

ABSTRACT
The RNA-dependent RNA polymerase (RdRp) of hepatitis C virus (HCV) is essential for viral genome replication. Crystal structures of the HCV RdRp reveal two C-terminal features, a β-loop and a C-terminal arm, suitably located for involvement in positioning components of the initiation complex. Here we show that these two elements intimately regulate template and nucleotide binding, initiation, and elongation. We constructed a series of β-loop and C-terminal arm mutants, which were used for in vitro analysis of RdRp de novo initiation and primer extension activities. All mutants showed a substantial decrease in initiation activities but a marked increase in primer extension activities, indicating an ability to form more stable elongation complexes with long primer-template RNAs. Structural studies of the mutants indicated that these enzyme properties might be attributed to an increased flexibility in the C-terminal features resulting in a more open polymerase cleft, which likely favors the elongation process but hampers the initiation steps. A UTP cocrystal structure of one mutant shows, in contrast to the wild-type protein, several alternate conformations of the substrate, confirming that even subtle changes in the C-terminal arm result in a more loosely organized active site and flexible binding modes of the nucleotide. We used a subgenomic replicon system to assess the effects of the same mutations on viral replication in cells. Even the subtlest mutations either severely impaired or completely abolished the ability of the replicon to replicate, further supporting the concept that the correct positioning of both the β-loop and C-terminal arm plays an essential role during initiation and in HCV replication in general.
IMPORTANCE HCV RNA polymerase is a key target for the development of directly acting agents to cure HCV infections, which necessitates a thorough understanding of the functional roles of the various structural features of the RdRp. Here we show that even highly conservative changes, e.g., Tyr→Phe or Asp→Glu, in these seemingly peripheral structural features have profound effects on the initiation and elongation properties of the HCV polymerase.
INTRODUCTION
Viral RNA-dependent RNA polymerases (RdRps) represent the catalytic core that facilitates genome replication of RNA viruses. In order to maintain genome integrity, all RdRps must have a mechanism for initiating replication effectively. In vivo, viral RdRps can initiate genome synthesis by using either a primer-dependent or a de novo (primer-independent) mechanism (1, 2). In the primer-dependent mechanism, a short oligonucleotide or a protein is used to provide an initial platform on which the nascent strand is built. In de novo initiation, the RdRp links the second incoming nucleotide to the already present nucleotide known as the initiating nucleotide (NTPi). The hepatitis C virus (HCV) RdRp and other related viral RdRps utilize such a mechanism in vivo (1, 2), although primer-dependent synthesis can be measured in vitro. Recently, short di- and trinucleotide primers have been used to produce stalled, highly stable elongation complexes (3).
Crystal structures of viral RdRps from ∼20 distinct positive-sense RNA virus families have been determined (4–25). These viral RdRps assume the prototypical “right-hand” architecture characteristic of all polymerases, with subdomains termed fingers, palm, and thumb. Additional features have been identified in viral RdRp structures, which are likely critical for priming, elongation, and processivity. For example, extended loops common to all viral RdRps interconnect the finger and thumb domains to encircle the active site. These loops have been shown to have a role in discrimination between double- and single-stranded templates (26). Although structurally diverse overall, a number of common active-site features have been identified among the de novo-initiating RdRps. These polymerases, including those of members of the Flaviviridae family (4–7, 13, 17, 18), bacteriophage φ6 (8), and members of the Reoviridae (9), possess an elaborate thumb domain as well as C-terminal features with loops that penetrate deep into the active site. Such features and extensions, which are absent in the prototypical (or “classical”) primer-dependent DNA polymerases and reverse transcriptases (RTs), could conceivably play a role in de novo initiation. The HCV RdRp, encoded by the viral NS5B gene, contains two such structural elements: a β-loop in the thumb domain and a long flexible C-terminal arm (4–7, 13, 17, 18). When superimposed onto the ternary complex of HIV RT—double-stranded DNA (dsDNA), the β-loop clashes with the bound duplex, and both elements block the natural exit from the NS5B active site. This finding has led to speculations that in HCV NS5B, one or both elements may regulate RNA binding and positioning.
Studies using recombinant HCV NS5B mutants with either the β-loop or the C-terminal arm removed have begun to define the roles of these elements. The removal of either element results in improved in vitro activity (27–29). However, this is accompanied by a relaxed specificity during initiation. It has been shown that mutants lacking the β-loop can initiate at internal sites on the template or use double-stranded RNA templates not used by the wild-type (WT) enzyme (1, 28). Furthermore, the removal of either element affects the mode of initiation used. In vitro, HCV NS5B can use either a de novo or a primer-dependent mode of initiation. An rGTP-dependent mechanism for discrimination between these methods of initiation has also been demonstrated, whereby rGTP binding at the NTPi site inhibits primer-dependent initiation (6, 30). This effect is dramatically decreased, however, in the absence of either the β-loop or the C-terminal arm.
In this work, we demonstrate that even the subtlest changes, such as Tyr-to-Phe or Asp-to-Glu changes, introduced by site-directed mutagenesis into the β-loop or C-terminal arm can have significant effects on in vitro activities and ultimately on viral replication. These effects are shown to culminate in altered rNTP binding, dramatically reduced initiation activities, and improved primer extension capabilities.
MATERIALS AND METHODS
Protein expression and purification.
NS5B of genotype 1b strain J4 was previously cloned into pET23a with a C-terminal His tag and a C-terminal 21-amino-acid truncation (NS5B-Δ21) (7). The protein was expressed in Escherichia coli and purified as described previously (7), with one additional purification step. After elution from Ni-nitrilotriacetic acid (NTA) Superflow resin, fractions containing NS5B were pooled and applied onto poly(U)-Sepharose 4B resin (Amersham Biosciences), preequilibrated with buffer A (150 mM NaCl, 10% glycerol, 5 mM dithiothreitol [DTT], 10 mM Tris [pH 7.5]). The column was washed with 20 column volumes of buffer A, and the protein was eluted with a 200-ml gradient of buffer B (1 M NaCl, 10% glycerol, 5 mM DTT, 10 mM Tris [pH 7.5]). Fractions were checked for purity by SDS-PAGE on a 12.5% acrylamide gel. Fractions of >95% purity were pooled and dialyzed against storage buffer (100 mM Tris [pH 8], 300 mM NaCl, 30% glycerol, and 5 mM DTT). Mutations were created in HCV J4 NS5B-Δ21 cDNA by using the QuikChange site-directed mutagenesis kit (Stratagene). The entire cDNA was then sequenced to confirm the presence of the mutation and that no additional changes had occurred inadvertently. The mutant plasmids were transformed into E. coli BL21(DE3) cells and expressed and purified in the same manner as for the wild-type protein.
Preparation of a 3′-NCR RNA template for RdRp activity assays.
The pGEM92f 3′-NCR plasmid, containing a 696-bp sequence encompassing the 3′ noncoding region (NCR) and part of NS5B, was linearized with ScaI and used for in vitro transcription reactions with a T7 RiboMAX large-scale RNA production kit (Promega). After treatment with DNase I, the RNA transcript was purified by phenol-chloroform extraction and ethanol precipitation and desalted by using a G-50 Nick spin column (Pharmacia), eluting the product into 10 mM Tris (pH 7.6). RNA was quantified by measurement of the optical density at 260 nm, and integrity was checked by agarose gel electrophoresis.
Primer extension assays.
Extension reactions were performed with 37-μl volumes containing 200 nM NS5B, 1.72 μg/ml 3′ NCR, 3.2 mM MgCl2, 2 μM UTP, 8 μCi/ml [α-33P]UTP (Amersham Bioscience), 500 μM GTP, 500 μM CTP, 500 μM ATP, 125 mM NaCl, 20 mM Tris (pH 7), 1 mM DTT, and 0.4 U/μl RNasin (Promega). Reaction mixtures were incubated at 30°C for the times indicated, and reactions were stopped by the addition of an equal volume of 0.1 M EDTA in phosphate-buffered saline (PBS). Aliquots of the reaction mix were spotted onto DEAE filter mats (PerkinElmer), and unincorporated nucleotides were removed by washing twice for 15 min in 2× saline-sodium citrate buffer (300 mM NaCl, 30 mM sodium citrate [pH 7.0]) and then once in 95% ethanol. Filter mats were dried, sealed in sample bags containing 10 ml Optiscint HiSafe scintillation fluid (PerkinElmer), and measured for radioactive counts with a Wallac 1205 MicroBeta counter. Radiolabel specific activity was determined by spotting an aliquot of the quenched reaction mix onto the filter mat after the washing steps. In single-cycle experiments, reaction mixtures included 8 μCi/ml [α-33P]GTP (PerkinElmer) and only 3 μM cold GTP. NS5B and 3′-NCR RNA were incubated for 20 min at 30°C, and reactions were started by the addition of nucleotides and 0.25 mg/ml heparin (Sigma-Aldrich).
