

Abstract
The cause of chronic pelvic pain in interstitial cystitis/painful bladder syndrome (IC/PBS) remains unclear; autoimmunity is a possible etiology. We have recently shown that injection of a single immunogenic peptide of uroplakin 3A (UPK3A 65-84) induces experimental autoimmune cystitis (EAC) in female BALB/cJ mice that is unique among experimental models in accurately reflecting both the urinary symptoms and pelvic pain of IC/PBS. The aim of this project was to identify the roles of mast cells and mast cell chemoattractant/activator monocyte chemoattractant protein-1 [chemokine (C-C motif) ligand 2 (CCL2)] in the allodynia in this model. We immunized 6- to 8-wk-old female BALB/cJ mice with UPK3A 65-84 peptide and, 5–40 days later, observed increased responses to stimulation of the suprapubic abdominal and hindpaw surfaces with von Frey monofilaments compared with mice injected with adjuvant alone. Suprapubic and hindpaw tactile allodynia responses by EAC mice were blocked by instillation of lidocaine into the bladder but not by lidocaine in the uterus, confirming the bladder as the source of the hypersensitivity. Markedly increased numbers of activated mast cells and expression of CCL2 were found in the bladder after immunization with UPK3A 65-84. Hypersensitive responses were inhibited by mast cell stabilizer cromolyn sodium and antagonists of histamine receptors 1 and 2. Furthermore, BALB/cJ mice with deletion of the Ccl2 or chemokine (C-C motif) receptor 2 gene exhibited markedly reduced allodynia and accumulation of mast cells after UPK3A 65-84 immunization. These results show that UPK3A 65-84 immunization causes chronic visceral allodynia and suggest that it is mediated by CCL2-driven mast cell accumulation in the bladder.
Keywords: uroplakin 3A, pelvic pain, chemokine (C-C motif) ligand 2-deficient mice, chemokine (C-C motif) receptor 2-deficient mice, mast cell inhibitors
interstitial cystitis/painful bladder syndrome (IC/PBS) is a chronic sterile bladder syndrome characterized by symptoms of urinary frequency, urgency, and pelvic pain/hypersensitivity (9). IC/PBS occurs mainly in women, with a prevalence estimated in different studies to be two to five times higher than in men (12, 35). The most recent National Institutes of Health-funded epidemiological study of IC/PBS in women in the United States (the Rand IC Epidemiology study) identified prevalences of 6.5% and 2.7% based on high sensitivity and high specificity criteria, respectively, for diagnosing IC/PBS (7). Those percentages translated into 3.3–7.9 million women 18 yr old or older with IC/PBS symptoms. IC/PBS patients suffer from pelvic pain over the course of their lives, often during their most productive years. Pelvic pain in IC/PBS patients is generally diffuse and involves hyperalgesia on the body surface “referred” from the bladder, due to the convergence of visceral and somatic sensory fibers in the central nervous system (51). Progression in the management of pain from this disease has been slow due to a lack of understanding of the underlying pathophysiology.
The etiopathogenesis of IC/PBS is still unclear; many theories have been proposed, relating mainly to increased urothelial permeability and/or neurogenic inflammation with bladder sensory nerve upregulation (58). Possible etiological stimuli include autoimmunity, toxic substances in the urine, defects in the glycosaminoglycan layer or uroepithelial cells of the bladder wall, previous infectious agents or chronic subclinical infection, psychological distress, genetic susceptibility, and neuroendocrine or angiogenic factors (58). All of those factors can stimulate pathways leading to an enhanced production of inflammatory mediators and cytokines. Histologically, bladder inflammation in IC/PBS patients ranges from severe, generally in patients with Hunner's ulcers, to negligible, with the majority of patients exhibiting mild inflammation (15).
Several small studies have found markedly increased numbers of mast cells in the bladder in IC/PBS patients, particularly in the detrusor muscle and in patients with Hunner's ulcers (for a review, see Ref. 54). More recently, mild mastocytosis (mean of 34.5 mast cells/mm2) was found in the lamina propria in 179 of 203 patients of the IC Data Base Study (32). However, in that study, mast cells were not counted in the detrusor muscle, where higher numbers of mast cells in IC/PBS have typically been found (54). In other studies, increased levels of the mast cell protease tryptase and metabolites of the mast cell product histamine have been found in the urine of IC/PBS patients (10, 19). In the bladder in IC/PBS patients and in other tissues in other inflammatory disorders, mast cells accumulate mostly at the tips of nerve endings (33, 41, 56). Mast cells have been shown to play an important role in mediating pain in animal models of diseases such as IC/PBS, irritable bowel syndrome, and neuropathic and postoperative pain (40, 47, 49, 57). Also, histamine released from mast cells has been shown to mediate pelvic pain in a virus-induced neurogenic cystitis model (45). Histamine depolarizes neurons (59), induces neuronal hypertrophy (28), and, in a positive feedback loop, induces increased mast cell density (28).
Chemokine (C-C motif) ligand 2 (CCL2), also referred to as monocyte chemotactic protein (MCP)-1, is a member of the C-C chemokine family; it has a chemotactic effect on mast cells and monocytes (16) and also induces degranulation of mast cells (14). In rat models of peripheral neuropathy, administration of a CCL2-neutralizing antibody reduced the neuropathic pain, whereas an intraspinal application of CCL2 to naïve rats caused tactile allodynia (53). Also, mice with genetic deletion of chemokine (C-C motif) receptor 2 (CCR2), the receptor on the surface of mast cells for CCL2 and other chemokines, exhibited reduced tactile pain responses after nerve injury (1). More recently, C57BL/6J mice with genetic deletion of either Ccl2 or Ccr2 were shown to be resistant to pelvic hyperalgesia associated with experimental autoimmune prostatitis (44). Increased levels of CCL2 have been found in urine and bladder tissue of IC/PBS patients (39), and CCL2 has been suggested as a biomarker for IC/PBS and chronic pelvic pain syndrome (17, 39).
We recently created an experimental autoimmune cystitis (EAC) model by injecting a bladder-specific uroplakin 3A-derived immunogenic peptide (UPK3A 65-84) into female BALB/cJ mice (27). The peptide induces CD4-positive T cell-mediated autoimmunity that manifests the urodynamic and pelvic pain phenotypes of human IC/PBS. In the present study, we clearly show that chronic tactile allodynia in EAC mice originates in the bladder and is driven by CCL2-mediated mast cell accumulation and release of histamine from mast cells. Visceral pain referred from the bladder was measured by applying von Frey filaments to the skin on the suprapubic abdominal and hindpaw regions, a widely accepted, noninvasive nociceptive stimulation technique used in other studies of visceral pain (31, 48).
MATERIALS AND METHODS
Ethics statement.
All mouse protocols were preapproved by the Institutional Animal Care and Use Committee of Case Western Reserve University (permit 2009-0131) in compliance with the Public Health Service policy on humane care and use of laboratory animals. All dissections were performed with mice under isoflurane anesthesia and were followed by euthanasia with an overdose of ketamine-xylazine. All efforts were made to minimize suffering.
Mice and immunization.
Female wild-type (WT) BALB/cJ mice were purchased from Jackson Laboratory (Bar Harbor, ME). Adult female and male BALB/cJ mice with homozygous deletion of the Ccl2 gene (Ccl2−/− mice) (13, 37) or the Ccr2 gene (Ccr2−/− mice) (6, 30) were obtained from Dr. B. Rollins (Harvard Medical School, Boston, MA) (13) and Dr. N. Mukaida (Kanazawa University, Kanazawa, Ishikawa, Japan) (6), respectively. In experiments with Ccl2−/− or Ccr2−/− mice, WT BALB/cJ mice from Jackson Laboratory were used as controls. Knockout mice were bred, and all mice were maintained on a regular 12:12-h light-dark cycle with ad libitum food and water in the Animal Resource Center of Case Western Reserve University. At 8–10 wk of age, mice were injected subcutaneously with 200 μg UPK3A 65-84 in 200 μl of an emulsion of equal volumes of water and complete Freund's adjuvant containing 400 μg Mycobacteria tuberculosis H37RA (CFA; Difco Laboratories, Detroit, MI) or with an emulsion of water and CFA alone, as previously described (27). Mice were euthanized by asphyxiation with CO2 followed by cervical dislocation on the number of days after immunization as indicated in the figures.
Tactile allodynia assessment.
