

Abstract
A substantial fraction of the human proteome encodes disordered proteins. Protein disorder is associated with a variety of cellular functions and misfunction, and is therefore of clear import to biological systems. However, disorder lends itself to conformational flexibility and heterogeneity, rendering proteins which feature prominent disorder difficult to study using conventional structural biology methods. Here we discuss a few examples of how single-molecule methods are providing new insight into the biophysics and complexity of these proteins by avoiding ensemble averaging, thereby providing direct information about the complex distributions and dynamics of this important class of proteins. Examples of note include characterization of isolated IDPs in solution as collapsed and dynamic species, detailed insight into complex IDP folding landscapes, and new information about how tunable regulation of structure-mediated binding cooperativity and consequent function can be achieved through protein disorder. With these exciting advances in view, we conclude with a discussion of a few complementary and emerging single-molecule efforts of particular promise, including complementary and enhanced methodologies for studying disorder in proteins, and experiments to investigate the potential role for IDP-induced phase separation as a critical functional element in biological systems.
Introduction
Proteins are involved in myriad cellular and developmental roles, including architecture, chemical reactions, selective transport across biological membranes, and interaction and regulation of biomolecular networks and signaling cascades. To date, static 3D structural characterization of large ensembles of highly ordered proteins has dominated structural biology, and has provided much insight into protein function. Despite this success, intrinsic disorder is now understood to be a critical and ubiquitous contributor to protein function, leading to a substantial revision in the classic 3D-structure-function paradigm, and highlighting the need for investigational approaches not limited to well-behaved and structurally robust proteins 1-6. Biophysicists have long recognized that to a greater or lesser extent, proteins are in general dynamic and flexible species. However, intrinsically disordered regions in proteins, whether local or global (IDRs and IDPs respectively), encode a much greater degree of these features, and require both new perspective and new tools for detailed investigation. The physics of this disorder could confer a number of biologically significant functional advantages on these systems, and therefore not only require, but merit careful study. Additionally, a number of disease-linked amyloid forming proteins are disordered in their monomeric-unbound states, suggesting a potential and important link between disorder and aberrant misfolding. Therefore, a detailed biophysical understanding of these paradigm-shifting proteins is important for both fundamental protein science and a more precise understanding of cellular function and disease, despite the inherent challenges in studying such conformationally complex and dynamic species.
Expanding the experimental potential for understanding IDP biophysics has been a significant opportunity afforded through some exciting advances in single-molecule detection methods over the past few decades 7-10. Capitalizing on improvements in relevant technologies, biophysical single-molecule experiments based on force, fluorescence and other methods began appearing in the 1980s 11-16. These methods fundamentally altered our views of molecular complexity and opened the door to more direct tests of mechanistic models by avoiding the averaging and loss of information that are necessary in ensemble experiments to achieve high signal-to-noise data. Single-molecule methods have already been used to probe the complex conformational distributions, dynamics, interactions, and aggregation propensities of IDPs, with much success. Early application of single-molecule techniques to IDPs began appearing in the literature in the mid-late 2000’s, with investigation of conformational features, dynamics and interactions of amyloidogenic IDPs. Also, and of particular note for aggregation-prone members of this protein class, single-molecule experiments utilize very low molecular concentrations, avoiding the confounding effect of unwanted aggregation or molecular interaction. Several studies have followed since on these and other types of IDPs, and have broadened our understanding of the biophysics of proteins and the systems in which they function.
Discussed below is a sampling of some of the important biological questions being answered with single-molecule experiments, presented in three broad classes of structural and functional complexity: (i) the conformational features and dynamics of monomeric IDPs, (ii) interaction of IDPs with binding partners and concomitant folding, and (iii) more complex behavior of IDPs, with a specific focus on binding-modulated function by interaction with multiple partners. Biophysical features at each of these levels are expected to offer critical insight into biological function, and single-molecule investigation is helping to shed light on each of these levels of molecular and folding complexity. Lastly, we also discuss a few complementary and emerging directions for the utility of single-molecule methods in the effort to study disordered protein systems.
Structural features and dynamics of monomeric IDPs
Investigation of protein disorder begins conceptually with intrinsic structural propensity in monomers, arising from a defining sequence of amino acids. Higher order interactions and structural features can be thought of as functions of this basic state.
In an elegant 2006 study, polyglutamine (“poly-Q”) was investigated by Crick and coworkers as a model of the Huntington’s disease-causing protein, huntingtin, using fluorescence correlation spectroscopy (FCS) to determine the scaling relationship between poly-Q chain length and molecular diffusion times 17. FCS is a near-single-molecule resolution method to measure and analyze fluorescence fluctuations in a subfemtoliter detection volume (achieved through confocal detection) from molecules diffusing freely in solution 18. Fluorescence intensity data are subjected to correlation analysis to identify molecular events ranging from molecular diffusion (as relatively slow decays) to conformational dynamics at very rapid time scales.
Poly-Q was studied by labeling the cysteine residue in Gly-GlnN-Cys-Lys2 peptides (where N = chain length) with the bright fluorescent dye Alexa 488 by a maleimide moiety. These experiments showed a monotonic increase of diffusion time with chain length, with no perceptible change in the trend at or around the disease-critical tract length of N=35. Even more significantly from a polymer physics perspective, the scaling of diffusion times as a function of chain length revealed a slope v of 0.32 ± 0.02 (Figure 1A), which indicated a polymer in poor solvent. From this result, the authors concluded that poly-Q is poorly solvated and compacted in aqueous solution, an unexpected result for a protein with minimal hydrophobicity.