De novo initiation assays.
Initiation assays were performed with 40-μl volumes containing freshly purified mutant and wild-type NS5B samples at 500 nM; 300 nM 3′-dideoxy-terminated RNA (5′-CCUUCUAUCUCGUAUACCdd-3′); 4 mM MgCl2; 300 nM each ATP, CTP, and UTP; 200 nM GTP, 0.5 μl of [α-33P]GTP (10 μCi/ml; MP Biomedicals); 25 mM NaCl; 25 mM Tris (pH 7.5); 1 mM DTT; 0.4 U/μl RNasin (Promega); and 0.25% Triton X-100. Reaction mixtures were preincubated for 15 min at 27°C, and reactions were started by the addition of cold and labeled rGTP. Assays were allowed to proceed for up to 120 min and stopped by adding formamide (90%, vol/vol), SDS (0.03%, wt/vol), and 100 mM EDTA to the mixtures. Assay samples were loaded and separated on a 20% acrylamide–7 M urea denaturing 1× Tris-borate-EDTA (TBE) gel. A Typhoon 9400 scanner (GE Healthcare) was used to image and analyze the gels.
Cell cultures.
Cell monolayers of the human hepatoma cell line Huh-7 (31) were routinely grown in Dulbecco's modified Eagle medium (DMEM; Invitrogen Life Technologies) lacking sodium pyruvate and supplemented with 4.5 mg/ml glucose, 2 mM l-glutamine, 0.1 mM nonessential amino acids, 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% fetal calf serum. Cell lines were incubated at 37°C in 5% CO2 and passaged twice weekly by treatment with 0.05% trypsin–0.02% EDTA and seeding at a dilution of 1:3 to 1:5.
Plasmid constructions.
Construction of pPI-ETSacIIJ4 was based on the genotype 1b strain Con1 transient luciferase replicon vector PI-ET (32), in which the Con1 NS5B region had been replaced by J4 sequences. Mutations were created in the pPI_ETSacIIJ4 cDNA by using the QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's protocol. The entire cDNA was then sequenced to confirm the presence of the mutation and that no additional change had taken place inadvertently.
Preparation of replicon RNA.
Runoff transcripts of HCV replicon RNA were generated by linearizing pPI_ETSacIIJ4 plasmid DNA with consecutive restriction digests using AseI and ScaI (New England BioLabs). The linearized DNA was extracted with phenol-chloroform, ethanol precipitated, and dissolved in RNase-free deionized water. In vitro transcription reactions were set up by using 1 μg of linearized DNA as a template, in 100-μl volumes, using the T7 RiboMAX large-scale RNA production system (Promega) according to the manufacturer's instructions. Reaction mixtures were incubated for 4 h at 30°C before the addition of a further 100 U of T7 RNA polymerase and further incubation at 30°C for 1.5 h. Reactions were terminated, and the template DNA was degraded by incubation with 20 U of RQ1 RNase-free DNase for 1 h at 37°C. The RNA product was purified by using RNeasy kits (Qiagen), by eluting the RNA in 50 μl of RNase-free deionized water. RNA was quantified by measurement of the optical density at 260 nm, and integrity was checked by agarose gel electrophoresis.
Transient-replication assays.
Subconfluent monolayers of Huh-7 cells were detached from the culture dish by treatment with trypsin and 0.02% EDTA, washed with 50 ml PBS, and resuspended in 1× Optimix solution (Equibio Ltd.) containing 2 mM ATP and 5 mM glutathione, to give a final concentration of 1 × 107 cells/ml. The cell suspension (400 μl) was mixed with 10 μg replicon RNA by gentle pipetting, transferred into a 0.4-cm electroporation cuvette, and subjected to an electric pulse at 950 μF and 270 V by using a Gene Pulser apparatus (Bio-Rad). Electroporated cells were mixed with 9 ml of DMEM (supplemented as described above for normal cell culture procedures), and 2 ml of the cell suspension was seeded into 32-mm-diameter wells in cell culture plates. Plates were incubated at 37°C in 5% CO2 and harvested at the given time points. Medium was removed from cells and replaced with 400 μl of Glo Lysis buffer (Promega). After incubation for 10 min at room temperature, cells were washed off the bottom of the well, and the cell suspension was removed. Duplicate 100-μl volumes of the cell suspension were mixed with 25 μl of luciferase reagent (Promega) in the wells of white opaque OptiPlate 96-well plates (PerkinElmer) and incubated in the dark for 5 min before measurement of luminescence by using a Packard TopCount NXT instrument. Values obtained for cells harvested at 4 h posttransfection were used to normalize the data for transfection efficiency.
Protein crystallization and X-ray data collection.
Purified wild-type and mutant NS5B proteins were concentrated to 7 to 10 mg/ml in a solution containing 600 mM NaCl, 10% glycerol, 5 mM DTT, and 10 mM Tris (pH 7.5). Crystals were grown by the hanging-drop vapor diffusion method at 18°C with mixing of 1:1 volumes of the protein and a reservoir solution containing 50 mM morpholineethanesulfonic acid (MES) (pH 5.0), 20% polyethylene glycol 4000 (PEG 4000), 10% glycerol, and 5 mM DTT (7). Wild-type NS5B-Δ21 and the W550A, D559E, and Y448F mutants produced crystals with dimensions of up to 60 μm by 60 μm by 400 μm under these conditions. Crystals of the W550N mutant, which were much smaller, were transferred into reservoir solution containing 22% PEG 4000 and crushed to produce crystal seeds. Seeds were streaked into hanging drops set up as described above and grew into crystals with maximum dimensions of 40 μm by 40 μm by 500 μm. Crystals were flash-frozen in liquid nitrogen after soaking for 10 to 30 s in cryoprotectant containing 50 mM MES (pH 5.1), 20% glycerol, 5 mM DTT, and 20% PEG 4000. For the W550N/UTP/Mn2+ complex, 12 mM UTP was soaked into crystals for 6 min before transferring them into cryoprotectant solution containing 15 mM MnCl2. Crystals were maintained at 100 K by using an Oxford Cryosystems cryostream, and diffraction data were collected with a MAR345 image plate detector and an ADSC Quantum 4R charge-coupled-device (CCD) detector at the Synchrotron Radiation Source, Daresbury Laboratory, on stations 14.1 and 14.2. Analysis of the diffraction data showed that all crystals were isomorphous to the previously determined wild-type crystal form, also belonging to space group P212121, with typical unit cell dimensions of a = 106 Å, b = 108 Å, and c = 134 Å (7). The Matthews parameter, VM, was 2.91 Å3 Da−1, assuming two molecules in the asymmetric unit. Further details are collated in Table 1.
TABLE 1.