Tactile sensitivities of the suprapubic and hindpaw regions of mice, with the former considered a surrogate for pelvic visceral pain (47), were measured using a series of 14 von Frey filaments with increasing calibrated forces from 0.008 to 10.0 g (Stoelting, Wood Dale, IL). These filaments provide an approximately logarithmic series of forces and a linear scale of perceived intensity. Beginning with the smallest filament, each filament was applied a total of 10 times for 3 s, with intervals of 8 s between each stimulus. The behaviors that were considered to be a positive response were as follows: 1) sharp retraction of the stimulated body part (suprapubic abdominal region or hindpaw), 2) instant licking and/or scratching of the stimulated area, or 3) jumping. The percentage of positive responses from 10 total trials (response frequency) was recorded for each monofilament. In addition, the von Frey force, defined as that which would elicit a response 50% of the time (50% threshold), was calculated for each mouse from the regression line drawn through the linear portion of a plot of response frequency versus log of the von Frey force using GraphPad Prism 6 (GraphPad Software, La Jolla, CA).
Lidocaine instillation.
On the 40th day after immunization with UPK3A 65-84, mice were anesthetized with isoflurane, and the local anesthetic lidocaine (Sigma-Aldrich, 50 μl of a 2% solution in sterile saline) or saline vehicle alone was instilled into the lumen of the bladder or uterus using a 24-gauge, 0.7 × 19-mm catheter (BD Angiocath Autoguard, BD Medical Systems, Sandy, UT). Saline alone was administered to CFA-injected control mice. Tactile hypersensitivity assessment using von Frey filaments was performed 1 h after drug administration.
Mast cell staining and counting.
Bladders harvested from UPK3A 65-84- and CFA-injected mice 10, 20, and 40 days after injection were fixed in 10% phosphate-buffered formalin, embedded in paraffin, and sectioned using a microtome. For staining of mast cells, bladder cross sections were deparaffinized, hydrated sequentially with xylene, alcohol, and distilled H2O, and stained in 0.1% toluidine blue working solution for 2–3 min. After being rinsed with distilled H2O and dehydration, slides were mounted with coverslips using Cytoseal XYL mounting medium solution (Richard-Allan Scientific, Kalamazoo, MI). Resting mast cells with intensely staining cytoplasmic granules and activated (degranulated) mast cells were distinguished under a light microscope at ×40 magnification and counted separately. For each mouse, mast cells were counted by two observers independently in eight different toluidine blue-stained sections of the bladder, and the average of the results of the two observers was used.
CCL2 and IgE ELISA.
Blood and whole bladders were harvested from UPK3A 65-84- and CFA-injected mice 10, 20, and 40 days after injection. Serum was separated and stored at −80°C for ELISA. Half of each bladder was stored at −80°C for ELISA, and the other half was used for RNA isolation (see Real-time quantitative RT-PCR below). In addition, six other tissues (the uterus, ovary, colon, liver, kidney, and lung) were harvested from UPK3A 65-84-immunized mice 40 days after immunization and stored at −80°C for ELISA. Frozen tissues were homogenized in RIPA buffer with protease inhibitor cocktail (EMD Millipore, Billerica, MA) using a PowerGen 125 homogenizer (Fisher Scientific, Pittsburgh, PA), and protein concentrations were determined by the method of Bradford (Bio-Rad Protein Assay, Bio-Rad Laboratories, Hercules, CA). CCL2 and IgE concentrations were measured in tissues and serum, respectively, using ELISA kits (CCL2, Ray Biotech, Norcross, GA; IgE, BioLegend, San Diego, CA) according to the manufacturers' instructions. Absorbance at 450 nm was read in a Versamax ELISA microplate reader (Molecular Devices, Sunnyvale, CA).
Real-time quantitative RT-PCR.
Bladder halves from UPK3A 65-84-immunized and CFA-injected control mice that were not frozen (see ELISA above) were immediately homogenized in TRIzol reagent (Invitrogen, Carlsbad, CA) followed by isolation of total RNA. cDNA was synthesized from the RNA using a Super Script III cDNA synthesis kit with random hexamer primers (Invitrogen). cDNAs were analyzed for the expression of the genes for Ccl2, Fc ε receptor 1 α-subunit (FceR1a), and β-actin (loading control) by quantitative PCR using gene-specific primer pairs designed with the online Universal Probe Library Assay Design Center (Roche, Mannheim, Germany) and purchased from Life Technologies (Grand Island, NY). The following sequences of primers were used: Ccl2, 5′-CACAGTTGCCGGCTGGAGCAT-3′ (sense) and 5′-GTAGCAGCAGGTGAGTGGGGC-3′ (antisense); FceR1a, 5′-TGTGTACTTGAATGTAACGCAAGA-3′ (sense) and 5′-GGACTAAGACCATGTCAGCAGAT-3′ (antisense); and β-actin, 5′-GGTCATCACTATTGGCAACG-3′ (sense) and 5′-ACGGATGTCAACGTCACACT-3′ (antisense). Quantitative PCR was performed using a SYBR Green PCR Master kit with an ABI Prism 7500 Sequence Detection System (Applied Biosystems, Foster City, CA). Ccl2 and FceR1a gene expression levels were normalized to the corresponding β-actin gene expression level and calculated relative to the average level in CFA-immunized mice (set to 1.0) by the comparative CT method (where CT is threshold cycle) (36) after we confirmed that the mean levels of β-actin mRNA did not differ significantly between EAC and CFA-injected mice.
Oral drug administration.
On the 40th day after immunization with UPK3A 65-84, the mast cell stabilizer cromolyn sodium (26), the histamine receptor H1 antagonist cetirizine, or the histamine receptor H2 antagonist ranitidine (47) (all from Sigma-Aldrich) were administered orally by gavage at 10 mg/kg in sterile saline using a Hamilton syringe with a 2.5-cm-long ball-tipped needle. Drugs were administered three times at 2-h intervals. The drug dosages were similar to those previously used in mice (26, 47), and the administration intervals were chosen to accommodate the short biological half-life of cromolyn sodium [∼1.3 h (39a)], the effectiveness of which depends on the clearance of histamine and other factors secreted by mast cells. Positive control UPK3A 65-84-immunized mice and negative control CFA-immunized mice received saline vehicle alone by gavage at the same volume and intervals as the drug solutions. Tactile allodynia assessment was performed using von Frey filaments 1 h after the final drug administration. Immediately after allodynia assessment, all mice were anesthetized, and bladders were harvested for mast cell counting.
Statistical analysis.
In experiments involving measurements at multiple time points, comparisons of tactile allodynia (log-transformed 50% threshold von Frey forces), mast cell numbers, and CCL2 protein levels between UPK3A 65-84-immunized and CFA-immunized mice (WT, Ccl2−/−, or Ccr2−/− mice), and allodynia in WT versus Ccl2−/− or Ccr2−/− mice were performed using two-way ANOVA with Bonferroni multiple comparisons with (allodynia) or without (mast cell numbers and CCL2 protein levels) repeated measures. Comparisons of allodynia among groups in experiments with lidocaine, cromolyn sodium, or antihistamines (Figs. 2 and 4) as well as comparisons of mast cell numbers among WT and knockout mice immunized with UPK3A 65-84 or CFA (Fig. 8) were performed using one-way ANOVA with Tukey's multiple-comparisons test. CCL2 protein levels in different tissues (Fig. 5C) were compared with levels in the bladder using one-way ANOVA with Dunnett's multiple-comparisons test. Comparisons of Ccl2 and FceR1a mRNA levels by quantitative RT-PCR were performed using an unpaired, two-tailed Student's t-test, due to significant interactions in two-way ANOVAs of those data. Welch's correction was used in the t-tests in cases where the variances were significantly different. P values of <0.05 were considered statistically significant.
Fig. 2.