Figure 1. Monomeric IDP structural features and dynamics.
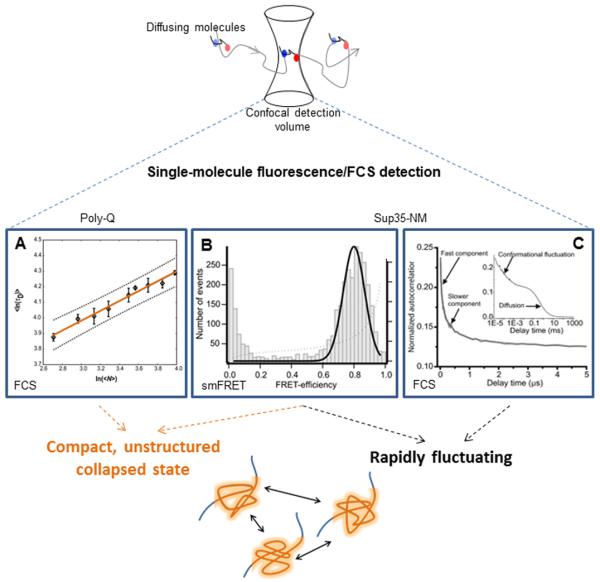
Figure top depicts the principle of diffusion smFRET and FCS detection. Data Panel A is adapted from Crick et al. Proc. Natl. Acad. Sci. (2006) 103:16764, and shows the scaling of diffusion times vs. chain length as measured by FCS. Panels B and C are from Mukhopadhyay et al. Proc. Natl. Acad. Sci. (2007) 104:2649, which show evidence for rapid IDP fluctuations via a relatively narrow smFRET peak on FRET efficiency vs. population (number of events) histogram (B) and rapid-timescale FCS decays (C).
Soon after these results were published, single-molecule investigation of the yeast protein Sup35 corroborated compaction as a feature in IDPs, but did so via direct measurement of intramolecular distance across its amyloid-determining NM region (for N-terminus and middle), which is disordered in the native state 19. Förster resonance energy transfer at single-molecule resolution (termed smFRET), is a powerful method for studying structural features in biological molecules, and is especially well-suited for investigating conformationally heterogeneous IDPs. In smFRET, energy is transferred in a non-radiative and distance-dependent manner between appropriate donor and acceptor fluorophores 9,20,21. In a diffusing format, fluorescence intensities are recorded for each molecule that traverses a subfemtoliter detection volume and FRET efficiency (EFRET) values are calculated, providing a sensitive measure of interdye distance across molecular distances, and an indication of the protein’s conformational state.
In the NM experiments, thousands of protein monomers labeled with amyloid region-flanking donor and acceptor dyes were examined individually using smFRET. The resulting EFRET values, plotted as histograms, revealed a population enriched in high EFRET states with a mean value of 0.8 (Figure 1B), which corresponds to an inter-dye distance of ~ 43 Å. Compared to denaturing conditions, where protein expansion resulted in a dye separation of ~ 63 Å (EFRET ≈ 0.3), these data provided direct evidence of proximity of the NM spanning dyes, consistent with a population of compact monomers.
Further, it was noted that the observed EFRET peak was quite narrowly focused around the peak center. Such a narrow peak indicates either of two possible scenarios: (i) a uniform and highly stable population of conformers (unexpected for an IDP, but consistent with a stably folded structure), or (ii) rapid fluctuation of monomers relative to the detection timescale of 0.5 ms. Additional smFRET experiments utilized guanidinium hydrocholoride to denature the protein molecules and showed a progressive, non-cooperative decrease in EFRET consistent with a population lacking stable structural elements, suggesting that rapid conformational rearrangement was likely responsible. To provide further support for the conclusions drawn from these single-molecule results, specific FCS experiments were designed and utilized, identifying fluorescence decays in the 20-300 ns time scale (Figure 1C), and confirming that the monomer population was indeed an ensemble of compact and rapidly fluctuating structures. In these NM experiments, single-molecule methods proved to be a powerful investigational tool for direct examination of a protein known to be structurally complex and heterogeneous, both in their ability to offer an information-rich snapshot into the population in various conditions, and also in their ability to inform and direct other biophysical methodologies (FCS).
Subsequent single-molecule studies have provided further insight into the polymer physics of IDPs 22-25. In one study using smFRET, Muller-Spath and Serrano et al. showed that two IDPs with high net charge show scaling behavior indicating expanded conformations as compared to unfolded states of folded proteins under the same conditions. In another study, Soranno and coworkers used smFRET, FCS and polymer physics theory to quantify rapid dynamics and internal friction in the same set of proteins, and found that the internal friction is a function of compaction in disordered/unfolded protein states 25. These findings extended the biophysical characterization of disorder in proteins, and led the authors to suggest that expansion may facilitate binding of these IDPs to their cellular partners.
While rapid fluctuations appear to be typical for many IDP monomers, slower IDP dynamics have also been observed at single-molecule resolution. In one set of studies, Lamboy and Kim et al. studied IκBα, inhibitor of the transcriptional factor NF-κB, whose disorder in the functionally important Ankyrin repeats (AR) 5 and 6 has been shown to play a key role in its degradation and regulation 26,27. Using smFRET to study immobilized proteins (which enabled long time-trajectories to be monitored), they observed that AR 5 and 6 are not always disordered in unbound IκBα, but rather stay in their folded conformation for the majority of time, with occasional dynamics to visit a disordered state 28. Moreover, they could observe that the disordered state almost completely disappeared upon binding to NF-κB, consistent with previous studies that demonstrated stabilization of IκBα upon NF-κB binding. A later smFRET study by the same authors showed that the AR 6 domain was more prone to disorder than the AR 5 domain. Thus, while the AR 5 domain did not show disorder at room temperature (requiring an increase to 37 °C to show disorder), the AR 6 domain demonstrated disordered even at room temperature 29.
