculated an approximate Bayes’ factor56 in favor of association, given by:
where βj is the allelic effect of the jth SNP, with corresponding standard error σj, and , which incorporates a N(0,0.22) prior for βj. This prior gives high probability to small effect sizes, and only small probability to large effect sizes. We then calculated the posterior probability that the jth SNP is causal by:
where the summation in the denominator is over all SNPs passing QC across the locus. We compared the meta-analysis results and credible sets of SNPs likely to contain the causal variant as described57. Assuming a single causal variant at each locus, a 95% credible set of variants was then constructed by: (i) ranking all SNPs according to their Bayes’ factor; and (ii) combining ranked SNPs until their cumulative posterior probability exceeded 0.95. For each locus, we calculated the number of SNPs contained within the 95% credible sets, and the length of the genomic interval covered by these SNPs.
Comparison of loci across traits
To determine whether the identified loci were also associated with any of 22 cardio-metabolic traits, we obtained association data from meta-analysis consortia DIAGRAM (T2D)58, CARDIoGRAM-C4D (CAD)59, ICBP (SBP, DBP)60, GIANT (BMI, height)36,37, GLGC (HDL, LDL, and TG)61, MAGIC (fasting glucose, fasting insulin, fasting insulin adjusted for BMI, and two-hour glucose)62-64, ADIPOGen (BMI-adjusted adiponectin)65, CKDgen (urine albumin-to-creatinine ratio (UACR), estimated glomerular filtration rate (eGFR), and overall CKD)66,67, ReproGen (age at menarche, age at menopause)68,69, and GEFOS (bone mineral density)70; others provided association data for IgA nephropathy71 (also Kiryluk K, Choi M, Lifton RP, Gharavi AG, unpublished data) and for endometriosis (stage B cases only)72. Proxies (r2>0.80 in CEU) were used when an index SNP was unavailable.
We also searched the National Human Genome Research Institute (NHGRI) GWAS Catalog for previous SNP-trait associations near our lead SNPs73. We supplemented the catalog with additional genome-wide significant SNP-trait associations from the literature13,70,74-80. We used PLINK to identify SNPs within 500 kb of lead SNPs using 1000 Genomes Project Pilot I genotype data and LD (r2) values from CEU81,82; for rs7759742, HapMap release 22 CEU data81,83 were used. All SNPs within the specified regions were compared with the NHGRI GWAS Catalog16.
Enrichment of concordant cross-trait associations and effects
To evaluate whether the alleles associated with increased WHRadjBMI at the 49 identified SNPs convey effects for any of the 22 cardiometabolic traits, we conducted meta-regression analyses of the beta-estimates on these metabolic outcomes from other consortia with the beta-estimates for WHRadjBMI in our data65.
Based on the association data across traits, we generated a matrix of Z-scores by dividing the association betas for each of the 49 WHRadjBMI SNPs for each of 22 traits by their respective standard errors. The traits did not include WHRadjBMI or nephropathy in Chinese subjects, but did include HIPadjBMI and WCadjBMI. Each Z-score was made positive if the original trait-increasing allele also increased the look-up trait and negative if not. Missing associations with were assigned a value of zero. We performed unsupervised hierarchical clustering of the Z score matrix in R using the default settings of the “heatplot” function from the made4 library (version 1.20.0), agglomerating clusters using average linkage and Pearson correlation metric distance. The rows and columns of matrix values were each automatically scaled to range from 3 to −3. Confidence in the hierarchical clustering was assessed by bootstrap analysis (10,000 resamplings) using the R package “pvclust”84.
Identification of candidate functional variants
The 1000 Genomes CEU pilot data were queried for SNPs within 500 kb and in LD (r2>0.7, an arbitrary threshold) with any index SNP. All identified variants were then annotated based on RefSeq transcripts using Annovar to identify potential nonsynonymous variants near identified association signals. The distance between each variant and the nearest transcription start site were calculated using gene annotations from GENCODE (v.12).
To investigate whether SNPs in LD with index SNPs are also in LD with common copy number variants (CNVs), we extracted waist trait association results for a list of SNP proxies that are in high LD (r2>0.8, CEU) with CNVs in European populations as described previously7. Altogether 6,200 CNV-tagging SNPs were used, which are estimated collectively to capture>40% of CNVs>1 kb in size.
Expression quantitative trait loci (eQTLs)
We examined our lead SNPs in eQTL datasets from several sources (Supplementary Note) for cis effects significant at P<10−5. We then checked if the trait-associated SNP also had the strongest association with the expression level of its corresponding transcript. If not, we identified a nearby SNP that had a stronger association with expression (peak transcript SNP) of that transcript. To check whether effects of the peak transcript SNP and waist trait-associated SNP overlapped, we conducted conditional analyses to estimate associations between the waist-associated SNP and transcript level when the peak transcript-associated SNP was also included in the model, and vice versa. If the association for the expression-associated SNP was not significant (P>0.05) when conditioned on the waist-associated SNP, we concluded that the waist-associated SNP is likely to explain a substantial proportion of the variance in gene transcript levels in the region. For SNPs that passed these criteria in either women or men eQTL datasets from deCODE, we investigated sex heterogeneity in gene transcript levels for whole blood (312 men, 435 women) and subcutaneous adipose tissue (252 men, 351 women) based on the sex-specific beta estimates and standard errors, while accounting for potential correlation between the sex-specific associations8.
Epigenomic regulatory element overlap with individual variants
We examined overlap of regulatory elements with the 68 trait-associated variants and variants in LD with them (r2>0.7, 1000 Genomes Phase 1 version 2 EUR85), totaling 1,547 variants. We obtained regulatory element data sets from the ENCODE Consortium24 and Roadmap Epigenomics Project25 corresponding to eight tissues selected based on a current understanding of WHRadjBMI pathways. The 226 regulatory element datasets included experimentally identified regions of open chromatin (DNase-seq, FAIRE-seq), histone modification (H3K4me1, H3K27ac, H3K4me3, H3K9ac, and H3K4me2), and transcription factor binding (Supplementary Table 17). When available, we downloaded data processed during the ENCODE Integrative Analysis24. We processed Roadmap Epigenomics sequencing data with multiple biological replicates using MACS286 and the same Irreproducible Discovery Rate pipeline used in the ENCODE Integrative Analysis. Roadmap Epigenomics data with only a single replicate was processed using MACS2 alone.