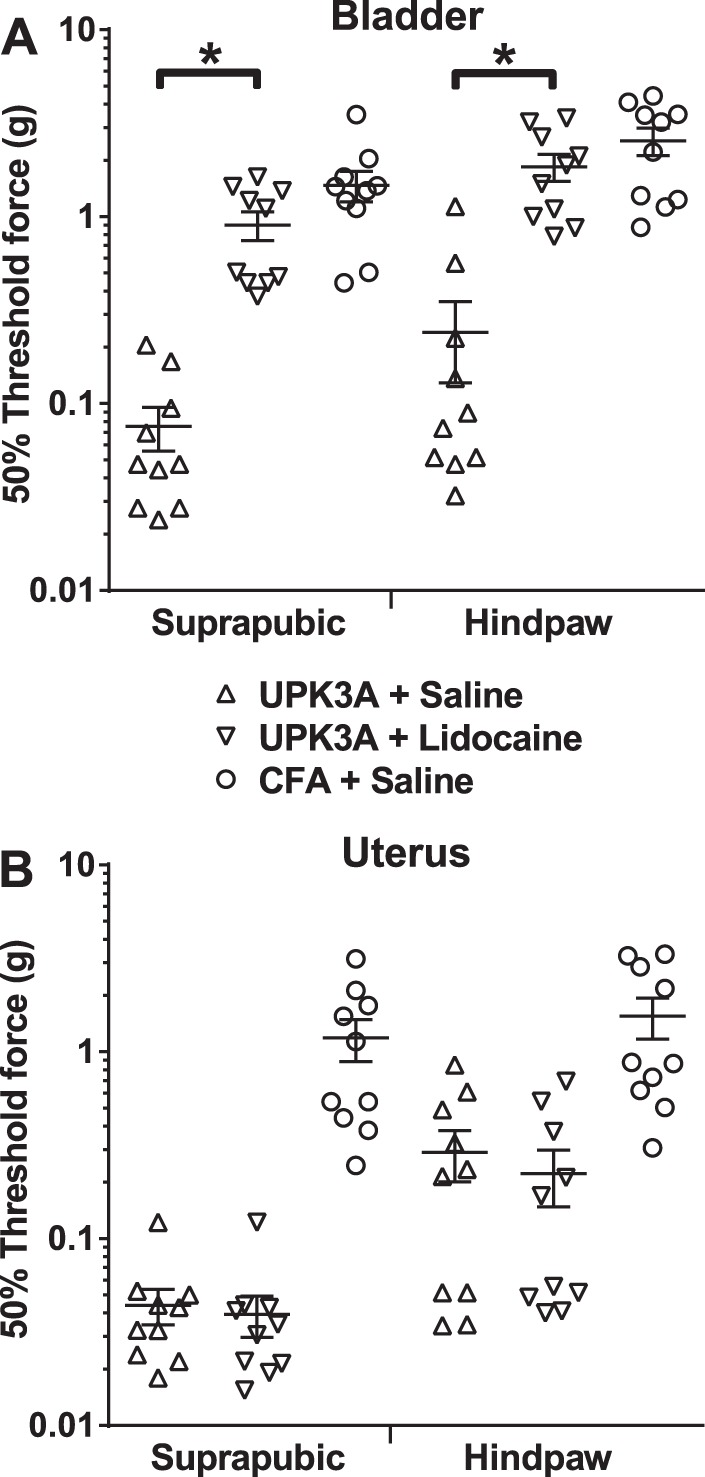
Lidocaine instilled into the bladder reduces tactile allodynia of experimental autoimmune cystitis (EAC) mice. Forty days after immunization of mice with UPK3A 65-84, lidocaine or saline vehicle was instilled into the lumen of the bladder or uterus. Negative control mice were immunized with CFA alone followed 40 days later by the instillation of saline vehicle into the bladder or uterus. One hour after the instillation of drug or vehicle, tactile allodynia assessment was performed using von Frey filaments, and 50% response thresholds were calculated for each mouse, as in Fig. 1. Data points indicate 50% response thresholds of individual mice (n = 10 mice/group); horizontal bars and error bars indicate means and SEs, respectively. A: instillation of lidocaine into bladders of EAC mice resulted in significantly reduced allodynia (higher 50% response thresholds) in response to the application of von Frey filaments to either suprapubic or hindpaw regions, to levels that were not significantly different from CFA-injected control mice administered saline vehicle (analyzed by one-way ANOVA of log-transformed 50% response thresholds with Tukey's multiple-comparisons test; *P < 0.0001). B: on the other hand, the instillation of lidocaine into the uterus of EAC mice had no significant effect on 50% response thresholds on either suprapubic or hindpaw regions compared with the instillation of saline alone.
Fig. 4.
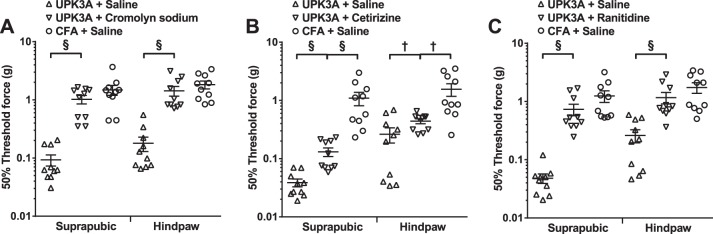
Mast cell stabilizer and antihistamines reduce tactile allodynia in EAC mice. A: oral administration of the mast cell stabilizer cromolyn sodium to mice 40 days after immunization with UPK3A 65-84 resulted in significantly reduced tactile allodynia (higher 50% response thresholds) in response to application of von Frey filaments to either suprapubic or hindpaw regions, to levels that were not significantly different from CFA-immunized control mice administered saline vehicle. Antagonists of histamine receptor H1 (cetirizine; B) and H2 (ranitidine; C) significantly increased 50% thresholds of von Frey filaments applied to suprapubic and hindpaw regions compared with saline vehicle-treated, UPK3A 65-84-immunized mice. However, whereas ranitidine reduced both suprapubic and hindpaw tactile allodynia responses (increased 50% response thresholds) to levels that were not significantly different from CFA + saline control mice (C), significant differences remained between cetirizine-treated, UPK3A 65-84-immunized mice and the respective CFA-injected control mice (C). Drug administration and tactile allodynia assessment were performed as described in materials and methods. Data points indicate the 50% response thresholds of individual mice (n = 10 mice/group); horizontal bars and error bars indicate means and SEs. Suprapubic and hindpaw data were analyzed separately by one-way ANOVA of log-transformed 50% response thresholds with Tukey's multiple-comparisons test. §P < 0.001; †P < 0.05.
Fig. 8.
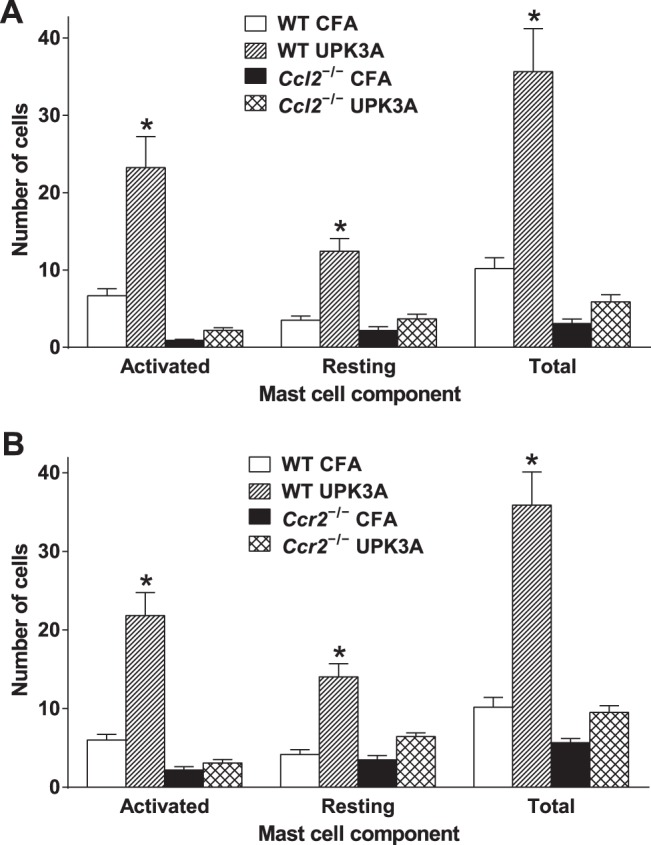
Mast cell accumulation and activation in the bladder are markedly reduced in Ccl2−/− mice and Ccr2−/− mice immunized with UPK3A 65-84. On the 40th day after immunization with UPK3A 65-84 or CFA alone, mice in which tactile allodynia was assessed in Figs. 6 and 7 were euthanized 1 h after assessment, and bladders were then harvested and processed for toluidine blue staining of mast cells as described in materials and methods. Activated (granular) and resting (nongranular) mast cells were counted in bladder cross sections from UPK3A 65-84- or CFA-immunized WT or Ccl2−/− mice (n = 10 mice/group). A: numbers of activated, resting, and total mast cells were significantly higher in bladders from UPK3A 65-84-immunized WT mice compared with UPK3A 65-84-immunized Ccl2−/− mice or with CFA-immunized WT or Ccl2−/− mice (*P < 0.0001 in comparisons of UPK3A 65-84-immunized WT mice with each other group by one-way ANOVA with Tukey's multiple-comparisons test; no other pairs of groups differed significantly from each other). B: similar results were found with Ccr2−/− mice (*P < 0.0001 in comparisons of UPK3A 65-84-immunized WT mice with each other group; no other pairs of groups differed significantly from each other). Bars are means ± SE.
Fig. 5.