In a different study, Choi et al. investigated disordered synaptic proteins by encapsulating them in tethered vesicles, a detection format that localizes single molecules close to a surface without direct tethering and any associated unwanted perturbations 30. They observed fluctuations on the second timescale for two disordered synaptic proteins, neuroligin and NMDAR-2B glutamate receptor, though they noted that the physical basis for these fluctuations was not clear. A follow-up study by the same group showed that proline residues in the cytoplasmic domain of NMDAR-2B were closely related to the observed fluctuations, and substitution of a few prolines with alanine significantly lowered the relative population of proteins exhibiting those fluctuations 31.
Through important studies of monomeric species such as these, a picture has emerged that is shaping our understanding of protein disorder as a mechanism for dynamic structure-mediated response in biological systems.
Coupling of IDP binding and folding
A number of IDPs have been shown to transition from a disordered state in isolation to a folded state in conjunction with binding partner interaction. Key mechanistic questions aimed at understanding the functional implications of this observation are whether folding or binding occurs first, and whether the reaction mechanism involves multiple states and folding intermediates. Many of these questions have been probed extensively in the protein α-synuclein (aS), which has served as an excellent tool in the exploration of these fundamental protein folding questions. With a substantial presence in neurons, aS has putative roles in synaptic vesicle fusion and vesicle transport, and its aggregation and mutation have been linked to Parkinson’s disease.
Prior to single-molecule investigation, biophysical and structural studies of aS had indicated that the protein acquires helical secondary structure upon binding to lipid mimics and membranes. However, there was disagreement about the particular structure of the helical conformation that resulted from binding-partner interaction. NMR data for aS in complex with small SDS micelles indicated that the protein populated a hairpin-like helical structure 32, whereas EPR data for aS in the presence of lipid vesicles indicated a more extended helical structure 33. Based on these studies, mechanistic possibilities to explain aS’s binding/folding behavior included (1) that only one of these structures was accessed upon binding, (2) that different structures were accessed upon binding to different partners, and (3) that both structures could were accessed even in the presence of one partner. In an effort to place these observations into a cohesive model, smFRET experiments (similar to those described above for Sup35) were carried out by Ferreon and coworkers to distinguish between each of the competing models for the relationship between these helical states, and eventually their role in normal cell function 34. For these studies, aS was labeled at two positions spanning the helical region, such that formation of the hairpin state (short inter-dye distance) would lead to high EFRET, while extended helix formation (long inter-dye distance) would lead to low EFRET. Histogram analysis of the smFRET data showed a complex series of transitions for a titration with SDS, which serves as a small-molecule binding partner (Figure 2A), revealing several interesting features of this protein’s structure acquisition.
Figure 2. IDP interactions with a small molecule partner by smFRET and microfluidic mixing.
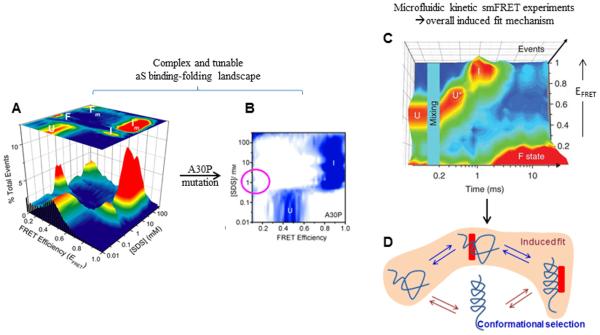
Panel A is adapted from Ferreon et al. Proc. Natl. Acad. Sci. (2009) 106:5645, and Panel B from Ferreon et al. Angew. Chem. Int. Ed. (2010) 49:3469, showing 3D accumulated smFRET histograms for SDS induced binding-folding of α-synuclein, for wildtype (A) and A30P mutant (B). Note the disappearance of the extended state on Panel B (magenta circle), as described in the text. Panel C is from Gambin et al. Nature Methods (2011) 8:239, and shows complex multistep SDS-induced folding of α-synuclein as detected by smFRET coupled with rapid mixing in a microfluidic device, for rapid kinetics study. Panel D depicts possible mechanistic pathways for IDP folding reactions mediated by small molecule binding, with the panel C data providing direct evidence for an induced folding pathway.
Beginning in a disordered state, the αS population switched initially to a hairpin conformation, and then into a predominantly extended state as SDS approached the critical micelle concentration (CMC). As the SDS concentration was increased beyond CMC, the protein switched back to a hairpin state (consistent with the prior NMR data collected at a similar SDS concentration), and finally switched yet again to an extended helical state at high SDS concentrations. Structural assignments for these smFRET data were made in conjunction with an extensive set of complementary CD data, as well as data from previously published reports in the literature. These results showed that rather than populating one or the other of the two structures, different supramolecular characteristics of the small molecule binding partner resulted in a complex and evolving mixture of species. A follow-up study by Ferreon and Moran et al. 35 showed that this complex folding pathway could be altered, by the Parkinson’s disease-related A30P mutation, which displayed a substantial destabilization of the extended helical state under sub-CMC conditions (Figure 2B). Interestingly, however, low surface-curvature environments (i.e. high SDS concentration and comparatively large lipid vesicles) seem to nevertheless “enforce” an extended state upon the protein, suggesting surface curvature as a substantial conformation-determining factor. This study revealed an important biophysical difference in the mutant protein that may link to biology and misfunction, a result that could have been obscured in a lower-resolution analysis of this complex folding behavior.
