

Abstract
Breast cancer is among the most commonly diagnosed cancer types in women worldwide and is the second leading cause of cancer-related disease in the USA. SH2 domains recruit signaling proteins to phosphotyrosine residues on aberrantly activated growth factor and cytokine receptors and contribute to cancer cell cycling, metastasis, angiogenesis and so on. Herein we review phosphopeptide mimetic and small-molecule approaches targeting the SH2 domains of Grb2, Grb7 and STAT3 that inhibit their targets and reduce proliferation in in vitro breast cancer models. Only STAT3 inhibitors have been evaluated in in vivo models and have led to tumor reduction. Taken together, these studies suggest that targeting SH2 domains is an important approach to the treatment of breast cancer.
Breast cancer is among the most commonly diagnosed types of cancer in women worldwide and is the second leading cause of cancer-related disease in the USA, accounting for more than 40,000 deaths annually [1]. Breast cancers have a high degree of genomic heterogeneity, which has a significant influence on treatment options, patient response to therapy, patterns of metastasis and patient survival [2,3]. Breast tumors are classified into specific intrinsic subtypes based on the presence or absence of estrogen receptor (ER), progesterone receptor, ERBB2/Her2 oncogene amplification, the proliferation marker Ki-67 and the level of claudins, proteins involved in formation of tight junctions [4]. Importantly, the diagnosis, treatment and outcome are dependent upon the intrinsic subtype of the individual breast tumor [5]. Breast tumors that express the ER, and/or PR, lack amplification of Her2 and have low Ki-67 are classified as luminal A [2] and represent approximately 40% of all breast tumors [6]. Patients with this subtype have the highest positive survival rates, in part due to the development of hormone therapies. Breast cancers that have amplification of the ERBB2/Her2 oncogene are another major subclass [2] and represent approximately 20% of all breast cancers [6]. This subtype of breast cancer has been the focus of significant efforts to develop targeted therapeutics. While there have been significant advances in the development of therapeutics that target ER and the Her2 oncogene [7–9], few targeted therapeutics have been developed to treat breast tumors that lack ER/progesterone receptor and Her2 oncogene, classified as triple-negative/basal-like subtype tumors (TNBC). Owing to the lack of hormone receptors and Her2 oncogene amplification, patients with TNBC have a less favorable prognosis than those with other subtypes of breast cancer [10–12], demonstrating the significant need to focus effort to identify therapeutic targets in this subtype.
Signaling pathways originating from growth factors or cytokines are aberrantly activated in all breast cancers and contribute to cancer cell cycling, metastasis, angiogenesis and so on, although these aberrations are not unique to breast cancer. A major mechanism of transmitting signals is by phosphorylation and dephosphorylation of tyrosine residues on key proteins, such as growth factor and cytokine receptors, adapter proteins and enzymes, such as non-receptor tyrosine kinases. Phosphorylation creates sites for protein–protein interactions between the phosphorylated protein and signaling molecules containing SH2 and phosphotyrosine binding domains that recognize phosphotyrosine [13]. In this review we will discuss efforts to develop agents that target SH2 domains that have been evaluated in breast cancer models.
SH2 domains are approximately 100 amino acid subunits that mediate the transduction of signals via formation of multiprotein complexes initiated by recognition and binding to select phosphotyrosine residues on receptors and other proteins [13]. SH2 domains were recognized early in the 1990s as potential targets for several diseases, including cancer (reviewed in [14]). Early crystal structures of SH2 domains complexed with phosphopeptide ligands provided valuable information on the specifics of phosphopeptide–protein interactions [15,16]. This led to intense efforts in the 1990s and early 2000s by the pharmaceutical industry, academic laboratories and government laboratories to develop small-molecule phosphopeptide mimetic inhibitors, mainly targeting Src kinase, the Src-family kinase Lck, p85, the regulatory subunit of PI3K and Grb2 and these efforts have been reviewed extensively [17–21]. More recently, the SH2 domains of Grb7 [22] and STAT3, STAT5 have been targeted with peptides and peptide mimetics [23,24].
Developing viable drugs targeting SH2 domains has significant challenges. The phosphotyrosine residue has been estimated to provide one half of the binding energy of phosphopeptides to the SH2 domain [25]. However, the negative charge of the phosphate is a significant barrier to cell penetration and phosphate groups can be removed by phosphatases. Additionally, peptides are subject to proteolytic cleavage and, as a rule, exhibit poor bioavailability and cell permeability. Overall the pharmaceutical industry was successful in converting phosphopeptides to non-peptide mimics. However, overcoming the negative charge requirement was so significantly problematic that SH2 domains were abandoned as undruggable targets. Efforts by academic laboratories are continuing and only recently reports of cell-permeable, SH2 domain-targeted inhibitors with in vivo activity have been reported [26,27].
Inhibitors targeting Grb2
Grb2 is a scaffold protein composed of two SH3 domains that flank a central SH2 domain. On binding of growth factors (e.g., EGF) to their receptors (e.g., Her-2), trans phosphorylation of tyrosine residues on intracellular domains by the kinase activity of the receptor occurs. Via its SH2 domain Grb2 is recruited to the receptor phosphotyrosine residues, at which time its SH3 domains recruit SOS. SOS binds to RAS and activates it leading to MEK pathway activation. Blocking the association of Grb2 and the growth factor with phosphopeptides or mimetics inhibits activation of RAS and the downstream MEK pathway. Readers are referred to excellent reviews on Grb2 as a cancer target [28] and on peptidomimetic inhibitor development [29].
The SH2 domain of Grb2 recognizes phosphotyrosine in the context of the sequence pTyr–Xaa–Asn (Figure 1). Using the peptidomimetic approach, researchers from Novartis (Basel, Switzerland) discovered that replacing the pY+1 residue with a 1-aminocyclohexylcarboxylate (Ac6c) and replacement of amino acids C-terminal to the required Asn with a naphthylpropyl group led to a lead phospho-tripeptide with low nM affinity [30,31]. To protect against cleavage of the phosphate by phosphatases, pTyr was replaced with 4-phosphonomethyl phenylalanine (Pmp) in peptidomimetic CGP78850 (Figure 1) [32]. Interestingly, in spite of the free phosphonate group with its negative charges, CGP78850 inhibited association of Grb2 with EGFR in intact serum-starved MDA-MB-468 triple-negative breast cancer cells stimulated with EGF in vitro. Significant inhibition was observed at 100 nM, while 10 μM brought cells to the non-EGF-stimulated state. Ras activation, as measured by interaction of Ras with a Ras binding domain–GST construct, and anchorage-independent growth, was also inhibited by treating cells with 50 μM inhibitor. To block the charge of the phosphonate, CGP78850 was converted into the prodrug CGP85793 using McQuigan’s bio-reversible phenyl phosphoramidate scheme (Figure 1) [33]. The prodrug inhibited Ras activation at 50-fold lower concentration than the free phosphonate and also inhibited proliferation of MDA-MB-468 cells. CGP85793 also stimulated increases in levels of p27Kip1 and p21Waf1/Cip1/CAP1, albeit at the high concentration of 100 μM. Both the free phosphonate and prodrug were found to inhibit tumor cell motility processes in A431 and MDCK cells [34]. Further development of these compounds was not reported.
Figure 1.
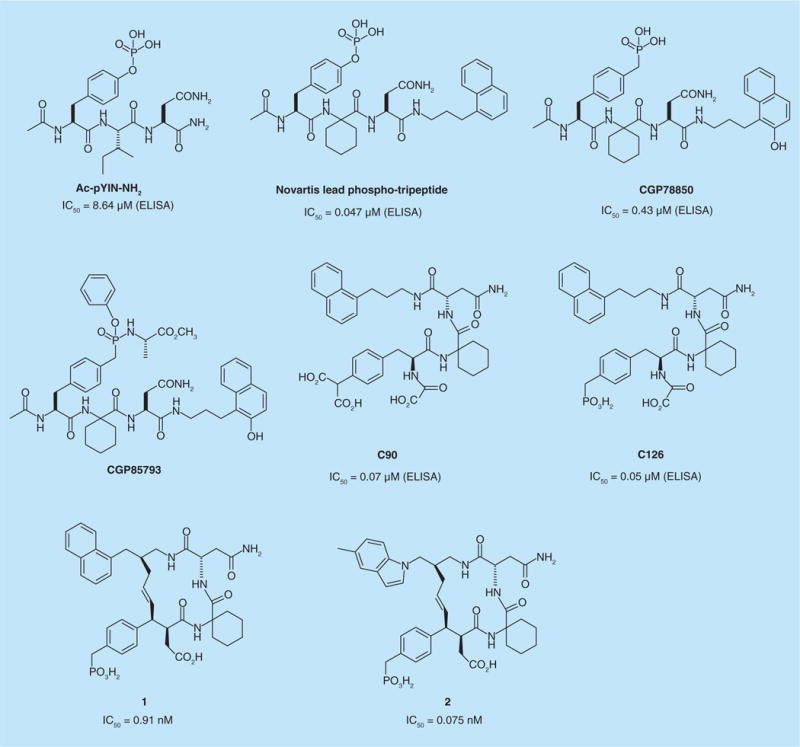
Peptidomimetic inhibitors of Grb2.
Terrence Burke and colleagues and the National Cancer Institute (NCI, Frederick, MD, USA) performed a series of structure–activity relationship studies on the Novartis lead phospho-tripeptide to probe interactions with the SH2 domain of Grb2. Phosphotyrosine was replaced with a series of non-hydrolyzable surrogates to prevent inactivation by phosphatases [35–37]. The phosphate was replaced with groups such as malonyloxy, malonyl, phosphonomethyl, phosphonodifluoromethyl, carboxy, carboxymethyl and others to test the interaction of different displays of negative charge with the positively charged Arg67 and Arg86 in the phosphate-binding pocket. Derivatization of the α-amino group as an oxalyl amide imparted greater affinity than as an acetamide, likely due to an extra ionic interaction with quanidinium group of Arg67, which lines the pTyr-binding pocket (C90 and C126, Figure 1). Taking a lead from Novartis, the naphthyl group was replaced with an indole group [35]. As part of the structure–activity relationship, inhibitors were evaluated for their ability to bind to Grb2 in extracellular ELISA or surface plasmon resonance (SPR) assays; inhibit the interaction of Grb2 and erbB-2 in MDA-MB-453 breast cancer cells; and, exert cytotoxicity against this line, which is dependent on Grb2 for proliferation. Interestingly, the negative charges on the phosphonate, malonate and oxalyl groups did not prevent cellular activity and some of these compounds potently inhibited proliferation of breast cancer cells, suggesting utility of this approach for breast cancer treatment. These studies led to two candidates, C90 and C126 (Figure 1), that were studied in greater detail. C90 incorporates para-malonylphenylalanine (Pmf) as a pTyr replacement and C126 employs Pmp. These two materials exhibited very high affinity for the SH2 domain of Grb2, with IC50 values of 70 and 50 nM in extracellular ELISA competition affinity measurements [35]. Both compounds inhibited intracellular association of Grb2 and erbB-2, as well as downstream MAPK activation in cultured MDA-MB-453 breast cancer cells [35]. C60 and C126 inhibited association of Grb2 and c-Met with IC50s of 30 nM. [38]. They were selective for Grb2 as levels of PI3K downstream protein pAkt, pErk1 and pErk2 were not affected. As tools, they provided evidence that Grb2 does not transduce mitogenic signals originating from HGF/c-Met [38]. C60 and C126 exhibited antiangiogenic properties in in vitro assays [39] and C60 showed ability to inhibit cell migration and metastasis when pretreated cells were implanted as xenografts in nude mice [40].
