

Abstract
The increasing prevalence of drug-resistant bacterial infections is driving the discovery and development not only of new antibiotics, but also of inhibitors of virulence factors that are crucial for in vivo pathogenicity. One such virulence factor is the type III secretion system (T3SS), which plays a critical role in the establishment and dissemination of Pseudomonas aeruginosa infections. We have recently described the discovery and characterization of a series of inhibitors of P. aeruginosa T3SS based on a phenoxyacetamide scaffold. To better characterize the factors involved in potent T3SS inhibition, we have conducted a systematic exploration of this structure, revealing several highly responsive structure-activity relationships indicative of interaction with a specific target. Most of the structural features contributing to potency were additive, and combination of those features produced optimized inhibitors with IC50 values <1 µM.
Keywords: type III secretion system, virulence factor, pseudomonas aeruginosa, phenoxyacetamide, Mitsunobu reaction
1. Introduction
Finding effective treatments for bacterial infections caused by antibiotic-resistant strains of pathogenic bacteria is a critical area of unmet medical need, and the emergence of these multi-drug-resistant pathogens has recently caused some to warn of an impending “post-antibiotic era” in which most current antibiotics will no longer be effective.1–3 The discovery and development of new classes of antibiotics has been a major strategy in combating drug resistance, but the difficulties of discovering new scaffolds capable of evading well-established resistance mechanisms such as efflux and target modification have limited the availability of new drugs primarily to analogs of existing classes. Additionally, many pharmaceutical companies have either stopped or slowed their antibacterial programs in favor of more profitable therapeutic areas, and new classes of antibiotics have been difficult to commercialize.4–8 Although bacterial genomics studies have identified many potential new targets, these have not yielded effective novel antibiotics thus far.9
Screening for inhibitors of novel targets may provide new scaffolds, but it is important to identify targets that are not dependent on traditional antibacterial mechanisms, and may thus be less susceptible to pre-existing resistance elements in the population. Virulence factors represent a class of novel targets that has been underexploited for antibacterial discovery. Although virulence factors are not essential for bacterial growth or viability in laboratory culture, they exhibit varying degrees of essentiality for establishment, dissemination, survival or pathogenicity of bacterial infections in the host.10, 11 Bacterial secretion systems are of particular interest because they usually contain some accessible extracellular components and are utilized by many bacterial pathogens to transport host cell-degrading proteins such as elastases and phospholipases, and drug-resistance elements such as β-lactamases across bacterial membranes; inhibition of these processes sensitizes the pathogen to drug and immune system attacks.12–14
The type III secretion system (T3SS) is found in many Gram-negative bacterial species, including the important pathogens Pseudomonas aeruginosa, Escherichia coli, Salmonella enterica, Shigella spp., Vibrio cholerae, and Chlamydia spp. It consists of a complex multiprotein assembly that spans the bacterial inner and outer membranes and the host cellular membrane to secrete effector toxins and translocate them directly into host cells.15 In P. aeruginosa, effector toxins (primarily ExoS and ExoU) impede the rapid innate immune response to colonizing bacteria by killing host phagocytic cells.16 The expression of T3SS in P. aeruginosa thus facilitates the establishment and dissemination of bacterial infection,17 and is ultimately associated with poor clinical outcomes.18
P. aeruginosa is an opportunistic pathogen in humans, but it is a common and extremely virulent cause of serious infections in immune-compromised/suppressed patients (e.g., HIV and cancer), cystic fibrosis patients, and those on mechanical ventilation or with burn wounds. Current antibiotic treatment strategies exhibit failure rates as high as 18%, even when the organism is susceptible to the antibiotic being administered.19, 20 Therefore, inhibitors of P. aeruginosa T3SS may be useful drugs, either alone or in combination with antibiotics, for enabling a robust innate immune response to block the establishment and dissemination of infection and to reduce persister cell levels.21 Indeed, recent studies with humanized monoclonal antibodies to the P. aeruginosa T3SS needle tip protein PcrV suggest that T3SS inhibition will be a useful clinical approach.22–24
Several groups have published structures of small molecule T3SS inhibitors.25 While numerous substructures have been identified, few are drug-like, and none of these small molecules has proceeded to clinical trials. Although members of the salicylidene acylhydrazide class26, 27 have been studied using in vivo models, no specific molecular target could be identified,28 thus reducing overall interest in this class. Our investigations have produced a set of promising scaffolds29 that we have used for hit-to-lead optimization. In particular, the phenoxyacetamides MBX 1641 and MBX 1642 (1, 2; Figure 1) are small, drug-like molecules with low micromolar activity against P. aeruginosa T3SS in assays of both T3SS-mediated secretion and translocation, and they possess a readily modifiable structure. The activity of the phenoxyacetamide scaffold in translocation assays compares favorably to the corresponding activity of the well-studied salicylidene acylhydrazide INP-007 (IC50 = 0.8 µM).30 Additionally, the recently reported finding that pscF mutations confer resistance to the phenoxyacetamides suggests that they bind in a specific manner to the T3SS needle protein PscF,30 which is an extracellular component distinct from the monoclonal antibody target PcrV. Beginning from this promising starting point, we conducted a rigorous analysis of the structure and activity of a large series of phenoxyacetamide T3SS inhibitors.
Figure 1.

T3SS hit compounds.
2. Results and discussion
2.1. General considerations
In the initial high-throughput screening campaign, we identified a series of closely related compounds29 that provided a starting point from which to systematically explore the structure-activity relationships (SARs) of the phenoxyacetamide scaffold. Initial data suggested that the substituents on both aromatic rings were important to the activity of the compounds, but little information was available regarding the important features in the central region of the molecule. We undertook a process by which different portions of the molecule were independently optimized, followed by the synthesis of highly active T3SS inhibitors made by combining the best features found in preceding optimization steps.
Once compounds were synthesized, they were tested for activity against T3SS in two related, but distinct assays. As described previously,29 the secretion assay uses an effector ExoS-β-lactamase (ExoS-βLA) fusion protein to test whether compounds inhibit T3SS-mediated secretion, as determined by the rate of hydrolysis of the chromogenic β-lactam nitrocefin by the externalized ExoS-βLA. Compounds that effectively inhibit the secretion assay were subjected to a second, confirmatory assay. That more clinically relevant translocation assay tests the ability of the compounds to inhibit intoxication of target CHO cells by infecting P. aeruginosa cells, which produce a complete T3SS apparatus, including the adaptor proteins PcrV and PopB/PopD. Compounds that effectively inhibit the translocation process prevent the death of target CHO cells as measured by standard LDH release assay.29 Furthermore, we determined the cytotoxicity of the compounds in the same assay but in the absence of P. aeruginosa cells to determine inhibitor selectivity in the translocation assay. Representative compounds were also tested for their antibacterial activity (see supplementary information); none of the potent T3SS inhibitory compounds significantly altered the doubling time for growth of P. aeruginosa strain PAO1 when compared to a DMSO control. This confirms that selective T3SS inhibition, and not retardation of bacterial growth, is responsible for the reduced secretion and translocation found in the assays above.