Global enrichment of WHRadjBMI-associated loci in epigenomic datasets
We performed permutation-based tests in a subset of 60 open chromatin (DNase-seq) and histone modification (H3K27ac, H3K4me1, H3K4me3, H3K9ac) datasets to identify global enrichment of the WHRadjBMI-associated loci. We matched the index SNP at each locus with 500 variants having no evidence of association (P>0.5, ~1.2 million total variants) with a similar distance to the nearest gene (±11,655 bp), number of variants in LD (±8 variants), and minor allele frequency. Using these pools, we created 10,000 sets of control variants for each of the 49 loci and identified variants in LD (r2>0.7) and within 1 Mb. For each SNP set, we calculated the number of loci with at least one variant located in a regulatory region under the assumption that one regulatory variant is responsible for each association signal. We initially calculated an enrichment P value by finding the proportion of control sets for which as many or more loci overlap a regulatory element than the set of associated loci. For increased P value accuracy, we estimated the P value assuming a sum of binomial distributions to represent the number of index SNPs or their LD proxies that overlap a regulatory dataset compared to the 500 matched control sets.
GRAIL
Gene Relationships Among Implicated Loci (GRAIL)19 is a text-mining algorithm that evaluates the degree of relatedness among genes within trait regions. Using PubMed abstracts, a subset of genes enriched for relatedness and a set of keywords that suggest putative pathways are identified. To avoid potential bias from papers investigating candidate genes stimulated by GWAS, we restricted our search to abstracts published prior to 2006. We tested for enrichment of connectivity in the independent SNPs that were significant in our study at P<10−5.
MAGENTA
To investigate if pathways including predefined sets of genes were enriched in the lower part of the gene P value distribution for WHRadjBMI, we performed a pathway analysis using Magenta 2.420 and SNPs present in both the Metabochip and GWAS meta-analyses. SNPs were assigned to a gene if within 110 kb upstream or 40 kb downstream of the transcript’s boundaries. The most significant SNP P value within this interval was adjusted for putative confounders (gene size, number of SNPs in a gene, LD pattern) using stepwise linear regression, creating a gene association score. If the same SNP was assigned to multiple genes, only the gene with the lowest gene score was kept. The HLA region was removed from further analyses due to its high LD structure and gene density. Each gene was then assigned pathway terms using Gene Ontology (GO), PANTHER, Ingenuity and Kyoto Encyclopedia of Genes and Genomes (KEGG)87-90. Finally, the genes were ranked based on their gene association score, and a modified gene-set enrichment analysis (GSEA) using MAGENTA was performed. This analysis tested for enrichment of gene association score ranks above a given rank cutoff (including 5% of all genes) in a gene-set belonging to a predefined pathway term, compared to multiple, equally sized gene-sets that were randomly sampled from all genes in the genome. 10,000-1,000,000 gene-set permutations were performed.
Data-driven Expression-Prioritized Integration for Complex Traits (DEPICT)
This method is described in detail elsewhere23,36. Briefly, DEPICT uses gene expression data derived from a panel of 77,840 expression arrays91, 5,984 molecular pathways (based on 169,810 high-confidence experimentally-derived protein-protein interactions92), 2,473 phenotypic gene sets (based on 211,882 gene-phenotype pairs from the Mouse Genetics Initiative93), 737 Reactome pathways94, 184 KEGG pathways95, and 5,083 GO terms19. DEPICT uses the expression data to reconstitute the protein-protein interaction gene sets, mouse phenotype gene sets, Reactome pathway gene sets, KEGG pathway gene sets, and GO term gene sets. To avoid biasing the identification of genes and pathways covered by SNPs on the Metabochip, analyses were restricted to GWAS cohort data and included 226 WHRadjBMI SNPs in 78 non-overlapping loci with sex-combined P<10−5. We used DEPICT to map genes to associated WHRadjBMI loci, which then allowed us to (1) systematically identify the most likely causal gene(s) in a given associated region, (2) identify reconstituted gene sets that were enriched in genes from associated regions, and (3) identify tissue and cell type annotations in which genes from associated regions were highly expressed. Associated regions were defined by all genes residing within LD (r2>0.5) distance of the WHRadjBMI-associated index SNPs. Overlapping regions were merged, and SNPs that mapped near to or within the HLA region were excluded. The 93 WHRadjBMI SNPs with P<10−5 (clumping thresholds: HapMap release 27 CEU r2=0.01, 500 kb) resulted in 78 non-overlapping regions. GWAS+Metabochip index SNPs were annotated with DEPICT-prioritized genes if the DEPICT (GWAS-only) SNP was located within 500 kb. To mark related gene sets, we first quantified significant gene sets’ pairwise overlap using a non-probabilistic version of the reconstituted gene sets and the Jaccard index measure. Groups of gene sets with mutual Jaccard indices >0.25 were subsequently referred to as meta gene sets and named by the most significant gene set in the group (Supplementary Table 18 and Fig. 2a). In Figures 2a-b, gene sets with similarities between 0.1-0.25 were connected by an edge that was scaled according to degree of similarity. The Cytoscape tool was used to construct parts of Figure 296. In Figure 2c, we show the significance of all cell type annotations and annotations that were categorized as “Tissues” at the outermost level of the Medical Subject Heading ontology.
Extended Data
Extended Data Figure 1. Overall WHRadjBMI meta-analysis study design.

Data (dashed lines) and analyses (solid lines) related to the genome-wide association study (GWAS) cohorts for waist-hip ratio adjusted for body mass index (WHRadjBMI) are colored red and those related to the Metabochip (MC) cohorts are colored blue. The two genomic control (λGC) corrections (within-study and among-studies) performed on associations from each dataset are represented by gray-outlined circles. The λGC corrections for the GWAS meta-analysis were based on all SNPs and the λGC corrections for the Metabochip meta-analysis were based on a null set of 4,319 SNPs previously associated with QT interval. The joint meta-analysis of the GWAS and MC datasets is colored purple. All SNP counts reflect a sample size filter of N ≥ 50,000 subjects. Additional WHRadjBMI meta-analyses included Metabochip data from up to 14,371 subjects of East Asian, South Asian, or African American ancestry from eight cohorts. Counts for the meta-analyses of waist circumference (WC), hip circumference (HIP), and their BMI-adjusted counterparts (WCadjBMI and HIPadjBMI) differ from those of WHRadjBMI because some cohorts only had phenotype data available for one type of body circumference measurement (see Supplementary Table 2).
Extended Data Figure 2. Female- and male-specific effects, phenotypic variances, and genetic correlations.