A fundamental question about aS’s transition from disordered to ordered monomer is whether binding was accomplished by a subset of transiently folded proteins already in the appropriate conformation (conformational selection), or whether the disordered IDP first bound and then folded within the context of a (semi-) disordered complex (induced fit). This question was investigated through development of an improved microfluidic device that combined rapid mixing with smFRET detection to study the time-dependent transition of aS to extended helical states in the presence of SDS 36. This particular design improved the time resolution over existing approaches by approximately two orders of magnitude by using buffer redirection to quickly decelerate a sample stream to accomplish both fast mixing in a rapid flow and slow flow through the detection volume for effective single-molecule detection, opening new doors for targeted and time-resolved smFRET analysis. This improved time-resolution revealed that rather than being directly accessed from the disordered state, the extended helix emerges subsequent to formation of the hairpin state, which itself is formed through a somewhat gradual transition (Figure 2C). In terms of model-distinguishing conclusions, these smFRET data showed clearly that both helical structures formed by an induced fit mechanism under these conditions (Figure 2D), via a series of binding and folding events occurring on the reaction pathway. Thus, although IDPs lack highly ordered globular structure found in classically folding proteins, they can still have inducible structural elements tightly coupled to small molecule binding, which may play key roles in cellular biology. In the next section, we describe studies where single-molecule methods have shed light on the interplay between coupled binding-folding IDP transitions and allosteric regulation.
Allosteric regulation in IDPs: a function for disorder in biology
Single-molecule methods have uncovered a number of physical characteristics of IDPs in isolation and in complex with small molecule partners, as discussed above. However, the broader biological context for these features is both a question of great significance and challenge. Here, we summarize briefly some additional layers of conformational complexity that have been observed at single-molecule resolution, with implications for allosteric regulation through IDP structure-modulation 37,38.
Allostery refers to the perturbation of a biochemical reaction (including binding or enzyme action) by physical changes in the molecule’s structure at positions distal to the site of action. Observations of allosteric effects are of great biophysical interest as they imply the ability to propagate a structural signal over a relatively long molecular range between two sites not immediately connected. In terms of broad biological significance, to understand allostery is to understand further the extent and flexibility of regulation mechanisms employed in biology, a critical goal in biomedical research.
As discussed above, binding to lipid membranes and the associated conformational changes in aS are believed to be important mediators/modulators of its function. Given that covalent modification of this protein (including nitration) is thought to play a role in PD, Sevcsik et al. sought to answer the question of whether tyrosine nitration would affect this protein’s lipid vesicle binding affinity, and discovered an interesting effect 39. Illustrating once again the utility of smFRET to study this flexible class of proteins, the authors observed that nitration at three tyrosines in the C-terminal region, which is believed not to be directly involved in the binding reaction, disrupted vesicle binding, despite being physically separated from the binding interface. Therefore, they concluded that the effect was by an allosteric mechanism that perturbs the conformational ensembles required for stable binding, providing evidence to suggest that protein disorder might allow for rather sensitive mediation of allosteric regulation.
In another recent example, the disordered viral oncoportein E1A was also found to demonstrate allosteric and binding-cooperativity modulation, with important implications for functional consequence40. E1A can dramatically alter the replication machinery in cells through interactions with a number of cellular partners, serving as a hub for a number of related protein interactions. Of note, protein disorder has been suggested to facilitate such a “hub” functionality, as structural plasticity might allow for and even facilitate interaction with a potentially broad spectrum of cellular partners, and has been observed as a central feature in several signaling networks and viral cell cycles.
Previous NMR studies of E1A had yielded some information about this IDP in complex with two of its key partners, TAZ2 and pRb41. However, the protein and its complexes are aggregation prone, limiting those studies to a short E1A fragment at the high concentrations necessary for study by NMR. In order to answer a broader range of important questions about this system, and to understand the role of different E1A sequence elements, Ferreon et al. used smFRET measurements (Figure 3A,B) of unbound and bound (folded) populations to measure dissociation constants for a series of truncation mutants of E1A in complex with TAZ2 and pRb 40. It is important to note that by performing these experiments at low-concentration single-molecule conditions, characterization of individual complexes of interest could be performed in spite of their propensity to aggregate. The smFRET data were used to construct binding phase diagrams for this ternary system (Figure 3C), which could be analyzed to understand the cooperativity of E1A interactions. In this format, it was observed that a short E1A construct lacking the N-terminal region showed negative cooperativity for binding of both partners (TAZ2 and pRb), wherein binding to one partner weakened binding to the other. In striking contrast, restoration of the N-terminal region resulted in a dramatic switch to positive binding cooperativity, with binding of one partner enhancing binding of the other (Figure 3C). The phase diagrams also showed that the two different cooperativity regimes changed the relative areas of the different E1A species (unbound, binary, ternary complexes), which revealed a shift in the overall population distribution of each state over a fixed set of conditions. Since these different complexes have different cellular activities, switching of the binding biophysics (cooperativity) through additional binding partners that can sequester particular sequence elements could have profound effects on the biology of the system. These results illustrated clearly a fundamental mechanism by which a limited length sequence in a hub IDP of relatively small size can still result in complex layers of regulation of multiple functions.
Figure 3. Allostery and regulation in IDPs.