2.2. Substituents at α-position
As we have previously observed,29 the α-position of the amide is sensitive to substituent variation, and we began our investigation of the SAR with that functionality. Thus, commercially available ethyl 2-bromoalkanoates were first reacted with 2,4-dichlorophenol (3) in the presence of base to provide the corresponding 2-(phenoxy)alkanoic acid esters 4b–e (Scheme 1). Saponification of the esters produced the acids 5b–e which, along with commercially available acid 5a, were coupled to piperonylamine to provide the amides 6a–e. To synthesize the chiral acids required to produce the enantiomers of MBX 1641 (1), we utilized a Mitsunobu reaction protocol starting from either enantiomer of ethyl lactate. To produce (R)-1, we first reacted 2,4-dichlorophenol (3) with ethyl l-lactate under standard Mitsunobu conditions to produce ester 7. Saponification of the ester, followed by peptide coupling produced the (R)-enantiomer of MBX 1641. The (S)-isomer was produced in the same manner from ethyl d-lactate (not shown).
Scheme 1.

Synthesis of phenoxyacetamide T3SS inhibitors with varying substituents at the α-position.
Reagents and conditions: (a) ethyl-2-bromopropionate, K2CO3, DMF, 50 °C; (b) KOH, MeOH/H2O, 20 °C; (c) piperonylamine, HATU, DIPEA, DMF, 20 °C; (d) ethyl l-lactate, DIAD, PPh3, THF, 20 °C.
The biological activity of this series confirmed our earlier findings that the nature of the substituent at the α-position of the amide is extremely important for activity (Table 1). Removing the substituent entirely abrogates all activity, as does adding a second small alkyl group (i.e., 6a and 6b, respectively). By increasing the size of the substituent, we noted that the ethyl group (6c) was optimal, while the incorporation of even larger groups (6d, 6e) was again highly detrimental. Furthermore, as a confirmation of our previous finding,29 the two enantiomers exhibited very different activities, with the (R)-enantiomer ((R)-1) showing excellent inhibition of both secretion and translocation, and the (S)-isomer ((S)-1) displaying no significant activity up to the maximum concentration tested. We surmise that this portion of the molecule is interacting with a well-defined hydrophobic pocket that can accommodate the methyl or ethyl group of 1 or 6c, but larger substituents cause steric clashes that lower the activity of the compounds. The reason for the low potency of compound 6a, which lacks any substituent at the α-position, is not currently clear, but the results suggest that an α-position substituent is critical for providing binding energy and possibly proper orientation of the inhibitor with its target protein.31
Table 1.
Activity and cytotoxicity of T3SS inhibitors with varying substituents at the α-position.
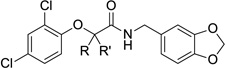 |
|||||
|---|---|---|---|---|---|
| Compound # |
R | R’ | secretion IC50 (µM) |
translocation IC50 (µM) |
translocation CC50 (µM) |
| 1 | −Me | −H | 7.8 ± 2.0 | 11 ± 2 | >100 |
| 6a | −H | −H | >100 | n.d. | n.d. |
| 6b | −Me | −Me | >100 | n.d. | n.d. |
| 6c | −Et | −H | 3.9 ± 1.3 | 3.0 ± 1.0 | 67 ± 18 |
| 6d | −nPr | −H | >100 | n.d. | n.d. |
| 6e | −iPr | −H | >100 | n.d. | n.d. |
| (R)-1 | −Me (R) | −H | 4.3 ± 1.5 | 8.9 ± 5.5 | 61 ± 19 |
| (S)-1 | −Me (S) | −H | >100 | n.d. | n.d. |
n.d.: value not determined
2.3. Substituents on phenoxide ring
Alterations of the substituents on the phenoxide ring were accomplished in a similar manner (Scheme 2) by treating ethyl 2-bromopropionate with the corresponding phenols in the presence of base to make the esters 10c–h, and then saponifying the esters to the corresponding acids (11c–h). The acids, along with commercially available acids 11a and 11b, were then coupled with piperonylamine to provide the target amides 12a–h. To synthesize the pyridyl analog 16, ethyl lactate was again used to directly displace the 2-chloro group of 2,3,5-trichloropyridine and form intermediate ester 14. Saponification and amide bond formation produced the desired target compound 16.
Scheme 2.

Synthesis of phenoxyacetamide T3SS inhibitors with varying substituents on the phenoxide ring.
Reagents and conditions: (a) ethyl-2-bromopropionate, K2CO3, DMF, 50 °C; (b) KOH, MeOH/H2O, 20 °C; (c) piperonylamine, HATU, DIPEA, DMF, 20 °C; (d) Na metal, ethyl lactate, diglyme, 200 °C.
The activity of the compounds was also quite sensitive to the nature of the substituents found on the phenoxide ring (Table 2). Although we individually removed each of the chlorine substituents, and replaced them with a variety of other substituents having a range of electronic properties, any alteration in the 2,4-dichlorophenoxy functionality met with significantly reduced potency. Even the modest change of the chloro groups for fluoro groups (i.e., 12c–e) produced compounds with, at best, five-fold lower activity in the secretion assay. In contrast to the immutability of the substituents, we successfully replaced the phenyl ring with the analogous pyridine system to provide compound 16. Not only was this compound more potent than the parent compound 1 in the secretion assay, but incorporating the nitrogen atom created a compound with lower overall lipophilicity.
Table 2.
Activity and cytotoxicity of T3SS inhibitors with varying substituents on the phenoxide ring.
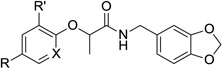 |
||||||
|---|---|---|---|---|---|---|
| Compound # |
X | R | R’ | secretion IC50 (µM) |
translocation IC50 (µM) |
translocation CC50 (µM) |
| 1 | CH | −Cl | −Cl | 7.8 ± 2.0 | 11 ± 2 | >100 |
| 12a | CH | −H | −Cl | 27.0 +/−4.2 | >100 | >100 |
| 12b | CH | −Cl | −H | 70.1 +/−0.7 | >65 | 65.0 +/−4.2 |
| 12c | CH | −F | −Cl | 34 ± 1 | >100 | >100 |
| 12d | CH | −Cl | −F | >100 | n.d. | n.d. |
| 12e | CH | −F | −F | >100 | n.d. | n.d. |
| 12f | CH | −Me | −Cl | 34.0 +/−4.4 | >100 | >100 |
| 12g | CH | −CN | −Cl | >100 | n.d. | n.d. |
| 12h | CH | −OMe | −Cl | >100 | n.d. | n.d. |
| 16 | N | −Cl | −Cl | 5.6 ± 0.2 | 22 ± 10 | >100 |
n.d.: value not determined
2.4. Phenoxide oxygen replacement
Replacing the phenoxyacetyl linkage (Scheme 3) with other related linker units required simple amide bond formation between either piperonylamine or 4-fluorobenzylamine and commercially available acids in the case of compounds 23a–b and 25a–b, but longer syntheses in the case of thioethers 20a and 20b and sulfones 21a and 21b. 2,4-Dichlorothiophenol 17 was thus used in a displacement reaction with ethyl 2-bromopropionate to form intermediate ester 18. Saponification followed by HATU-based coupling provided amides 20a and 20b. To form the corresponding sulfones 21a and 21b, the thioethers 20a and 20b were both oxidized with m-CPBA in moderate yield.
Scheme 3.

Synthesis of phenoxyacetamide-like T3SS inhibitors with alternative linkers.