Macrocyclization led to extremely high-affinity inhibitors with IC50s in the low nM range, with one very potent candidate in the low pM range [41–43]. In addition to the presumed reduction in the binding entropy penalty due to cyclization, affinity was enhanced by inclusion of a carboxymethyl substituent at the α-position of the pTyr mimic, again allowing ionic interaction with the side chain of Arg67. Inhibitor 1 (Figure 1) had an IC50 of 910 pM in extracellular SPR assays [42]. It was able to inhibit Grb2–erbB-2 interaction with an IC50 of approximately 1 μM in MDA-MB-453 cells. Inhibition of proliferation of this cell line occurred with an IC50 of 1.7 μM, whereas with MDA-MB-231, TNBC cells that are not dependent on Grb2, the IC50 was >10 μM. Replacing the naphthyl group with the 5-methylindole group, inhibitor 2, resulted in a great increase in affinity with the IC50 of 75 pM (SPR). The high affinity was reflected in the ability to inhibit Grb2–erbB-2 interaction (IC50 ≤10 nM) and proliferation (IC50 = 0.63 μM) in MDA-MB-453 breast cancer cells. Selectivity was demonstrated by the lack of inhibition of proliferation of MDA-MB-231 cells [42].
Although the peptidomimetics reported by the Novartis and Burke groups were not advanced to preclinical in vivo cancer models, with the exception of the limited study of Burkes’s C60, the results reported by these groups in cultured cells demonstrate the potential of blocking the SH2 domain of Grb2 as a modality for the treatment of breast cancer.
Inhibitors targeting Grb7
Grb7 is the prototype member of the Grb7 adapter protein family that also includes Grb10 and Grb14. These multidomain cytoplasmic proteins comprise an N-terminal proline-rich region, a central region known as the GM region and a C-terminal SH2 domain [44]. The SH2 domain of Grb7 binds numerous signaling molecules and receptors including the ErbB family of tyrosine kinase receptors and tyrosine-phosphorylated signaling molecules associated with cell migration and invasion, such as the FAK [44]. Grb7 is co-amplified with ErbB2 in a panel of breast cancer subtypes [45] and clinical data showed the association between high Grb7 expression and poor outcomes in TNBC patients [22,46]. Recent studies using proteomic analysis revealed activation of Grb7 as part of EGFR signaling activity in inflammatory breast cancer [47]. Therefore, targeting the SH2 domain is a potentially promising therapeutic strategy for the treatment of tumors overexpressing Grb7.
Using phage-display technology, the group of David N Krag identified a series of non-phosphorylated peptide inhibitors binding the SH2 domain of Grb7. All of these peptides contained a Tyr–X–Asn motif as part of constrained cyclic peptides containing 11 amino acids [48]. Note that this is the recognition site for Grb2. One of the leads, G7–18NATE (Figure 2) inhibited the association of Grb7 with members of the ErbB tyrosine-kinase family at approximately 100 μm in cellular extracts of SK-BR-3 breast cancer cells. Later, Krag and coworkers synthesized cell-permeable derivatives of G7–18NATE, where a cell-penetrating peptide, Penetratin or Tat, was covalently attached to the C-terminal carboxyl group of Cys11. Both adducts, G7–18NATE– Penetratin (G7–18NATE-P) and G7–18NATE–Tat (G7–18NATE-T) inhibited proliferation of SK-BR-3, ZR-75–30, MDA-MB-361 and MDA-MB-231 cell lines with EC50 values of 7.5–8 μM [49]. G7–18NATE-P was also shown to improve the inhibitory effect on SK-BR-3 cell proliferation when given in combination with doxorubicin and herceptin [49].
Figure 2. Non-phosphorylated cyclic peptide inhibitors targeting the SH2 domain of Grb7.
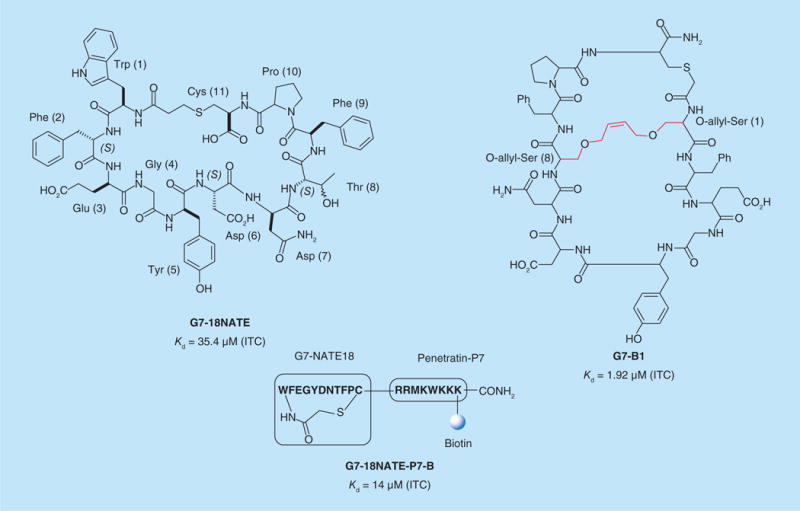
ITC: Isothermal calorimetry.
A novel cell-permeable derivative of G7–18NATE was reported by the group of J Wilce [50]. G7–18NATE was ligated to the last seven residues of Penetratin and a biotin tag allowing intracellular visualization of the inhibitor by confocal microscopy. G7–18NATE-P7–biotin (G7–18NATE-P7-B) was obtained by solid-phase peptide synthesis using conventional Fmoc chemistry. Biotin was selectively installed on the C-terminal Lys side chain of the shortened Penetratin sequence prior to cyclization of the final peptide via thioether bond formation between the side chain of Cys11 and N-terminal Trp1 of the G7–18NATE moiety (Figure 2). G7–18NATE-P7-B was shown to be efficiently taken up by MDA-MB-468 breast cancer cells after 30 min exposure to 10 μM inhibitor [50]. Isothermal calorimetry (ITC) experiments demonstrated that G7–18NATE-P7-B exhibited a twofold higher binding affinity, compared with G7–18NATE, thus indicating that the biotinylated Penetratin sequence did not negatively impact the ability of G7–18NATE to bind the Grb7-SH2 domain [50]. The same research group recently reported a second generation bicyclic peptide inhibitor derived from G7–18NATE with improved binding affinity for the SH2 domain of Grb7 [51]. The crystal structure of a G7–18NATE–Grb7-SH2 domain complex showed that Trp 1 and Thr 8 of the lead were not directly involved in binding interactions [52]. Based on this observation, Wilce and coworkers explored the hypothesis that constricting the inhibitor in its bound conformation would result in an improved affinity for the Grb7-SH2 domain. To this aim, they synthesized G7-B1 (Figure 2), a bicyclic version of G7–18NATE where Trp1 and Thr8 were replaced with O-allyl-serine residues allowing intramolecular linkage by metathesis reaction. The constrained bicyclic peptide resulted in a two- to three-fold improvement in binding affinity compared with the monocylic precursor. Although G7-B1 still awaits evaluation in cellular assays, it appears a promising Grb7 inhibitor. These cyclic peptides demonstrate proof-of-principle that inhibiting the SH2 domain of Grb7 has potential for breast cancer treatment.
Using phosphopeptide Grb2 inhibitors to derive a 3D structural template, Ambaye et al. searched the NCI database and identified a series of benzopyrazine inhibitors of Grb7 [53]. Compounds were found to bind to the SH2 domain of Grb7 by both thermofluor and ITC. Lead NSC642056 (Figure 3) exhibited somewhat modest affinity with a Kd of 17.2 μM measured by ITC, and inhibited proliferation of MDA-MB-468 TNBC cells with an IC50 of 86 μM. In a slightly different approach, a second screen using G7–18NATE as a structural template identified a series of benzamides as potential inhibitors [54]. Again thermofluor and ITC provided proof of binding to the SH2 domain of Grb7 of the lead, NSC104999 (Figure 3), and a series of analogs. The Kd of the lead was found to be 32.3 μM for isolated Grb7 SH2 domain and the IC50 for growth inhibition of cultured MDA-MB-468 cells was 39.9 μM. For both series, ITC revealed significant differences in enthalpic and entropic contributions that were largely canceled out leading to similar free energies of binding and Kd ranges of 1–32 μM. The authors point out that for both series selectivity studies are required to determine if growth inhibition is due to Grb7 inhibition or off-target effects.
Figure 3. Non-peptidic inhibitors of Grb7 discovered by computational screening.
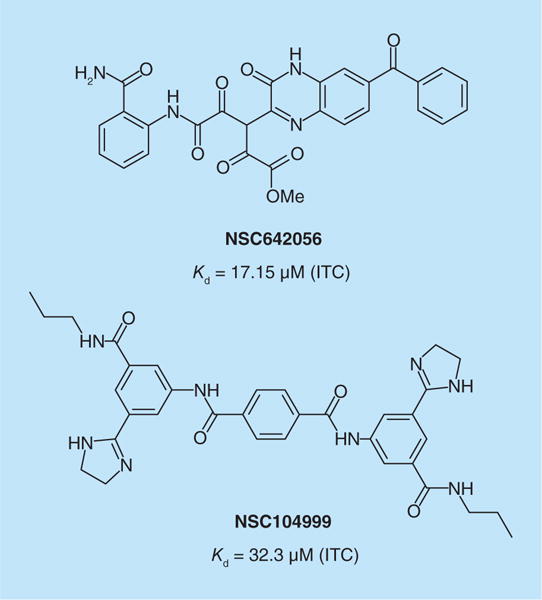
ITC: Isothermal calorimetry.
Specificity of inhibitors for Grb2 & Grb7
Both Grb2 and Grb7 recognize Tyr–Xaa–Asn as the recruiting motif. Thus, selectivity for individual proteins may be difficult to achieve. The non-phosphorylated cyclic peptide, G7–18NATE, was found to be selective for Grb7 over Grb2, Grb10 and Grb14 [48]. However, the second virtual screen conducted by Ambaye et al. found NSC708238 among the first set of hits [54]. This material is C90 reported by the Burke laboratory (Figure 1). Thus, small phosphopeptide and related mimics based on pTyr–Xaa–Asn have the potential to ligate to both Grb2 and Grb7 and biological results must be interpreted with this in mind.