Crystallographic statistics for HCV RNA polymerase mutants
| Parameter | Value for HCV RNA polymerase mutanta |
|||
|---|---|---|---|---|
| Y448F | W550N | W550A | D559E | |
| Data collection statistics | ||||
| a (Å) | 105.59 | 105.49 | 106.22 | 106.00 |
| b (Å) | 108.35 | 107.95 | 108.42 | 107.65 |
| c (Å) | 134.25 | 133.25 | 133.95 | 133.75 |
| Resolution range (Å) | 29.96–2.59 (2.63–2.59) | 21.88–2.71 (2.76–2.71) | 49.90–2.52 (2.56–2.52) | 53.8–3.00 (3.05–3.00) |
| No. of reflections observed | 125,800 | 215,760 | 201,221 | 126,511 |
| No. of unique (possible) reflections | 43,737 (48,705) | 40,622 (42,052) | 49,043 (52,734) | 29,813 (31,382) |
| Rmerge (%)b | 6.7 (23.9) | 13.8 (46.0) | 19.0 (68.4) | 12.3 (32.0) |
| Completeness (%) | 89.8 (91.4) | 96.6 (94.0) | 93.0 (80.9) | 95.2 (84.3) |
| I/σI | 18.7 (3.65) | 12.2 (3.7) | 12.7 (3.5) | 9.3 (2.8) |
| Refinement statistics | ||||
| RMS deviations | ||||
| Bond lengths (Å) | 0.009 | 0.009 | 0.009 | 0.011 |
| Bond angles (°) | 1.346 | 1.270 | 1.267 | 1.447 |
| Rwork (%) | 17.7 | 16.6 | 18.6 | 17.3 |
| Rfree (%)c | 24.9 | 24.8 | 26.6 | 25.8 |
| Asymmetric unit content | ||||
| No. of amino acid residues | 1,130 | 1,132 | 1,136 | 1,136 |
| No. of solvent molecules | 406 | 424 | 465 | 487 |
| PDB accession no. | 4RY4 | 4RY5 | 4RY6 | 4RY7 |
All structures are isomorphous to the P212121 orthorhombic crystal form of WT NS5B (PDB accession number 1NB4). Values in parentheses refer to the highest-resolution shell.
Rmerge = 100 × ΣhΣi|Ih,i − 〈Ih〉|/ΣhΣiIh,i, where 〈Ih〉 is the mean intensity of symmetry-related reflections, Ih,i.
The size of test set was ≤7.0%.
Structure determination and refinement.
Data were indexed, scaled, and reduced by using the programs DENZO and SCALEPACK (33). Phases were derived from the structure of wild-type apo-NS5B-Δ21 (PDB accession number 1NB4) (7). Iterative refinement and manual rebuilding of the respective models were performed by using the programs COOT and PHENIX (34, 35). After initial rigid body refinement of the W550N/Mn2+/UTP complex, electron density was apparent in an Fo−Fc difference map in the region of the catalytic site. By using COOT, two Mn2+ and two UTP molecules were modeled into this density, with the triphosphate occupying alternate conformations. The program PROCHECK (36) was used for geometrical and stereochemical verification of the models. A summary of diffraction data and refinement statistics is shown in Table 1.
Analytical ultracentrifugation (AUC).
Sedimentation velocity experiments were performed on a Beckman model XL-I analytical ultracentrifuge. Samples were dialyzed into a solution containing 10 mM Tris (pH 7.5), 600 mM NaCl, 10% glycerol, and 5 mM β-mercaptoethanol and concentrated to a final concentration of 2 μM. The partial specific volume of the proteins was calculated based on the amino acid composition, and the temperature-corrected density of the solvent was calculated from the chemical composition of the buffer by using the computer program SEDNTERP V 1.08. Sedimentation velocity runs were carried out with 400 μl of protein samples and 400 μl of matching reference buffer loaded into two-channel aluminum centerpieces in an An-60 Ti rotor. Data were collected for 320 min at 7°C, using a rotor speed of 60,000 rpm. Sample sedimentation was detected by using Rayleigh interference optics. Multiple scans at different time points were fitted to a continuous size distribution by using the program SEDFIT V 8.8 (37).
RESULTS
In order to study in detail the functional roles of the NS5B β-loop and C-terminal arm, single point mutations were designed to disrupt either element without introducing drastic structural changes. We previously determined the structure of HCV NS5B from the HCV J4 strain with a C-terminal 21-residue truncation (PDB accession number 1NB4) (7), and by using our model as a guide, three sites were selected for mutation: Y448 in the β-loop and W550 and D559 in the C-terminal arm. All the residues chosen are highly conserved among HCV sequences and are involved in stabilizing interactions with the three major domains of NS5B: palm, thumb, and fingers (Fig. 1A). Tyrosine 448 is located on the tip of the β-loop and stabilizes the loop by forming a hydrogen bond with the Y195 main chain in the palm domain (Fig. 1B). This residue is of additional interest because when HCV NS5B is superimposed onto the reovirus polymerase λ3/RNA complex or the φ6 polymerase/RNA complex (8, 9), NS5B Y448 is in the position equivalent to that of the φ6 tyrosine residue, which forms a stacking platform for the rNTPi. Tryptophan 550 is at the center of a string of completely conserved hydrophobic residues (L547, W550, and F551), which form tight interactions with thumb domain residues, tethering the C-terminal arm to the putative RNA binding cleft (Fig. 1C). Aspartate 559 is situated further along and tightly connects the C-terminal arm with the finger domain by forming charged interactions with three separate residues in this domain (S96, R168, and K172) (Fig. 1D).
FIG 1.

(A) Overall structure of HCV NS5B highlighting some of the stabilizing interactions of the C-terminal domains. The β-loop and C-terminal arm regions are highlighted in black, residue Y448 is represented in blue, W550 is indicated in green, and D559 is shown as blue sticks and spheres. “DD” indicates the NS5B active site aspartate residues. (B) Closeup view of Y448 and the surrounding NS5B residues. The backbone oxygen of Y195 (atom colors) in the palm domain forms a hydrogen bond with Y448 OH1 (shown in blue). (C) Conserved residues on the C-terminal arm (L547 and F551 [shown in dark gray]) and on the thumb domain (I413, M414, I462, and L466 [shown in light gray]) form a deep hydrophobic pocket around W550. (D) Closeup view of interactions around D559. Residues S96, R168, and K172 in the finger domain, which form hydrogen bonds with D559, are shown in black. All figures were rendered with PyMol.
Role of residue 448 in the β-loop and residues 550 and 559 in the C-terminal arm.
Y448A, Y448F, W550A, W550N, and D559E mutations were introduced into our previously established HCV J4 NS5B-Δ21 construct (7). Mutant and wild-type (WT) NS5B proteins were all expressed, purified, assayed, and crystallized in an identical manner. In vitro enzyme activities were studied in detail with de novo initiation and primer extension assays. De novo initiation experiments were carried out with a 3′-dideoxy-terminated 18-mer RNA template, which is incapable of “copy-back” or self-priming. In order to collect more biologically relevant data, the template RNA chosen for the extension assays was a subgenomic sequence with a length of 407 nucleotides (nt) containing the entire HCV 3′ noncoding region (NCR) and part of the coding sequence for NS5B.
The results of in vitro RdRp activity assays performed over a 60-min time course are summarized in Fig. 2A. Mutations at all three positions resulted in increased nucleotide incorporation activity compared to that of the parent enzyme. This finding suggests that even subtle local changes in the β-loop and C-terminal arm, and not just large truncations (27–29, 47), regulate overall RdRp activity. At positions where additional mutations were introduced, greater alterations in residue properties resulted in more pronounced effects. For example, the subtle mutation of tyrosine to phenylalanine in β-loop residue 448 gave only a slight increase in nucleotide incorporation at the 60-min time point, whereas the complete removal of the aromatic ring in the Y448A mutant resulted in a 1.7-fold increase. Similarly, C-terminal arm W550A and W550N mutants gave 1.5-fold and 2.53-fold increases, respectively, over WT NS5B incorporation at the 60-min time point (Fig. 2A). Several NS5B activity studies have focused on the importance of hydrophobic forces in the C-terminal interfaces. However, the 1.5-fold increase in activity shown by the D559E mutant after 60 min demonstrates that polar interactions are also important for definitive C-terminal arm positioning (Fig. 1A).
FIG 2.

(A) Time course of nucleotide incorporation by the wild-type (WT) NS5B and C-terminal domain mutants. Note the distinct lag with wild-type NS5B at the beginning of the time course (0 to 10 min). All assays were carried out in triplicate, and errors were negligible. Reaction conditions are detailed in Materials and Methods. (B) Single-cycle activity assays with NS5B-Δ21 and C-terminal domain mutants. All assays were carried out in triplicate, and errors were negligible. NMP, nucleotide monophosphate.
β-Loop and C-terminal arm regulation occurs during template binding.