Reagents and conditions: (a) ethyl-2-bromopropionate, K2CO3, DMF, 50 °C; (b) KOH, MeOH/H2O, 20 °C; (c) piperonylamine or 4-fluorobenzylamine, HATU, DIPEA, DMF, 20 °C; (d) m-CPBA, CHCl3, 20 °C.
Unlike the previous modifications, replacing the oxygen linker with other heteroatoms or a methylene unit resulted in a range of activities (Table 3). The thioether compounds (20a and 20b) had a slightly greater potency than the parent compounds in the secretion assay, but surprisingly, the results in the translocation assay were quite different, and the compounds were not active up to the maximum dose tested. This differentiation between inhibition of T3SS-mediated secretion and translocation is surprising and of interest biologically. However, it does make the thioether compounds substantially less attractive for drug development, and no further analogs in this series were pursued. Although oxidation of the thioethers to the corresponding sulfones did decrease the lipophilicity of the compounds, the change also severely decreased activity in both assays of T3SS inhibition. The amines 23a and 23b showed an intermediate level of activity with 3–4 fold decreases in potency in both the secretion and translocation assays. The methylene-linked compounds 25a and 25b, like the sulfones, showed no activity and were not pursued further. We also noted at this point the similarity between the behavior of the 3,4-methylenedioxy analogs (i.e., 20a, 21a, 23a, and 25a), and the corresponding 4-fluorophenyl analogs (i.e., 20b, 21b, 23b, and 25b); although some differences were evident (e.g., 2 was more cytotoxic than 1 and 23b was more active then 23a in the translocation assay), the overall trends were consistent. We subsequently concentrated our efforts on synthesizing analogs with the 4-fluorophenyl moiety because of the greater availability of requisite starting materials and the higher microsomal stability of the 4-fluorophenyl analogs synthesized to date (data not shown).
Table 3.
Activity and cytotoxicity of T3SS inhibitors with alternative linkers.
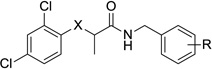 |
|||||
|---|---|---|---|---|---|
| Compound # |
X | R | secretion IC50 (µM) |
translocation IC50 (µM) |
translocation CC50 (µM) |
| 1 | O | 3,4-methylenedioxy | 7.8 ± 2.0 | 11 ± 2 | >100 |
| 2 | O | 4-F | 9.8 ± 2.9 | 6.7 ± 1.0 | 59 ± 16 |
| 20a | S | 3,4-methylenedioxy | 7.8 ± 2.0 | >100 | >100 |
| 20b | S | 4-F | 7.8 ± 2.0 | >100 | >100 |
| 21a | SO2 | 3,4-methylenedioxy | >100 | n.d. | n.d. |
| 21b | SO2 | 4-F | >100 | n.d. | n.d. |
| 23a | NH | 3,4-methylenedioxy | 45 ± 4 | >100 | >100 |
| 23b | NH | 4-F | 34 ± 8 | 32 ± 5 | >100 |
| 25a | CH2 | 3,4-methylenedioxy | >100 | n.d. | n.d. |
| 25b | CH2 | 4-F | >100 | n.d. | n.d. |
n.d.: value not determined
2.5. Linker analogs and benzyl substituents
Once we had determined the best fragment to use in subsequent amide bond formation reactions, we used the commercially available 2-(2,4-dichlorophenoxy)propionic acid (26) to synthesize a large number of target analogs. Acid 26 was directly coupled with different amines to produce the amides 27a–o (Scheme 4), 28a–d (Scheme 5), and 34a–y (Scheme 6). Additionally, the esters 29a and 29b, the hydrazides 30a and 30b, and the hydroxamate 31 were all synthesized by using acid 29 as the coupling partner and HATU as the coupling agent (Scheme 5). The synthesis of ketone 33, however, required the synthesis of Weinreb amide 32 as an intermediate. The Weinreb amide was then treated with a Grignard reagent derived from 4-fluorophenethyl bromide to produce the desired ketone 33.
Scheme 4.

Synthesis of phenoxyacetamide T3SS inhibitors with varying substituents at the benzylic position.
Reagents and conditions: (a) amine, HATU, DIPEA, DMF, 20 °C.
Scheme 5.

Synthesis of phenoxyacetamide T3SS inhibitors with amide linker modifications.
Reagents and conditions: (a) amine, HATU, DIPEA, DMF, 20 °C; (b) alcohol, HATU, DIPEA, DMF, 20 °C; (c) 4-fluorophenylhydrazine or 4-flurorbenzylhydrazine, HATU, DIPEA, DMF, 20 °C; (d) O-benzylhydroxylamie, HATU, DIPEA, DMF, 20 °C; (e) N,O-dimethylhydroxylamine, HATU, DIPEA, DMF, 20 °C; (f) 4-fluorophenethylmagnesium bromide, THF, 0 °C.
Scheme 6.

Synthesis of phenoxyacetamide T3SS inhibitors with varying benzylamine substituents.
Reagents and conditions: (a) amine, HATU, DIPEA, DMF, 20 °C.
Unlike the previously discussed portions of the phenoxyacetamide T3SS inhibitors, where few changes were tolerated, many more changes could be effected on the benzyl amide side of the scaffold without loss of potency (Table 4). We started by investigating the influence of different substituents at the benzylic methylene position. The incorporation of small alkyl groups (resulting in racemic mixtures of diastereomers) improved the overall potency by up to 3.5 fold, with propyl and butyl groups showing the greatest gains in activity in the secretion assay. The effect of the benzylic chiral center was much less pronounced than the phenoxide chiral center; with compound made from (R)-methylbenzylamine and chiral (R)-acid (27c) being only about 60% more active than the compound synthesized from (S)-methylbenzylamine and chiral (R)-acid (27b). The benzylic methylene group could also be disubstituted; the gem-dimethyl compound 27d and spiro-cyclopropyl analog 27e both had excellent activity in the secretion assay. Interestingly, most of the alkylated analogs (with the exception of 27i) had much lower activity in the translocation assay. The lack of activity in the translocation assay, as was the case for the thioether analogs discussed previously, is difficult to rationalize in terms of mechanism, but was sufficient to preclude additional optimization. Although larger hydrophobic substituents were not pursued, the steric requirements were well-defined by this range of compounds. In contrast, incorporating heteroatom-containing substituents (e.g., 27l–o; also as mixtures of diastereomers) into the side-chain did provide potent compounds with better results in the translocation assay. In particular, the hydroxymethyl-substituted compound 27l and the nitrile 27m displayed a good balance of activity and lowered hydrophobicity, although we could not determine an exact value for the translocation assay in the case of 27l because of cytotoxicity. It is very interesting to note that the acid 27o, however, was completely devoid of activity, even though the corresponding ester 27n performed well in the secretion assay.
Table 4.
Activity and cytotoxicity of T3SS inhibitors with varying substituents at the benzylic position.