Inhibitors targeting STAT3
STAT3 transmits signals from IL-6 family cytokines and growth factors, such as EGF, PDGF, ALK and VEGF, directly from the receptor to the nucleus where it is involved in the transcription of downstream genes. It is involved in cell cycling, metastasis, angiogenesis and immune cell evasion in most human cancers, including breast cancer, and its activity has been reviewed extensively [55–57]. On activation of the cytokine or growth factor receptor, STAT3, via its SH2 domain, is recruited to pTyr residues on the cytoplasmic domains of the receptor. Upon receptor docking, Tyr705 of STAT3 becomes phosphorylated by associated JAK, Src kinase or the kinase activity of the receptor. Tyr705 phosphorylation, termed activation, results in STAT3 dimerization by reciprocal pTyr-SH2 domain interactions. The dimer is translated to the nucleus where STAT3 behaves as a transcription factor and participates in the expression of various genes including those in the acute phase response, angiogenesis and cell cycling. STAT3 has been shown to be activated in a large percentage of breast cancer tumor samples from patients and is constitutively activated in several breast cancer cell lines, both human and murine [58,59]. Several groups have developed small-molecule and phosphopeptide-based compounds targeting the SH2 domain of STAT3 and these have been recently reviewed [23,60–62].
Small molecules targeting the SH2 domain
Several groups have screened compound libraries to find small molecules that have conventional drug-like properties to target the SH2 domains of STAT3. One of the first publications was that of Song et al., who reported the discovery of STA-21 (Figure 4) using a virtual library docking to the SH2 domain of STAT3 [63]. This compound inhibited STAT3-dependant luciferase activity in MDA-MB-435 cells, as well as the expression of canonical STAT3 downstream genes. Preferential growth inhibition was also observed in breast cancer cell lines harboring constitutively active STAT3 phosphorylation.
Figure 4. Small-molecule inhibitors proposed to target the SH2 domain of STAT3.
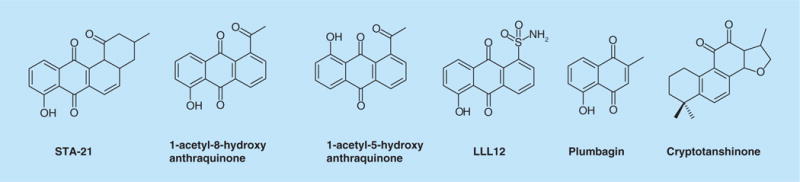
Binding affinities for these compounds were not reported.
No data showing actual binding to the SH2 domain were presented in this study. Computer-based docking models are useful for the development of hypotheses about the interactions between compounds and their target proteins. To test the hypothesis put forward by Song et al. that STA-21 binds directly to the SH2 domain, our laboratory acquired this compound and found that at concentrations up to 100 μM it did not compete with a fluorescein-tagged phosphopeptide, FAM–Ala–pTyr–Leu–Pro–Gln–Thr–Val–NH2 (FAM = 5-carboxyfluorescein), in our fluorescence polarization assay (10 nM peptide, 80 nM STAT3) (Figure 5) [McMurray JS et al., Unpublished Data] [64]. This experiment strongly suggests that STA-21 does not bind to the SH2 domain of STAT3 and that the reported activity in cells was likely due to indirect perturbation of STAT3 phosphorylation.
Figure 5. Neither STA-21 nor cryptotanshinone exhibit affinity for the SH2 domain of STAT3.
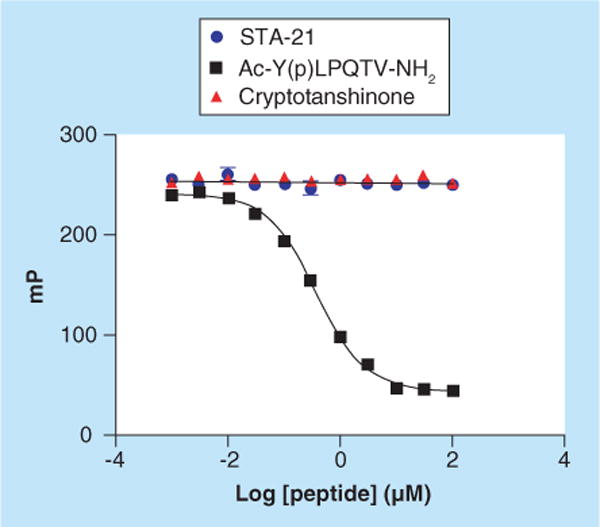
Competition of the binding of FAM–Ala–pTyr–Leu–Gln–Thr–Val– NH2 to STAT3 measured by fluorescence polarization using the conditions of Coleman et al. [64] (FAM = 5-carboxygluorescein). Fluorescent-peptide (10 nM) and full-length STAT3 (80 nM) were incubated with increasing concentrations of phosphopeptide, STA-21, or cryptotanshinone and polarization was read.
Subsequently reported analogs of STA-21, 1-acetyl-5-hyroxyanthraquinone, 1-acetyl-8-hyroxyanthraquinone and LLL12, are quite effective at inhibiting cancer cell proliferation, inducing apoptosis and so on, concomitantly with reductions in pSTAT3 levels in cultured cells [65,66]. These analogs were also docked to the SH2 domain of STAT3 using computational methods, but no biochemical evidence was presented to show that they bind to the SH2 domain of STAT3. STA-21 and its analogs are benzoquinones and anthroquinones, which are known to exert cytotoxic effects through redox recycling and arylation of essential nucleophiles, such as glutathione and protein thiols [67]. These inhibitors are structural analogs of plumbagin, a well known phytochemical that has been shown to have antibacterial properties, alter redox potential in cells, inhibit NF-κB, alter mitotic spindles and chelate heavy metals, among other effects [68]. Since no appreciable binding to the SH2 domain of STAT3 was measured, the off-target activities of the hydroxyquinone moiety may be responsible for the antiproliferative activity of STA-21.
As STAT3 both transmits VEGF signals and is involved in the transcription of the VEGF gene [69,70], LLL12 was assayed for antiangiogenic activity. It inhibited proliferation, tube formation and migration of human umbilical vascular endothelial cells in vitro. LLL12 inhibited the growth of OS-1 osteosarcoma xenografts, which was accompanied by an extensive reduction in microvessel density and STAT3 phosphorylation levels. Growth inhibition was the result of inhibition of VEGF signaling as well as the inhibition of the production of angiogenic factors, VEGF, MMP-9, angiopoetin, tissue factor and FGF1, which was attributed to the near complete inhibition of pSTAT3 measured in the tumors [71]. Although this was not a breast cancer model, these results suggest that LLL12 is a novel antiangiogenic agent.
Another small-molecule natural product, cryptotanshinone, was reported to inhibit STAT3 activity in tumor cells [72]. A molecular model generated by computational docking reported by the authors suggested that this molecule binds to the phosphotyrosine binding site on the SH2 domain of STAT3. To test this hypothesis, we also acquired this material and found that at concentrations as high as 100 μM it does not bind to STAT3 in our fluorescence polarization assay [McMurray JS et al., Unpublished Data]. Therefore, it is unlikely that cryptotanshinone binds to the SH2 domain of STAT3 in cultured cells. It was recently reported that cryptotanshinone reduced the phosphorylation of JAK2, which phosphorylates STAT3 [73]. The mechanism is likely related to the increased expression of Src homology region 2 domain-containing phosphatase-1 on addition of this material to cells [73]. Cryptotanshinone, an orthoquinone, not surprisingly has several other activities associated with it, such as inhibition of NF-κB and COX-2 and activation of the PI3K/Akt pathway [74,75]. Thus, the activities of this material leading to cell death will be difficult to precisely enumerate.
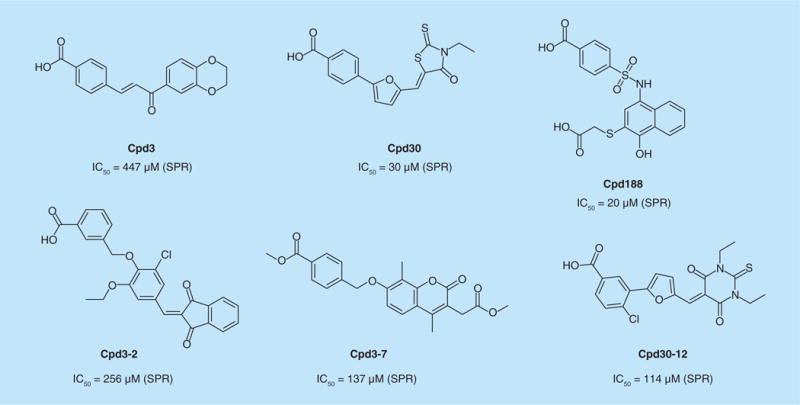
David Tweardy and colleagues carried out a virtual screen of small molecules to find candidates that bind to the SH2 domain of STAT3. Three leads were identified and were found to compete with a phosphopeptide derived from EGF pTyr1068 in a SPR assay for binding to STAT3, thus, providing evidence that they actually bind to the SH2 domain [76]. Cpd3, Cpd30 and Cpd188 (Figure 6) exhibited IC50 values of 447, 30 and 20 μM, respectively, in the SPR assay and inhibited IL-6-stimulated STAT3 phosphorylation in HepG2 cells with IC50 values of 91, 18 and 73 μM. Similarity screening of other compound databases revealed that Cpd3–7, Cpd3–12 and Cpd30–20 were capable of inhibiting phosphopeptide binding to STAT3, as well as IL-6-stimulated STAT3 phosphorylation. This panel was assayed for the ability to induce apoptosis in a panel of breast cancer lines. Compounds Cpd3 and Cpd30 were selective for cell lines with constitutively phosphorylated STAT3, MDA-MB-468, MDA-MB-231 and MDA-MB-435. No effect was observed on the control lines MDA-MB-453 and MCF7 not harboring constitutively phosphorylated STAT3. Of all the compounds, Cpd188 was the most potent, causing apoptosis in cell lines expressing high levels of phospho-STAT3 with IC50 values of 0.73–7.01 μM. Cross-reactivity with the control lines was observed, albeit with reduced potencies: IC50 values were 15 and 17 μM.
Figure 6. Small-molecule STAT3 inhibitors.
SPR: Surface plasmon resonance.
TNBC and Her2+ breast cancers are enriched for cells defined as cancer stem cells (CSCs) or tumor-initiating cells [77,78], which play a role in development of resistance to chemotherapeutic agents, and their persistence following primary systemic chemotherapy has been associated with poor prognosis [79,80]. CD44+/CD24low/− CSCs in breast tumors require the JAK2/STAT3 signaling pathway for growth [81]. Inflammatory breast cancer, the most metastatic form of breast cancer, has been demonstrated to be enriched for CSCs [82], which is associated with activation of the JAK/STAT3 pathway [82]. Cpd188 in combination with docetaxel reduced CSC populations in human breast cancer xenograft models in mice, resulting in improved recurrence-free survival [60].