A close examination of data from nucleotide incorporation assays showed that the difference between WT and mutant activities was more pronounced in the earlier stages of the reaction (range, 0 to 20 min) (Fig. 2A), indicating that the WT enzyme has a longer lag phase than the mutants. This raised the question as to whether the NS5B mutants are simply able to bind template RNA more rapidly than the WT. In order to address this point, single-cycle RdRp assays were performed, in which each enzyme molecule can undergo only one round of nucleotide incorporation (Fig. 2B). In these assays, enzymes were incubated with template RNA prior to the start of the reaction, to allow the formation of a preinitiation complex. After 20 min, reactions were started by simultaneously adding nucleotides and heparin to the mixture. Heparin sequesters any unbound enzyme at the start of the reaction as well as any that falls off the template RNA subsequently during a reaction. Hence, the amount of activity observed is directly proportional to the amount of complex formation during the initial incubation stage. In this type of assay, all mutants were more active than the WT, although the most conservative mutant, Y448F, essentially behaved like the parent enzyme (Fig. 2B). Furthermore, the difference between WT and mutant activities was even more pronounced than under conditions where all rNTPs were supplied and heparin was not present. At the 60-min time point, the Y448A and W550A mutants had twice the amount of label incorporated, the D559E mutant had more than 5 times the amount of label incorporated, and the W550N mutant had nearly 13 times the amount of label incorporated by the WT. These in vitro activity assays show that the mutants more readily form catalytically competent complexes with the template RNA during the preincubation period.
Effect of residue 448 in the β-loop and residues 550 and 559 in the C-terminal arm on de novo initiation.
Wild-type NS5B and the Y448F, W550N, and D559E mutants were tested in de novo initiation assays to determine the potential effects that subtle alterations in the β-loop and C-terminal arm might have on the assembly of a competent initiation complex. Analysis of the resulting RNA products on an acrylamide-urea denaturing gel showed that WT NS5B is capable of forming the expected 18-mer RNA copies albeit in a distributive fashion (Fig. 3). Under the chosen reaction conditions, n + 1 products (19-mers) were also observed, presumably due to the slight molar excess of enzyme over substrates and the known terminal transferase properties of HCV NS5B. However, the three mutant RdRps displayed significantly altered initiation behaviors. The Y448F mutant showed residual activity and faint bands for the expected product (Fig. 3). The W550N and D559E mutants were essentially unable to initiate de novo, which may indicate that the NS5B active-site cavity of these two mutants was sufficiently altered and that a stable initiation complex could not be formed. Crystallographic and hydrodynamic studies were carried out to address this question at the molecular level.
FIG 3.
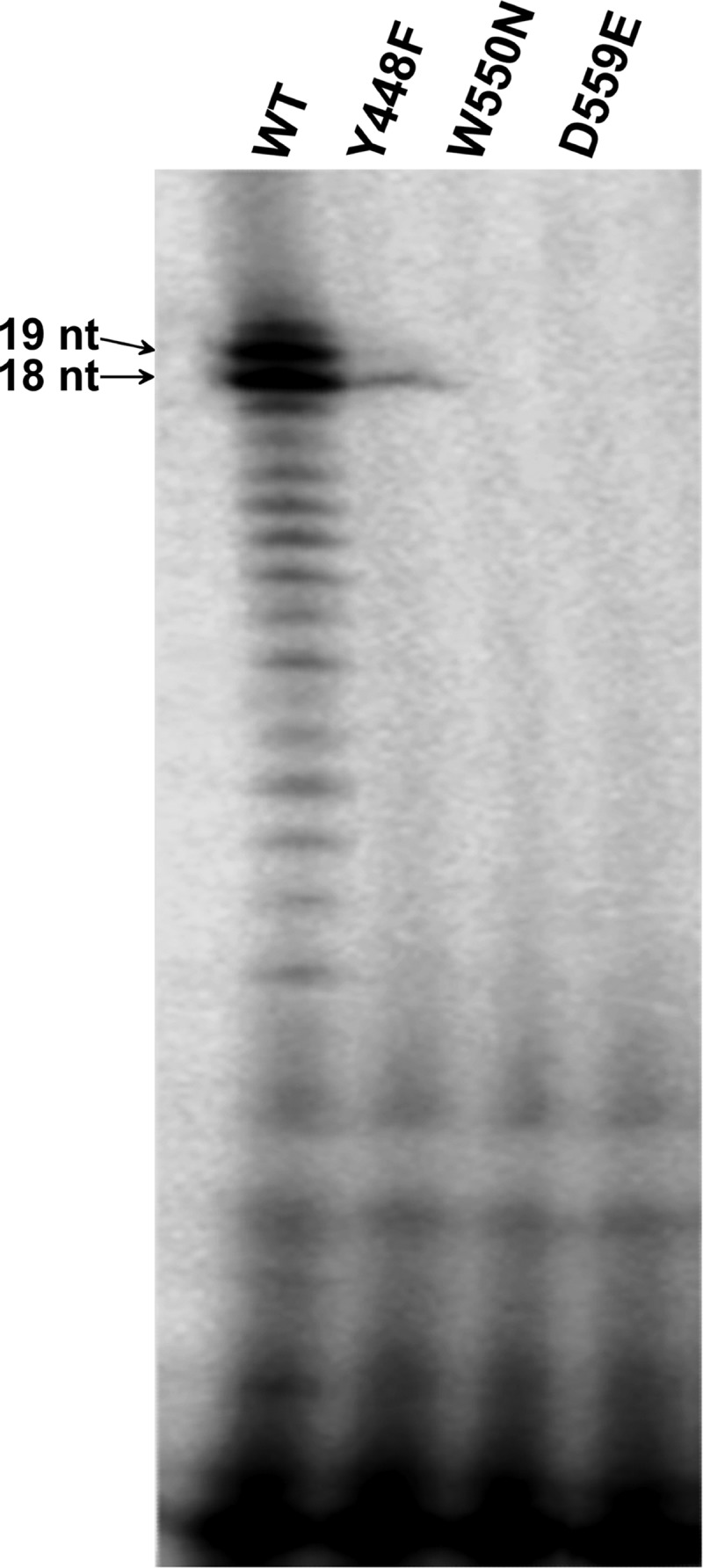
HCV NS5B de novo initiation assays. WT NS5B and the Y448F, W550N, and D559E mutants (500 nM each) were incubated with 300 nM 3′-dideoxy-terminated RNA; 300 nM ATP, CTP, and UTP; 200 nM GTP; and 0.5 μl of [α-33P]GTP (10 μCi/ml). Reactions were stopped after 90 min by adding formamide-SDS-EDTA buffer to the mixture, and fractions were separated by denaturing PAGE and analyzed on a Typhoon phosphorimager.
Additional flexibility in the C-terminal arm affects the mode of nucleotide binding and impedes initiation.
In order to dissect the underlying structural basis for the altered de novo priming activities, the shortened lag phase, and the faster RNA template binding exhibited by the mutants, the crystal structures of the W550A, W550N, D559E, and Y448F mutants were determined. With the exception of the W550N mutant, all the mutants crystallized under exactly the same conditions as those for the Δ21 wild-type protein (7). The W550N mutant precipitated rapidly under native growth conditions, yielding only small crystals. However, larger crystals were grown successfully from seeds created from first-generation crystals under these conditions. All mutant crystals were isomorphous to the P212121 crystal form of WT NS5B (7), with two independent NS5B molecules per asymmetric unit. In all of the structures, residues 1 to 565 could be placed unambiguously and refined for both copies within the asymmetric unit, before the His6-tagged C terminus of NS5B became completely disordered. At position 448, the density corresponding to the phenylalanine is discontinuous between the main chain and the aromatic side chain, possibly indicating rapid movements due to the lacking hydrogen bond in WT NS5B (Fig. 4A). The electron density corresponding to the main-chain and side-chain atoms of residues 538 to 550 is less well defined than that in the corresponding region of the wild-type NS5B density maps, suggesting increased flexibility of the C-terminal arm (Fig. 4B). The electron density around residue 559 in the D559E mutant indicates that, unlike the internal “salt-bridged” aspartate in wild-type NS5B, the longer glutamate side chain is pointing away from the finger domain in an external orientation (Fig. 4C). In this position, the glutamate does not interact with the protein matrix, thereby causing destabilization and an increased flexibility of the C-terminal arm in the D559E protein. Crystallographic details and refinement statistics are summarized in Table 1.