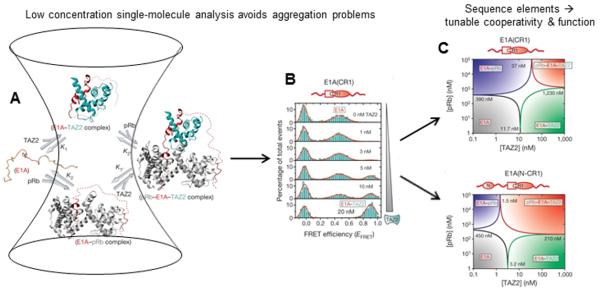
Panel A depicts single-molecule detection of binding for the case of hub IDP E1A binding to two partners, with smFRET data shown in panel B. Panel C shows protein binding phase diagrams assembled from smFRET data, that demonstrate a switching of binding cooperativity depending on available E1A sequence elements. Panels A, B and C are adapted from Ferreon et al. Nature (2013) 498:390.
The above studies uncovered new aspects of allosteric effects in IDPs, in part by leveraging the strengths of single-molecule methods to directly monitor structural distributions for aggregation-prone species by avoiding complications from aggregation. We anticipate that these and other examples of allosteric regulation, both of IDPs and more classically folding proteins, will continue to benefit from single-molecule investigation.
Complementary and Emerging directions
Several examples have been discussed above to show some of the new insights gained from single-molecule fluorescence methods into the biological physics and chemistry of IDPs. The remainder of this review will focus on complementary or emerging areas in single-molecule studies of protein disorder and associated complexity.
Protein phase separation and cellular granules
An exciting area that has emerged over the past few years is the cellular role of phase separation and corresponding formation of liquid droplets or particles, and the possibility that protein disorder may play an essential and formative role in these biochemically distinct centers 22,42-46. The cellular volume is differentiated into various compartments that serve different functions. The best-known compartments are membrane-bound organelles such as the nucleus and Golgi body. Unlike these organelles, phase separated granules are cellular organelles that are not enclosed by membranes, yet distinguish themselves from the rest of the cytosol via phase separation, and are often visible using even low-resolution light microscopy 44. These droplet-like structures are in liquid form, yet separated from the cytosolic space by energetically driven demixing 47. The high local concentrations and distinct chemical environments of these droplets may facilitate interactions and biochemical reactions, while excluding unwanted cellular components and potentially enriching others 42-44,46. Interestingly, protein disorder has been shown to be important for such phase separation 22,42-45,46 and some studies have even reported structures similar to amyloid fibrils in close relationship with phase-separated granules 42,43.
Given the anticipated complexity in distributions of species involved in the phase separation process, single-molecule methods could provide a powerful means to understand several important but unexplored aspects of the physics and chemistry within droplets. Methods such as smFRET and FCS can be used to probe the early binding and structural changes that nucleate and lead to formation, growth and other dynamic changes in droplets; features important in regulating droplets and their activities in cells. Other important issues that could be probed include the structural features of IDPs and other proteins in the altered interior environment of droplets, access of molecules from outside to inside droplets, and how and why reaction rates are altered within these “microreactors”.
Force-based methods for detailed characterization of IDP structural landscapes
Although IDPs are intrinsically “disordered,” the disordered states, as we have discussed above, are not like typical denatured globular proteins. Therefore, understanding the free energy landscapes and the mechanical properties of these proteins, including transient or persistent structure(s), is an important challenge to meet. While these questions are intriguing in their own right from a biophysical perspective, they also relate critically to biology, especially where IDP structure relates critically to neurodegenerative and other disease 48.
Force methods such as optical tweezers and atomic force microscopy (AFM) permit high-resolution measurement in “pulling” experiments, where protein molecules are extended as a function of applied force, and the resulting force profiles reveal information about the protein’s energy landscape in a particular conformation 49 (Figure 4A50, C50, B51). Once again, the protein aS has proven to be a target of great interest and utility, playing a central role in demonstrating yet another unique and powerful single-molecule method for studying protein disorder. Early AFM studies on aS indicated that it could access “β-like structure” that is resistant to mechanical unfolding, requiring >100pN to be extended 48,52 (Figure 4D). Given that aggregation and concomitant acquisition of well-defined β-sheet structure is a prominent feature in the protein’s link to PD, these AFM data suggested that this nascent β-sheet structure might represent a significant precursor to the more stable and disease-associated structures. However, evidence of this conformation has not been observed by other experimental methods, leading the authors to suggest later that their results may reflect a confounding impact of the unique polyprotein construct used in the AFM studies, which may make it more susceptible to β-sheet formation, although accentuation of a more modest but existing propensity may also be a possibility 8.
Figure 4. Mechanical measurements of IDPs.