Peptidomimetic inhibitors targeting the SH2 domain of STAT3
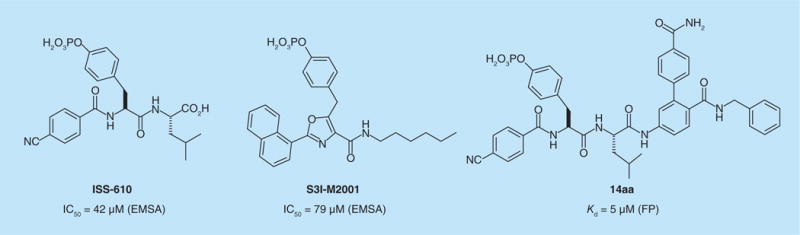
James Turkson and colleagues developed both peptidomimetic and small-molecule inhibitors of STAT3. They first described tripeptide mimetics derived from Tyr705, Pro–pTyr–Leu–Lys–Thr–Lys and pTyr SH2 domain-binding sequence in STAT3 dimers [83]. Pro–pTyr–Leu and Ala–pTyr–Leu showed inhibition of STAT3 activation at approximately 200 μM in cellular extracts of EGF-stimulated fibroblasts [84]. Further modifications of the lead tripeptides by substitution of the Y-1 residue with aromatic groups led to a library of synthetic derivatives, one of which, ISS-610 (Figure 7), showed preferential inhibition of STAT3 at 100 μM in Src-transformed mouse fibroblasts (NIH3T3/v-Src) and human breast cancer cells MDA-MB-231 and MDA-MB-435 with constitutive STAT3 activation. Computational analysis of the interactions between ISS-610 and the SH2 domain of STAT3 combined with structural information derived from the x-ray crystal structure of STAT3β led Siddiquee et al. to the development of S3I-M2001 (Figure 7), an oxazole derivative of ISS-610 [85]. This compound inhibited STAT3 phosphorylation at 100 μM in NIH 3T3/v-Src and MDA-435-MB cells and tumor growth in human breast tumor xenografts [85]. In a continuing effort to develop more potent and selective inhibitors targeting the SH2 domain of STAT3, Patrick Gunning and co-workers reported a library of hybrid peptidomimetic inhibitors derived from ISS-610 and peptide 1.6, one of the STAT3 inhibitors developed in the McMurray laboratory [86]. In vitro evaluation of the lead compound 14aa (Figure 7) on NIH-3T3/v-Src cells showed inhibition of STAT3 at 100 μM. Interestingly, a non-phosphorylated version of 14aa inhibited STAT3 phosphorylation in cultured cells.
Figure 7. Peptidomimetic inhibitors of STAT3 derived from Pro–pTyr–Leu–Lys–Thr–Lys.
FP: Fluorescence polarization.
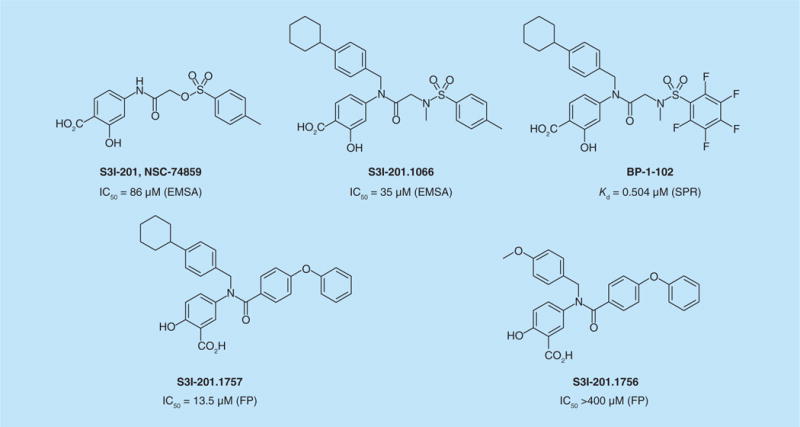
A computer-aided screening approach of compound libraries from the NCI, identified S3I-201 (NSC-74859, Figure 8), which inhibited STAT3– DNA complex formation and decreased survival of Src-transformed mouse fibroblasts and human breast cancer cell lines MDA-MB-231, MDA-MB-468 and MDA-MB-435 that harbor constitutive STAT3 activation. Inhibition of tumor growth was also observed in MDA-231-MB xenografts in mice [85]. Molecular models suggested that the salicylic acid group docked to the phosphotyrosine binding site. Modifications of the glycolic acid scaffold of S3I-201 were made to improve binding affinity, paying particular attention to the third hydrophobic subpocket of the SH2 domain of STAT3. In addition to breaking up STAT3–DNA complexes, binding to the SH2 domain was demonstrated by competition of a high affinity fluorescently labeled phosphopeptide using fluorescence polarization [87]. One of these derivatives, SF-1–066 (or S3I-201.1066, Figure 8), showed inhibition of STAT3 at 50 μM in NIH-3T3/v-Src, Panc-1 and MDA-231-MB cells as well as reduction of tumor growth in human MDA-231-MB breast cancer xenografts [88]. Recently, Zhang et al. reported BP-1–102, an orally bioavailable analog of SF-1–066 (8) [27]. The authors provided extensive SPR, fluorescence polarization (FP) and ITC evidence of binding to the phosphotyrosine binding pocket of the SH2 domain of STAT3. Interestingly the Kd determined by SPR was 504 nM, whereas that determined by FP was 4.1 μM. Unfortunately intrinsic binding was not studied directly by ITC. BP-102 inhibited both anchorage-dependent and independent proliferation of cell lines exhibiting constitutive phosphorylation of STAT3, including the TNBC line, MDA-MB-231. Those lines not expressing pSTAT3 were not growth inhibited. BP-102 inhibited the growth of MDA-MB-231 xenografts in mice, which was accompanied by reduction in canonical downstream genes c-Myc, cyclin D1, Bcl-XL, survivin, and VEGF. Evidence of antiangiogeneic activity was not reported. BP-102 displayed favorable oral and intravenous pharmacokinetic characteristics and was found at concentrations of 55 and 35 μg/g tumor tissue from iv. and oral delivery, respectively. BP-102 provides strong evidence that inhibition of STAT3 phosphorylation is a promising strategy for treatment of breast cancer.
Figure 8. Small-molecule STAT3 inhibitors derived from S3I-201/NSC-74859.
FP: Fluorescence polarization; SPR: Surface plasmon resonance.
Page et al. developed further analogs of BP-1–102 and SF-1–066 to explore alkyl substitution on the sulfonamide nitrogen [89]. Although several analogs were cytotoxic to MDA-MB-468 cells, no major improvements in potency were found over the N-methyl leads. Structure–activity relationship studies starting with S3I-201 was carried out independently by Urlam et al. [90]. In contrast to the 4-aminosalicylates in BP-1–102 and SF-1–066, the authors found that 5-aminosalicylates were quite effective as pTyr mimics. The sulfonylglycolamide was replaced by benzamides and again a 4-cyclohexylbenzyl substituent on the nitrogen provided both binding energy and cellular potency. This work led to S3I-1757 (Figure 8), reported by Zhang et al. [91], which selectively inhibited the phosphorylation of STAT3 over AKT1 and ERK1/2, nuclear accumulation of phospho-STAT3, STAT3–DNA binding and transcriptional activation and suppressed expression STAT3 target genes, such as Bcl-xL (BCL2L1), survivin (BIRC5), cyclin D1 (CCND1) and MMP-9. Furthermore, S3I-1757 inhibited anchorage-dependent and independent growth, migration and invasion of human breast cancer cells, which depend on STAT3. The importance of the cyclohexyl substituent of the benzyl group was illustrated by S3I-1756, which had no impact on any of the intracellular or extracellular parameters inhibited by S3I-1757.
To find a lead for the development of phosphopeptide ligands targeting the SH2 domain of STAT3, our group screened a series of phosphopeptides derived from known receptor docking sites [92]. The lead, Peptide 1.6, Ac–pTyr–Leu–Pro–Gln–Thr–Val–NH2 (Figure 9), possessed Tyr–Xaa–Yaa–Gln, reported to be the recognition determinant for STAT3 [93,94]. Similar sequences were discovered independently using a combinatorial peptide library [95]. Extensive studies were carried out to probe the phosphopeptide binding site [64,96–101]. These studies revealed that phosphotyrosine could be replaced by the conformationally constrained 4-phosphocinnamate [99] (e.g., PM-66F, Figure 9) and inclusion of a methyl group on the β-carbon of the cinnamate increased affinity [97]. The central Leu–Pro dipeptide had a trans peptide bond [96] and could be replaced with the tricyclic lactam Haic [99]. The C-terminal Thr–Val dipeptide could be replaced [64], but the side chain of carboxamide of glutamine was essential for high affinity.
Figure 9. Peptidomimetic inhibitors of STAT3 derived from gp130 Tyr904.
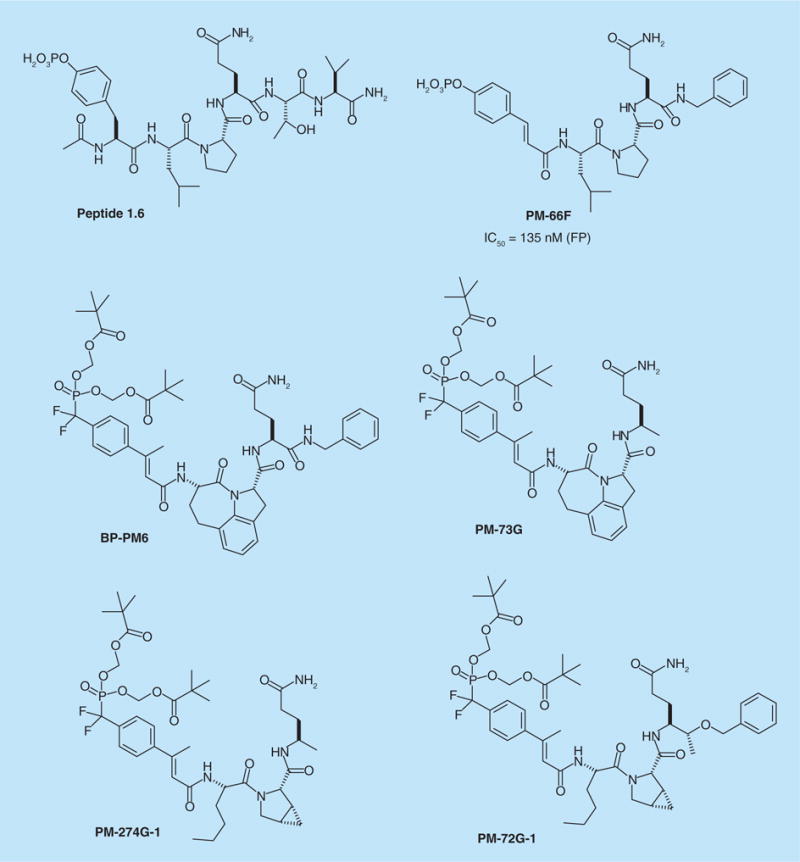
FP: Fluorescence polarization.
To prepare these phosphopeptide mimetics for cellular studies, the phosphate group was replaced with the phosphatase-stable phosphonodifluoromethyl group [97,102]. To block the negative charges of the phosphonate and allow cell penetration, the oxygens were capped with carboxyesterase-labile pivaloyloxymethyl groups [97,102]. Cellular potency was influenced by structural features that had little or opposite effects on affinity for the protein [97,100]. For example, replacement of the C-terminal CONHCH2C6H5 group of BP-PM6 (Figure 9) with a simple methyl group (PM-73G) brought the concentration for complete inhibition of STAT3 phosphorylation from 10 μM to 500 nM, a 20-fold increase in potency. Substitution of proline with cis-3,4-methanoproline (PM-272G-1), 4,4-difluoroproline or 4,4-dimethylp-roline increased cellular potency by 20-fold over the parent proline-containing inhibitor [100].