FIG 4.

SigmaA-weighted 2mFo−DFc electron density maps contoured at 1.2 RMS deviations above the mean density corresponding to Y448F, W550N, and D559E mutations. All models were refined and density maps were calculated with Phenix1.9 (see Table 1 for details). The figures were rendered with PyMol. (A) Phenylalanine 448 and surrounding residues in the finger (blue), palm (red), and thumb (green) domains. Note how the densities corresponding to the F448 and M414 side chains are significantly lower than those for adjacent residues (residues 193, 197, 447, and 551). (B) In the W550N mutant, the hydrophobic pocket (*) originally filled by the indole ring of W550 in WT NS5B is no longer occupied. The W550N mutation causes substantial motions near the site of mutation, and thus, features in the density maps around residues G549 and L548 are less well defined. (C) The side chain of E559 has moved away from the original D559 location near K172 and R168 (faint in the background) and is now oriented more toward the bulk solvent. G557 is situated immediately next to the NTPi site, which explains why slight movements in this region can directly affect catalysis. (D) Bias-free electron density omit map superimposed on the W550A/UTP/Mn complex (light gray carbons, W550N mutant; dark gray carbons, UTP; green, Mn2+). The phosphate moieties adopt two alternate conformations with approximately equal occupancies. In complex “UTPa,” the phosphates are in close contact with both metal ions, resembling the classical binding mode first described for T7 DNA polymerase and HIV-1 reverse transcriptase. “UTPb” more closely resembles the binding mode found for wild-type HCV J4 NS5B (46), where the γ-phosphate interacts with metal B on the left and basic residues on the finger domain (e.g., R48, K51, and R158).
It was suspected that the accelerated template binding exhibited by the C-terminal arm mutants might be a result of increased interior movements, thus creating more space in the RNA binding cleft. Structural comparisons, however, showed few substantial differences in α-carbon positions between any of the mutants and the WT NS5B structure, in terms of overall structural changes around the site of the mutation. In fact, the maximum root mean square (RMS) deviation between the positions of Cα atoms in any mutant NS5B molecule and the equivalent WT molecule was 0.473 Å, which is only slightly above the estimated coordinate error (34, 35).
More significant local movements, however, were observed for the W550N/UTP complex than for the WT/UTP complex determined at a very similar resolution. Comparison of α-carbon positions revealed that there were concerted movements in the β-flap, the C-terminal arm, and, significantly, helix R (residues 458 to 467). The movements in this region were ∼0.5 Å in the β-flap, ∼1.0 Å in the C-terminal arm (residues 530 to 565), and up to 1.2 Å in helix R. Furthermore, careful analysis of either relative intramolecular temperature factor indicated that the mutations had created more flexibility within the mutated elements and surrounding regions. This was most significant for the W550N structure, where higher relative temperature factor values and, presumably, a higher degree of flexibility are associated with the hydrophobic residues, which occupy the RNA binding cleft. The ratios of the mean B-factor values of residues 546 to 551 to those of the entire protein are 2.29 and 3.07 for molecules A and B, respectively. These values are significantly higher than those obtained for the WT structure, 1.72 and 1.81 for molecules A and B, respectively.
In order to examine the effect of C-terminal arm flexibility on substrate binding, the structure of the W550N mutant in complex with UTP and Mn2+ ions was determined. The structure of HCV J4 NS5B in complex with UTP and Mn2+ ions was previously determined (PDB accession number 1NB6) (7). In this complex, a single UTP molecule is present in the NTPi +1 (catalytic) site but not in the NTPi (priming) site. When the W550N structure was refined by using the apo-Δ21 structure (accession number 1NB4) as a starting model, examination of an Fo−Fc map showed several electron density peaks at least 4 times higher than the RMS density at the catalytic GDD motif (residues 317 to 319) in both NS5B copies. Initially, two catalytic metal ions and a triphosphate moiety were modeled into the density in both molecules (Fig. 4D). Superposition of the W550N complex onto that of the corresponding wild-type NS5B structure, using palm domain residues, placed equivalent Mn2+ ions within 0.5 Å of one another. Further inspection of Fo−Fc density maps revealed additional features near the nucleoside and the triphosphate moieties of each UTP molecule. In contrast to wild-type NS5B, the W550N mutant appears to bind the triphosphate in two alternate conformations, as shown in Fig. 4. Careful refinement in PHENIX (35) revealed that the W550N mutant binds UTP in two conformations, with approximate occupancies of 0.6 and 0.4, respectively. While the positions of the ribose moiety and the α-phosphate group in the two alternate UTP conformations in the W550N mutant are in good agreement, greater differences were found for the positions of the β- and γ-phosphates. Conformation A is stabilized by interactions with metal A (near D319) and R158 (in motif F) and roughly corresponds to the “curled” binding mode reported for T7 DNA polymerase and HIV-1 RT (38, 39). Conformation B, however, is more extended and very closely resembles that described previously for wild-type HCV J4 NS5B (7). As in the wild-type NS5B structure, the UTP γ-phosphate in the W550N mutant is stabilized by main-chain interactions with C223 and interactions with basic residues on the fingertips (R48). Interestingly, the mutation of W550 to Asn appears to introduce a greater degree of flexibility into the C-terminal arm as well as the RdRp active site. Consequently, the position of the triphosphate moiety is able to shift relative to the metal ion positions in both the wild-type and the mutant structures. Interestingly, the triphosphate moiety in molecule A of the W550N mutant superimposes well onto that of the UTP/NS5B complex of the BK strain of HCV (PDB accession number 1GX6) (6). The increase in flexibility and repositioning of the rNTP in the W550N complex could possibly lead to an occasional shortening of the distance between the P- and C-site rNTP positions and, thus, facilitate the formation of the first phosphodiester bond during de novo initiation, thereby providing a plausible structural explanation for the noticeable lack of a lag phase in the W550 mutants.
C-terminal arm mutations allow a more open structure in solution.
Pairwise superpositions of structures were restricted to matching the catalytically relevant RdRp regions (encompassing motifs A, B, C, and E), while “peripheral” regions, such as the finger, fingertip, and thumb domains, were omitted from these superpositions. Interestingly, the most obvious differences in these comparisons were observed for the W550N mutant and WT NS5B. The resulting distance plot (Fig. 5A) shows small deviations of <0.5 Å for residues 1 to 380 and larger movements of >0.5 Å for the remaining residues in the thumb domain. Not surprisingly, the C-terminal arm shows deviations of well above 1 Å (Fig. 5A). However, the subtlest differences in the comparison of α-carbon positions can be found between the Y448F mutant structure and the parent structure. Except for a few sharp peaks corresponding to both fingertip regions (residues 18 to 40 and 145 to 155) and 2 surface loops in the finger and thumb domains, the deviations are consistently around or less than 0.5 Å (Fig. 5B).
FIG 5.

Relative motions in the thumb domain and C-terminal arm of the Y448F and W550N mutants. (A) The superposition of the W550N mutant and WT NS5B (top) and the corresponding deviations in α-carbon positions (bottom) show that the mutant structure adopts a more open conformation. This is particularly evident for the thumb domain (residues 370 to 528) and even more so for the C-terminal arm (residues 528 to 566). The large deviations around positions 25 and 150 correspond to the flexible fingertips, long extensions within the NS5B finger domain. (B) Comparison of the Y448F mutant and WT NS5B shows smaller structural differences, with consistently around or less than a 0.5-Å difference in α-carbon positions, providing some structural underpinning toward the more wild-type-like phenotype of the Y448F mutant.
Given the subtlety of these overall differences between the WT and the β-loop and C-terminal arm mutants, it cannot be completely ruled out that the restraints imposed by the crystal lattice prevent larger domain motions. Therefore, two of the mutants (W550A and W550N) were analyzed alongside the WT by sedimentation velocity analytical ultracentrifugation (AUC) to determine whether any structural differences occur in solution. Experiments were performed at 7°C with >600 mM NaCl and with a glycerol content of 10% to maintain protein monodispersity. Under these conditions, the W550A and W550N mutants had observed sedimentation coefficients of 1.66 and 1.65, respectively, values significantly lower than that for the wild-type protein, 1.72 (Fig. 6). This finding suggests that in solution, the mutants have a less spherical shape than the wild-type enzyme, likely due to C-terminal arm movement or partial detachment from the remainder of the NS5B structure, as seen tentatively for the W550N/UTP complex (Fig. 4D and 5A). It is also possible that the movement of the thumb domain may be a contributing factor, as crystal structures of NS5B with up to 55 of the residues removed from the C terminus (NS5B-Δ55) show an 8° rotation of the thumb domain compared to our His-tagged NS5B-Δ21 structures (6, 7).