a, Figure showing effect beta estimates for the 20 WHRadjBMI SNPs showing significant evidence of sexual dimorphism. Sex-specific effect betas and 95% confidence intervals for SNPs associated with waist-hip ratio adjusted for body mass index (WHRadjBMI) are shown as red circles and blue squares for women and men, respectively. The SNPs are classified into three categories: (i) those showing a female-specific effect (“Women SSE”), namely a significant effect in women and no effect in men (Pwomen < 5 × 10−8, Pmen ≥ 0.05), (ii) those showing a pronounced female effect (“Women CED”), namely a significant effect in women and a less significant but directionally consistent effect in men (Pwomen < 5 × 10−8, 5 × 10−8 < Pmen ≤ 0.05); and (iii) those showing a male-specific effect (“Men SSE”), namely a significant effect in men and no effect in women (Pmen < 5 × 10−8, Pwomen ≥ 0.05). Within each of the three categories, the loci were sorted by increasing P value of sex-based heterogeneity in the effect betas. b, Figure showing standardized sex-specific phenotypic variance components for six waist-related traits. Values are shown in men (M) and women (W) from the Swedish Twin Registry (N = 11,875). The ACE models are decomposed into additive genetic components (A) shown in black, common environmental components (C) in gray, and non-shared environmental components (E) in white. Components are shown for waist circumference (WC), hip circumference (HIP), waist-hip ratio (WHR), and their body mass index (BMI)-adjusted counterparts (WCadjBMI, HIPadjBMI, and WHRadjBMI). When the A component is different in men and women with P < 0.05 for a given trait, its name is marked with an asterisk. c, Table showing genetic correlations of waist-related traits with height, adjusted for age and body mass index. Genetic correlations of three traits with height were based on variance component models in the Framingham Heart Study and TWINGENE study (see Online Methods). WCadjBMI, waist circumference adjusted for BMI; WHRadjBMI, waist-hip ratio adjusted for BMI; HIPadjBMI, hip circumference adjusted for BMI.
Extended Data Figure 3. Cumulative genetic risk scores for WHRadjBMI applied to the KORA study cohort.
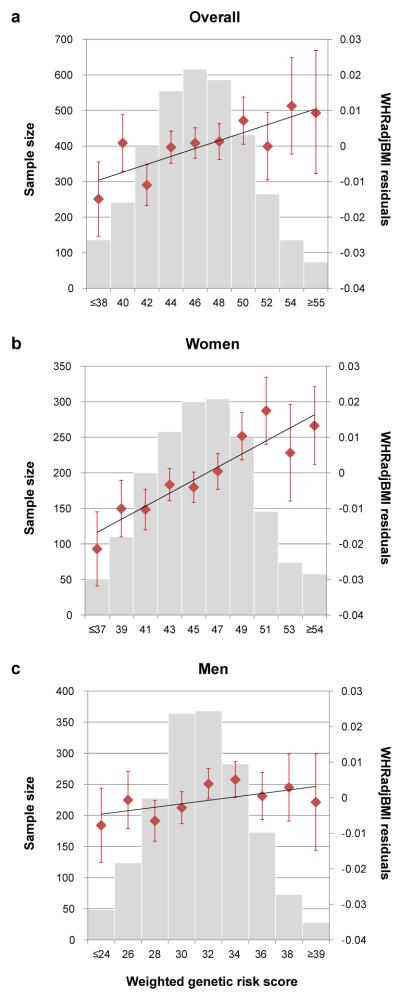
a, All subjects (N = 3,440, Ptrend = 6.7 × 10−4). b, Only women (N = 1,750, Ptrend = 1.0 × 10−11). c, Only men (N = 1,690, Ptrend = 0.02). Each genetic risk score (GRS) illustrates the joint effect of the waist-hip ratio adjusted for body mass index (WHRadjBMI)-increasing alleles of the 49 identified variants from Table 1 weighted by the relative effect sizes from the applicable sex-combined or sex-specific meta-analysis. The mean WHRadjBMI residual and 95% confidence interval is plotted for each GRS category (red dots). The histograms show each GRS is normally distributed in KORA (gray bars).
Extended Data Figure 4. Heat map of unsupervised hierarchical clustering of the effects of 49 WHRadjBMI SNPs on 22 anthropometric and metabolic traits and diseases.

The matrix of Z-scores representing the set of associations was scaled by row (locus name) and by column (trait) to range from −3 to 3. Negative values (blue) indicate that the waist-hip ratio adjusted for body mass index (WHRadjBMI)-increasing allele was associated with decreased values of the trait and positive values (red) indicate that this allele was associated with increased values of the trait. Dendrograms indicating the clustering relationships are shown to the left and above the heat map. The WHRadjBMI-increasing alleles at the 49 lead SNPs segregate into three major clusters comprised of alleles that associate with: 1) larger waist circumference adjusted for BMI (WCadjBMI) and smaller hip circumference adjusted for BMI (HIPadjBMI) (n = 30 SNPs); 2) taller stature and larger WCadjBMI (n = 8 SNPs); and 3) shorter stature and smaller HIPadjBMI (n = 11 SNPs). The three visually identified SNP clusters could be statistically distinguished with >90% confidence. Alleles of the first cluster were predominantly associated with lower high density lipoprotein (HDL) cholesterol and with higher triglycerides and fasting insulin adjusted for BMI (FIadjBMI). eGFRcrea, estimated glomerular filtration rate based on creatinine; LDL cholesterol, low-density lipoprotein cholesterol; UACR, urine albumin-to-creatinine ratio; BMD, bone mineral density.
Extended Data Figure 5. Regulatory element overlap with WHRadjBMI-associated loci.

a, Five variants associated with waist-hip ratio adjusted for body mass index (WHRadjBMI) and located ~77 kb upstream of the first CALCRL transcription start site overlap regions with genomic evidence of regulatory activity in endothelial cells. b, Five WHRadjBMI variants, including rs8817452, in a 1.1 kb region (box) ~250 kb downstream of the first LEKR1 transcription start site overlap evidence of active enhancer activity in adipose nuclei. Signal enrichment tracks are from the ENCODE Integrative Analysis and the Roadmap Epigenomics track hubs on the UCSC Genome Browser. Transcripts are from the GENCODE basic annotation.
Extended Data Table 1. WHRadjBMI loci with multiple association signals in the sex-combined and/or sex-specific approximate conditional meta-analyses.
P values and β coefficients for the association with WHRadjBMI from the joint model in the approximate conditional analysis of combined GWAS and Metabochip studies. SNPs selected by conditional analyses as independently associated with WHRadjBMI in a meta-analysis (sex-combined, women- or men-specific) have their respective summary statistics for these analyses marked in black and bold. SNPs not selected by a particular conditional analysis as independently associated are marked in gray and show the association analysis results for the SNP conditioned on the locus SNPs selected by GCTA. Sample sizes are from the unconditioned meta-analysis.