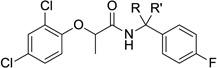 |
|||||
|---|---|---|---|---|---|
| Compound # |
R | R’ | secretion IC50 (µM) |
translocation IC50 (µM) |
translocation CC50 (µM) |
| 2 | −H | −H | 9.8 ± 2.9 | 6.7 ± 1.0 | 59 ± 16 |
| 27a | −Me | −H | 6.0 ± 1.2 | 23 ± 1 | >100 |
| 27b* | −Me (S) | −H | 4.2 ± 0.2 | >32 | 32 ± 1 |
| 27c* | −Me (R) | −H | 2.6 ± 0.9 | >97 | 97 ± 9 |
| 27d | −Me | −Me | 3.3 ± 0.6 | >100 | >100 |
| 27e | −CH2CH2- | 5.4 ± 0.2 | >28 | 28 ± 14 | |
| 27f | −Et | −H | 4.4 ± 0.6 | 25 ± 1 | >100 |
| 27g | −nPr | −H | 3.9 ± 0.7 | >100 | >100 |
| 27h | −iPr | −H | 3.5 ± 1.9 | >100 | >100 |
| 27i | −cPr | −H | 3.0 ± 1.8 | 8.3 ± 2.3 | >100 |
| 27j | −nBu | −H | 4.5 ± 0.4 | >100 | >100 |
| 27k | −cBu | −H | 2.7 ± 0.7 | >100 | >100 |
| 27l | −CH2OH | −H | 6.0 ± 1.5 | >18 | 18 ± 2 |
| 27m | −CN | −H | 5.8 ± 1.0 | 12 ± 2 | >100 |
| 27n | −COOMe | −H | 8.1 ± 2.0 | >100 | >100 |
| 27o | −COOH | −H | >100 | n.d. | n.d. |
n.d.: value not determined
Compounds synthesized using (R)-acid
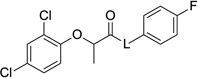
By altering the length of the amide linker in the parent compounds, we also determined an optimal length for this half of the molecule (Table 5). Thus, the benzyl (2) and phenethyl (28b) compounds were equipotent in the secretion assay, but the anilide 28a and the phenylpropyl compound 28c were inactive. Interestingly, alkylating the amide nitrogen provided a compound (28d) that was somewhat less active than the parent, but the change was not completely unfavorable, suggesting that hydrogen-bonding between amide nitrogen and the target may not be necessary for activity. Incorporation of other linker constituents, such as those found in the esters 29a and 29b and the ketone 33 produced compounds devoid of activity, but the hydrazides 30a and 30b and the hydroxamate 31 were active, although more cytotoxic than optimal.
Table 5.
Activity and cytotoxicity of T3SS inhibitors with amide linker modifications.
 |
||||
|---|---|---|---|---|
| Compound # |
L | secretion IC50 (µM) |
translocation IC50 (µM) |
translocation CC50 (µM) |
| 2 | −NHCH2- | 9.8 ± 2.9 | 6.7 ± 1.0 | 59 ± 16 |
| 28a | −NH- | >100 | n.d. | n.d. |
| 28b | −NHCH2CH2- | 12 ± 4 | >17 | 17 ± 11 |
| 28c | −NHCH2CH2CH2- | >100 | n.d. | n.d. |
| 28d | −N(Me)CH2- | 28.5 +/−10.6 | n.d. | n.d. |
| 29a | −OCH2- | >100 | n.d. | n.d. |
| 29b | −OCH2CH2- | >100 | n.d. | n.d. |
| 30a | −NHNH- | 47 ± 7 | 23 ± 4 | 35 ± 2 |
| 30b | −NHNHCH2- | 4.3 ± 0.6 | 8.3 ± 2.3 | 31 ± 1 |
| 31 | −NHOCH2- | 4.0 ± 0.3 | 24 ± 7 | 59 ± 15 |
| 33 | −CH2CH2- | >100 | n.d. | n.d. |
n.d.: value not determined
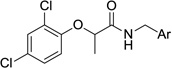
By synthesizing a range of analogs with different substituents on the benzyl aromatic ring, we discovered that many different functionalities could be accommodated (Table 6). We systematically synthesized compounds with no substituents (34a), with chloro, fluoro, methyl and methoxy monosubstituents at all three positions (i.e., 34b–l), and with two methoxy substituents (34m–n). Although the methoxy-substituted compounds were somewhat less potent, the compounds with other substituents were almost equally efficient at inhibiting T3SS in the secretion assay. The only compounds from this group, however, with good activity in the translocation assay and low cytotoxicity, were the 2-fluoro and 2-chloro analogs 34c and 34f. In contrast, incorporation of 5:6 fused heterocyclic systems provided a number of even more potent analogs. Thus, compounds with benzothiophene (34o), benzimidazole (34q) and indole (34r–v) heterocycles had potent activity in the secretion and translocation assays. Interestingly, the attachment site for these fused systems did not seem to influence the activity significantly, as seen by the range of indole analogs tested, but did seem to affect the cytotoxicity (e.g., note the difference in cytotoxicity between indoles 34r and 34s). The addition of nitrogen atoms to the 6-membered aromatic ring (i.e., azaindole 34w and pyridines 34x and 34y), interestingly, yielded analogs with reduced activity with respect to the corresponding compounds lacking that nitrogen. The lower level of substituent discrimination at this end of the molecule suggests that the benzylamine portion does not fit into as well-defined a binding pocket as does the phenoxide ring. This aromatic system may participate in a π-π stacking interaction, and this feature might provide future opportunities to tune the properties of the scaffold by incorporating various substituents on the aromatic rings.
Table 6.
Activity and cytotoxicity of T3SS inhibitors with varying substituents on the benzylamine.
 |
||||
|---|---|---|---|---|
| Compound # |
Ar | secretion IC50 (µM) |
translocation IC50 (µM) |
translocation CC50 (µM) |
| 1 | 3,4-methylenedioxyphenyl | 7.8 ± 2.0 | 11.1 ± 2.3 | >100 |
| 2 | 4-fluorophenyl | 9.8 ± 2.9 | 6.7 ± 1.0 | 59 ± 16 |
| 34a | phenyl | 11 ± 3 | 12 ± 7 | 95 ± 21 |
| 34b | 3-fluorophenyl | 15 ± 3 | >27 | 27 ± 1 |
| 34c | 2-fluorophenyl | 6.3 ± 1.4 | 8.3 ± 3.0 | 47± 2 |
| 34d | 4-chlorophenyl | 8.5 ± 2.6 | >39 | 39 ± 8 |
| 34e | 3-chlorophenyl | 9.6 ± 4.1 | >19 | 19 ± 10 |
| 34f | 2-chlorophenyl | 9.2 ± 2.6 | 9.7 ± 4.2 | >100 |
| 34g | 4-methylphenyl | 8.1 ± 1.8 | >25 | 25 ± 7 |
| 34h | 3-methylphenyl | 8.5 ± 0.1 | >45 | 45 ± 7 |
| 34i | 2-methylphenyl | 11 ± 1 | >100 | >100 |
| 34j | 4-methoxyphenyl | 12 ± 5 | 23 ± 10 | 71 ± 2 |
| 34k | 3-methoxyphenyl | 16 ± 7 | >100 | >100 |
| 34l | 2-methoxyphenyl | 28 ± 5 | n.d. | n.d. |
| 34m | 3,4-dimethoxyphenyl | 22 ± 2 | >36 | 36 ± 6 |
| 34n | 2,4-dimethoxyphenyl | 19 ± 4 | >100 | >100 |
| 34o | benzothiophene-5-yl | 6.0 ± 1.6 | 5.3 ± 3.7 | 30 ± 11 |
| 34p | benzofuran-5-yl | 3.7 ± 1.6 | >11 | 11 ± 5 |
| 34q | benzimidazole-5-yl | 11 ± 5 | 33 ± 8 | 62 ± 7 |
| 34r | indole-5-yl | 2.1 ± 0.6 | 4.8 ± 0.4 | >100 |
| 34s | indole-6-yl | 1.9 ± 0.3 | 5.7 ± 0.6 | 9.4 ± 3.6 |
| 34t | indole-4-yl | 4.8 ± 1.2 | 4.3 ± 2.3 | 24 ± 2 |
| 34u | indole-2-yl | 8.9 ± 0.2 | >12 | 12 ± 4 |
| 34v | 1-methylindole-5-yl | 5.1 ± 1.4 | >6.3 | 6.3 ± 2.6 |
| 34w | pyrrolo[2,3-b]pyridin-5-yl | 9.5 ± 1.3 | >21 | 21 ± 4 |
| 34x | 6-fluoropyridin-3-yl | 58 ± 20 | >100 | >100 |
| 34y | 5-fluoropyridin-2-yl | 41 ± 18 | >100 | >100 |
n.d.: value not determined
2.6. Optimized inhibitors
After having defined beneficial substituent patterns for each position in the molecule, we sought to synthesize optimized compounds that contained the best features into single inhibitors. It was clear that the acid portion of the molecule was relatively selective with regard to the choice of potential functionalities, while the amine portion was much more flexible. Thus, we chose to make a small series of compounds that had the common features of an (R)-ethyl α-substituent, oxygen linker, and a 3,5-dichloropyridyl aromatic system. Three of the best amine moieties (i.e., the aminomethylbenzothiophene, aminomethylindole, and fluorophenylethanolamine,) were coupled to the optimized acid to make second-generation inhibitors.