A set of prodrugs, which included PM-73G, PM-72G-1 and PM-274G-1, was tested for the ability to inhibit the phosphorylation of STAT3 in several cancer cell lines, including the MDA-MB-468 TNBC line. Overall the prodrugs were very potent and specific, exhibiting inhibition of constitutive STAT3 phosphorylation significantly at 100 nM, with complete inhibition observed at 500 nM. At 5 μM, the compounds were selective for STAT3 over EGF- induced STAT5, PI3K and Src, and were ten-fold more selective for STAT3 over STAT1. In contrast to the dogma, daily dosing of 5 μM, ten-times the concentration that completely inhibited STAT3 phosphorylation, resulted in no significant cytotoxicity for breast cancer lines MDA-MB-468 and MCF7, nor for the lung cancer line, HCC827, nor the ovarian cancer line, SKOV3-ip. The IC50 for MDA-MB-468 cells was between 25 and 50 μM. However, at 25 μM, 2 h treatment showed significant inhibition of STAT5, pAkt and pFAKTyr861. Thus, high concentrations of inhibitor resulted in off-target effects that correlated with cytotoxicity of cancer cells.
Treatment of MDA-MB-468 orthotopic xenografts by intratumoral injection led to significant reduction in tumor growth, which was accompanied by significant inhibition of tumor vascularization and VEGF protein [26]. Daily intraperitoneal (systemic) injection led to significant tumor reduction with inhibition of angiogenesis. Thus, it appears that selective inhibition of STAT3, while not cytotoxic to cancer cells, leads to impaired VEGF signaling and to inhibition of tumor angiogenesis [26].
JAK inhibition
Although the emphasis of this article is on agents that target SH2 domains in breast cancer, it is worthwhile to discuss inhibitors of JAKs, which prevent the phosphorylation of STAT proteins. A dogma developed over time that suggests that STAT3 is required for tumor cell growth and proliferation [56,103]. In contrast to the evidence provided for BP-102 [27], research with JAK inhibitors is showing that this hypothesis is not correct. Pyridone P6, first reported by Merck (Rahway, NJ, USA) [104], is an inhibitor of all of the JAKs. Kreiss et al. observed that treatment of a panel of melanoma cell lines with this compound brought pSTAT3 levels to below detectable levels, but this had no effect on proliferation in vitro [105]. As is the case with most kinase inhibitors, Pyridone P6 inhibits other kinases, notably PDK1 [106], but these off-target effects are not cytotoxic. AZ1480, introduced by researchers at AstraZeneca (Waltham, MA, USA) [107], preferentially inhibits JAK2 over the other JAKs [108]. At concentrations that completely inhibited STAT3 phosphorylation, Hedvat et al. showed that this material had no effect on the proliferation of MDA-MB-468 (breast), DU145 (prostate) and MDAH2774 (ovarian) cancer cells in vitro [108]. This finding was recapitulated in subsequent publications from this group [109,110] and others [111]. The JAK1/2 inhibitor, ruxolitinib, US FDA-approved for myelodysplastic disorders, inhibits pSTAT3 and did not affect growth of lung cancer cell lines in 2D cultures, but it did inhibit anchorage-independent colony formation in soft agar [112]. These reports with the JAK inhibitors and the phosphopeptide mimics targeting the SH2 domain [97] show that inhibiting the phosphorylation of Tyr705 of STAT3 is not cytotoxic to cancer cells, which is also supported by a report that treatment of ovarian cancer cells with siltuximab, the anti-IL-6 monoclonal antibody, resulted in pSTAT3 inhibition with no effect on proliferation [113]. By inference, if a compound inhibits STAT3 phosphorylation and it kills cells, it must be doing the latter by off-target effects.
Future perspective
SH2 domains play key roles in signaling pathways that contribute to the pathologies of the many variants of breast cancer. Early Grb2 inhibitors provided proof-of-principle that SH2 domains could be inhibited in intact cells. As certain variants of breast cancer are sensitive to Grb2 inhibition and others are not, personalized therapy strategies would have to be employed with this target. The work with the Grb2 inhibitors and recent progress with the derivatized aminosalicylate inhibitors of STAT3(BP-102) shows that negatively charged compounds can enter cells and tissues in vivo and inhibit SH2 domain targets. The more traditional phosphonate prodrug approach has also been employed to inhibit the SH2 domain of STAT3 both in cultured cells and in vivo, suggesting that this technology may lead to agents to treat breast cancer.
STAT3 has received considerable attention as a target for breast cancer. A caveat is emerging that inhibition of its activation, either by selectively targeting the SH2 domain, by inhibition of upstream cytokine binding or by inhibition of JAK activity, indicates that this protein is not a target for direct cancer cell killing. Rather, tumor reduction is likely achieved by inhibiting supporting processes, such as angiogenesis. This has to be kept in mind when evaluating new agents targeting the SH2 domain of STAT3. If a new inhibitor expresses cytotoxicity in 2D cultures, it likely has off-target effects in addition to, or that contribute to, inhibition of pSTAT3. These off-target activities could be helpful or a liability when it comes to treating patients. One successful example of a cytotoxic compound that also inhibits STAT3 phosphorylation is LLL12, which displayed promising antiangiogenic activity in vivo. Overall, several compounds discussed herein demonstrate that inhibition of the SH2 domain of STAT3 could be a useful strategy for the treatment of breast cancer.
The pharmaceutical industry essentially abandoned the SH2 domain as a viable target. The steady improvement in the potency in vivo of SH2 domain inhibitors, notably STAT3, in recent years by academic laboratories suggests that this domain is indeed druggable. Other SH2 domains, for example, p85, the regulatory subunit of PI3K, SH2-containing phosphatases, phospholipase C-γ or STAT5, are potentially useful targets for the treatment of one or more forms of breast cancer. Given the progress reported herein, it is expected that these proteins will be targeted in the future.
Executive summary.
Background
Several types of breast cancer, including triple-negative and inflammatory breast cancer have dysregulated growth and metastatic signaling pathways in which SH2 domains play integral parts.
SH2 domains are involved in key protein–protein interactions that transmit aberrant cell growth signals in several molecular pathways in most human cancers, including breast cancer.
Targeting SH2 domains is an untapped potential modality for treating breast cancers.
Grb2, Grb7 and STAT3 contribute to aberrant signaling pathways in breast cancer and the SH2 domains of these proteins have been targeted by industrial, government and academic laboratories.
SH2 domains recognize phosphotyrosine residues and have been challenging to target in cells and tissues because the required dense negative charge on ligands or inhibitors has been difficult to deliver. The pharmaceutical industry was not successful in developing clinical candidates targeting SH2 domains.
Inhibitors targeting Grb2
Research on peptidomimetic inhibitors of Grb2 indicates that free phosphonates are capable of crossing cell membranes and inhibiting protein–protein interactions mediated by the SH2 domain. As phosphonates, these materials have a charge of -1 at physiological pH.
Certain breast cancer cell lines are sensitive to Grb2 inhibition. Thus, patients will have to be stratified as to which will likely respond to this treatment.
Inhibitors targeting Grb7
Cyclic peptide inhibitors provided proof-of-principle that inhibition of Grb7 is a potential treatment modality for breast cancer. Furthermore, they served as structural templates for the identification of small-molecule inhibitors.
Low-affinity small-molecule inhibitors have been identified in initial screens but optimization is needed to properly assess their potential.
Specificity of inhibitors for Grb2 & Grb7
Grb2 and Grb7 both recognize pTyr–Xaa–Asn motifs. Selective inhibition of one over the other must be demonstrated to fully understand the consequences of blocking their SH2 domains on the biology of breast cancer cells in vitro as well as in in vivo breast cancer models.
Inhibitors targeting STAT3
Research with upstream blockers of IL-6 and JAK inhibitors, as well as phosphopeptide mimetic prodrugs targeting the SH2 domain, shows that selective inhibition of Tyr705 phosphorylation of STAT3, the activation event for this protein, does not lead to direct killing of cancer cells, including breast cancer cells. In tumor models in vivo, tumor growth inhibition appears to correlate with inhibition of angiogenesis and possibly impaired tumor immune evasion.
By inference, if a small molecule inhibits tumor cell proliferation, it likely perturbs off-targets pathways leading to cell death.
Two small-molecule inhibitors (i.e., STA-21 and cryptotanshinone) do not bind to the SH2 domain of STAT3. Given their quinonoid structures, it is likely that they inhibit cell growth by off-target effects.
Research with hydroxysalicylate inhibitors of STAT3 also show that a negatively charged compound can enter cells and disrupt the function of the SH2 domain of this target.
Peptide mimetic prodrugs that selectively target the SH2 domain of STAT3 reduce the growth of human breast tumor xenografts in murine models, accompanied by a significant reduction of angiogenesis.
Compounds with both cytotoxic and STAT3 inhibitory properties will likely be effective therapeutics due to both cancer cell killing and reduction of supporting angiogenesis, provided that overall toxicity is acceptable.
STAT3 inhibitors represent a novel class of antiangiogenic agents.
Acknowledgments
Financial disclosure
The authors are grateful to the NCI for grant CA096652, the CTT/TI-3D Chemistry and Molecularly-Targeted Therapeutic Development Grant Program, and the MDACC IRG program for funding this work.
No writing assistance was utilized in the production of this manuscript.
Key terms
- SH2 domain
An ~100 amino acid subunit of important signaling proteins that recruits these molecules to specific multiprotein complexes to propagate growth factor and cytokine signals. SH2 domains bind to specific phosphotyrosine residues and are partners in key protein–protein interactions required for cell cycling, angiogenesis, immunity, inflammation and so on in normal physiology. Inhibiting these interactions is hypothesized to reverse the pathophysiology of diseases such as cancer.
- Phosphopeptide
A peptide in which a tyrosine, serine, threonine or histidine are phosphorylated, typically on the side chain oxygen or imidazole nitrogen.
- Grb2
Growth factor receptor-bound protein 2. On binding of growth factors, such as EGFR, Grb2 binds to phosphotyrosine residues on the receptor via its SH2 domain and recruits other proteins, such as SOS and RAS, to transmit growth signals to the cell nucleus. Certain breast cancers are dependent on Grb2 while others are not.
- Grb7
Growth factor receptor-bound protein 7. Similar to Grb2, in certain contexts Grb7 also transmits growth factor signals to the nucleus, via initial binding of it SH2 domain to key phosphotyrosine residues on the growth factor receptor. Grb7 activity is elevated in breast cancers.
- STAT3
Signal transducer and activator of transcription 3. STAT3 is recruited to specific phosphotyrosine residues on receptors of growth factors, notably VEGF and EGF. More importantly STAT3 transmits signals from the cytokine IL-6, which is elevated in most human cancers. STAT3 is activated in most human cancers and is considered a target for cancer therapy.