| Sex-combined | Women | Men | Sex diff. P‡ | CEU r2 with lead SNP | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||||
| Locus* | SNP | Position (bp) | Nearest gene(s) | EA† | EAF | β | P | N | β | P | N | β | P | N | ||
| TBX15- | rs2645294 | 119,376,110 | WARS2 | T | 0,6 | 0.031 | 7.60E-19 | 209,808 | 0.035 | 1.50E-14 | 116,596 | 0.014 | 2.20E-02 | 93,346 | 4.90E-03 | Same |
| WARS2 | rs1106529 | 119,333,020 | TBX15 | A | 0.8 | 0.016 | 1.40E-03 | 209,930 | 0.021 | 1.10E-03 | 116,663 | 0.034 | 4.80E-09 | 93,401 | 1.10E-01 | 0.43 |
| [chr 1] | rs12143789 | 119,298,677 | TBX15 | C | 0.2 | 0.026 | 1.00E-09 | 209,874 | 0.022 | 1.30E-04 | 116,640 | 0.019 | 2.30E-03 | 93,369 | 7.10E-01 | 0.06 |
| rs12731372 | 118,654,498 | SPAG17 | C | 0.8 | 0.024 | 1.30E-09 | 209,856 | 0.02 | 1.10E-04 | 116,636 | 0.028 | 3.40E-06 | 93,354 | 2.80E-01 | >500 kb | |
|
| ||||||||||||||||
| GRB14- | rs1128249∥ | 165,236,870 | COBLL1 | G | 0.6 | 0.062 | 8.60E-19 | 209,414 | 0.093 | 1.00E-24 | 116,348 | −0.002 | 7.10E-01 | 93,200 | 8.60E-22 | 0.93 |
| COBLL1 | rs12692737 | 165,262,555 | COBLL1 | A | 0.3 | 0.043 | 1.60E-08 | 203,265 | 0.134 | 2.70E-26 | 112,317 | 0.003 | 5.70E-01 | 91,082 | 2.80E-21 | 0.71 |
| [chr 2] | rs12692738 | 165,266,498 | COBLL1 | T | 0.8 | 0 021 | 5.90E-05 | 209,551 | 0.092 | 3.80E-20 | 116,474 | −0.005 | 4. 10E-01 | 93,211 | 4.70E-18 | 0.3 |
| rs17185198 | 165,268,482 | COBLL1 | A | 0.8 | 0.002 | 7.40E-01 | 207,702 | 0.072 | 8.50E-13 | 115,657 | −0.004 | 5.80E-01 | 92,179 | 8.00E-11 | 0.15 | |
|
| ||||||||||||||||
| PRBM1 | rs13083798 | 52,624,788 | PRBM1 | A | 0.5 | 0.023 | 4.10E-11 | 209,128 | 0.013 | 1.20E-0l | 115,974 | 0.016 | 1.10E-03 | 93,288 | 7. 40E-01 | 0.88 |
| [chr3] | rs12489828 | 52,542,054 | NT5DC2 | T | 0.6 | 0.011 | 6.50E-02 | 204,485 | 0.029 | 2.60E-10 | 112,633 | −0.015 | 2.90E-03 | 91,986 | 7.20E-11 | 0.57 |
|
| ||||||||||||||||
| MAP3K1 | rs3936510 | 55,896,623 | MAP3K1 | T | 0.2 | 0.022 | 1.50E-06 | 207,896 | 0.042 | 6.00E-12 | 115,645 | −0.002 | 8.20E-0l | 92,386 | 5.90E-07 | 0.88 |
| [chr 5] | rs459193 | 55,842,508 | ANKRD55 | A | 0.3 | 0.026 | 1.60E-11 | 209,952 | 0.016 | 1.90E-03 | 116,677 | 0.033 | 6.70E-09 | 93,410 | 2.30E-02 | 0.06 |
|
| ||||||||||||||||
| VEGFA | rs998584§ | 43,865,874 | VEGFA | A | 0.5 | 0.043 | 1.10E-29 | 189,620 | 0.065 | 1.00E-35 | 106,771 | 0.018 | 8.20E-04 | 82,983 | 3.10E-10 | 0.84 |
| [chr 6] | rs4714699 | 43,910,541 | VEGFA | C | 0.4 | 0.019 | 3.50E-07 | 193,327 | 0.028 | 1.00E-08 | 107,987 | 0.007 | 1.90E-01 | 85,475 | 4.90E-03 | 0.01 |
|
| ||||||||||||||||
| RSP03 | rs1936805§ | 127,493,809 | RSP03 | T | 0.5 | 0.038 | 2.00E-28 | 209,859 | 0.071 | 6.40E-37 | 116,602 | 0.031 | 3.30E-10 | 93,392 | 8.40E-08 | Same |
| [chr 6] | rs11961815 | 127,477,288 | RSP03 | A | 0.8 | 0.022 | 5.00E-06 | 209,679 | 0.037 | 6.50E-09 | 116,503 | 0.021 | 3.60E-03 | 93,310 | 6.90E-02 | 0.32 |
| rs72959041∥ | 127,496,586 | RSP03 | A | 0.1 | 0.101 | 8.70E-15 | 72,472 | - | - | - | - | - | - | - | 0.05 | |
|
| ||||||||||||||||
| NFE2L3, | rs1534696 | 26,363,764 | SNX10 | C | 0.4 | 0.011 | 2.00E-03 | 198,194 | 0.028 | 2.00E-08 | 111,643 | −0.007 | 1.90E-0l | 86,685 | 2.20E-07 | Same |
| SNX10 ¶ | rs10245353 | 25,825,139 | NFE2L3 | A | 0.2 | 0.035 | 8.40E-16 | 210,008 | 0 016 | 1 30E-01 | 116,704 | 0.027 | 1.40E-05 | 93,438 | 3.60E-01 | Same |
| [chr 7] | rs3902751 | 25,828,164 | NFE2L3 | A | 0.3 | 0.009 | 2.00E-0l | 209,969 | 0.039 | 4.20E-14 | 116,676 | 0.019 | 8.40E-04 | 93,427 | 7.40E-03 | 0.608¶ |
|
| ||||||||||||||||
| HOXC13 | rs1443512 | 52,628,951 | HOXC13 | A | 0.2 | 0.016 | 2.70E-03 | 209,980 | 0.04 | 1. 10E-14 | 116,688 | 0.012 | 3.00E-02 | 93,425 | 1.80E-04 | Same |
| [chr 12] | rs10783615 | 52,636,040 | HOXC12 | G | 0.1 | 0.037 | 6.70E-14 | 209,368 | 0.023 | 8.50E-03 | 116,356 | 0.022 | 1.80E-03 | 93,146 | 9.30E-01 | 0.59 |
| rs2071449§ | 52,714,278 | HOXC4/5/6 | A | 0.4 | 0.028 | 5.00E-15 | 206,953 | 0.026 | 4.60E-08 | 114,259 | 0.029 | 3.40E-08 | 92,829 | 6.60E-01 | 0 | |
|
| ||||||||||||||||
| CCDC92 | rs4765219 | 123,006,063 | CCDC92 | C | 0.7 | 0.025 | 6.90E-12 | 209,807 | 0.032 | 2.50E-11 | 116,592 | 0.018 | 5.30E-04 | 93,350 | 3.80E-02 | Same |
| [chr 12] | rs863750 | 123,071,397 | ZNF664 | T | 0 6 | 0.022 | 3.90E-10 | 209,371 | 0.031 | 1.60E-11 | 116,367 | 0.015 | 4.00E-03 | 93,138 | 1.80E-02 | 0.02 |
Locus and lead SNPs are defined by Table 1
The effect allele is the WHRadjBMI-increasing allele in the sex-combined analysis.