Preparation of these optimized inhibitors was accomplished by first constructing the required acid using Mitsunobu reaction conditions. Thus, 3,5-dichloro-2-pyridone (13) and methyl (S)-2-hydroxybutanoate were coupled in the presence of triphenylphosphine and diisopropylazodicarboxylate (DIAD) to provide the ester 35 as the (R)-enantiomer. Although some of the N-alkylated product was observed,32 the desired O-alkylated ester 35 was the major component and was isolated in good yield. Subsequent saponification of ester 35 to acid 36 was again followed by a HATU-mediated coupling reaction with different amine partners to provide optimized compounds 37a–c.
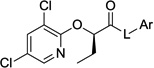
All of the optimized compounds (37a–c) demonstrated varying degrees of improved anti-T3SS activity or cytotoxicity (Table 7). The most improved compound, benzothiophene 37a, demonstrated a 6-fold total potency increase in the β-lactamase secretion assay over the unoptimized compound 34o (Table 6), and also less cytotoxicity. However, the structurally similar indole 37b showed only about half as much improvement (2.6-fold in the secretion assay and 2.5-fold in the translocation assay) over the corresponding 34r (Table 6). Surprisingly, the difference in potency for the hydroxymethyl-substituted analog 37c and its parent compound, 27l (Table 4), was not statistically significant. However, reductions in the cytotoxicity of optimized compound 37c did result in a clear therapeutic index for the translocation assay, unlike 27l, for which the activity in the translocation assay could not be measured due to cytotoxic effects. Importantly, although not all of the compounds were improved to an equal degree by the scaffold optimization, two target compounds (37a and 37b) did exhibit submicromolar activity in the β-lactamase secretion assay and low micromolar activity in the translocation assay with low (>100 µM) cytotoxicity.
Table 7.
Activity and cytotoxicity of T3SS inhibitors with multiple optimizations.
 |
|||||
|---|---|---|---|---|---|
| Compound # |
L | Ar | secretion IC50 (µM) |
translocation IC50 (µM) |
translocation CC50 (µM) |
| 1 | −NHCH2- | 3,4-methylenedioxyphenyl | 7.8 ± 2.0 | 11 ± 2 | >100 |
| 2 | −NHCH2- | 4-fluorophenyl | 9.8 ± 2.9 | 6.7 ± 1.0 | 59 ± 16 |
| 37a | −NHCH2- | benzothiophene-5-yl | 0.9 ± 0.1 | 3.6 ± 0.1 | >100 |
| 37b | −NHCH2- | indole-5-yl | 0.8 ±-0.1 | 1.9 ± 0.2 | >100 |
| 37c | −NHCH(CH2OH)- | 4-fluorophenyl | 6.9 ± 0.2 | 29.3 ± 3.2 | 75 ± 21 |
3. Conclusion
A large library of T3SS inhibitors was synthesized and the structure-activity relationships in this phenoxyacetamide scaffold were defined. The SAR of the system is quite responsive and reproducible, consistent with a very specific interaction of the ligand with a discrete protein target, which appears to be the T3SS needle protein PscF according to the recent identification and characterization of pscF mutations conferring resistance to the phenoxyacetamides.30 The dichlorophenyl portion of the molecule is quite sensitive to changes in structure, as is the substituent at the α-position of the amide. We have defined (R)-2-(3,5-dichloropyridin-2-yloxy)butanoic acid as the optimal fragment for further optimization. In contrast, many different substituents on the amine portion of the molecule provide opportunities to improve potency and selectivity of the compounds. Two compounds, 37a and 37b, were the most potent (IC50 <1 µM) of the current study; both also had low cytotoxicity (>100 µM) in a standard LDH release assay. Further exploration will be required to define a preclinical candidate, and this work will be reported in the future.
4. Experimental Section
4.1. Chemistry
4.1.1. General
Reagents and solvents were obtained from commercial sources and used without additional purification. Evaporation of solvents was accomplished under reduced pressure (40–60 mmHg), at less than 40 °C, unless otherwise noted. Chromatography solvent systems are expressed in v/v ratios or as % vol. Melting points were taken on EZ-Melt automated melting point apparatus (Stanford Research Systems, Inc.) in manual mode, and are uncorrected. Thin-layer chromatography was performed on silica gel GHLF plates from Analtech (Newark, DE), and the chromatograms were visualized under UV light at 254 nm. 1H NMR spectra were obtained at 300 MHz on a Bruker DPX300 spectrometer; chemical shift values for 1H were determined relative to an internal tetramethylsilane standard (0.00 ppm). Mass spectrometry was performed by CreaGen Biosciences (Woburn, MA). Analytical HPLC was performed by Averica Discovery Services (Marlborough, MA). All target compounds were found to be ≥95% pure by analytical HPLC unless otherwise noted.
4.1.1.1. General synthesis (A) of 2-phenoxypropanamides 1, 2, 6a, (R)-1, (S)-1
To solutions of the corresponding 2-phenoxyalkylcarboxylic acids in DMF (5 mL/mmol) were added 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI; 1.1 eq.) and 1-hydroxy-7-azabenzotriazole (1.1 eq.). The solutions were stirred at room temperature for 30 min, then the corresponding benzylamines (1.2 eq.) and diisopropylethylamine (3.0 eq.) were added. The resulting mixtures were stirred at room temperature for an additional 16 h, then poured into 10% aqueous citric acid solutions. The aqueous mixtures were extracted with EtOAc, and the extracts were washed with 5% aqueous NaHCO3 and brine, then dried over MgSO4, filtered, and evaporated to provide white solids. The solids were crystallized from EtOAc/hexane to provide the products 1, 2, and 6a.