FIG 6.

Sedimentation velocity analytical ultracentrifugation analysis of WT NS5B and the W550A and W550N C-terminal arm mutants. Experiments were performed with 2 μM samples in a solution containing 10 mM Tris (pH 7.5), 600 mM NaCl, 10% glycerol, and 5 mM β-mercaptoethanol at 7°C.
β-Loop and C-terminal arm functions are essential for replication in cells.
In order to investigate further how subtle β-loop and C-terminal arm mutations might affect complete viral replication, the same mutations were tested in a transient HCV replicon assay. The respective mutations were introduced into an HCV subgenomic replicon clone, pPI_ETSacIIJ4, a construct containing NS3-NS5B genes from the J4 strain of HCV and a firefly luciferase reporter gene. As a negative control, a completely replication-deficient construct was also engineered by mutating one of the essential catalytic aspartates (D318N). Modified replicon RNAs and wild-type RNA were transfected into Huh-7 cells by electroporation. Luciferase activity was measured in cells harvested at 4 h posttransfection and subsequently at daily intervals. Due to differences in transfection efficiency, values were normalized by expressing them as a percentage of the luciferase activity determined at 4 h posttransfection (Fig. 7). All mutations substantially affected the level of viral replication in the replicon system, suggesting that both the β-hairpin and C-terminal arm are critical for HCV replication in vivo. In general, there was an inverse correlation between in vitro activities and viral replication in cells, whereas the mutants that were most active in in vitro elongation assays (W550N and D559E) (Fig. 2B) were effectively as inactive in the replicon system as the negative control (D318N). The three other mutants that were less active in extension assays showed some degree of replication in cells (Fig. 7). Despite the minimal structural alteration at position 448 (Y448F) and the minor changes in in vitro activities, within the replicon system, the mutant showed an almost 10-fold drop in viral RNA levels after 3 days. These results further confirm that not only the presence but also the precise positioning of the β-loop and the C-terminal arm is crucial for NS5B to initiate and replicate properly in cells.
FIG 7.

Transient-replicon analysis of WT NS5B and C-terminal domain mutants. Huh-7 cells were transfected with 10 μg replicon RNA by electroporation. After 4, 24, 48, 72, and 96 h, cells were lysed, and luciferase activities were determined. Values are corrected for transfection efficiency, as determined by measuring the luciferase activity 4 h after transfection.
DISCUSSION
De novo initiation during HCV genome replication is an intricate process that involves the correct positioning and stabilization of template RNA, two individual nucleotides, and two metal ions within the initiation complex. In this study, we provide experimental evidence that the loss of a single hydrogen bond or a single Coulomb interaction affects the precise positioning of two prominent C-terminal features, the β-loop and C-terminal arm. The slightest perturbations in this important region appear to control the productive binding of both the template RNA and nucleotides and, thus, have major downstream consequences.
It was previously shown that the complete removal of either the β-loop or C-terminal arm results in a dramatic change in enzyme activity (1, 2, 27–29). In this study, we found that single-residue substitutions designed to disrupt the positioning of these loops have similarly drastic effects: increased primer extension activity in vitro and a severe loss of de novo initiation. The removal of a single hydroxyl group from the β-loop or a salt bridge within the C-terminal arm has profound effects on various steps of the NS5B catalytic cycle. However, the phenotypes of the mutants discussed here changed to a much lesser extent, with up to a 2.5-fold increase in activities, compared to other studies, where increases from 5-fold to 150-fold have been reported for complete C-terminal arm or β-loop deletants (1, 27, 29). Previous studies in which the C-terminal arm was gradually shortened suggested that the three conserved hydrophobic residues L547, W550, and F551 are the main stabilizing residues of this region, as in vitro activity increases significantly with the removal of residues 545 to 552 (27). Additionally, our studies demonstrate that not only hydrophobic interaction surfaces along the C-terminal arm but also electrostatic interactions, namely, those at Asp559 or Glu559, can play an important role in the regulation of initiation and elongation activities. Our crystallographic and hydrodynamic data suggested that this could be due to the mutants adopting a more open structure where C-terminal elements move away from the finger and palm subdomains. Indeed, comparisons of various wild-type NS5B structures demonstrate that the protein can move from a closed to a slightly more open conformation in which the thumb domain is rotated by up to 7° (6, 7, 14). The concerted movements observed in the thumb domains of the W550A structure and the W550N/UTP complex are on a somewhat smaller but still significant scale. It is likely that by destabilizing either the β-loop or the C-terminal arm, the mutations favor transition into the open, elongating RdRp conformation. Adachi et al. showed previously that alanine mutations of hydrophobic C-terminal arm residues (L547A to F551A) alter the structure adopted by NS5B in crystals from a closed form to a more open form (27). In the study presented here, our more “conservative” mutants crystallized mostly in the closed form, but localized, concerted movements near the sites of mutation and adjacent regions were still observed (Fig. 5).
When C-terminal domain mutants were tested in a subgenomic replicon system, the respective RNAs showed marked decreases in translation and replication despite having increased RdRp activity in vitro. Similar phenotypes have been found for mutants completely lacking the β-loop (28). As this assay system involves the translation and replication of viral RNA, the effect of any mutation on RNA structure and function has to be considered, in addition to the effects on protein structure. Six regions within the NS5B coding region of the HCV genome have been identified as having conserved elements of secondary structure, which may have functional importance (40–43). The region coding for the β-loop does not overlap any of these regions; hence, the effects observed are likely to be due to alterations of protein structure. However, the codons for W550 and D559 fall within two of the structured cis-replicating elements identified, 5BSL3.1 and 5BSL3.2, respectively (41). Previous studies in which these structures were disrupted by synonymous mutations have shown that 5BSL3.2 is absolutely required for efficient replication (41, 42). Therefore, the distinct lack of replicon activity shown by the D559E mutant might be not only a consequence of poor initiation capabilities but also a result of the disruption of local RNA structures or vital life cycle functions. Previous studies investigating the importance of 5BSL3.1 in replication have provided conflicting results. Two groups have shown that the disruption of these structures has no effect (32, 41), while another group showed that it was lethal to viral replication (42). In our study, the W550A mutant showed some replicon activity although at a lower level than that of the wild-type system, and therefore, any change in the RNA structure incurred did not appear to abolish RNA replication completely. As both the W550A and the W550N mutations involved the changing of all three nucleotides within the codon, they probably had similarly pronounced effects on RNA structure. Therefore, the complete lack of W550N replicon activity over W550A or WT replicons must be due at least partially to significant changes in protein structure.
These results indicate that both the β-loop and the C-terminal arm are vital for HCV replication and that the loss of even a single hydrogen bond (as in the Y448F mutant) can have detrimental effects. Previous studies suggested that only the presence of the β-loop and not the amino acid composition is important for de novo initiation or discrimination against primed templates (30). However, our data clearly indicate that the primary sequence and the intricate local network of side-chain interactions are essential for the regulation of initiation, elongation, and viral replication in vivo. The somewhat conflicting results from in vitro assays and replicon assays can be reconciled by the fact that these two C-terminal features are vital for the correct selection and proper positioning of the initiation complex components.