Test for sex difference in conditional analysis based on the effect correlation estimate from primary analyses; values significant at the table-wise Bonferroni threshold of 0.05 / 25=2×10−3 are marked in bold.
SNPs selected by conditional analysis in the sex-combined analysis; proxies were selected by joint conditional analysis in the women- and/or men-specific analyses.
SNP not present in the sex-specific meta-analyses due to sample size filter requiring N ≥ 50,000; sample size from GCTA.
At NFE2L3-SNX10, different lead SNPs were identified in the European and all-ancestry analyses but LD is reported with respect to rs10245353. Chr, chromosome; EA, effect allele; EAF, effect allele frequency.
Extended Data Table 2. Enrichments of 49 WHRadjBMI signal SNPs with metabolic and anthropometric traits.
The 49 waist-hip ratio adjusted for body mass index (WHRadjBMI) SNPs were tested for association with other traits by GWAS meta-analyses performed by other groups (see Online Methods). The maximum sample size available is shown overall or separately for 61 cases/controls. N indicates the number of the total SNPs for which the WHRadjBMI-increasing allele is associated with the trait in the concordant direction (increased levels, except for HDL-C, adiponectin, and BMI). One-sided binomial P values test whether this number is greater than expected by chance (null P = 0.5 and null P = 0.025, respectively). The tests do not account for correlation between WHRadjBMI and the tested traits. P values representing significant column-wise enrichment (P < 0.05 / 23 tests) are marked in red and bold.
| SNPs in concordant direction | SNPs in concordant direction with P < 0.05 | ||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Trait | Max. sample size | N | Total | P | N | Total | P |
| Type 2 diabetes (T2D) | 86,200 | 37 | 49 | 2.35E-04 | 16 | 49 | 3.56E-14 |
| Fasting glucose (FG) | 132,996 | 35 | 49 | 1.90E-03 | 8 | 49 | 2.75E-05 |
| Fasting insulin adjusted for BMI (FladjBMI) | 103,496 | 45 | 49 | 4.11E-10 | 36 | 49 | 4.04E-47 |
| 2-hour glucose (G120) | 42,853 | 33 | 49 | 1.06E-02 | 7 | 49 | 2.09E-04 |
| Diastolic blood pressure (DBP) | 69,760 | 34 | 49 | 4.70E-03 | 10 | 49 | 3.21 E-07 |
| Systolic blood pressure (SBP) | 69,774 | 38 | 49 | 7.10E-05 | 6 | 49 | 1.36E-03 |
| Body mass index (BMI) | 322,120 | 40 | 49 | 4.63E-06 | 23 | 49 | 4.42E-24 |
| Height | 253,209 | 25 | 49 | 5.00E-01 | 14 | 49 | 1.10E-11 |
| High-density lipoprotein cholesterol (HDL-C) | 187,142 | 45 | 49 | 4.11E-10 | 24 | 49 | 1.22E-25 |
| Low-density lipoprotein cholesterol (LDL-C) | 173,067 | 33 | 49 | 1.06E-02 | 12 | 49 | 2.32E-09 |
| Triglycerides (TG) | 177,838 | 46 | 49 | 3.49E-11 | 29 | 49 | 6.02E-34 |
| Adiponectin | 29,347 | 41 | 49 | 9.82E-07 | 20 | 49 | 1.28E-19 |
| Endometriosis | 1,364/7,060 | 24 | 45 | 3.83E-01 | 4 | 45 | 2.58E-02 |
| Nephropathy (in Chinese subjects) | 1,194/902 | 18 | 43 | 8.89E-01 | 0 | 43 | 1.00E+00 |
| Nephropathy (in Italian subjects) | 1,045/1,340 | 20 | 43 | 7.29E-01 | 1 | 43 | 6.63E-01 |
| Estimated glomerular filtration rate of creatinine (eGFRcrea) | 74,354 | 29 | 49 | 1.26E-01 | 3 | 49 | 1.24E-01 |
| Chronic kidney disease (CKD) | 74,354 | 17 | 49 | 9.89E-01 | 2 | 49 | 3.47E-01 |
| Urine albumin-to-creatinine ratio (UACR) | 31,580 | 22 | 49 | 8.04E-01 | 2 | 49 | 3.47E-01 |
| Menopause | 87,802 | 28 | 49 | 1.96E-01 | 1 | 49 | 7.11E-01 |
| Menarche | 38,968 | 23 | 49 | 7.16E-01 | 2 | 49 | 3.47E-01 |
| Coronary artery disease (CAD) | 191,198 | 27 | 48 | 2.35E-01 | 9 | 48 | 2.64E-06 |
| Femoral neck bone mineral density (FN-BMD) | 32,960 | 25 | 49 | 5.00E-01 | 4 | 49 | 3.40E-02 |
| Lumbar spine bone mineral density (LS-BMD) | 31,798 | 28 | 49 | 1.96E-01 | 3 | 49 | 1.24E-01 |
Extended Data Table 3. Enrichment of 49 WHRadjBMI-associated loci in epigenomic datasets.
Enrichment of waist-hip ratio adjusted for BMI (WHRadjBMI)-associated loci in regulatory elements from selected WHRadjBMI-relevant tissues. P values are derived using a sum of binomial distributions (see Methods). P values below a Bonferroni-corrected threshold for 60 tests of 8.3 × 10−4 are indicated in bold font. The binomial based P values are similar to P values generated from 10,000 permutation tests. Dashes indicate that datasets were not available.
| Sample | Tissue | DNase I HS | H3K4me1 | H3K27ac | H3K4me3 | H3K9ac |
|---|---|---|---|---|---|---|
| Adipose Nuclei | Adipose | - | 9.6E-06 | 1.2E-13 | 0.0051 | 0.0010 |
| GM12878 | Blood | 0.029 | 0.032 | 0.32 | 0.050 | 0.030 |
| Osteoblasts | Bone | 0.082 | 4.1E-06 | 1.8E-04 | 9.9E-04 | - |
| Astrocytes | Brain | 0.013 | 0.0044 | 0.0077 | 0.0047 | - |
| Anterior Caudate | Brain | - | 2.9E-04 | 0.026 | 0.018 | 0.015 |
| Mid Frontal Lobe | Brain | - | 0.029 | 0.023 | 0.023 | 0.036 |
| Substantia Nigra | Brain | - | 0.047 | - | 0.023 | 0.045 |
| Cerebellum | Brain | 0.048 | - | - | - | - |
| Cerebrum Frontal | Brain | 0.054 | - | - | - | - |
| Frontal Cortex | Brain | 0.022 | - | - | - | - |
| HUVEC | Endothelial | 5.0E-05 | 0.011 | 0.0011 | 0.023 | 0.040 |
| Adult Liver | Liver | - | 0.0057 | - | 0.15 | 0.29 |
| HepG2 | Liver | 0.015 | 7.7E-05 | 0.023 | 5.0E-04 | 0.085 |
| Hepatocytes | Liver | 0.59 | - | - | - | - |
| Huh-7 | Liver | 0.0024 | - | - | - | - |
| Myoctye | Muscle | 2.9E-04 | 1.3E-04 | 0.0026 | 0.015 | 0.0041 |
| PSOAS | Muscle | 0.0012 | - | - | - | - |
| Skeletal Muscle | Muscle | - | 7.3E-04 | 7.8E-05 | 0.0075 | 0.25 |
| Pancreatic Islet | Pancreatic Islets | 0.40 | 0.68 | - | 0.37 | 0.61 |
Extended Data Table 4. Candidate genes at new loci associated with additional waist and hip-related traits.