4.1.1.2. General synthesis (B) of 2-phenoxypropanamides 6b–e
To solutions of the corresponding 2-phenoxyalkylcarboxylic acids in DMF (5 mL/mmol) were added 2-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate (HATU; 1.2 eq.), the corresponding benzylamines (1.2 eq.), and diisopropylethylamine (1.3 eq.). The resulting mixtures were stirred at room temperature for 16 h, then poured into water (10 × volume). The aqueous mixtures were then stirred at room temperature until solids precipitate. The solids were filtered, rinsed with water, and dried to provide solids that were recrystallized from CH2Cl2/hexane to provide the products 6b–e.
4.1.1.3. General synthesis (C) of 2-phenoxypropanoic acids (5b–e)
To stirred solutions of 2,4-dichlorophenol (2.50 g, 15.0 mmol) in DMF (25 mL) were added K2CO3 (2.60 g, 18.8 mmol) and the corresponding ethyl 2-bromoalkyl esters (18.8 mmol). The mixtures were stirred at 50 °C for 16 h, then cooled to room temperature and filtered to remove the inorganic solids. The filtrates were evaporated under high vacuum (<1 mmHg) to provide clear oils that were dissolved in absolute ethanol (25 mL). Aqueous solutions of NaOH (2.0 M, 12 mL, 24 mmol) were added, and the combined solutions heated to reflux for 4 h. The resulting mixtures were cooled to room temperature and evaporated under reduced pressure to provide residues that were dissolved in water (75 mL). The aqueous solutions were acidified (pH 2) with aqueous HCl (2.0 M) to provide precipitates that were filtered, rinsed with water (20 mL), and dried under vacuum to provide the products 5b–e.
4.1.1.4. General synthesis (D) of 2-phenoxypropanoic acids 11c–h
To stirred solutions of the corresponding phenol (15.0 mmol) in DMF (25 mL) are added K2CO3 (2.60 g, 19 mmol) and ethyl 2-bromopropanoate (2.5 mL, 19 mmol). The mixtures were stirred at 50 °C for 16 h, then cooled to room temperature and filtered to remove the inorganic solids. The filtrates were evaporated under high vacuum (<1 mmHg) to provide clear oils that were dissolved in absolute ethanol (25 mL). Aqueous solutions of NaOH (2.0 M, 12 mL, 24 mmol) were added, and the combined solutions heated to reflux for 4 h. The resulting mixtures were cooled to room temperature and evaporated under reduced pressure to provide residues that were dissolved in water (75 mL). The aqueous solutions were acidified (pH 2) with aqueous HCl (2.0 M) to provide precipitates that were filtered, rinsed with water (20 mL), and dried under vacuum to provide the products 11c–h.
4.1.1.5. General synthesis (E) of 2-phenoxypropanamides 12a–h
To solutions of the corresponding 2-phenoxypropanoic acids (0.43 mmol) in DMF (2 mL) were added 2-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate (HATU; 200 mg, 0.53 mmol), piperonylamine (86 mg, 0.57 mmol), and diisopropylethylamine (100 µL, 0.57 mmol). The resulting mixtures were stirred at room temperature for 16 h, then poured into water (20 mL). The aqueous mixtures were then stirred at room temperature until solids precipitate. The solids were filtered, rinsed with water, and dried to provide solids that were recrystallized from CH2Cl2/hexane to provide the products 12a–h.
4.1.1.6. General synthesis (F) of 2-phenoxypropanamides 27a–p
To solutions of 2-(2,4-dichlorophenoxy)propanoic acid (100 mg, 0.43 mmol) in DMF (2 mL) were added 2-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate (HATU; 200 mg, 0.53 mmol), the corresponding benzylamines (0.50 mmol), and diisopropylethylamine (100 µL, 0.57 mmol). The resulting mixtures were stirred at room temperature for 16 h, then poured into water (20 mL). The aqueous mixtures were then stirred at room temperature until solids precipitate. The solids were filtered, rinsed with water, and dried to provide solids that were recrystallized from CH2Cl2/hexane to provide the products 27a–p. With the exception of 27d and 27e, the remaining compounds were isolated as inseparable mixtures of diastereomers; where possible, the matched pairs of NMR signals are noted.
4.1.1.7. General synthesis (G) of 2-phenoxypropanamides 28a–e
To solutions of 2-(2,4-dichlorophenoxy)propanoic acid (100 mg, 0.43 mmol) in DMF (2 mL) were added 2-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate (HATU; 200 mg, 0.53 mmol), the corresponding amines (0.50 mmol), and diisopropylethylamine (100 µL, 0.57 mmol). The resulting mixtures were stirred at room temperature for 16 h, then poured into water (20 mL). The aqueous mixtures were then stirred at room temperature until solids precipitate. The solids were filtered, rinsed with water, and dried to provide solids that were recrystallized from CH2Cl2/hexane to provide the products 27a–e.
4.1.1.8. General synthesis (H) of 2-phenoxypropanamides 34a–y
To solutions of 2-(2,4-dichlorophenoxy)propanoic acid (100 mg, 0.43 mmol) in DMF (2 mL) were added 2-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate (HATU; 200 mg, 0.53 mmol), the corresponding benzylamines (0.50 mmol), and diisopropylethylamine (100 µL, 0.57 mmol). The resulting mixtures were stirred at room temperature for 16 h, then poured into water (20 mL). The aqueous mixtures were then stirred at room temperature until solids precipitate. The solids were filtered, rinsed with water, and dried to provide solids that were recrystallized from CH2Cl2/hexane to provide the products 34a–y.
4.1.1.9. General synthesis (I) of (R)-2-pyridyloxybutanamides 37a–d
To solutions of (R)-2-[(3,5-dichloropyridin-2-yl)oxy]butanoic acid (125 mg, 0.50 mmol) in DMF (2 mL) were added 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI; 125 mg, 0.65 mmol), hydroxybenzotriazole (HOBt; 88 mg, 0.65 mmol), the corresponding benzylamines (0.60 mmol), and triethylamine (182 mg, 1.5 mmol). The resulting mixtures were stirred at room temperature for 16 h, then poured into 5% aqueous NaHCO3 (20 mL). The aqueous mixtures were then extracted with EtOAc (20 mL). The extracts were washed with water (20 mL) and brine (20 mL), dried over Na2SO4, filtered, and evaporated. The filtrates were evaporated, and subjected to flash chromatography on silica gel with 0–70% EtOAc/hexane. Product-containing fractions were pooled and evaporated to provide crude mixtures that were recrystallized from CH2Cl2/hexane to provide the products 37a–d.
4.1.2. Syntheses
4.1.2.1. N-[3,4-(Methylenedioxy)benzyl]-2-(2,4-dichlorophenoxy)propanamide (MBX 1641; 1)
General synthesis A was followed using 2-(2,4-dichlorophenoxy)propanoic acid (1.28 g, 5.5 mmol) and 4-fluorobenzylamine (0.75 mL, 6.5 mmol) to provide 1.70 g (91%) of product as a white solid: mp 109–111 °C; Rf 0.57 (1:1 hexane:EtOAc); 1H-NMR (CDCl3) δ 7.39 (d, 1H), 7.22–7.17 (m, 3H), 7.03–6.97 (m, 3H), 6.86 (d, 1H), 4.74 (q, 1H), 4.47–4.44 (m, 2H), 1.64 (d, 3H); m/z expected 367.0 found 368.0 (M+H)+.