- Peptide mimetic
A non-peptide chemical compound that has structural features allowing it to express the same biological activity as a particular peptide.
Footnotes
Competing interests disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.American Cancer Society. www.cancer.org/research/cancerfactsfigures/cancerfactsfigures/cancer-facts-and-figures-2010.
- 2.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martin M, Romero A, Cheang MC, et al. Genomic predictors of response to doxorubicin versus docetaxel in primary breast cancer. Breast Cancer Res Treat. 2011;128(1):127–136. doi: 10.1007/s10549-011-1461-y. [DOI] [PubMed] [Google Scholar]
- 4.Prat A, Perou CM. Deconstructing the molecular portraits of breast cancer. Mol Oncol. 2011;5(1):5–23. doi: 10.1016/j.molonc.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parker JS, Mullins M, Cheang MC, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27(8):1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burke WM, Jin X, Lin HJ, et al. Inhibition of constitutively active Stat3 suppresses growth of human ovarian and breast cancer cells. Oncogene. 2001;20(55):7925–7934. doi: 10.1038/sj.onc.1204990. [DOI] [PubMed] [Google Scholar]
- 7.Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F, Gianni L. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol. 2011;9(1):16–32. doi: 10.1038/nrclinonc.2011.177. [DOI] [PubMed] [Google Scholar]
- 8.Lin NU, Winer EP. Advances in adjuvant endocrine therapy for postmenopausal women. J Clin Oncol. 2008;26(5):798–805. doi: 10.1200/JCO.2007.15.0946. [DOI] [PubMed] [Google Scholar]
- 9.Peng J, Sengupta S, Jordan VC. Potential of selective estrogen receptor modulators as treatments and preventives of breast cancer. Anticancer Agents Med Chem. 2009;9(5):481–499. doi: 10.2174/187152009788451833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carey LA, Perou CM, Livasy CA, et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA. 2006;295(21):2492–2502. doi: 10.1001/jama.295.21.2492. [DOI] [PubMed] [Google Scholar]
- 11.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363(20):1938–1948. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 12.Onitilo AA, Engel JM, Greenlee RT, Mukesh BN. Breast cancer subtypes based on ER/PR and Her2 expression: comparison of clinicopathologic features and survival. Clin Med Res. 2009;7(1–2):4–13. doi: 10.3121/cmr.2009.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pawson T. Protein modules and signalling networks. Nature. 1995;373:573–580. doi: 10.1038/373573a0. [DOI] [PubMed] [Google Scholar]
- 14.Waksman G, Kumaran S, Lubman O. SH2 domains: role, structure and implications for molecular medicine. Exp Rev Mol Med. 2004;6(03):1–18. doi: 10.1017/S1462399404007331. [DOI] [PubMed] [Google Scholar]
- 15.Kuriyan J, Cowburn D. Modular peptide recognition domains in eukaryotic signaling. Annu Rev Biophys Biomol Struct. 1997;26:259–288. doi: 10.1146/annurev.biophys.26.1.259. [DOI] [PubMed] [Google Scholar]
- 16.Najmudin S, Bax B, Guillory RJ. Structural analysis of the SH2 and SH3 domains: modules that regulate protein interactions. J Biochem Mol Biol Biophys. 1997;1(2):73–88. [Google Scholar]
- 17.Garcia-Echeverria C. Antagonists of the homology 2 (SH2) domains of Grb2, Src, Lck and ZAP-70. Curr Med Chem. 2001;8(13):1589–1604. doi: 10.2174/0929867013371905. [DOI] [PubMed] [Google Scholar]
- 18.Garcia-Echeverria C. Inhibitors of signaling interfaces: targeting Src homology 2 domains in drug discovery. In: Fabbro D, McCormick F, editors. Protein Tyrosine Kinases: From Inhibitors to Useful Drugs. Humana Press Inc; Totowa, NJ, USA: 2006. pp. 31–52. [Google Scholar]
- 19.Muller G. Peptidomimetic SH2 domain antagonists for targeting signal transduction. Top Curr Chem. 2001;211:17–59. [Google Scholar]
- 20.Sawyer TK. Src homology-2 domains: structure, mechanisms, and drug discovery. Biopolymers. 1998;47(3):243–261. doi: 10.1002/(SICI)1097-0282(1998)47:3<243::AID-BIP4>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 21.Shakespeare WC. SH2 domain inhibition: a problem solved? Curr Opin Chem Biol. 2001;5(4):409–415. doi: 10.1016/s1367-5931(00)00222-2. [DOI] [PubMed] [Google Scholar]
- 22.Giricz O, Calvo V, Pero SC, Krag DN, Sparano JA, Kenny PA. GRB7 is required for triple-negative breast cancer cell invasion and survival. Breast Cancer Res Treat. 2012;133(2):607–615. doi: 10.1007/s10549-011-1822-6. [DOI] [PubMed] [Google Scholar]
- 23.Haftchenary S, Avadisian M, Gunning PT. Inhibiting aberrant Stat3 function with molecular therapeutics: a progress report. Anticancer Drugs. 2011;22(2):115–127. doi: 10.1097/CAD.0b013e328341185b. [DOI] [PubMed] [Google Scholar]
- 24.Matsuno K, Masuda Y, Uehara Y, et al. Identification of a new series of STAT3 inhibitors by virtual screening. ACS Med Chem Lett. 2010;1(8):371–375. doi: 10.1021/ml1000273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bradshaw JM, Mitaxov V, Waksman G. Investigation of phosphotyrosine recognition by the SH2 domain of the Src kinase. J Mol Biol. 1999;293(4):971–985. doi: 10.1006/jmbi.1999.3190. [DOI] [PubMed] [Google Scholar]
- 26••.Auzenne EJ, Klostergaard J, Mandal PK, et al. A phosphopeptide mimetic prodrug targeting the SH2 domain of STAT3 inhibits tumor growth and angiogenesis. J Exp Ther Oncol. 2012;10(2):155–162. First example of a phosphopeptide mimic prodrug targeting an SH2 domain in vivo with systemic administration. Inhibition of STAT3 phosphorylation led to tumor growth inhibition accompanied by reduced angiogenesis. [PMC free article] [PubMed] [Google Scholar]
- 27••.Zhang X, Yue P, Page BD, et al. Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts. Proc Natl Acad Sci USA. 2012;109(24):9623–9628. doi: 10.1073/pnas.1121606109. In-depth report of the development of a potent, orally available, non-peptide inhibitor of the SH2 domain of STAT3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giubellino A, Burke TR, Jr, Bottaro DP. Grb2 signaling in cell motility and cancer. Expert Opin Ther Targets. 2008;12(8):1021–1033. doi: 10.1517/14728222.12.8.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burke TR. Development of Grb2 SH2 domain signaling antagonists: a potential new class of antiproliferative agents. Int J Pept Res Ther. 2006;12(1):33–48. doi: 10.1007/s10989-006-9014-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Furet P, Gay B, Caravatti G, et al. Structure-based design and synthesis of high affinity tripeptide ligands of the Grb2-SH2 domain. J Med Chem. 1998;41(18):3442–3449. doi: 10.1021/jm980159a. [DOI] [PubMed] [Google Scholar]
- 31.Garcia-Echeverria C, Furet P, Gay B, et al. Potent antagonists of the SH2 domain of Grb2: optimization of the X+1 position of 3-amino-Z-Tyr(PO3H2)-X+1-Asn-NH2. J Med Chem. 1998;41(11):1741–1744. doi: 10.1021/jm970856n. [DOI] [PubMed] [Google Scholar]
- 32•.Gay B, Suarez S, Caravatti G, Furet P, Meyer T, Schoepfer J. Selective GRB2 SH2 inhibitors as anti-Ras therapy. Int J Cancer. 1999;83(2):235–241. doi: 10.1002/(sici)1097-0215(19991008)83:2<235::aid-ijc15>3.0.co;2-b. Demonstrates the utility of phosphopeptide mimics targeting an SH2 domain in intact cells. Both a free phosphonate and a prodrug demonstrated inhibition of Grb2 in breast cancer cells. [DOI] [PubMed] [Google Scholar]
- 33.McGuigan C, Pathirana RN, Balzarini J, De Clercq E. Intracellular delivery of bioactive AZT nucleotides by aryl phosphate derivatives of AZT. J Med Chem. 1993;36(8):1048–1052. doi: 10.1021/jm00060a013. [DOI] [PubMed] [Google Scholar]
- 34•.Gay B, Suarez S, Weber C, et al. Effect of potent and selective inhibitors of the Grb2 SH2 domain on cell motility. J Biol Chem. 1999;274(33):23311–23315. doi: 10.1074/jbc.274.33.23311. Demonstrates disruption of cellular activity with a phosphopeptide mimic targeting an SH2 domain; that of Grb2. [DOI] [PubMed] [Google Scholar]
- 35.Gao Y, Luo J, Yao ZJ, et al. Inhibition of Grb2 SH2 domain binding by non-phosphate-containing ligands. 2. 4-(2-Malonyl)phenylalanine as a potent phosphotyrosyl mimetic. J Med Chem. 2000;43(5):911–920. doi: 10.1021/jm9904248. [DOI] [PubMed] [Google Scholar]
- 36.Yao ZJ, King CR, Cao T, et al. Potent inhibition of Grb2 SH2 domain binding by non-phosphate-containing ligands. J Med Chem. 1999;42(1):25–35. doi: 10.1021/jm980388x. [DOI] [PubMed] [Google Scholar]
- 37.Kang SU, Worthy KM, Bindu LK, et al. Design and synthesis of 4-(alpha-hydroxymalonyl)phenylalanine as a new phosphotyrosyl mimetic and its use in growth factor receptor bound 2 src-homology 2 (Grb2 SH2) domain-binding peptides. J Med Chem. 2005;48(16):5369–5372. doi: 10.1021/jm050154v. [DOI] [PubMed] [Google Scholar]
- 38.Atabey N, Gao Y, Yao ZJ, et al. Potent blockade of hepatocyte growth factor-stimulated cell motility, matrix invasion and branching morphogenesis by antagonists of Grb2 Src homology 2 domain interactions. J Biol Chem. 2001;276(17):14308–14314. doi: 10.1074/jbc.M010202200. [DOI] [PubMed] [Google Scholar]
- 39.Soriano JV, Liu N, Gao Y, et al. Inhibition of angiogenesis by growth factor receptor bound protein 2-Src homology 2 domain bound antagonists. Mol Cancer Ther. 2004;3(10):1289–1299. [PubMed] [Google Scholar]
- 40.Giubellino A, Gao Y, Lee S, et al. Inhibition of tumor metastasis by a growth factor receptor bound protein 2 Src homology 2 domain-binding antagonist. Cancer Res. 2007;67(13):6012–6016. doi: 10.1158/0008-5472.CAN-07-0022. [DOI] [PubMed] [Google Scholar]
- 41.Gao Y, Voigt J, Wu JX, Yang D, Burke TR., Jr Macrocyclization in the design of a conformationally constrained Grb2 SH2 domain inhibitor. Bioorg Med Chem Lett. 2001;11(14):1889–1892. doi: 10.1016/s0960-894x(01)00316-x. [DOI] [PubMed] [Google Scholar]
- 42.Shi ZD, Lee K, Liu H, et al. A novel macrocyclic tetrapeptide mimetic that exhibits low-picomolar Grb2 SH2 domain-binding affinity. Biochem Biophys Res Commun. 2003;310(2):378–383. doi: 10.1016/j.bbrc.2003.09.029. [DOI] [PubMed] [Google Scholar]