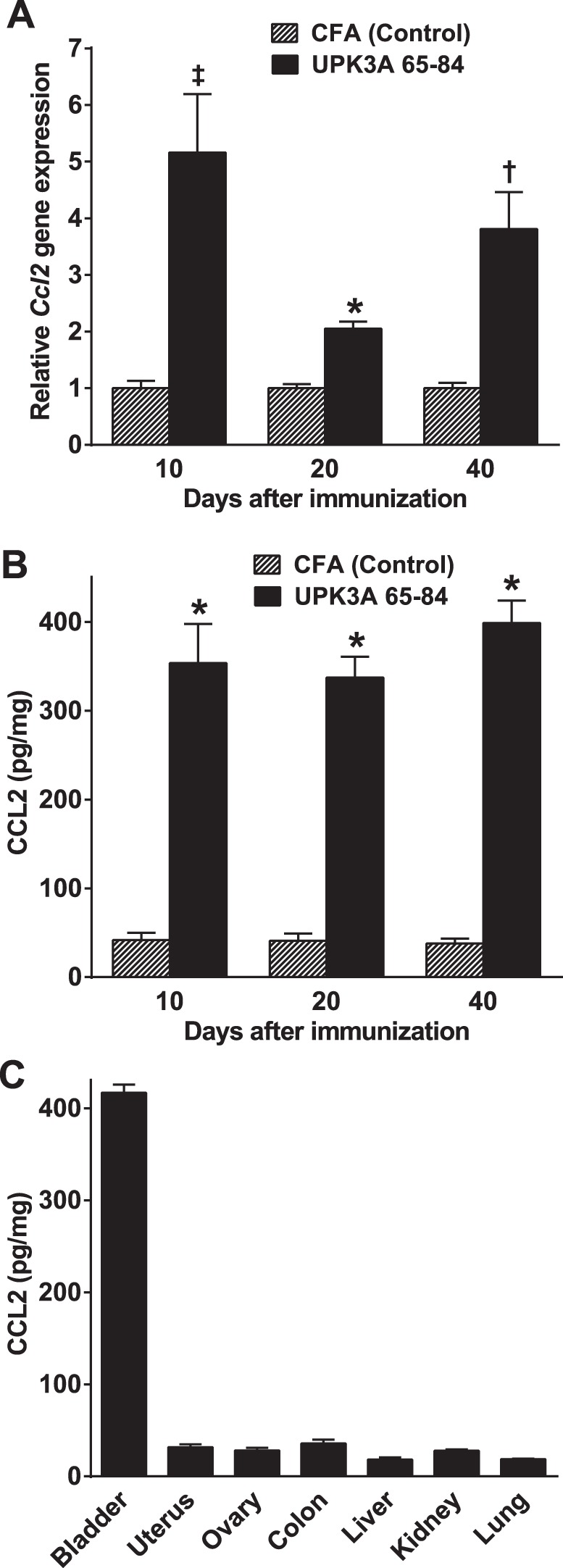
Increased chemokine (C-C motif) ligand 2 (CCL2) production in the bladder in EAC mice. UPK3A 65-84-immunized and CFA-injected control mice were euthanized 10, 20, and 40 days after immunization, and the bladder and other organs were harvested and processed as described in materials and methods. A: Ccl2 mRNA levels measured by quantitative RT-PCR were significantly higher in UPK3A 65-84-immunized mice compared with CFA-injected control mice at all three time points (n = 8 mice/group at 10 and 20 days; n = 5 mice/group at 40 days), as analyzed by unpaired t-tests using Welch's correction for 10 and 40 days. *P < 0.0001; ‡P < 0.01; †P < 0.05. B: CCL2 protein levels measured by ELISA were significantly higher in UPK3A 65-84-immunized mice at all three time points (n = 10 mice/group at each time point), as analyzed by two-way ANOVA with Bonferroni multiple-comparisons test. *P < 0.0001. C: the level of CCL2 protein was substantially higher in the bladder compared with six other organs in EAC mice 40 days after immunization with UPK3A 65-84 (n = 3, P < 0.0001 for each organ compared with the bladder by one-way ANOVA with Dunnett's multiple-comparison test). Bars are means ± SE.
RESULTS
Early onset and persistence of tactile allodynia in EAC mice.
In our previously created model of EAC in female BALB/cJ mice, we showed that bladder-specific UPK3A 65-84 peptide induced a CD4-positive T cell-mediated autoimmune response that manifested the major symptoms of human IC/PBS, including increased urinary frequency, decreased urine output per void, and increased referred pelvic pain responses (27). In the present study, we first evaluated the onset and duration of the allodynia induced by UPK3A 65-84 compared with CFA-immunized mice (control) and uninjected naïve mice (baseline). Mechanical stimulation of UPK3A 65-84-immunized mice with von Frey filaments of increasing size on both the suprapubic region (Fig. 1A) and hindpaw (Fig. 1D) resulted in significantly greater tactile sensitivity relative to naïve mice within 5 days of immunization and persisting through 40 days of immunization, the last time point examined. Much smaller increases in tactile sensitivity relative to naïve mice were observed in CFA-immunized mice stimulated on both the suprapubic region (Fig. 1B) and hindpaw (Fig. 1E) 5–40 days after injection. Calculation of 50% threshold forces revealed significantly lower tactile response thresholds in UPK3A 65-84-immunized mice stimulated on both the suprapubic region (Fig. 1C) and hindpaw (Fig. 1F) relative to CFA-immunized mice at every time point. These results demonstrate the persistence of allodynia in our EAC model, mimicking human IC/PBS disease and confirming that it is an appropriate model for investigation of the pelvic pain mechanism.
Fig. 1.

Immunization with bladder-specific uroplakin 3A-derived immunogenic peptide (UPK3A 65-84) causes early and chronic tactile allodynia in female BALB/cJ mice. Mice were assessed for referred visceral hyperalgesia by applying a series of 14 von Frey filaments of increasing forces to the suprapubic (A–C) and hindpaw (D–F) regions. Each data point in A, B, D, and E indicates the mean response frequency (percentage of responses out of 10 trials) ± SE on the indicated number of days after immunization with UPK3A 65-84 (A and D) or complete Freund's adjuvant containing 400 μg Mycobacteria tuberculosis H37RA (CFA; B and E) or in naïve mice (n = 10 mice/group). Data points in C and F indicate mean 50% threshold forces ± SE for the different groups, as determined by linear regression of response frequency versus log of the filament force for each mouse at the indicated time points. At every time point, the mean 50% threshold in mice immunized with UPK3A 65-84 was significantly lower than in CFA-injected mice in both suprapubic (C) and hindpaw (F) regions, as determined by two-way ANOVA of log-transformed 50% response thresholds with Bonferroni multiple-comparisons test. *P < 0.0001; ‡P < 0.01.
Chronic tactile allodynia in EAC mice emanates from the bladder.
To determine whether or not the suprapubic and hindpaw tactile allodynia in EAC mice emanated specifically from the bladder, lidocaine was instilled directly into the bladder or uterus of mice 40 days after immunization with UPK3A 65-84 followed 1 h later by tactile response assessment using von Frey filaments. Lidocaine instillation into the bladder resulted in significantly increased 50% response thresholds of von Frey filaments applied to the suprapubic or hindpaw region (Fig. 2A) compared with EAC mice with saline vehicle alone instilled into the bladder. Suprapubic and hindpaw 50% thresholds of UPK3A 65-84-immunized mice with lidocaine instilled into the bladder were not significantly different from CFA-injected control mice with saline instilled into the bladder (Fig. 2A). On the other hand, instillation of lidocaine into the uterus of EAC mice had no effect on 50% response thresholds in either suprapubic or hindpaw regions compared with instillation of saline alone (Fig. 2B). These results confirm the bladder-specific origin of the tactile allodynia in EAC mice.
EAC mice have increased mast cell numbers in the bladder.
To determine if mast cells accumulate in the bladder in EAC mice, as in many human IC/PBS patients, bladders were harvested from UPK3A 65-84-immunized mice and CFA-injected control mice 10, 20, and 40 days after immunization, and mast cells were stained with toluidine blue. Figure 3B shows representative toluidine blue-stained bladder cross sections of UPK3A 65-84-immunized and CFA-injected control mice, with mast cells (active or resting) indicated by arrows. Resting mast cells containing densely packed, intensely stained cytoplasmic granules and activated mast cells with dispersed granules within and beyond the cell boundary were counted separately (Fig. 3A). Numbers of activated, resting, and total mast cells were significantly higher in UPK3A 65-84-immunized mice compared with CFA-immunized mice at all three time points, with the exception of resting mast cells on day 20 (Fig. 3, C–E). In both EAC and control mice, most mast cells were found in the detrusor, whereas none were detected in the urothelium. The latter is probably a consequence of formalin fixation, which has been reported to inhibit detection of mast cells in the urothelium with toluidine blue (3, 20). We also measured levels in the whole bladder of mRNA for the high-affinity IgE receptor FceR1a, the expression of which is limited to mast cells and basophils in mice (29). FceR1a expression was significantly increased 10, 20, and 40 days after immunization with UPK3A 65-84 compared with CFA, with the greatest increase on day 10 (Fig. 3F). Levels of IgE in serum determined by ELISA 40 days after immunization were not significantly different in EAC mice relative to CFA-injected control mice (data not shown).