4.1.2.2. N-(4-Fluorobenzyl)-2-(2,4-dichlorophenoxy)propanamide (MBX 1642; 2)
General synthesis A was followed using 2-(2,4-dichlorophenoxy)propanoic acid (1.28 g, 5.5 mmol) and piperonylamine (0.81 mL, 6.5 mmol) to provide 1.84 g (92%) of product as a white solid: mp 120–121 °C; Rf 0.52 (1:1 hexane:EtOAc); 1H-NMR (CDCl3) δ 7.38 (d, 1H), 7.19 (dd, 1H), 6.91 (s, 1H), 6.85 (d, 1H), 6.76–6.68 (m, 3H), 5.95 (s, 2H), 4.73 (q, 1H), 4.40–4.37 (m, 2H), 1.64 (d, 3H); m/z expected 341.0 found 342.0 (M+H)+.
4.1.2.3. N-[3,4-(Methylenedioxy)benzyl]-2-(2,4-dichlorophenoxy)acetamide (6a)
General synthesis A was followed using 2-(2,4-dichlorophenoxy)acetic acid (5a; 1.0 g, 4.5 mmol) and piperonylamine (0.68 mL, 5.4 mmol) to provide 0.84 g (53%) of product as a white solid: mp 117–119 °C; Rf 0.58 (1:1 hexane:EtOAc); 1H-NMR (CDCl3) δ 7.39 (d, 1H), 7.21 (dd, 1H), 7.01 (s, br, 1H), 6.83 (d, 1H), 6.78–6.76 (m, 3H), 5.95 (s, 2H), 4.55 (s, 2H), 4.45 (d, 2H); m/z expected 353.0 found 354.0 (M+H)+.
4.1.2.5. 2-(2,4-Dichlorophenoxy)-2-methylpropanoic acid (5b)
General synthesis C was followed using ethyl 2-bromo-2-methylpropanoate (3.67 g, 18.8 mmol), with the exception that the product was extracted from the acidified aqueous suspension, dried over MgSO4, filtered, and evaporated, to provide 3.33 g (89%) of product as an clear oil that was used without further purification: 1H-NMR (CDCl3) δ 7.41 (d, 1H), 7.19–7.15 (m, 1H), 7.02 (d, 1H), 1.64 (s, 6H).
4.1.2.6 N-[3,4-(Methylenedioxy)benzyl]-2-(2,4-dichlorophenoxy)2-methylpropanamide (6b)
General synthesis B was followed using 2-(2,4-dichlorophenoxy)-2-methylpropanoic acid (5b; 136 mg, 0.55 mmol) and piperonylamine (85 µL, 0.66 mmol) to provide 0.84 g (53%) of product as an off-white powder: mp 92–94 °C; Rf 0.60 (1:1 hexane:EtOAc); 1H-NMR (CDCl3) δ 7.37 (d, 1H), 7.26 (s, 1H), 7.13 (d, 1H), 6.94 (d, 1H), 6.76 (m, 3H), 5.95 (s, 2H), 4.41 (s, 2H), 1.57 (s, 6H); m/z expected 381.1 found 382.0 (M+H)+.
4.1.2.7. 2-(2,4-Dichlorophenoxy)butanoic acid (5c)
General synthesis C was followed using ethyl 2-bromo-2-butanoate (3.65 g, 18.8 mmol) to provide 2.95 g (79%) of product as an off-white powder that was used without further purification: 1H-NMR (CDCl3) δ 7.39 (d, 1H), 7.15 (dd, 1H), 6.79 (d, 1H), 6.10 (br, 1H), 4.62 (t, 1H), 2.09 (quint, 2H), 1.69 (t, 3H).
4.1.2.8. N-[3,4-(Methylenedioxy)benzyl]-2-(2,4-dichlorophenoxy)butanamide (6c)
General synthesis B was followed using 2-(2,4-dichlorophenoxy)butanoic acid (5c; 107 mg, 0.43 mmol) and piperonylamine (65 µL, 0.52 mmol) to provide 55 mg (36%) of product as a light brown powder: mp 110–114 °C; Rf 0.62 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.37 (d, 1H), 7.17 (dd, 1H), 6.82 (d, 1H), 6.72 (d, 2H), 6.67–6.65 (m, 2H), 5.94 (s, 2H), 4.61 (t, 1H) 4.32 (d, 2H), 2.08–1.99 (m, 2H), 1.04 (t, 3H); m/z expected 381.1 found 382.1 (M+H)+.
4.1.2.9. 2-(2,4-Dichlorophenoxy)pentanoic acid (5d)
General synthesis C was followed using ethyl 2-bromo-2-pentanoate (3.93 g, 18.8 mmol) to provide 3.91 g (99%) of product as an off-white powder that was used without further purification: 1H-NMR (CDCl3) δ 7.87 (br, 1H), 7.39 (d, 1H), 7.15 (dd, 1H), 6.76 (d, 1H), 4.65 (q, 1H), 2.12–1.92 (m, 2H), 1.67–1.54 (m, 2H), 0.99 (t, 3H).
4.1.2.10. N-[3,4-(Methylenedioxy)benzyl]-2-(2,4-dichlorophenoxy)pentanamide (6d)
General synthesis B was followed using 2-(2,4-dichlorophenoxy)pentanoic acid (5d; 113 mg, 0.43 mmol) and piperonylamine (65 µL, 0.52 mmol) to provide 26 mg (15%) of product as a light brown powder: mp 91–94 °C; Rf 0.72 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.36 (d, 1H), 7.16 (dd, 1H), 6.83–6.71 (m, 3H), 6.65–6.63 (m, 2H), 5.94 (s, 2H), 4.63 (t, 1H) 4.35 (d, 2H), 2.10–1.93 (m, 2H), 1.59–1.48 (m, 2H), 0.95 (t, 3H); m/z expected 395.1 found 396.1 (M+H)+.
4.1.2.11. 2-(2,4-Dichlorophenoxy)3-methylbutanoic acid (5e)
General synthesis C was followed using ethyl 2-bromo-3-methylbutanoate (3.95 g, 18.8 mmol), with the exception that the product was extracted from the acidified aqueous suspension, dried over MgSO4, filtered, and evaporated, to provide 3.33 g (89%) of product as a slightly yellow oil that was used without further purification: 1H-NMR (CDCl3) δ 7.37 (d, 1H), 7.14 (dd, 1H), 6.74 (d, 1H), 4.44 (d, 1H), 2.42–2.36 (m, 1H), 1.16–1.12 (m, 6H).