Correct initiation opposite the 3′ terminus of the template RNA is essential for maintaining genome integrity. Mutations in the β-loop and C-terminal arm increase flexibility and allow the protein to assume a more open structure, which in turn may allow the template to pass through the active site, resulting in internal initiation. Internal initiation has indeed been observed for NS5B mutants lacking the β-loop (1). Furthermore, increased flexibility creating space in the active-site cleft may decrease the selection of the correct over an incorrect nucleotide. The W550N/UTP cocrystal structure showed that the mutation causes multiple UTP binding modes and a displacement of the catalytic nucleotide from the native binding position. Both incorrect positioning and unfaithful nucleotide incorporation could have a detrimental effect on the quality of HCV genomes synthesized in subgenomic replicon assays and, hence, significantly affect viral replication. In the context of the current work, we have not been able to show whether the C-terminal regions directly interact with and stabilize NTPi, as UTP binds only at the catalytic site within this crystal form and under the conditions used. The structures of two other de novo-initiating polymerases have shown different modes for stabilizing the NTPi (8, 13). The structure of an initiation complex of φ6 polymerase shows a tyrosine residue in the C-terminal blocking element acting as a stacking platform for the priming nucleotide (8). However, the structure of bovine viral diarrhea virus (BVDV) polymerase in complex with GTP shows the nucleotide binding in a location adjacent to the catalytic or “C” site (13). It has been suggested that GTP binds in this position during initiation to stabilize the NTPi. Ferron et al. modeled GTP in a similar position in a model of GB virus C (GBV-C) polymerase (44) and in the HCV NS5B crystal structure and proposed that both of these polymerases use a similar mechanism of nucleotide stabilization. However, the HCV NS5B crystal structure (strain BK, residues 1 to 531) used for GBV-C modeling lacks 55 residues at the C terminus (6), which in turn may cause a significant outward rotation of the thumb domain, as demonstrated by X-ray solution scattering and crystallography (45). However, the shape of the binding site for the remote “priming” GTP in NS5B-Δ55 (BK) (45) is altered compared to the closed conformation of the NS5B-Δ21/UTP complex (7), in part explaining why the latter structures show only a single rNTP bound to the initiation site. An atomic-resolution crystal structure of the complete ternary complex with template RNA, two rNTPs, and two (or more) metal ions is desperately needed to obtained a more complete picture and a deeper understanding of the intricacies of de novo initiation and RNA elongation.
ACKNOWLEDGMENTS
We gratefully acknowledge support by the personnel at SRS 14.2, NSLS X4C, and the Wadsworth Center Biochemistry Core facilities. We thank J. D. Pata for experimental advice, C. M. Rice for fruitful discussions, and D. J. Rowlands and J. D. Pata for critical reading of the manuscript.
This work was supported in part by an MRC CASE studentship award to A.L.C., a University Research Fellowship to J.J., and Health Research Inc./Ordway Research Institute funds to C.A.D. and J.J.
REFERENCES
- 1.Hong Z, Cameron CE, Walker MP, Castro C, Yao N, Lau JYN, Zhong W. 2001. A novel mechanism to ensure terminal initiation by hepatitis C virus NS5B polymerase. Virology 285:6–11. doi: 10.1006/viro.2001.0948. [DOI] [PubMed] [Google Scholar]
- 2.Kao CC, Singh P, Eckert DJ. 2001. De novo initiation of viral RNA-dependent RNA synthesis. Virology 287:251–260. doi: 10.1006/viro.2001.1039. [DOI] [PubMed] [Google Scholar]
- 3.Jin Z, Leveque V, Ma H, Johnson KA, Klumpp K. 2012. Assembly, purification, and pre-steady-state kinetic analysis of active RNA-dependent RNA polymerase elongation complex. J Biol Chem 287:10674–10683. doi: 10.1074/jbc.M111.325530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ago H, Adachi T, Yoshida A, Yamamoto M, Habuka N, Yatsunami K, Miyano M. 1999. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Structure 7:1417–1426. doi: 10.1016/S0969-2126(00)80031-3. [DOI] [PubMed] [Google Scholar]
- 5.Lesburg CA, Cable MB, Ferrari E, Hong Z, Mannarino AF, Weber PC. 1999. Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat Struct Biol 6:937–934. doi: 10.1038/13305. [DOI] [PubMed] [Google Scholar]
- 6.Bressanelli S, Tomei L, Rey FA, De Francesco R. 2002. Structural analysis of the hepatitis C virus RNA polymerase in complex with ribonucleotides. J Virol 76:3482–3492. doi: 10.1128/JVI.76.7.3482-3492.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O'Farrell D, Trowbridge R, Rowlands D, Jager J. 2003. Substrate complexes of hepatitis C virus RNA polymerase (HCV-J4): structural evidence for nucleotide import and de novo initiation. J Mol Biol 326:1025–1035. doi: 10.1016/S0022-2836(02)01439-0. [DOI] [PubMed] [Google Scholar]
- 8.Butcher SJ, Grimes JM, Makeyev EV, Bamford DH, Stuart DI. 2001. A mechanism for initiation RNA-dependent RNA polymerisation. Nature 410:235–240. doi: 10.1038/35065653. [DOI] [PubMed] [Google Scholar]
- 9.Tao Y, Farsetta DL, Nibert ML, Harrison SC. 2002. RNA synthesis in a cage—structural studies of reovirus polymerase lambda3. Cell 111:733–745. doi: 10.1016/S0092-8674(02)01110-8. [DOI] [PubMed] [Google Scholar]
- 10.Ng KK, Cherney MM, Vazquez AL, Machin A, Alonso JM, Parra F, James MN. 2002. Crystal structures of active and inactive conformations of a caliciviral RNA-dependent RNA polymerase. J Biol Chem 277:1381–1387. doi: 10.1074/jbc.M109261200. [DOI] [PubMed] [Google Scholar]
- 11.Ferrer-Orta C, Arias A, Perez-Luque R, Escarmis C, Domingo E, Verdaguer N. 2004. Structure of foot-and-mouth disease virus RNA-dependent RNA polymerase and its complex with a template-primer RNA. J Biol Chem 279:47212–47221. doi: 10.1074/jbc.M405465200. [DOI] [PubMed] [Google Scholar]
- 12.Love RA, Maegley KA, Yu X, Ferre RA, Lingardo LK, Diehl W, Parge HE, Dragovich PS, Fuhrman SA. 2004. The crystal structure of the RNA-dependent RNA polymerase from human rhinovirus: a dual function target for common cold antiviral therapy. Structure 12:1533–1544. doi: 10.1016/j.str.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 13.Choi KH, Groarke JM, Young DC, Kuhn RJ, Smith JL, Pevear DC, Rossmann MG. 2004. The structure of the RNA-dependent RNA polymerase from bovine viral diarrhea virus establishes the role of GTP in de novo initiation. Proc Natl Acad Sci U S A 101:4425–4430. doi: 10.1073/pnas.0400660101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Biswal BK, Cherney MM, Wang M, Chan L, Yannopoulos CG, Bilimoria D, Nicolas O, Bedard J, James MNG. 2005. Crystal structures of the RNA-dependent RNA polymerase genotype 2a of hepatitis C virus reveal two conformations and suggest mechanisms of inhibition by non-nucleoside inhibitors. J Biol Chem 280:18202–18210. doi: 10.1074/jbc.M413410200. [DOI] [PubMed] [Google Scholar]
- 15.Fullerton SWB, Blaschke M, Coutard B, Gebhardt J, Gorbalenya A, Canard B, Tucker PA, Rohayem J. 2007. Structural and functional characterization of sapovirus RNA-dependent RNA polymerase. J Virol 81:1858–1871. doi: 10.1128/JVI.01462-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ng KK, Pendas-Franco N, Rojo J, Boga JA, Machin A, Alonso JM, Parra F. 2004. Crystal structure of Norwalk virus polymerase reveals the carboxyl terminus in the active site cleft. J Biol Chem 279:16638–16645. doi: 10.1074/jbc.M400584200. [DOI] [PubMed] [Google Scholar]
- 17.Malet H, Egloff MP, Selisko B, Butcher RE, Wright PJ, Roberts M, Gruez A, Sulzenbacher G, Vonrhein C, Bricogne G, Mackenzie JM, Khromykh AA, Davidson AD, Canard B. 2007. Crystal structure of the RNA polymerase domain of the West Nile virus non-structural protein 5. J Biol Chem 282:10678–10689. doi: 10.1074/jbc.M607273200. [DOI] [PubMed] [Google Scholar]