Fig. 3.

Mast cell accumulation in bladders of EAC mice. Mast cells in bladder cross sections from UPK3A 65-84- and CFA-immunized mice were stained with toluidine blue (A; ×40 magnification), and activated (degranulated) mast cells and resting mast cells with cytoplasmic granules were then counted separately under a light microscope at ×40 magnification in eight different sections of each bladder. B: total mast cells, as indicated by arrows, in representative bladder cross sections (×3 magnification) from UPK3A 65-84- and CFA-immunized mice. Bladders from UPK3A 65-84-immunized mice contained significantly more activated (C), resting (D), and total (E) mast cells than CFA-injected mice at 10, 20, and 40 days after immunization, with the exception of resting mast cells on 20 days after immunization, when analyzed by two-way ANOVA with Bonferroni multiple-comparisons test (n = 10 mice/group at each time point). F: also, Fc ε receptor 1 α-subunit (FceR1a) gene expression by quantitative RT-PCR was significantly higher in the whole bladder of UPK3A 65-84-immunized mice relative to CFA-injected mice 10, 20, and 40 days after immunization (average values in CFA-injected mice were set at 1.0; results were analyzed by unpaired t-tests due to significant interactions in two-way ANOVAs, with Welch's correction at 10 and 40 days; n = 8 mice/group at each time point). Bars are means ± SE. *P < 0.0001; ‡P < 0.01; †P < 0.05.
Administration of ranitidine, cetirizine, and cromolyn sodium attenuated chronic tactile allodynia in the EAC model.
Mast cell effects can be inhibited in vivo by blocking degranulation with the mast cell stabilizer cromolyn sodium or by blocking specific histamine receptor classes. Oral administration of cromolyn sodium to mice 40 days after immunization with UPK3A 65-84 resulted in significantly reduced tactile allodynia (higher 50% response thresholds) in response to application of von Frey filaments to either suprapubic or hindpaw regions, to levels that were not significantly different from CFA-immunized control mice administered saline vehicle (Fig. 4A). Antagonists of histamine receptor H1 (cetirizine; Fig. 4B) and H2 (ranitidine; Fig. 4C) also significantly increased the thresholds of responses to von Frey filaments applied to suprapubic and hindpaw regions compared with saline vehicle-treated, UPK3A 65-84-immunized mice. However, the 50% threshold forces for allodynia responses on both body surfaces in cetirizine-treated, UPK3A 65-84-immunized mice remained significantly lower than in saline-treated CFA-injected control mice (Fig. 4B), whereas in ranitidine-treated, UPK3A 65-84-immunized mice, the 50% response thresholds on both body surfaces were at levels that were not significantly different from saline-treated CFA-injected control mice (Fig. 4C), similar to the results with the mast cell stabilizer cromolyn sodium. These results show that mast cells and the mast cell effector histamine have critical roles in the chronic tactile allodynia of EAC mice and that histamine receptor H2 appears to be a stronger mediator of the allodynia than histamine receptor H1.
Increased CCL2 in the bladder in EAC mice.
To assess expression of the mast cell chemoattractant/activator CCL2 in the bladder in EAC mice, bladders were harvested from UPK3A 65-84-immunized mice and CFA-injected control mice 10, 20, and 40 days after immunization. Half of each bladder was processed for ELISA, whereas the other half was processed for quantitative RT-PCR, to quantify CCL2 at transcriptional and translational levels, respectively. Both Ccl2 mRNA and CCL2 protein levels were significantly higher in UPK3A 65-84-immunized mice compared with CFA-immunized mice on all three time points (Fig. 5, A and B), demonstrating consistent elevation of CCL2 in conjunction with mast cell accumulation in the bladder and chronic tactile allodynia in EAC mice. Furthermore, the increased level of CCL2 protein in EAC mice 40 days after immunization was restricted to the bladder compared with six other organs (Fig. 5C).
Ccl2−/− mice develop markedly less tactile allodynia than WT mice after immunization with UPK3A 65–84.
BALB/cJ mice with deletion of the Ccl2 gene were used to establish the role of CCL2 in the chronic tactile allodynia of EAC mice. Immunization of female Ccl2−/− BALB/cJ mice with UPK3A 65-84 resulted in markedly less allodynia (higher 50% response thresholds) in both suprapubic (Fig. 6A) and hindpaw (Fig. 6B) regions compared with UPK3A 65-84-immunized WT mice at all time points from 5 to 40 days after immunization. No significant differences were found at any time point between Ccl2−/− and WT mice injected with CFA alone (suprapubic, Fig. 6C; hindpaw, Fig. 6D). In addition, comparisons of 50% thresholds in UPK3A 65-84-immunized Ccl2−/− mice versus CFA-injected Ccl2−/− mice revealed no differences in suprapubic (compare Ccl2−/− lines in Fig. 6, A and C) or hindpaw (compare Ccl2−/− lines in Fig. 6, B and D) regions except for transient allodynia in the hindpaw region on day 5 due to UPK3A 65-84 (P < 0.05 only on day 5 by Bonferroni posttest after two-way ANOVA of log-transformed 50% response thresholds). These data show that CCL2 plays a critical role in the development and persistence of chronic tactile allodynia in BALB/cJ mice immunized with UPK3A 65-84, although the peptide-induced allodynia may include a minor, transient CCL2-independent component.
Fig. 6.

Ccl2−/− BALB/cJ mice exhibit significantly less tactile allodynia than wild-type (WT) mice after immunization with UPK3A 65-84. Mice were assessed for referred visceral hyperalgesia 5, 10, 20, 30, and 40 days after immunization with UPK3A 65-84 or CFA by applying von Frey filaments of increasing forces to suprapubic and hindpaw regions as described in materials and methods. 50% response thresholds were calculated by linear regression of response frequencies versus log von Frey forces, and mean 50% thresholds ± SE at the different time points were plotted. A and B: suprapubic (A) and hindpaw (B) 50% thresholds of naïve and UPK3A 65-84-immunized WT and Ccl2−/− mice (n = 10 mice/group). C and D: suprapubic (C) and hindpaw (D) 50% thresholds of naïve and CFA-injected WT and Ccl2−/− mice (n = 7 mice/group). UPK3A 65-84 immunization yielded significantly less suprapubic (A) and hindpaw (B) allodynia (higher 50% thresholds) in Ccl2−/− mice than in WT mice at all time points (*P < 0.0001 and §P < 0.001 by two-way ANOVA of log-transformed 50% response thresholds with Bonferroni multiple-comparisons test). No significant differences in suprapubic (C) or hindpaw (D) allodynia were found between CFA-injected WT and Ccl2−/− mice.
Ccr2−/− mice develop markedly less tactile allodynia than WT mice after immunization with UPK3A 65–84.
BALB/cJ mice with deletion of the Ccr2 gene, the receptor for CCL2, were used to establish the role of CCR2 in the chronic tactile allodynia of EAC mice. Immunization of female Ccr2−/− BALB/cJ mice with UPK3A 65-84 resulted in markedly less allodynia (higher 50% response thresholds) in both suprapubic (Fig. 7A) and hindpaw (Fig. 7B) regions compared with UPK3A 65-84-immunized WT mice at all time points from 5 to 40 days after immunization. In contrast to the comparison of Ccl2−/− and WT mice injected with CFA alone (Fig. 6, C and D), CFA-injected WT mice in this experiment were found to have significant tactile allodynia at multiple early time points compared with CFA-injected Ccr2−/− mice in suprapubic (Fig. 7C) and hindpaw (Fig. 7D) regions. The significance of that minor, CCR2-dependent effect of CFA on the WT mice is unclear; it may reflect transient inflammation caused by the Mycobacteria tuberculosis antigen or another CFA component. UPK3A 65-84-immunized Ccr2−/− mice and CFA-injected Ccr2−/− mice had similar 50% response thresholds at all time points in suprapubic (compare Ccr2−/− lines in Fig. 7, A and C) and hindpaw (compare Ccr2−/− lines in Fig. 7, B and D) regions. These data show that CCR2, the receptor for CCL2 and several other chemokines, is necessary for the development and persistence of chronic tactile allodynia in BALB/cJ mice immunized with UPK3A 65-84.