- 43.Wei CQ, Gao Y, Lee K, et al. Macrocyclization in the design of Grb2 SH2 domain-binding ligands exhibiting high potency in whole-cell systems. J Med Chem. 2003;46(2):244–254. doi: 10.1021/jm0203635. [DOI] [PubMed] [Google Scholar]
- 44.Han DC, Shen TL, Guan JL. The Grb7 family proteins. Structure, interactions with other signaling molecules and potential cellular functions. Oncogene. 2001;20(44):6315–6321. doi: 10.1038/sj.onc.1204775. [DOI] [PubMed] [Google Scholar]
- 45.Stein D, Wu J, Fuqua SA, et al. The SH2 domain protein GRB7 is co-amplified, overexpressed and in a tight complex with HER2 in breast cancer. EMBO J. 1994;13(6):1331–1340. doi: 10.1002/j.1460-2075.1994.tb06386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sparano JA, Goldstein LJ, Childs BH, et al. Relationship between quantitative GRB7 RNA expression and recurrence after adjuvant anthracycline chemotherapy in triple-negative breast cancer. Clin Cancer Res. 2011;17(22):7194–7203. doi: 10.1158/1078-0432.CCR-10-3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang EY, Cristofanilli M, Robertson F, et al. Genome wide proteomics of ERBB2 and EGFR and other oncogenic pathways in inflammatory breast cancer. J Proteome Res. 2013;12(6):2805–2817. doi: 10.1021/pr4001527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pero SC, Oligino L, Daly RJ, et al. Identification of novel non-phosphorylated ligands, which bind selectively to the SH2 domain of Grb7. J Biol Chem. 2002;277(14):11918–11926. doi: 10.1074/jbc.M111816200. [DOI] [PubMed] [Google Scholar]
- 49.Pero SC, Shukla GS, Cookson MM, Flemer S, Jr, Krag DN. Combination treatment with Grb7 peptide and doxorubicin or trastuzumab (Herceptin) results in cooperative cell growth inhibition in breast cancer cells. Br J Cancer. 2007;96(10):1520–1525. doi: 10.1038/sj.bjc.6603732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ambaye ND, Lim RC, Clayton DJ, et al. Uptake of a cell permeable G7–18NATE contruct into cells and binding with the Grb-7-SH2 domain. Biopolymers. 2011;96(2):181–188. doi: 10.1002/bip.21403. [DOI] [PubMed] [Google Scholar]
- 51.Gunzburg MJ, Ambaye ND, Del BMP, Perlmutter P, Wilce JA. Design and testing of bicyclic inhibitors of Grb7-are two cycles better than one? Biopolymers. 2013;100(5):543–549. doi: 10.1002/bip.22237. [DOI] [PubMed] [Google Scholar]
- 52.Ambaye ND, Pero SC, Gunzburg MJ, et al. Structural basis of binding by cyclic nonphosphorylated peptide antagonists of Grb7 implicated in breast cancer progression. J Mol Biol. 2011;412(3):397–411. doi: 10.1016/j.jmb.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 53.Ambaye ND, Gunzburg MJ, Lim RCC, Price JT, Wilce MCJ, Wilce JA. Benzopyrazine derivatives: a novel class of growth factor receptor bound protein 7 antagonists. Bioorg Med Chem. 2011;19(1):693–701. doi: 10.1016/j.bmc.2010.10.030. [DOI] [PubMed] [Google Scholar]
- 54.Ambaye ND, Gunzburg MJ, Lim RCC, Price JT, Wilce MCJ, Wilce JA. The discovery of phenylbenzamide derivatives as Grb7-based antitumor agents. ChemMedChem. 2013;8(2):280–288. doi: 10.1002/cmdc.201200400. [DOI] [PubMed] [Google Scholar]
- 55.Huang S. Regulation of metastases by signal transducer and activator of transcription 3 signaling pathway: clinical implications. Clin Cancer Res. 2007;13(5):1362–1366. doi: 10.1158/1078-0432.CCR-06-2313. [DOI] [PubMed] [Google Scholar]
- 56.Yu H, Jove R. The STATs of cancer – new molecular targets come of age. Nat Rev Cancer. 2004;4(2):97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 57.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(11):798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Diaz N, Minton S, Cox C, et al. Activation of Stat3 in primary tumors from high-risk breast cancer patients is associated with elevated levels of activated Src and survivin expression. Clin Cancer Res. 2006;12(1):20–28. doi: 10.1158/1078-0432.CCR-04-1749. [DOI] [PubMed] [Google Scholar]
- 59.Berishaj M, Gao SP, Ahmed S, et al. Stat3 is tyrosine-phosphorylated through the interleukin-6/glycoprotein 130/Janus kinase pathway in breast cancer. Breast Cancer Res. 2007;9(3):R32. doi: 10.1186/bcr1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60•.Dave B, Landis MD, Tweardy DJ, et al. Selective small molecule Stat3 inhibitor reduces breast cancer tumor-initiating cells and improves recurrence free survival in a human-xenograft model. PLoS ONE. 2012;7(8):e30207. doi: 10.1371/journal.pone.0030207. Cancer stem cells are hypothesized to regenerate tumors and to promote metastasis. This paper suggests that inhibition of STAT3 phosphorylation in inhibitory to cancer stem cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lavecchia A, Di Giovanni C, Novellino E. STAT-3 inhibitors: state of the art and new horizons for cancer treatment. Curr Med Chem. 2011;18(16):2359–2375. doi: 10.2174/092986711795843218. [DOI] [PubMed] [Google Scholar]
- 62.Miklossy G, Hilliard TS, Turkson J. Therapeutic modulators of STAT signalling for human diseases. Nat Rev Drug Discov. 2013;12(8):611–629. doi: 10.1038/nrd4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Song H, Wang R, Wang S, Lin J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc Natl Acad Sci USA. 2005;102(13):4700–4705. doi: 10.1073/pnas.0409894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Coleman DR, IV, Ren Z, Mandal PK, et al. Investigation of the binding determinants of phosphopeptides targeted to the SRC homology 2 domain of the signal transducer and activator of transcription 3. Development of a high-affinity peptide inhibitor. J Med Chem. 2005;48(21):6661–6670. doi: 10.1021/jm050513m. [DOI] [PubMed] [Google Scholar]
- 65••.Wei C-C, Ball S, Lin L, et al. Two small molecule compounds, LLL12 and FLLL32, exhibit potent inhibitory activity on STAT3 in human rhabdomyosarcoma cells. Int J Oncol. 2011;38(1):279–285. Correlates STAT3 inhibition with reduction in angiogenesis. These materials are both cytotoxic and novel antiangiogenic agents. [PubMed] [Google Scholar]
- 66.Bhasin D, Cisek K, Pandharkar T, et al. Design, synthesis, and studies of small molecule STAT3 inhibitors. Bioorg Med Chem Lett. 2008;18(1):391–395. doi: 10.1016/j.bmcl.2007.10.031. [DOI] [PubMed] [Google Scholar]
- 67.O’Brien PJ. Molecular mechanisms of quinone cytotoxicity. Chem Biol Interact. 1991;80(1):1–41. doi: 10.1016/0009-2797(91)90029-7. [DOI] [PubMed] [Google Scholar]
- 68.Padhye S, Dandawate P, Yusufi M, Ahmad A, Sarkar FH. Perspectives on medicinal properties of plumbagin and its analogs. Med Res Rev. 2012;32(6):1131–1158. doi: 10.1002/med.20235. [DOI] [PubMed] [Google Scholar]
- 69.Niu G, Wright KL, Huang M, et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene. 2002;21(13):2000–2008. doi: 10.1038/sj.onc.1205260. [DOI] [PubMed] [Google Scholar]
- 70.Gray MJ, Zhang J, Ellis LM, et al. HIF-1alpha, STAT3, CBP/p300 and Ref-1/APE are components of a transcriptional complex that regulates Src-dependent hypoxia-induced expression of VEGF in pancreatic and prostate carcinomas. Oncogene. 2005;24(19):3110–3120. doi: 10.1038/sj.onc.1208513. [DOI] [PubMed] [Google Scholar]
- 71.Bid HK, Oswald D, Li C, London CA, Lin J, Houghton PJ. Anti-angiogenic activity of a small molecule STAT3 inhibitor LLL12. PLoS ONE. 2012;7(4):e35513. doi: 10.1371/journal.pone.0035513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shin DS, Kim HN, Shin KD, et al. Cryptotanshinone inhibits constitutive signal transducer and activator of transcription 3 function through blocking the dimerization in DU145 prostate cancer cells. Cancer Res. 2009;69(1):193–202. doi: 10.1158/0008-5472.CAN-08-2575. [DOI] [PubMed] [Google Scholar]
- 73.Jung JH, Kwon TR, Jeong SJ, et al. Apoptosis induced by tanshinone IIA and cryptotanshinone is mediated by distinct JAK/STAT3/5 and SHP1/2 signaling in chronic myeloid leukemia K562 cells. Evid Based Complement Alternat Med. 2013;2013:805639. doi: 10.1155/2013/805639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jin YC, Kim CW, Kim YM, et al. Cryptotanshinone, a lipophilic compound of Salvia miltiorrriza root, inhibits TNF-alpha-induced expression of adhesion molecules in HUVEC and attenuates rat myocardial ischemia/reperfusion injury in vivo. Eur J Pharmacol. 2009;614(1–3):91–97. doi: 10.1016/j.ejphar.2009.04.038. [DOI] [PubMed] [Google Scholar]
- 75.Zhang F, Zheng W, Pi R, et al. Cryptotanshinone protects primary rat cortical neurons from glutamate-induced neurotoxicity via the activation of the phosphatidylinositol 3-kinase/Akt signaling pathway. Exp Brain Res. 2009;193(1):109–118. doi: 10.1007/s00221-008-1600-9. [DOI] [PubMed] [Google Scholar]
- 76.Xu X, Kasembeli MM, Jiang X, Tweardy BJ, Tweardy DJ. Chemical probes that competitively and selectively inhibit Stat3 activation. PLoS ONE. 2009;4(3):e4783. doi: 10.1371/journal.pone.0004783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Park SY, Lee HE, Li H, Shipitsin M, Gelman R, Polyak K. Heterogeneity for stem cell-related markers according to tumor subtype and histologic stage in breast cancer. Clin Cancer Res. 2010;16(3):876–887. doi: 10.1158/1078-0432.CCR-09-1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ricardo S, Vieira AF, Gerhard R, et al. Breast cancer stem cell markers CD44, CD24 and ALDH1: expression distribution within intrinsic molecular subtype. J Clin Pathol. 2011;64(11):937–946. doi: 10.1136/jcp.2011.090456. [DOI] [PubMed] [Google Scholar]