Candidate genes for loci shown on Table 3 based on secondary analyses or literature review. Further details are provided in other Supplementary Tables and the Supplementary Note. Loci are shown in order of chromosome and position.
| SNP | Trait | Chr | Locus | Expression QTL (P < 10−5)* | GRAIL (P < 0.05)† | Literature‡ | Other GWAS signals§ | nsSNPs and CNVs (r2> 0.7)∥ |
|---|---|---|---|---|---|---|---|---|
| rs10925060 | WCadjBMI | 1 | OR2W5-NRLP3 | - | - | NLRP3 | - | - |
| rs10929925 | HIP | 2 | SOX11 | - | SOX11 | SOX11 | - | - |
| rs2124969 | WCadjBMI | 2 | ITGB6 | PLA2R1 (SAT) | ITGB6 | - | Idiopathic membranous nephropathy (PLA2R1, LY75, ITGB6: RBMS1) | - |
| rs1664789 | WCadjBMI | 5 | ARL15 | - | - | ARL15 | - | - |
| rs 17472426 | WCadjBMI | 5 | CCNJL | - | - | FABP6 | - | - |
| rs722585 | HIPadjBMI | 6 | GMDS | - | - | - | - | - |
| rs7739232 | HIPadjBMI | 6 | KLHL31 | KLHL31 (SAT) | - | KLHL31-GCLC-ELVOL | - | - |
| rs1144 | WCadjBMI | 7 | SRPK2 | SRPK2 (LCL), MLL5 (Omental) | - | - | - | - |
| rs13241538 | HIPadjBMI | 7 | KLF14 | KLF14 (SAT) | - | KLF14 | HDL cholesterol, Triglycerides, Type 2 diabetes: KLF14 | - |
| rs2398893 | WHR | 9 | PTPDC1 | - | BARX1 | - | - | - |
| rs7044106 | HIPadjBMI | 9 | C5 | - | - | - | - | - |
| rs11607976 | HIP | 11 | MYEOV | - | CCND1 | FGF19-FGF4-FGF3 | - | - |
| rs1784203 | WCadjBMI | 11 | KIAA1731 | - | - | - | - | - |
| rs1394461 | WHR | 11 | CNTN5 | - | - | - | - | - |
| rs319564 | WHR | 13 | GPC6 | - | - | GPC6 | - | - |
| rs4985155 | HIP | 16 | PDXDC1 | PDXDC1 (SAT) | - | PLA2G10-NTAN1 | Femoral neck bone mineral density, Lumbar spine bone mineral density, Plasma phospholipid levels, Metabolic traits, Height: PDXDC1, NTAN1 | NTAN1 (S287P), NTAN1 (H283N) |
| rs2047937 | WCadjBMI | 16 | ZNF423 | - | - | ZNF423-CNEP1R1 | - | - |
| rs2034088 | HIPadjBMI | 17 | VPS53 | VPS53 (Liver, SAT), FAM101B (Omental, SAT) | - | - | - | - |
| rs1053593 | HIPadjBMI | 22 | HMGXB4 | TOM1 (PBMC), HMGXB4 (Blood, SAT) | - | HMGXB4 | - |
HMGXB4 (G165V), CNVR8147.1 |
Gene transcript levels associated with SNP genotype (expression QTL) in the indicated tissue(s).
Genes in pathways identified as enriched by GRAIL analysis.
Strongest candidate genes identified based on manual literature review.
Traits associated at P < 5 × 10−8 in GWAS lookups or in the GWAS catalog using the index SNP or a proxy in high linkage disequilibrium (LD) (r2 > 0.7), and the genes(s) named in those reports.
Nonsynonymous variants (nsSNPs) and copy number variants (CNVs) with tag SNPs in high LD with index SNP based on a 1000 Genomes CEU reference panel. DEPICT analysis was not performed for loci associated with these traits. Chr, Chromosome; WCadjBMI, waist circumference adjusted for body mass index (BMI); HIPadjBMI, hip circumference adjusted for BMI; WHR, waist-to-hip ratio.
Supplementary Material
Acknowledgments
We thank the more than 224,000 volunteers who participated in this study. Detailed acknowledgment of funding sources is provided in the Supplementary Note.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
REFERENCES
- 1.Pischon T, et al. General and abdominal adiposity and risk of death in Europe. N. Engl. J. Med. 2008;359:2105–2120. doi: 10.1056/NEJMoa0801891. [DOI] [PubMed] [Google Scholar]
- 2.Wang Y, Rimm EB, Stampfer MJ, Willett WC, Hu FB. Comparison of abdominal adiposity and overall obesity in predicting risk of type 2 diabetes among men. Am. J. Clin. Nutr. 2005;81:555–563. doi: 10.1093/ajcn/81.3.555. [DOI] [PubMed] [Google Scholar]
- 3.Canoy D. Distribution of body fat and risk of coronary heart disease in men and women. Curr. Opin. Cardiol. 2008;23:591–598. doi: 10.1097/HCO.0b013e328313133a. [DOI] [PubMed] [Google Scholar]
- 4.Snijder MB, et al. Associations of hip and thigh circumferences independent of waist circumference with the incidence of type 2 diabetes: the Hoorn Study. Am. J. Clin. Nutr. 2003;77:1192–1197. doi: 10.1093/ajcn/77.5.1192. [DOI] [PubMed] [Google Scholar]
- 5.Yusuf S, et al. Obesity and the risk of myocardial infarction in 27,000 participants from 52 countries: a case-control study. Lancet. 2005;366:1640–1649. doi: 10.1016/S0140-6736(05)67663-5. [DOI] [PubMed] [Google Scholar]
- 6.Mason C, Craig CL, Katzmarzyk PT. Influence of central and extremity circumferences on all-cause mortality in men and women. Obesity (Silver Spring) 2008;16:2690–2695. doi: 10.1038/oby.2008.438. [DOI] [PubMed] [Google Scholar]
- 7.Heid IM, et al. Meta-analysis identifies 13 new loci associated with waist-hip ratio and reveals sexual dimorphism in the genetic basis of fat distribution. Nat. Genet. 2010;42:949–960. doi: 10.1038/ng.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Randall JC, et al. Sex-stratified genome-wide association studies including 270,000 individuals show sexual dimorphism in genetic loci for anthropometric traits. PLoS Genet. 2013;9:e1003500. doi: 10.1371/journal.pgen.1003500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fox CS, et al. Genome-wide association of pericardial fat identifies a unique locus for ectopic fat. PLoS Genet. 2012;8:e1002705. doi: 10.1371/journal.pgen.1002705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fox CS, et al. Genome-wide association for abdominal subcutaneous and visceral adipose reveals a novel locus for visceral fat in women. PLoS Genet. 2012;8:e1002695. doi: 10.1371/journal.pgen.1002695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Voight BF, et al. The metabochip, a custom genotyping array for genetic studies of metabolic, cardiovascular, and anthropometric traits. PLoS Genet. 2012;8:e1002793. doi: 10.1371/journal.pgen.1002793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanna S, et al. Common variants in the GDF5-UQCC region are associated with variation in human height. Nat. Genet. 2008;40:198–203. doi: 10.1038/ng.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lango Allen H, et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature. 2010;467:832–838. doi: 10.1038/nature09410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang J, et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 2012;44:369–375. doi: 10.1038/ng.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang J, et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hindorff LA, et al. [Accessed 31 Jan 2013];A Catalog of Published Genome-Wide Association Studies. Available at http://www.genome.gov/gwastudies.