Fig. 7.

Chemokine (C-C motif) receptor 2 (Ccr2)−/− BALB/cJ mice exhibit significantly less tactile allodynia than WT mice after immunization with UPK3A 65-84. Mice were assessed for referred visceral hyperalgesia 5, 10, 20, 30, and 40 days after immunization with UPK3A 65-84 or CFA by applying von Frey filaments of increasing forces to suprapubic and hindpaw regions as described in materials and methods. 50% response thresholds were calculated by linear regression of response frequencies versus log von Frey forces, and mean 50% thresholds ± SE at the different time points were plotted. A and B: suprapubic (A) and hindpaw (B) 50% thresholds of naïve and UPK3A 65-84-immunized WT and Ccr2−/− mice (n = 10 mice/group). C and D: suprapubic (C) and hindpaw (D) 50% thresholds of naïve and CFA-injected WT and Ccr2−/− mice (n = 7 mice/group). UPK3A 65-84 immunization yielded significantly less suprapubic (A) and hindpaw (B) allodynia (higher 50% thresholds) in Ccr2−/− mice than in WT mice at all time points (*P < 0.0001 by two-way ANOVA of log-transformed 50% response thresholds with Bonferroni multiple comparisons test). In addition, CFA-injected WT mice had significantly lower 50% response thresholds than CFA-injected Ccr2−/− mice in the suprapubic region 5, 10, and 20 days after immunization (C; ‡P < 0.01, †P < 0.05) and in the hindpaw region 5 and 20 days after immunization (D; ‡P < 0.01).
CCL2 and CCR2 are required for mast cell accumulation in the bladder in EAC mice.
To determine if CCL2 and CCR2 are also required for the accumulation of mast cells in the bladder in EAC mice, mast cells were counted in Ccl2−/− mice and Ccr2−/− mice relative to WT mice 40 days after immunization with UPK3A 65-84 or CFA alone, using the mice in which tactile allodynia was assessed in Figs. 6 and 7. UPK3A 65-84-immunized WT mice had significantly higher numbers of toluidine blue-stained activated, resting, and total mast cells in the bladder compared with UPK3A 65-84-immunized Ccl2−/− mice, CFA-immunized WT mice, or CFA-immunized Ccl2−/− mice (P < 0.0001 in comparisons of UPK3A 65-84-immunized WT mice with each other group by one-way ANOVA with Tukey's multiple-comparisons test; Fig. 8A). No other significant differences were found in posttest comparisons of all other pairs of groups, including UPK3A 65-84-immunized versus CFA-injected Ccl2−/− mice. Similar results for found with Ccr2−/− mice (P < 0.0001 in comparisons of UPK3A 65-84-immunized WT mice vs. each other group; Fig. 8B).
DISCUSSION
The importance of translational models for uncovering the elusive pathophysiology of IC/PBS is well recognized (8). While numerous animal models have been developed to investigate this disease, our current murine EAC model, generated by bladder-specific UPK3A 65-84, is the first model that manifests both of the major features of IC/PBS, namely, increased urinary frequency (27) and chronic pelvic pain, as assessed by measurement of tactile allodynia. In our previous EAC model, immunization of mice with bladder-specific mouse uroplakin 2 protein induced a proinflammatory type 1 cytokine response in the bladder and increased urinary frequency but without detectable tactile allodynia (4). In the present study, we focused on the allodynia phenotype in the UPK3A 65-84 model, because it is considered the most distressing symptom of IC/PBS (55). By assessing tactile allodynia at multiple time points, we showed that our EAC mice develop early and chronic pain-like behavior in the pelvic (suprapubic) and hindpaw regions. The diffuse nature of chronic pain is common in IC/PBS patients (43), likely owing to central nervous system processing of visceral sensory signals (51). Here, we showed that both the pelvic and hindpaw allodynia in EAC mice arose specifically in the bladder, as both were alleviated by instillation of lidocaine into the bladder but were unaffected when lidocaine was instilled into the uterus. Localization of the source of the pain to the bladder is higher specificity than found by Rudick et al. (46), who reported that pain in a model of virus-induced neurogenic cystitis in female C57BL6/J mice was alleviated by the instillation of lidocaine into either the bladder or colon.
The persistent increase in numbers of activated and total mast cells in the bladder in EAC mice is consistent with findings of increased mast cells in the bladder and mast cell effectors in urine in many clinical studies and in animal models of IC/PBS and chronic prostatitis/chronic pelvic pain syndrome (10, 18, 19, 54). Inhibition of UPK3A 65-84-induced tactile allodynia by cromolyn sodium confirmed the requirement for mast cell activation. Mast cells accumulate mostly at the tips of nerve endings in inflamed tissues, including the bladder in IC/PBS (33, 41), and increased numbers of nerve endings have been found in the bladder submucosa and detrusor muscle in patients with IC, especially ulcerative IC, compared with control subjects (11, 24, 38). Histamine released from mast cells can activate neurons (59) and induce neuronal hypertrophy (28). Our finding that the histamine receptor H2 antagonist ranitidine inhibited allodynia in EAC mice to a greater extent than the histamine receptor H1 antagonist cetirizine is in agreement with Rudick et al. (45), who reported greater inhibition of pelvic allodynia by histamine receptor H2 gene deletion or the histamine receptor H2 antagonist ranitidine than by histamine receptor H1 gene deletion or the histamine receptor H1 antagonist diphenhydramine in their murine model of virus-induced neurogenic cystitis. That group also showed similar levels of mRNAs for histamine receptors H1–H4 in the bladder in female C57BL6/J mice (45). Cetirizine, used in our study, is a second-generation histamine receptor H1 antagonist that lacks sedative effects due to its minimal ability to cross the blood-brain barrier, unlike first-generation histamine receptor H1 antagonists such as diphenhydramine and hydroxyzine (34). In a pilot randomized clinical trial for IC/PBS, hydroxyzine yielded a slightly but not significantly higher response rate compared with placebo (50). Better results may have been precluded by the lack of activity of hydroxyzine against the histamine receptor H2 and/or an insufficient sample size to assess activity in a subgroup with bladder mastocytosis.
It is still not clear why mast cells migrate to, proliferate, and degranulate in the bladder in IC/PBS patients. One possible explanation is that damaged urothelial cells or other bladder cells secrete chemokines such as CCL2 that cause migration of mast cells to the bladder and their activation (16, 42, 49). In autoimmune models, it is not unusual for chemokines to be secreted by cells distant from the site of antigen presentation (23). Levels of CCL2 have been found to be elevated in urine and bladder tissue of IC patients, including in the urothelium, stroma, and detrusor muscle (39) as well as in the urothelium and detrusor muscle of mice with cyclophosphamide-induced cystitis (5). We found persistent elevations of Ccl2 mRNA and protein in the bladder in our EAC mice; however, identifying the precise cell sources of CCL2 production will require additional experiments.
Our results showing almost complete prevention of UPK3A 65-84-induced tactile allodynia by genetic deletion of either CCL2 or its receptor CCR2 are similar to the findings of Quick et al. (44) in C57BL/6J mice with experimental autoimmune prostatitis, suggesting a common mechanism in bladder-generated and prostate-generated pelvic allodynia. Furthermore, we showed that accumulations of activated, resting, and total mast cells in the bladder in UPK3A 65-84-immunized Ccl2−/− or Ccr2−/− mice were not significantly different from CFA-injected WT mice, confirming the necessity of CCL2 and its receptor for the accumulation of mast cells in the bladder in EAC mice. A remaining question is whether the primary effects of Ccl2 and Ccr2 gene deletions in this EAC model were in mast cell recruitment and allodynia or in the initial immune response to UPK3A 65-84 immunization. In the experimental autoimmune prostatitis model of Quick et al. (44), male Ccr2−/− mice immunized with rat prostate lysate were completely protected from pelvic allodynia and exhibited only small, nonsignificant reductions in numbers of prostate-infiltrating CD4-positive and CD8-positive T cells compared with WT mice. In separate studies (21, 25) of myelin oligodendrocyte glycoprotein peptide-induced experimental autoimmune encephalomyelitis, Ccl2−/− mice and Ccr2−/− mice on the C57BL/6 background exhibited less motor impairment than WT mice, but adoptive transfer of antigen primed T cells from the knockout mice to naïve WT recipients induced the disease phenotype. Furthermore, induction of experimental autoimmune encephalomyelitis and uveitis in Ccr2−/− BALB/c and C57BL/6 mice, respectively, yielded intact T cell responses and disease phenotypes that were as severe as in WT mice (22, 52). Based on those results, it is likely that the primary role of the CCL2-CCR2 pathway in our EAC model is in mast cell recruitment and allodynia rather than in the initial immune response, although clarification of the role of that pathway will require additional studies.