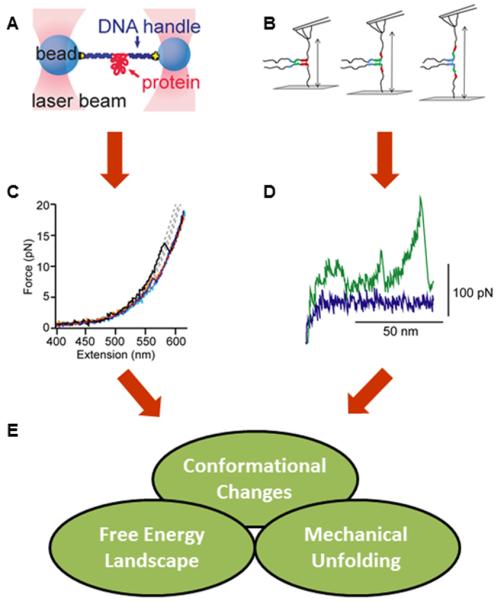
Panels A and B show schematic experiments (A: optical tweezers B: AFM. Both techniques require attachment of the protein to the probes (beads for optical tweezers, surface and cantilever tip for AFM) through tethers. Panels C and D show typical sets of data obtained from these methods, presented as force-extension curves (FEC) (C: optical tweezers (adapted from 50) D: AFM (adapted from 52)). Optical tweezers FEC is showing extension of an aS monomer by force, where “sawtooth” like patterns depict mechanical conformational changes. The FEC in Panel D shows the same type of data acquired using AFM. Here, the middle peak was interpreted to report on a conformational change of aS monomer via mechanically “weak” interaction. Panel E shows the types information that can be obtained from these experiments. Panels A and C are adapted from Neupane and Solanki et al. PLoS ONE (2014) 9:e86495 [50]. Panel B is adapted from Krasnoslobodtsev et al. PLoS ONE (2012) 7:e38099 [51]. Panel D is adapted from Sandal and Valle et al. PLoS Biol (2008) 6:e6 [52].
Recently, Solanki et al. assessed the free energy landscape of aS under low force (<10 pN) using optical tweezers, and reported that aS may populate three distinct, yet marginally stable structures under native condition 53. The data showed three minima where the energy barriers between different minima were much lower than those found in globular proteins, consistent with the less stable nature of an IDP in comparison with globular proteins.
In addition to monomer experiments, the energy landscapes of IDP oligomers have also been explored, with some early success and much promise for the future. In one study, Yu and coworkers studied interactions between aS monomers immobilized on both a surface and an AFM tip in an effort to investigate intermolecular aS interactions. Their data showed a long-lived mechanically stable structure that they postulate might be an important species in nucleation of amyloid fibril formation 54. More recently, Neupane et al. showed that multimeric aS may exhibit more complexity in terms of structures and force required to modify them, by using covalently linked dimers and tetramers 50. Finally, in a different experimental format providing complementary information, Miyagi and coworkers used high-speed AFM imaging to directly visualize structural changes in disordered regions in the protein FACT 55.
These examples hopefully provide a clear picture of how many important and fascinating questions might be asked and answered about the mechanical properties of IDPs, from residual structure in monomers to complexes or oligomers of varied stability, and further to the mechanical properties of IDP structures, aberrant and otherwise (Figure 4E). Improvements in these methodologies will no doubt be coupled with broader applications in the future.
Further advances in single-molecule detection capabilities
Above, we have discussed examples of interactions of IDPs with partners that are linked to functional conformational changes in these molecules. The studies discussed above represent relatively simple, although illuminating, demonstrations of some strengths of single-molecule methods. However, more complex multicomponent interactions are likely quite ubiquitous representatives of biological action by typical IDPs. Hence, the ability to probe multiple physical parameters simultaneously will ultimately be of great value in the collective effort to understand biological function and consequence. For example, newer multicolor FRET technologies 56 will be useful for correlating discrete conformational changes and/or binding events, or to understand the mechanistic basis of competitive binding or negative cooperativity. As another example, combined force and fluorescence methods 57 could be useful for understanding the effects of partner binding on mechanical stabilities of IDP structural features. These types of methods in conjunction with microfluidic mixing or sorting methods and multiparameter detection 58 could also be invaluable in understanding the mechanistic pathways of partner binding, or oligomer and amyloid fibril formation. Finally, to study larger features and phenomena such as the case of phase separation discussed above, it will be important to study how interacting ensembles of molecules give rise to mesoscale properties that may be important in cellular function. Hence, an exciting future lies ahead for the detection not merely of single molecules, but in the detection of multiple single molecules simultaneously in a system of interacting components.
Concluding remarks
As we have seen in this review, single-molecule methods have proven to be powerful tools, lending themselves extensively to efforts to understand the complex characteristics of protein disorder and complexity. From a single-molecule observational perspective, IDPs lacked the common stable structures of the globular proteins as was previously expected, yet could have characteristics unlike states derived from the denaturation of globular proteins. While this class of proteins may indeed be “disordered” in terms of conventional secondary and tertiary structure, the available data suggest perhaps a different “order” from globular proteins, tunable by sequence and interactions. These biophysical “rules” and features provide for a regulatable structural sensitivity that allows for singular and important function in cells. Now that we are past the initial stages of probing the capabilities of single-molecule methods and initiating studies on the basic biophysics of IDPs, new challenges await: deeper and more thorough understanding of the unique biophysics of IDPs, more direct investigation of the functional roles of IDP biophysics, and understanding how molecular interactions with other cellular elements result in structure and function on the mesoscopic cellular scale. These and other areas of single-molecule investigation promise to better illuminate the critical roles of the biophysics of protein disorder in cell and developmental biology.