- 17.Freathy RM, et al. Variants in ADCY5 and near CCNL1 are associated with fetal growth and birth weight. Nat. Genet. 2010;42:430–435. doi: 10.1038/ng.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoopes SL, Willcockson HH, Caron KM. Characteristics of multi-organ lymphangiectasia resulting from temporal deletion of calcitonin receptor-like receptor in adult mice. PLoS ONE. 2012;7:e45261. doi: 10.1371/journal.pone.0045261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raychaudhuri S, et al. Identifying relationships among genomic disease regions: predicting genes at pathogenic SNP associations and rare deletions. PLoS Genet. 2009;5:e1000534. doi: 10.1371/journal.pgen.1000534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Segre AV, Groop L, Mootha VK, Daly MJ, Altshuler D. Common inherited variation in mitochondrial genes is not enriched for associations with type 2 diabetes or related glycemic traits. PLoS Genet. 2010;6:e1001058. doi: 10.1371/journal.pgen.1001058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elias I, Franckhauser S, Bosch F. New insights into adipose tissue VEGF-A actions in the control of obesity and insulin resistance. Adipocyte. 2013;2:109–112. doi: 10.4161/adip.22880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pal A, et al. PTEN mutations as a cause of constitutive insulin sensitivity and obesity. N. Engl. J. Med. 2012;367:1002–1011. doi: 10.1056/NEJMoa1113966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pers T, et al. Biological interpretation of genome-wide association studies using predicted gene functions. 2014. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bernstein BE, et al. The NIH Roadmap Epigenomics Mapping Consortium Nat. Biotechnol. 2010;28:1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakagami H. The mechanism of white and brown adipocyte differentiation. Diabetes Metab J. 2013;37:85–90. doi: 10.4093/dmj.2013.37.2.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, et al. miR-17-5p and miR-106a are involved in the balance between osteogenic and adipogenic differentiation of adipose-derived mesenchymal stem cells. Stem Cell Res. 2013;10:313–324. doi: 10.1016/j.scr.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 28.Mori M, Nakagami H, Rodriguez-Araujo G, Nimura K, Kaneda Y. Essential role for miR-196a in brown adipogenesis of white fat progenitor cells. PLoS Biol. 2012;10:e1001314. doi: 10.1371/journal.pbio.1001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cao Y. Angiogenesis and vascular functions in modulation of obesity, adipose metabolism, and insulin sensitivity. Cell Metab. 2013;18:478–489. doi: 10.1016/j.cmet.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 30.Hagberg CE, et al. Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature. 2010;464:917–921. doi: 10.1038/nature08945. [DOI] [PubMed] [Google Scholar]
- 31.Zygmunt T, et al. Semaphorin-PlexinD1 signaling limits angiogenic potential via the VEGF decoy receptor sFlt1. Dev. Cell. 2011;21:301–314. doi: 10.1016/j.devcel.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shimizu I, et al. Semaphorin3E-Induced Inflammation Contributes to Insulin Resistance in Dietary Obesity. Cell Metab. 2013;18:491–504. doi: 10.1016/j.cmet.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 33.Hanada R, et al. Neuromedin U has a novel anorexigenic effect independent of the leptin signaling pathway. Nat. Med. 2004;10:1067–1073. doi: 10.1038/nm1106. [DOI] [PubMed] [Google Scholar]
- 34.Huang X, et al. FGFR4 prevents hyperlipidemia and insulin resistance but underlies high-fat diet induced fatty liver. Diabetes. 2007;56:2501–2510. doi: 10.2337/db07-0648. [DOI] [PubMed] [Google Scholar]
- 35.Foti D, et al. Lack of the architectural factor HMGA1 causes insulin resistance and diabetes in humans and mice. Nat. Med. 2005;11:765–773. doi: 10.1038/nm1254. [DOI] [PubMed] [Google Scholar]
- 36.Locke AE, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2014 doi: 10.1038/nature14177. Accompanying manuscript. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wood AR, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat. Genet. 2014 doi: 10.1038/ng.3097. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jaager K, Neuman T. Human dermal fibroblasts exhibit delayed adipogenic differentiation compared with mesenchymal stem cells. Stem Cells Dev. 2011;20:1327–1336. doi: 10.1089/scd.2010.0258. [DOI] [PubMed] [Google Scholar]
- 39.Goossens GH, et al. Expression of NLRP3 inflammasome and T cell population markers in adipose tissue are associated with insulin resistance and impaired glucose metabolism in humans. Mol. Immunol. 2012;50:142–149. doi: 10.1016/j.molimm.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 40.Maynard LM, et al. Childhood body composition in relation to body mass index. Pediatrics. 2001;107:344–350. doi: 10.1542/peds.107.2.344. [DOI] [PubMed] [Google Scholar]
- 41.Wells JC. Sexual dimorphism of body composition. Best Pract. Res. Clin. Endocrinol. Metab. 2007;21:415–430. doi: 10.1016/j.beem.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 42.Lovejoy JC, Champagne CM, de Jonge L, Xie H, Smith SR. Increased visceral fat and decreased energy expenditure during the menopausal transition. Int. J. Obes. 2008;32:949–958. doi: 10.1038/ijo.2008.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okada Y, et al. A genome-wide association study in 19,633 Japanese subjects identified LHX3-QSOX2 and IGF1 as adult height loci. Hum. Mol. Genet. 2010;19:2303–2312. doi: 10.1093/hmg/ddq091. [DOI] [PubMed] [Google Scholar]
- 44.Winkler TW, et al. Quality control and conduct of genome-wide association meta-analyses. Nat. Protoc. 2014;9:1192–1212. doi: 10.1038/nprot.2014.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 46.Buyske S, et al. Evaluation of the metabochip genotyping array in African Americans and implications for fine mapping of GWAS-identified loci: the PAGE study. PLoS ONE. 2012;7:e35651. doi: 10.1371/journal.pone.0035651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289–300. [Google Scholar]
- 49.Higgins JP, Thompson SG. Quantifying heterogeneity in a meta-analysis. Stat. Med. 2002;21:1539–1558. doi: 10.1002/sim.1186. [DOI] [PubMed] [Google Scholar]
- 50.Neale MC, Cardon LR, North Atlantic Treaty Organization . Methodology for genetic studies of twins and families. Kluwer Academic Publishers; 1992. Scientific Affairs Division. [Google Scholar]
- 51.Falconer DS. Introduction to Quantitative Genetics. 3rd edn Oliver and Boyd; 1990. [Google Scholar]
- 52.Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am. J. Hum. Genet. 1998;62:1198–1211. doi: 10.1086/301844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Neale MC. MX: Statistical Modeling. 4th edn Department of Psychiatry, Medical College of Virginia; 1997. [Google Scholar]