In conclusion, we have shown that chronic tactile allodynia in our murine EAC model originates in the bladder and is mediated by CCL2, at least in part through the recruitment and activation of mast cells. Although the bladder is a site of CCL2-dependent accumulation and activation of mast cells in our model, we cannot rule out that the accumulation of mast cells at another site, such as a higher nervous system site, contributes to the allodynia. Along with our previous characterization of this model, these results further establish UPK3A 65-84 immunization of female BALB/cJ mice as a robust EAC model that can be used for further mechanistic studies of the pathogenesis of IC/PBS and therapeutic approaches for the disease.
GRANTS
This work was supported by National Institutes of Health (NIH) Grant R03-HD-061825 (to F. Daneshgari). The authors acknowledge the use of the Leica SCN400 Slide Scanner in the Genetics Department Imaging Facility at Case Western Reserve University, made available through NIH Shared Instrumentation Grant 1-S10-RR-031845.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: F.B., C.Z.A., V.K.T., and F.D. conception and design of research; F.B., C.Z.A., K.I., A.O., and M.K. performed experiments; F.B., C.Z.A., K.I., A.O., V.K.T., and F.D. analyzed data; F.B., C.Z.A., K.I., A.O., V.K.T., and F.D. interpreted results of experiments; F.B. and C.Z.A. prepared figures; F.B., C.Z.A., V.K.T., and F.D. drafted manuscript; F.B., C.Z.A., K.I., A.O., M.K., V.K.T., and F.D. approved final version of manuscript; V.K.T. and F.D. edited and revised manuscript.
ACKNOWLEDGMENTS
The authors thank Amad Awadallah for assistance in tissue sectioning and preparation of histological samples and C. Thomas Powell for editorial assistance with this manuscript.
Present address of C. Z. Altuntas: Texas Institute of Biotechnology Education and Research, North American Univ., 10555 Stella Link Rd., Houston, TX 77025 (e-mail: cza@na.edu).
Present address of K. Izgi: Dept. of Medical Biochemistry, School of Medicine, Erciyes Univ., Melikgazi, Kayseri 38039, Turkey (e-mail: kenanizgi@erciyes.edu.tr).
REFERENCES
- 1.Abbadie C, Lindia JA, Cumiskey AM, Peterson LB, Mudgett JS, Bayne EK, DeMartino JA, MacIntyre DE, Forrest MJ. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc Natl Acad Sci USA 100: 7947–7952, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aldenborg F, Fall M, Enerback L. Proliferation and trans-epithelial migration of mucosal mast-cells in interstitial cystitis. Immunology 58: 411–416, 1986. [PMC free article] [PubMed] [Google Scholar]
- 4.Altuntas CZ, Daneshgari F, Sakalar C, Goksoy E, Gulen MF, Kavran M, Qin J, Li X, Tuohy VK. Autoimmunity to uroplakin II causes cystitis in mice: a novel model of interstitial cystitis. Eur Urol 61: 193–200, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arms L, Girard BM, Malley SE, Vizzard MA. Expression and function of CCL2/CCR2 in rat micturition reflexes and somatic sensitivity with urinary bladder inflammation. Am J Physiol Renal Physiol 305: F111–F122, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baba T, Nakamoto Y, Mukaida N. Crucial contribution of thymic Sirp α+ conventional dendritic cells to central tolerance against blood-borne antigens in a CCR2-dependent manner. J Immunol 183: 3053–3063, 2009. [DOI] [PubMed] [Google Scholar]
- 7.Berry SH, Elliott MN, Suttorp M, Bogart LM, Stoto MA, Eggers P, Nyberg L, Clemens JQ. Prevalence of symptoms of bladder pain syndrome/interstitial cystitis among adult females in the United States. J Urol 186: 540–544, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bjorling DE, Wang ZY, Bushman W. Models of inflammation of the lower urinary tract. Neurourol Urodyn 30: 673–682, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bogart LM, Berry SH, Clemens JQ. Symptoms of interstitial cystitis, painful bladder syndrome and similar diseases in women: a systematic review. J Urol 177: 450–456, 2007. [DOI] [PubMed] [Google Scholar]
- 10.Boucher W, el-Mansoury M, Pang X, Sant GR, Theoharides TC. Elevated mast cell tryptase in the urine of patients with interstitial cystitis. Br J Urol 76: 94–100, 1995. [DOI] [PubMed] [Google Scholar]
- 11.Christmas TJ, Rode J, Chapple CR, Milroy EJ, Turner-Warwick RT. Nerve fibre proliferation in interstitial cystitis. Virchows Arch A Pathol Anat Histopathol 416: 447–451, 1990. [DOI] [PubMed] [Google Scholar]
- 12.Clemens JQ, Meenan RT, Rosetti MC, Gao SY, Calhoun EA. Prevalence and incidence of interstitial cystitis in a managed care population. J Urol 173: 98–102, 2005. [DOI] [PubMed] [Google Scholar]
- 13.Collington SJ, Hallgren J, Pease JE, Jones TG, Rollins BJ, Westwick J, Austen KF, Williams TJ, Gurish MF, Weller CL. The role of the CCL2/CCR2 axis in mouse mast cell migration in vitro and in vivo. J Immunol 184: 6114–6123, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Conti P, Boucher W, Letourneau R, Feliciani C, Reale M, Barbacane RC, Vlagopoulos P, Bruneau G, Thibault J, Theoharides TC. Monocyte chemotactic protein-1 provokes mast cell aggregation and [3H]5HT release. Immunology 86: 434–440, 1995. [PMC free article] [PubMed] [Google Scholar]
- 15.Denson MA, Griebling TL, Cohen MB, Kreder KJ. Comparison of cystoscopic and histological findings in patients with suspected interstitial cystitis. J Urol 164: 1908–1911, 2000. [PubMed] [Google Scholar]
- 16.Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res 29: 313–326, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Desireddi NV, Campbell PL, Stern JA, Sobkoviak R, Chuai S, Shahrara S, Thumbikat P, Pope RM, Landis JR, Koch AE, Schaeffer AJ. Monocyte chemoattractant protein-1 and macrophage inflammatory protein-1α as possible biomarkers for the chronic pelvic pain syndrome. J Urol 179: 1852–1861, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Done JD, Rudick CN, Quick ML, Schaeffer AJ, Thumbikat P. Role of mast cells in male chronic pelvic pain. J Urol 187: 1473–1482, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.el-Mansoury M, Boucher W, Sant GR, Theoharides TC. Increased urine histamine and methylhistamine in interstitial cystitis. J Urol 152: 350–353, 1994. [DOI] [PubMed] [Google Scholar]
- 20.Fall M, Johansson SL, Aldenborg F. Chronic interstitial cystitis: a heterogeneous syndrome. J Urol 137: 35–38, 1987. [DOI] [PubMed] [Google Scholar]
- 21.Fife BT, Huffnagle GB, Kuziel WA, Karpus WJ. CC chemokine receptor 2 is critical for induction of experimental autoimmune encephalomyelitis. J Exp Med 192: 899–905, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gaupp S, Pitt D, Kuziel WA, Cannella B, Raine CS. Experimental autoimmune encephalomyelitis (EAE) in CCR2(−/−) mice: susceptibility in multiple strains. Am J Pathol 162: 139–150, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Glabinski AR, Tani M, Tuohy VK, Tuthill RJ, Ransohoff RM. Central nervous system chemokine mRNA accumulation follows initial leukocyte entry at the onset of acute murine experimental autoimmune encephalomyelitis. Brain Behav Immun 9: 315–330, 1995. [DOI] [PubMed] [Google Scholar]
- 24.Hohenfellner M, Nunes L, Schmidt RA, Lampel A, Thuroff JW, Tanagho EA. Interstitial cystitis: increased sympathetic innervation and related neuropeptide synthesis. J Urol 147: 587–591, 1992. [DOI] [PubMed] [Google Scholar]