- 79.Ginestier C, Hur MH, Charafe-Jauffret E, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1(5):555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee SJ, Ghosh SC, Han HD, et al. Metronomic activity of CD44-targeted hyaluronic acid-paclitaxel in ovarian carcinoma. Clin Cancer Res. 2012;18(15):4114–4121. doi: 10.1158/1078-0432.CCR-11-3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Marotta LL, Almendro V, Marusyk A, et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(−) stem cell-like breast cancer cells in human tumors. J Clin Invest. 2011;121(7):2723–2735. doi: 10.1172/JCI44745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Charafe-Jauffret E, Ginestier C, Iovino F, et al. Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clin Cancer Res. 2010;16(1):45–55. doi: 10.1158/1078-0432.CCR-09-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Becker S, Groner B, Muller CW. Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature. 1998;394(6689):145–151. doi: 10.1038/28101. [DOI] [PubMed] [Google Scholar]
- 84.Turkson J, Kim JS, Zhang S, et al. Novel peptidomimetic inhibitors of signal transducer and activator of transcription 3 dimerization and biological activity. Mol Cancer Ther. 2004;3(3):261–269. [PubMed] [Google Scholar]
- 85.Siddiquee KA, Gunning PT, Glenn M, et al. An oxazole-based small-molecule Stat3 inhibitor modulates Stat3 stability and processing and induces antitumor cell effects. ACS Chem Biol. 2007;2(12):787–798. doi: 10.1021/cb7001973. [DOI] [PubMed] [Google Scholar]
- 86.Shahani VM, Yue P, Fletcher S, et al. Design, synthesis, and in vitro characterization of novel hybrid peptidomimetic inhibitors of STAT3 protein. Bioorg Med Chem. 2011;19(5):1823–1838. doi: 10.1016/j.bmc.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fletcher S, Singh J, Zhang X, et al. Disruption of transcriptionally active Stat3 dimers with non-phosphorylated, salicylic acid-based small molecules: potent in vitro and tumor cell activities. ChemBioChem. 2009;10(12):1959–1964. doi: 10.1002/cbic.200900172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang X, Yue P, Fletcher S, Zhao W, Gunning PT, Turkson J. A novel small-molecule disrupts Stat3 SH2 domain-phosphotyrosine interactions and Stat3-dependent tumor processes. Biochem Pharmacol. 2010;79(10):1398–1409. doi: 10.1016/j.bcp.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Page BD, Croucher DC, Li ZH, et al. Inhibiting aberrant signal transducer and activator of transcription protein activation with tetrapodal, small molecule Src homology 2 domain binders: promising agents against multiple myeloma. J Med Chem. 2013;56(18):7190–7200. doi: 10.1021/jm3017255. [DOI] [PubMed] [Google Scholar]
- 90.Urlam MK, Pireddu R, Ge Y, et al. Development of new N-arylbenzamides as STAT3 dimerization inhibitors. MedChemComm. 2013;4(6):932–941. doi: 10.1039/C3MD20323A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91•.Zhang X, Sun Y, Pireddu R, et al. A novel inhibitor of STAT3 homodimerization selectively suppresses STAT3 activity and malignant transformation. Cancer Res. 2013;73(6):1922–1933. doi: 10.1158/0008-5472.CAN-12-3175. Another paper showing that mononegatively charged compounds can enter cells and inhibit STAT3 function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ren Z, Cabell LA, Schaefer TS, McMurray JS. Identification of a high-affinity phosphopeptide inhibitor of Stat3. Bioorg Med Chem Lett. 2003;13(4):633–636. doi: 10.1016/s0960-894x(02)01050-8. [DOI] [PubMed] [Google Scholar]
- 93.Stahl N, Farruggella TJ, Boulton TG, Zhong Z, Darnell JE, Jr, Yancopoulos GD. Choice of STATs and other substrates specified by modular tyrosine-based motifs in cytokine receptors. Science. 1995;267(5202):1349–1353. doi: 10.1126/science.7871433. [DOI] [PubMed] [Google Scholar]
- 94.Gerhartz C, Heesel B, Sasse J, et al. Differential activation of acute phase response factor/STAT3 and STAT1 via the cytoplasmic domain of the interleukin 6 signal transducer gp130. I. Definition of a novel phosphotyrosine motif mediating STAT1 activation. J Biol Chem. 1996;271(22):12991–12998. doi: 10.1074/jbc.271.22.12991. [DOI] [PubMed] [Google Scholar]
- 95.Wiederkehr-Adam M, Ernst P, Muller K, et al. Characterization of phosphopeptide motifs specific for the Src homology 2 domains of signal transducer and activator of transcription 1 (STAT1) and STAT3. J Biol Chem. 2003;278(18):16117–16128. doi: 10.1074/jbc.M300261200. [DOI] [PubMed] [Google Scholar]
- 96.Coleman DR, Kaluarachchi K, Ren Z, Chen X, McMurray JS. Solid phase synthesis of phosphopeptides incorporating 2,2-dimethyloxazolidine pseudoproline analogs: evidence for trans Leu-Pro peptide bonds in Stat3 inhibitors. Int J Pept Res Ther. 2008;14(1):1–9. [Google Scholar]
- 97.Mandal PK, Gao F, Lu Z, et al. Potent and selective phosphopeptide mimetic prodrugs targeted to the Src Homology 2 (SH2) domain of Signal Transducer and Activator of Transcription 3. J Med Chem. 2011;54(10):3549–5463. doi: 10.1021/jm2000882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mandal PK, Heard PA, Ren Z, Chen X, McMurray JS. Solid-phase synthesis of Stat3 inhibitors incorporating O-carbamoylserine and O-carbamoylthreonine as glutamine mimics. Bioorg Med Chem Lett. 2007;17(3):654–656. doi: 10.1016/j.bmcl.2006.10.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mandal PK, Limbrick D, Coleman DR, et al. Conformationally constrained peptidomimetic inhibitors of signal transducer and activator of transcription 3: evaluation and molecular modeling. J Med Chem. 2009;52(8):2429–2442. doi: 10.1021/jm801491w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100•.Mandal PK, Ren Z, Chen X, Kaluarachchi K, Liao WSL, McMurray JS. Structure–activity studies of phosphopeptidomimetic prodrugs targeting the Src homology 2 (SH2) domain of signal transducer and activator of transcription 3 (Stat3) Int J Pept Res Ther. 2013;19(1):3–12. doi: 10.1007/s10989-012-9313-0. Shows that subtle structural modifications have significant impact on the cellular potency of phosphopeptide mimics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mandal PK, Ren Z, Chen X, Xiong C, McMurray JS. Structure–affinity relationships of glutamine mimics incorporated into phosphopeptides targeted to the SH2 domain of signal transducer and activator of transcription 3. J Med Chem. 2009;52(19):6126–6141. doi: 10.1021/jm901105k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mandal PK, Liao WS, McMurray JS. Synthesis of phosphatase-stable, cell-permeable peptidomimetic prodrugs that target the SH2 domain of Stat3. Org Lett. 2009;11(15):3394–3397. doi: 10.1021/ol9012662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Costantino L, Barlocco D. STAT3 as a target for cancer drug discovery. Curr Med Chem. 2008;15:834–843. doi: 10.2174/092986708783955464. [DOI] [PubMed] [Google Scholar]
- 104.Thompson JE, Cubbon RM, Cummings RT, et al. Photochemical preparation of a pyridone containing tetracycle: a Jak protein kinase inhibitor. Bioorg Med Chem Lett. 2002;12(8):1219–1223. doi: 10.1016/s0960-894x(02)00106-3. [DOI] [PubMed] [Google Scholar]
- 105••.Kreis S, Munz GA, Haan S, Heinrich PC, Behrmann I. Cell density dependent increase of constitutive signal transducers and activators of transcription 3 activity in melanoma cells is mediated by Janus kinases. Mol Cancer Res. 2007;5(12):1331–1341. doi: 10.1158/1541-7786.MCR-07-0317. First paper showing that inhibition of STAT3 phosphorylation by JAK inhibition is not cytotoxic to cancer cells. [DOI] [PubMed] [Google Scholar]
- 106.Bobkova EV, Weber MJ, Xu Z, et al. Discovery of PDK1 kinase inhibitors with a novel mechanism of action by ultrahigh throughput screening. J Biol Chem. 2010;285(24):18838–18846. doi: 10.1074/jbc.M109.089946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ioannidis S, Lamb ML, Wang T, et al. Discovery of 5-Chloro-N(2)-[(1S)-1-(5-fluoropyrimidin-2-yl)ethyl]-N(4)-(5-methyl-1H-pyr azol-3-yl)pyrimidine-2,4-diamine (AZD1480) as a novel inhibitor of the Jak/Stat pathway. J Med Chem. 2011;54(1):262–276. doi: 10.1021/jm1011319. [DOI] [PubMed] [Google Scholar]
- 108••.Hedvat M, Huszar D, Herrmann A, et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009;16(6):487–497. doi: 10.1016/j.ccr.2009.10.015. Showed that selective inhibition of STAT3 is not cytotoxic to tumor cells in culture but results in tumor growth inhibition in vivo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Scuto A, Krejci P, Popplewell L, et al. The novel JAK inhibitor AZD1480 blocks STAT3 and FGFR3 signaling, resulting in suppression of human myeloma cell growth and survival. Leukemia. 2011;25(3):538–550. doi: 10.1038/leu.2010.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110•.Xin H, Herrmann A, Reckamp K, et al. Antiangiogenic and antimetastatic activity of JAK inhibitor AZD1480. Cancer Res. 2011;71(21):6601–6610. doi: 10.1158/0008-5472.CAN-11-1217. Shows that JAK inhibition leads to reductions in angiogenesis and metastasis, two of the hallmarks of STAT3 signaling. Furthermore, immune cell infiltration into tumors was inhibited. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Loveless ME, Lawson D, Collins M, et al. Comparisons of the efficacy of a Jak1/2 inhibitor (AZD1480) with a VEGF signaling inhibitor (cediranib) and sham treatments in mouse tumors using DCE-MRI, DW-MRI, and histology. Neoplasia. 2012;14(1):54–64. doi: 10.1593/neo.111478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Looyenga BD, Hutchings D, Cherni I, Kingsley C, Weiss GJ, Mackeigan JP. STAT3 is activated by JAK2 independent of key oncogenic driver mutations in non-small cell lung carcinoma. PLoS ONE. 2012;7(2):e30820. doi: 10.1371/journal.pone.0030820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113•.Coward J, Kulbe H, Chakravarty P, et al. Interleukin-6 as a therapeutic target in human ovarian cancer. Clin Cancer Res. 2011;17(18):6083–6096. doi: 10.1158/1078-0432.CCR-11-0945. IL-6 antibodies have entered clinical trials. This paper shows that blockade of IL-6 inhibits STAT3 phosphorylation as expected, but is not cytotoxic to cultured tumor cells. [DOI] [PMC free article] [PubMed] [Google Scholar]