Acknowledgements
We gratefully acknowledge funding from NIGMS/National Institutes of Health (grant 066833 to A.A.D.), NSF (grant MCB 1121959 to A.A.D.), and a scholarship from the Kwanjeong Educational Foundation, South Korea (Award Number 11AmB52G) to T.L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- (1).Wright PE, Dyson HJ. J Mol Biol. 1999;293:321–31. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- (2).Babu MM, Kriwacki RW, Pappu RV. Science. 2012;337:1460–1. doi: 10.1126/science.1228775. [DOI] [PubMed] [Google Scholar]
- (3).Uversky VN, Gillespie JR, Fink AL. Proteins. 2000;41:415–27. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- (4).Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, Ausio J, Nissen MS, Reeves R, Kang C, Kissinger CR, Bailey RW, Griswold MD, Chiu W, Garner EC, Obradovic Z. J Mol Graph Model. 2001;19:26–59. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- (5).Tompa P. Trends Biochem Sci. 2002;27:527–33. doi: 10.1016/s0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- (6).Forman-Kay JD, Mittag T. Structure. 2013;21:1492–9. doi: 10.1016/j.str.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ferreon ACM, Moran CR, Gambin Y, Deniz AA. In: Methods in Enzymology, Vol 472: Single Molecule Tools, Pt A: Fluorescence Based Approaches. Walter NG, editor. Vol. 472. Elsevier Academic Press Inc; San Diego: 2010. pp. 179–204. [Google Scholar]
- (8).Brucale M, Schuler B, Samori B. Chem Rev. 2014;114:3281–317. doi: 10.1021/cr400297g. [DOI] [PubMed] [Google Scholar]
- (9).Deniz AA, Mukhopadhyay S, Lemke EA. J. R. Soc. Interface. 2008;5:15–45. doi: 10.1098/rsif.2007.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Orrit M, Ha T, Sandoghdar V. Chem Soc Rev. 2014;43:973–6. doi: 10.1039/c4cs90001d. [DOI] [PubMed] [Google Scholar]
- (11).Lindsay SM, Thundat T, Nagahara L, Knipping U, Rill RL. Science. 1989;244:1063–4. doi: 10.1126/science.2727694. [DOI] [PubMed] [Google Scholar]
- (12).Block SM, Goldstein LS, Schnapp BJ. Nature. 1990;348:348–52. doi: 10.1038/348348a0. [DOI] [PubMed] [Google Scholar]
- (13).Moerner WE, Kador L. Physical review letters. 1989;62:2535–2538. doi: 10.1103/PhysRevLett.62.2535. [DOI] [PubMed] [Google Scholar]
- (14).Orrit M, Bernard J. Physical review letters. 1990;65:2716–2719. doi: 10.1103/PhysRevLett.65.2716. [DOI] [PubMed] [Google Scholar]
- (15).Howard J, Hudspeth AJ, Vale RD. Nature. 1989;342:154–8. doi: 10.1038/342154a0. [DOI] [PubMed] [Google Scholar]
- (16).Lyubchenko Y, Shlyakhtenko L, Harrington R, Oden P, Lindsay S. Proc Natl Acad Sci U S A. 1993;90:2137–40. doi: 10.1073/pnas.90.6.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Crick SL, Jayaraman M, Frieden C, Wetzel R, Pappu RV. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:16764–16769. doi: 10.1073/pnas.0608175103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Haustein E, Schwille P. In Annual review of biophysics and biomolecular structure. 2007;36:151–169. doi: 10.1146/annurev.biophys.36.040306.132612. [DOI] [PubMed] [Google Scholar]
- (19).Mukhopadhyay S, Krishnan R, Lemke EA, Lindquist S, Deniz AA. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:2649–2654. doi: 10.1073/pnas.0611503104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Cornish PV, Ha T. ACS Chem Biol. 2007;2:53–61. doi: 10.1021/cb600342a. [DOI] [PubMed] [Google Scholar]
- (21).Schuler B, Eaton WA. Curr Opin Struct Biol. 2008;18:16–26. doi: 10.1016/j.sbi.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Milles S, Lemke EA. Biophys. J. 2011;101:1710–9. doi: 10.1016/j.bpj.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Soranno A, Koenig I, Borgia MB, Hofmann H, Zosel F, Nettels D, Schuler B. Proc Natl Acad Sci U S A. 2014;111:4874–9. doi: 10.1073/pnas.1322611111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Muller-Spath S, Soranno A, Hirschfeld V, Hofmann H, Ruegger S, Reymond L, Nettels D, Schuler B. Proc Natl Acad Sci U S A. 2010;107:14609–14. doi: 10.1073/pnas.1001743107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Soranno A, Buchli B, Nettels D, Cheng RR, Muller-Spath S, Pfeil SH, Hoffmann A, Lipman EA, Makarov DE, Schuler B. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:17800–17806. doi: 10.1073/pnas.1117368109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Dea EL, Barken D, Peralta RQ, Tran KT, Werner SL, Kearns JD, Levchenko A, Hoffmann A. Molecular systems biology. 2007;3:111. doi: 10.1038/msb4100148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Mathes E, O’Dea EL, Hoffmann A, Ghosh G. The EMBO journal. 2008;27:1357–67. doi: 10.1038/emboj.2008.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Lamboy JA, Kim H, Lee KS, Ha T, Komives EA. Proc Natl Acad Sci U S A. 2011;108:10178–83. doi: 10.1073/pnas.1102226108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Lamboy JA, Kim H, Dembinski H, Ha T, Komives EA. J Mol Biol. 2013;425:2578–90. doi: 10.1016/j.jmb.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Choi UB, McCann JJ, Weninger KR, Bowen ME. Structure. 2011;19:566–76. doi: 10.1016/j.str.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Choi UB, Kazi R, Stenzoski N, Wollmuth LP, Uversky VN, Bowen ME. The Journal of biological chemistry. 2013;288:22506–15. doi: 10.1074/jbc.M113.477810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Ulmer TS, Bax A, Cole NB, Nussbaum RL. The Journal of biological chemistry. 2005;280:9595–603. doi: 10.1074/jbc.M411805200. [DOI] [PubMed] [Google Scholar]