- 18.Yap TL, Xu T, Chen Y-L, Malet H, Egloff M-P, Canard B, Vasudevan SG, Lescar J. 2007. Crystal structure of the dengue virus RNA-dependent RNA polymerase catalytic domain at 1.85-angstrom resolution. J Virol 81:4753–4765. doi: 10.1128/JVI.02283-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Campagnola G, Weygandt M, Scoggin K, Peersen O. 2008. Crystal structure of coxsackievirus B3 3Dpol highlights functional importance of residue 5 in picornaviral polymerases. J Virol 82:9458–9464. doi: 10.1128/JVI.00647-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gong P, Peersen OB. 2010. Structural basis for active site closure by the poliovirus RNA-dependent RNA polymerase. Proc Natl Acad Sci U S A 107:22505–22510. doi: 10.1073/pnas.1007626107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alam I, Lee JH, Cho KJ, Han KR, Yang JM, Chung MS, Kim KH. 2012. Crystal structures of murine norovirus-1 RNA-dependent RNA polymerase in complex with 2-thiouridine or ribavirin. Virology 426:143–151. doi: 10.1016/j.virol.2012.01.016. [DOI] [PubMed] [Google Scholar]
- 22.Takeshita D, Tomita K. 2012. Molecular basis for RNA polymerization by Qβ replicase. Nat Struct Mol Biol 19:229–237. doi: 10.1038/nsmb.2204. [DOI] [PubMed] [Google Scholar]
- 23.Wright S, Poranen MM, Bamford DH, Stuart DI, Grimes JM. 2012. Noncatalytic ions direct the RNA-dependent RNA polymerase of bacterial double-stranded RNA virus ϕ6 from de novo initiation to elongation. J Virol 86:2837–2849. doi: 10.1128/JVI.05168-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Noble CG, Lim SP, Chen YL, Liew CW, Yap L, Lescar J, Shi P-Y. 2013. Conformational flexibility of the dengue virus RNA-dependent RNA polymerase revealed by a complex with an inhibitor. J Virol 87:5291–5295. doi: 10.1128/JVI.00045-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bahar MW, Sarin LP, Graham SC, Pang J, Bamford DH, Stuart DI, Grimes JM. 2013. Structure of a VP1-VP3 complex suggests how birnaviruses package the VP1 polymerase. J Virol 87:3229–3236. doi: 10.1128/JVI.02939-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chinnaswamy S, Yarbrough I, Palaninathan S, Ranjith-Kumar CT, Vijayaraghavan V, Demeler B, Lemon SM, Sacchettini JC, Kao CC. 2008. A locking mechanism regulates RNA synthesis and host protein interaction by the hepatitis C virus polymerase. J Biol Chem 283:20535–20546. doi: 10.1074/jbc.M801490200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adachi T, Ago H, Habuka N, Okuda K, Komatsu M, Ikeda S, Yatsunami K. 2002. The essential role of C-terminal residues in regulating the activity of hepatitis C virus RNA-dependent RNA polymerase. Biochim Biophys Acta 1601:38–48. doi: 10.1016/S1570-9639(02)00433-8. [DOI] [PubMed] [Google Scholar]
- 28.Cheney IW, Naim S, Lai VCH, Dempsey S, Bellows D, Walker MP, Shim JH, Horscroft N, Hong Z, Zhong W. 2002. Mutations in NS5B polymerase of hepatitis C virus: impacts on in vitro enzymatic activity and viral RNA replication in the subgenomic replicon cell culture. Virology 297:298–306. doi: 10.1006/viro.2002.1461. [DOI] [PubMed] [Google Scholar]
- 29.Leveque VJ-P, Johnson RB, Parsons S, Ren J, Xie C, Zhang F, Wang QM. 2003. Identification of a C-terminal regulatory motif in hepatitis C virus RNA-dependent RNA polymerase: structural and biochemical analysis. J Virol 77:9020–9028. doi: 10.1128/JVI.77.16.9020-9028.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ranjith-Kumar CT, Gutshall L, Sarisky RT, Kao CC. 2003. Multiple interactions within the hepatitis C virus RNA polymerase repress primer-dependent RNA synthesis. J Mol Biol 330:675–685. doi: 10.1016/S0022-2836(03)00613-2. [DOI] [PubMed] [Google Scholar]
- 31.Nakabayashi H, Taketa K, Miyano K, Yamane T, Sato J. 1982. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res 42:3858–3863. [PubMed] [Google Scholar]
- 32.Friebe P, Lohmann V, Krieger N, Bartenschlager R. 2001. Sequences in the 5′ nontranslated region of hepatitis C virus required for RNA replication. J Virol 75:12047–12057. doi: 10.1128/JVI.75.24.12047-12057.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. [DOI] [PubMed] [Google Scholar]
- 34.CCP4. 1994. The CCP4 Suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 35.Zwart PH, Afonine PV, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, McKee E, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Storoni LC, Terwilliger TC, Adams PD. 2008. Automated structure solution with the PHENIX suite. Methods Mol Biol 426:419–435. doi: 10.1007/978-1-60327-058-8_28. [DOI] [PubMed] [Google Scholar]
- 36.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26:283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- 37.Schuck P. 2000. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys J 78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Doublié S, Tabor S, Long AM, Richardson CC, Ellenberger T. 1998. Crystal structure of a bacteriophage T7 DNA replication complex. Nature 391:251–258. doi: 10.1038/34593. [DOI] [PubMed] [Google Scholar]
- 39.Huang H, Chopra R, Verdine GL, Harrison SC. 1998. Structure of a covalently trapped catalytic complex of HIV reverse transcriptase: implications for drug resistance. Science 282:1669–1675. doi: 10.1126/science.282.5394.1669. [DOI] [PubMed] [Google Scholar]
- 40.Tuplin A, Evans DJ, Simmonds P. 2004. Detailed mapping of RNA secondary structures in core and NS5B-encoding region sequences of hepatitis C virus by RNase cleavage and novel bioinformatic prediction methods. J Gen Virol 85:3037–3047. doi: 10.1099/vir.0.80141-0. [DOI] [PubMed] [Google Scholar]
- 41.You S, Stump DD, Branch AD, Rice CM. 2004. A cis-acting replication element in the sequence encoding the NS5B RNA-dependent RNA polymerase is required for hepatitis C virus RNA replication. J Virol 78:1352–1366. doi: 10.1128/JVI.78.3.1352-1366.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee H, Shin H, Wimmer E, Paul AV. 2004. cis-Acting RNA signals in the NS5B C-terminal coding sequence of the hepatitis C virus genome. J Virol 78:10865–10877. doi: 10.1128/JVI.78.20.10865-10877.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith DB, Simmonds P. 1997. Characteristics of nucleotide substitution in the hepatitis C virus genome: constraints on sequence change in coding regions at both ends of the genome. J Mol Evol 45:238–246. doi: 10.1007/PL00006226. [DOI] [PubMed] [Google Scholar]
- 44.Ferron F, Bussetta C, Dutartre H, Canard B. 2005. The modeled structure of the RNA dependent RNA polymerase of GBV-C virus suggests a role for motif E in Flaviviridae RNA polymerases. BMC Bioinformatics 6:255. doi: 10.1186/1471-2105-6-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harrus D, Ahmed-El-Sayed N, Simister PC, Miller S, Triconnet M, Hagedorn CH, Mahias K, Rey FA, Astier-Gin T, Bressanelli S. 2010. Further insights into the roles of GTP and the C terminus of the hepatitis C virus polymerase in the initiation of RNA synthesis. J Biol Chem 285:32906–32918. doi: 10.1074/jbc.M110.151316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vives-Adrian L, Lujan C, Oliva B, van der Linden L, Selisko B, Coutard B, Canard B, van Kuppeveld FJ, Ferrer-Orta C, Verdaguer N. 2014. The crystal structure of a cardiovirus RNA-dependent RNA polymerase reveals an unusual conformation of the polymerase active site. J Virol 88:5595–5607. doi: 10.1128/JVI.03502-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boyce SE, Tirunagari N, Niedziela-Majka A, Perry J, Wong M, Kan E, Lagpacan L, Barauskas O, Hung M, Fenaux M, Appleby T, Watkins WJ, Schmitz U, Sakowicz R. 2014. Structural and regulatory elements of HCV NS5B polymerase—β-loop and C-terminal tail—are required for activity of allosteric thumb site II inhibitors. PLoS One 9:e84808. doi: 10.1371/journal.pone.0084808. [DOI] [PMC free article] [PubMed] [Google Scholar]