- 54.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Frazer KA, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wakefield J. A Bayesian measure of the probability of false discovery in genetic epidemiology studies. Am. J. Hum. Genet. 2007;81:208–227. doi: 10.1086/519024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wellcome Trust Case Control Consortium Bayesian refinement of association signals for 14 loci in 3 common diseases. Nat. Genet. 2012;44:1294–1301. doi: 10.1038/ng.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morris AP, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat. Genet. 2012;44:981–990. doi: 10.1038/ng.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Deloukas P, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat. Genet. 2013;45:25–33. doi: 10.1038/ng.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ehret GB, et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011;478:103–109. doi: 10.1038/nature10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Global Lipids Genetics Consortium Discovery and refinement of loci associated with lipid levels. Nat. Genet. 2013;45:1274–1283. doi: 10.1038/ng.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Scott RA, et al. Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat. Genet. 2012;44:991–1005. doi: 10.1038/ng.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Manning AK, et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat. Genet. 2012;44:659–669. doi: 10.1038/ng.2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Saxena R, et al. Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat. Genet. 2010;42:142–148. doi: 10.1038/ng.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dastani Z, et al. Novel loci for adiponectin levels and their influence on type 2 diabetes and metabolic traits: a multi-ethnic meta-analysis of 45,891 individuals. PLoS Genet. 2012;8:e1002607. doi: 10.1371/journal.pgen.1002607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pattaro C, et al. Genome-wide association and functional follow-up reveals new loci for kidney function. PLoS Genet. 2012;8:e1002584. doi: 10.1371/journal.pgen.1002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Boger CA, et al. CUBN is a gene locus for albuminuria. J. Am. Soc. Nephrol. 2011;22:555–570. doi: 10.1681/ASN.2010060598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stolk L, et al. Meta-analyses identify 13 loci associated with age at menopause and highlight DNA repair and immune pathways. Nat. Genet. 2012;44:260–268. doi: 10.1038/ng.1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Elks CE, et al. Thirty new loci for age at menarche identified by a meta-analysis of genome-wide association studies. Nat. Genet. 2010;42:1077–1085. doi: 10.1038/ng.714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Estrada K, et al. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat. Genet. 2012;44:491–501. doi: 10.1038/ng.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gharavi AG, et al. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat. Genet. 2011;43:321–327. doi: 10.1038/ng.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Painter JN, et al. Genome-wide association study identifies a locus at 7p15.2 associated with endometriosis. Nat. Genet. 2011;43:51–54. doi: 10.1038/ng.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hindorff LA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. U. S. A. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kamatani Y, et al. Genome-wide association study of hematological and biochemical traits in a Japanese population. Nat. Genet. 2010;42:210–215. doi: 10.1038/ng.531. [DOI] [PubMed] [Google Scholar]
- 75.Franke A, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sawcer S, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–219. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang KS, Liu XF, Aragam N. A genome-wide meta-analysis identifies novel loci associated with schizophrenia and bipolar disorder. Schizophr. Res. 2010;124:192–199. doi: 10.1016/j.schres.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 78.Cirulli ET, et al. Common genetic variation and performance on standardized cognitive tests. Eur. J. Hum. Genet. 2010;18:815–820. doi: 10.1038/ejhg.2010.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gieger C, et al. New gene functions in megakaryopoiesis and platelet formation. Nature. 2011;480:201–208. doi: 10.1038/nature10659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Need AC, et al. A genome-wide study of common SNPs and CNVs in cognitive performance in the CANTAB. Hum. Mol. Genet. 2009;18:4650–4661. doi: 10.1093/hmg/ddp413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Abecasis GR, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.The International HapMap Project Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 84.Suzuki R, Shimodaira H. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics. 2006;22:1540–1542. doi: 10.1093/bioinformatics/btl117. [DOI] [PubMed] [Google Scholar]
- 85.1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Feng J, Liu T, Qin B, Zhang Y, Liu XS. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 2012;7:1728–1740. doi: 10.1038/nprot.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ashburner M, et al. The Gene Ontology Consortium Gene ontology: tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mi H, Thomas P. PANTHER pathway: an ontology-based pathway database coupled with data analysis tools. Methods Mol. Biol. 2009;563:123–140. doi: 10.1007/978-1-60761-175-2_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jimenez-Marin A, Collado-Romero M, Ramirez-Boo M, Arce C, Garrido JJ. Biological pathway analysis by ArrayUnlock and Ingenuity Pathway Analysis. BMC Proc. 2009;3(Suppl 4):S6. doi: 10.1186/1753-6561-3-S4-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cvejic A, et al. SMIM1 underlies the Vel blood group and influences red blood cell traits. Nat. Genet. 2013;45:542–545. doi: 10.1038/ng.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lage K, et al. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat. Biotechnol. 2007;25:309–316. doi: 10.1038/nbt1295. [DOI] [PubMed] [Google Scholar]
- 93.Bult CJ, Blake JA, Kadin JA, Ringwald M, Eppig JT, Mouse Genome Database Group . IEEE International Symposium on Bio-Informatics and Biomedical Engineering. pp. 29–32. R. J. [Google Scholar]
- 94.Croft D, et al. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res. 2011;39:D691–697. doi: 10.1093/nar/gkq1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40:D109–114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Saito R, et al. A travel guide to Cytoscape plugins. Nat. Methods. 2012;9:1069–1076. doi: 10.1038/nmeth.2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.