- (33).Jao CC, Der-Sarkissian A, Chen J, Langen R. Proc Natl Acad Sci U S A. 2004;101:8331–6. doi: 10.1073/pnas.0400553101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Ferreon AC, Gambin Y, Lemke EA, Deniz AA. Proc Natl Acad Sci U S A. 2009;106:5645–50. doi: 10.1073/pnas.0809232106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Ferreon AC, Moran CR, Ferreon JC, Deniz AA. Angew Chem Int Ed Engl. 2010;49:3469–72. doi: 10.1002/anie.201000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Gambin Y, VanDelinder V, Ferreon AC, Lemke EA, Groisman A, Deniz AA. Nat Methods. 2011;8:239–41. doi: 10.1038/nmeth.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hilser VJ, Thompson EB. Proc Natl Acad Sci U S A. 2007;104:8311–5. doi: 10.1073/pnas.0700329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Garcia-Pino A, Balasubramanian S, Wyns L, Gazit E, De Greve H, Magnuson RD, Charlier D, van Nuland NA, Loris R. Cell. 2010;142:101–11. doi: 10.1016/j.cell.2010.05.039. [DOI] [PubMed] [Google Scholar]
- (39).Sevcsik E, Trexler AJ, Dunn JM, Rhoades E. J Am Chem Soc. 2011;133:7152–8. doi: 10.1021/ja2009554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Ferreon AC, Ferreon JC, Wright PE, Deniz AA. Nature. 2013;498:390–4. doi: 10.1038/nature12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Ferreon JC, Martinez-Yamout MA, Dyson HJ, Wright PE. Proc Natl Acad Sci U S A. 2009;106:13260–5. doi: 10.1073/pnas.0906770106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Aggarwal S, Snaidero N, Pahler G, Frey S, Sanchez P, Zweckstetter M, Janshoff A, Schneider A, Weil MT, Schaap IA, Gorlich D, Simons M. PLoS biology. 2013;11:e1001577. doi: 10.1371/journal.pbio.1001577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Kato M, Han TW, Xie S, Shi K, Du X, Wu LC, Mirzaei H, Goldsmith EJ, Longgood J, Pei J, Grishin NV, Frantz DE, Schneider JW, Chen S, Li L, Sawaya MR, Eisenberg D, Tycko R, McKnight SL. Cell. 2012;149:753–67. doi: 10.1016/j.cell.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Brangwynne CP. The Journal of cell biology. 2013;203:875–81. doi: 10.1083/jcb.201308087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Toretsky JA, Wright PE. Journal of Cell Biology. 2014;206:579–588. doi: 10.1083/jcb.201404124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Li P, Banjade S, Cheng HC, Kim S, Chen B, Guo L, Llaguno M, Hollingsworth JV, King DS, Banani SF, Russo PS, Jiang QX, Nixon BT, Rosen MK. Nature. 2012;483:336–40. doi: 10.1038/nature10879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Lee CF, Brangwynne CP, Gharakhani J, Hyman AA, Julicher F. Physical review letters. 2013;111:088101. doi: 10.1103/PhysRevLett.111.088101. [DOI] [PubMed] [Google Scholar]
- (48).Hervas R, Oroz J, Galera-Prat A, Goni O, Valbuena A, Vera AM, Gomez-Sicilia A, Losada-Urzaiz F, Uversky VN, Menendez M, Laurents DV, Bruix M, Carrion-Vazquez M. PLoS biology. 2012;10:e1001335. doi: 10.1371/journal.pbio.1001335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Greenleaf WJ, Woodside MT, Block SM. Annual review of biophysics and biomolecular structure. 2007;36:171–90. doi: 10.1146/annurev.biophys.36.101106.101451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Neupane K, Solanki A, Sosova I, Belov M, Woodside MT. PloS one. 2014;9:e86495. doi: 10.1371/journal.pone.0086495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Krasnoslobodtsev AV, Peng J, Asiago JM, Hindupur J, Rochet JC, Lyubchenko YL. PloS one. 2012;7:e38099. doi: 10.1371/journal.pone.0038099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Sandal M, Valle F, Tessari I, Mammi S, Bergantino E, Musiani F, Brucale M, Bubacco L, Samori B. PLoS biology. 2008;6:e6. doi: 10.1371/journal.pbio.0060006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Solanki A, Neupane K, Woodside MT. Physical review letters. 2014;112:158103. doi: 10.1103/PhysRevLett.112.158103. [DOI] [PubMed] [Google Scholar]
- (54).Yu J, Malkova S, Lyubchenko YL. J Mol Biol. 2008;384:992–1001. doi: 10.1016/j.jmb.2008.10.006. [DOI] [PubMed] [Google Scholar]
- (55).Miyagi A, Tsunaka Y, Uchihashi T, Mayanagi K, Hirose S, Morikawa K, Ando T. Chemphyschem : a European journal of chemical physics and physical chemistry. 2008;9:1859–66. doi: 10.1002/cphc.200800210. [DOI] [PubMed] [Google Scholar]
- (56).Gambin Y, Deniz AA. Mol Biosyst. 2010;6:1540–7. doi: 10.1039/c003024d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Lee KS, Marciel AB, Kozlov AG, Schroeder CM, Lohman TM, Ha T. J Mol Biol. 2014;426:2413–21. doi: 10.1016/j.jmb.2014.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Sisamakis E, Valeri A, Kalinin S, Rothwell PJ, Seidel CA. Methods Enzymol. 2010;475:455–514. doi: 10.1016/S0076-6879(10)75018-7. [DOI] [PubMed] [Google Scholar]