- 25.Huang DR, Wang J, Kivisakk P, Rollins BJ, Ransohoff RM. Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J Exp Med 193: 713–726, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang M, Pang XH, Karalis K, Theoharides TC. Stress-induced interleukin-6 release in mice is mast cell-dependent and more pronounced in apolipoprotein E knockout mice. Cardiovasc Res 59: 241–249, 2003. [DOI] [PubMed] [Google Scholar]
- 27.Izgi K, Altuntas CZ, Bicer F, Ozer A, Sakalar C, Li X, Tuohy VK, Daneshgari F. Uroplakin peptide-specific autoimmunity initiates interstitial cystitis/painful bladder syndrome in mice. PLOS ONE 8: e72067, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keles N, Yavuz Arican R, Coskun M, Elpek GO. Histamine induces the neuronal hypertrophy and increases the mast cell density in gastrointestinal tract. Exp Toxicol Pathol 64: 713–716, 2012. [DOI] [PubMed] [Google Scholar]
- 29.Kinet JP. The high-affinity IgE receptor (Fc εRI): from physiology to pathology. Annu Rev Immunol 17: 931–972, 1999. [DOI] [PubMed] [Google Scholar]
- 30.Kuziel WA, Morgan SJ, Dawson TC, Griffin S, Smithies O, Ley K, Maeda N. Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2. Proc Natl Acad Sci USA 94: 12053–12058, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laird JM, Martinez-Caro L, Garcia-Nicas E, Cervero F. A new model of visceral pain and referred hyperalgesia in the mouse. Pain 92: 335–342, 2001. [DOI] [PubMed] [Google Scholar]
- 32.Leiby BE, Landis JR, Propert KJ, Tomaszewski JE. Discovery of morphological subgroups that correlate with severity of symptoms in interstitial cystitis: a proposed biopsy classification system. J Urol 177: 142–148, 2007. [DOI] [PubMed] [Google Scholar]
- 33.Letourneau R, Pang X, Sant GR, Theoharides TC. Intragranular activation of bladder mast cells and their association with nerve processes in interstitial cystitis. Br J Urol 77: 41–54, 1996. [DOI] [PubMed] [Google Scholar]
- 34.Lieberman P. Histamine, antihistamines, and the central nervous system. Allergy Asthma Proc 30: 482–486, 2009. [DOI] [PubMed] [Google Scholar]
- 35.Link CL, Pulliam SJ, Hanno PM, Hall SA, Eggers PW, Kusek JW, McKinlay JB. Prevalence and psychosocial correlates of symptoms suggestive of painful bladder syndrome: results from the Boston area community health survey. J Urol 180: 599–606, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25: 402–408, 2001. [DOI] [PubMed] [Google Scholar]
- 37.Lu B, Rutledge BJ, Gu L, Fiorillo J, Lukacs NW, Kunkel SL, North R, Gerard C, Rollins BJ. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J Exp Med 187: 601–608, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lundeberg T, Liedberg H, Nordling L, Theodorsson E, Owzarski A, Ekman P. Interstitial cystitis: correlation with nerve fibres, mast cells and histamine. Br J Urol 71: 427–429, 1993. [DOI] [PubMed] [Google Scholar]
- 39.Lv JW, Luo Y, Leng J, Xue W, Liu DM, Huang YR. Aberrant expression of monocyte chemoattractant protein-1 (MCP-1) in interstitial cystitis patients. Sci Res Essays 5: 663–667, 2010. [Google Scholar]
- 39a.National Center for Biotechnology Information. PubChem Compound Database. Compound Summary for: CID 27503. Cromolyn Sodium (online). http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?cid=27503&loc=ec_rcs [24September2014]. [Google Scholar]
- 40.Oliveira SM, Drewes CC, Silva CR, Trevisan G, Boschen SL, Moreira CG, de Almeida Cabrini D, Da Cunha C, Ferreira J. Involvement of mast cells in a mouse model of postoperative pain. Eur J Pharmacol 672: 88–95, 2011. [DOI] [PubMed] [Google Scholar]
- 41.Pang X, Marchand J, Sant GR, Kream RM, Theoharides TC. Increased number of substance P positive nerve fibres in interstitial cystitis. Br J Urol 75: 744–750, 1995. [DOI] [PubMed] [Google Scholar]
- 42.Peeker R, Enerback L, Fall M, Aldenborg F. Recruitment, distribution and phenotypes of mast cells in interstitial cystitis. J Urol 163: 1009–1015, 2000. [PubMed] [Google Scholar]
- 43.Phillips K, Clauw DJ. Central pain mechanisms in chronic pain states–maybe it is all in their head. Best Pract Res Clin Rheumatol 25: 141–154, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Quick ML, Mukherjee S, Rudick CN, Done JD, Schaeffer AJ, Thumbikat P. CCL2 and CCL3 are essential mediators of pelvic pain in experimental autoimmune prostatitis. Am J Physiol Regul Integr Comp Physiol 303: R580–R589, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rudick CN, Bryce PJ, Guichelaar LA, Berry RE, Klumpp DJ. Mast cell-derived histamine mediates cystitis pain. PLOS ONE 3: e2096, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rudick CN, Chen MC, Mongiu AK, Klumpp DJ. Organ cross talk modulates pelvic pain. Am J Physiol Regul Integr Comp Physiol 293: R1191–R1198, 2007. [DOI] [PubMed] [Google Scholar]
- 47.Rudick CN, Schaeffer AJ, Klumpp DJ. Pharmacologic attenuation of pelvic pain in a murine model of interstitial cystitis. BMC Urol 9: 16, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rudick CN, Schaeffer AJ, Thumbikat P. Experimental autoimmune prostatitis induces chronic pelvic pain. Am J Physiol Regul Integr Comp Physiol 294: R1268–R1275, 2008. [DOI] [PubMed] [Google Scholar]
- 49.Sant GR, Kempuraj D, Marchand JE, Theoharides TC. The mast cell in interstitial cystitis: role in pathophysiology and pathogenesis. Urology 69: 34–40, 2007. [DOI] [PubMed] [Google Scholar]
- 50.Sant GR, Propert KJ, Hanno PM, Burks D, Culkin D, Diokno AC, Hardy C, Landis JR, Mayer R, Madigan R, Messing EM, Peters K, Theoharides TC, Warren J, Wein AJ, Steers W, Kusek JW, Nyberg LM; Interstitial Cystitis Clinical Trials Group. A pilot clinical trial of oral pentosan polysulfate and oral hydroxyzine in patients with interstitial cystitis. J Urol 170: 810–815, 2003. [DOI] [PubMed] [Google Scholar]
- 51.Sengupta JN. Visceral pain: the neurophysiological mechanism. Handb Exp Pharmacol 31–74, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sonoda KH, Yoshimura T, Egashira K, Charo IF, Ishibashi T. Neutrophil-dominant experimental autoimmune uveitis in CC-chemokine receptor 2 knockout mice. Acta Ophthalmol 89: e180–188, 2011. [DOI] [PubMed] [Google Scholar]
- 53.Thacker MA, Clark AK, Bishop T, Grist J, Yip PK, Moon LD, Thompson SW, Marchand F, McMahon SB. CCL2 is a key mediator of microglia activation in neuropathic pain states. Eur J Pain 13: 263–272, 2009. [DOI] [PubMed] [Google Scholar]
- 54.Theoharides TC, Kempuraj D, Sant GR. Mast cell involvement in interstitial cystitis: a review of human and experimental evidence. Urology 57: 47–55, 2001. [DOI] [PubMed] [Google Scholar]
- 55.Warren JW, Hanno PM. Symptoms of bladder pain syndrome. In: Bladder Pain Syndrome: a Guide for Clinicians, edited by Nordling J, Wyndaele JJ, van de Merwe JP, Bouchelouche P, Cervigni M, Fall M. New York: Springer Science+Business Media, 2013, p. 177–188. [Google Scholar]
- 56.Weller CL, Collington SJ, Williams T, Lamb JR. Mast cells in health and disease. Clin Sci 120: 473–484, 2011. [DOI] [PubMed] [Google Scholar]
- 57.Wood JD. Visceral pain: spinal afferents, enteric mast cells, enteric nervous system and stress. Curr Pharm Des 17: 1573–1575, 2011. [DOI] [PubMed] [Google Scholar]
- 58.Yoshimura N, Birder LA. Interstitial cystitis and related painful bladder syndromes: pathophysiology. In: Chronic Abdominal and Visceral Pain: Theory and Practice, edited by Pasricha PJ, Willis WD, Gebhart GF. New York: Informa Healthcare USA, 2006, p. 495–520. [Google Scholar]
- 59.Zhou J, Lee AW, Devidze N, Zhang Q, Kow LM, Pfaff DW. Histamine-induced excitatory responses in mouse ventromedial hypothalamic neurons: ionic mechanisms and estrogenic regulation. J Neurophysiol 98: 3143–3152, 2007. [DOI] [PubMed] [Google Scholar]