4.1.2.12. N-[3,4-(Methylenedioxy)benzyl]-2-(2,4-dichlorophenoxy)3-methylbutanamide (6e)
General synthesis B was followed using 2-(2,4-dichlorophenoxy)3-methylbutanoic acid (5e; 113 mg, 0.43 mmol) and piperonylamine (65 µL, 0.52 mmol) to provide 75 mg (44%) of product as white powder: mp 73–75 °C; Rf 0.80 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.36 (d, 1H), 7.15 (dd, 1H), 6.80 (d, 1H), 6.71 (d, 1H), 6.64–6.62 (m, 3H), 5.94 (s, 2H), 4.43 (d, 1H), 4.36–4.33 (m, 2H), 2.38–2.32 (m, 1H), 1.10 (s, 3H), 1.07 (s, 3H); m/z expected 395.1 found 396.1 (M+H)+.
4.1.2.13. Ethyl (R)-2-(2,4-dichlorophenoxy)propanoate (7)
To a solution of ethyl (S)-lactate (2.39 g, 20.2 mmol), 2,4-dichlorophenol (3.00 g, 18.4 mmol), and triphenyl phosphine (7.23 g, 27.6 mmol) in anhydrous THF (50 mL) was added a solution of diisopropylazodicarboxylate (5.46 g, 27.6 mmol) in anhydrous THF (50 mL). The combined solution was stirred at room temperature for 16 h, then the solvent was removed under vacuum. The residual oil was subjected to flash chromatography on silica gel with 0–10% EtOAc/hexane. Product-containing fractions were pooled and evaporated to provide 4.26 g (88%) of product as a clear oil: Rf 0.66 (4:1 hexane:EtOAc); 1H-NMR (CDCl3) δ 7.31 (d, 1H), 7.06 (dd, 1H), 6.76 (d, 1H), 4.70 (q, 1H), 4.20–4.13 (m, 2H), 1.62 (d, 3H), 1.20 (t, 3H)
4.1.2.14. (R)-2-(2,4-Dichlorophenoxy)propanoic acid (8)
To a solution of ethyl (R)-2-(2,4-dichlorophenoxy)propanoate (7; 0.80 g, 3.0 mmol) in absolute EtOH (12 mL) was added a solution of potassium hydroxide (2.48 g, 44.2 mmol) in water (12 mL). The combined solution was stirred at room temperature for 4 h, then acidified (pH 3) with conc. aq. HCl. The resulting precipitated solid was filtered, rinsed with water (20 mL), and dried to provide 0.46 g (64%) of product as a white solid, that was used without further purification: 1H-NMR (CDCl3) δ 7.40 (d, 1H), 7.16 (dd, 1H), 6.82 (d, 1H), 4.77 (q, 1H), 1.72 (d, 3H).
4.1.2.15. (R)-N-[3,4-(Methylenedioxy)benzyl]-2-(2,4-dichlorophenoxy)propanamide ((R)-1)
General synthesis A was followed using (R)-2-(2,4-dichlorophenoxy)propanoic acid (8; 128 mg, 0.55 mmol) and piperonylamine (0.81 mL, 0.65 mmol) to provide 91 mg (45%) of product as a white solid: mp 136–138 °C; Rf 0.52 (1:1 hexane:EtOAc); 1H-NMR (CDCl3) δ 7.38 (d, 1H), 7.19 (dd, 1H), 6.91 (s, 1H), 6.85 (d, 1H), 6.76–6.68 (m, 3H), 5.95 (s, 2h), 4.73 (q, 1h), 4.40–4.37 ( m, 2H), 1.64 (d, 3H); m/z expected 367.0 found 368.0 (M+H)+.
4.1.2.16. Ethyl (S)-2-(2,4-dichlorophenoxy)propanoate
To a solution of ethyl (R)-lactate (2.39 g, 20.2 mmol), 2,4-dichlorophenol (3.00 g, 18.4 mmol), and triphenyl phosphine (7.23 g, 27.6 mmol) in anhydrous THF (50 mL) was added a solution of diisopropylazodicarboxylate (5.46 g, 27.6 mmol) in anhydrous THF (50 mL). The combined solution was stirred at room temperature for 16 h, then the solvent was removed under vacuum. The residual oil was subjected to flash chromatography on silica gel with 0–10% EtOAc/hexane. Product-containing fractions were pooled and evaporated to provide 4.04 g (84%) of product as a clear oil: Rf 0.66 (4:1 hexane:EtOAc); 1H-NMR (CDCl3) δ 7.31 (d, 1H), 7.06 (dd, 1H), 6.76 (d, 1H), 4.70 (q, 1H), 4.20–4.13 (m, 2H), 1.62 (d, 3H), 1.20 (t, 3H)
4.1.2.17. (S)-2-(2,4-Dichlorophenoxy)propanoic acid
To a solution of ethyl (S)-2-(2,4-dichlorophenoxy)propanoate (0.80 g, 3.0 mmol) in absolute EtOH (12 mL) was added a solution of potassium hydroxide (2.48 g, 44.2 mmol) in water (12 mL). The combined solution was stirred at room temperature for 4 h, then acidified (pH 3) with conc. aq. HCl. The resulting precipitated solid was filtered, rinsed with water (20 mL), and dried to provide 0.52 g (73%) of product as a white solid, that was used without further purification: 1H-NMR (CDCl3) δ 7.40 (d, 1H), 7.16 (dd, 1H), 6.82 (d, 1H), 4.77 (q, 1H), 1.72 (d, 3H).
4.1.2.18. (S)-N-[3,4-(Methylenedioxy)benzyl]-2-(2,4-dichlorophenoxy)propanamide ((S)-1)
General synthesis A was followed using (S)-2-(2,4-dichlorophenoxy)propanoic acid (128 mg, 0.55 mmol) and piperonylamine (0.81 mL, 0.65 mmol) to provide 98 mg (49%) of product as a white solid: mp 140–142 °C; Rf 0.52 (1:1 hexane:EtOAc); 1H-NMR (CDCl3) δ 7.38 (d, 1H), 7.19 (dd, 1H), 6.91 (s, 1H), 6.85 (d, 1H), 6.76–6.68 (m, 3H), 5.95 (s, 2h), 4.73 (q, 1h), 4.40–4.37 ( m, 2H), 1.64 (d, 3H); m/z expected 367.0 found 368.0 (M+H)+.
4.1.2.19. N-[3,4-(Methylenedioxy)benzyl]-2-(2-chlorophenoxy)propanamide (12a)
General synthesis E was followed using 2-(2-chlorophenoxy)propanoic acid (11a; 100 mg, 0.50 mmol) to provide 155 mg (81%) of product as an off-white powder: mp 84–86 °C; Rf 0.53 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.87 (dd, 1H), 7.26–7.19 (m, 1H), 7.05 (s, br, 1H), 6.99–6.91 (m, 2H), 6.75–6.68 (m, 3H), 5.94 (s, 2H), 4.77 (q, 1H), 4.39 (d, 2H), 1.65 (d, 3H); m/z expected 333.1 found 334.3 (M+H)+.
4.1.2.20. N-[3,4-(Methylenedioxy)benzyl]-2-(4-chlorophenoxy)propanamide (12b)
General synthesis E was followed using 2-(4-chlorophenoxy)propanoic acid (11b; 100 mg, 0.50 mmol) to provide 121 mg (73%) of product as a fluffy white solid: mp 101–104 °C; Rf 0.55 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.26–7.21 (m, 2H), 6.84–6.79 (m, 2H), 6.71 (d, 1H), 6.64–6.61 (m, 3H), 5.94 (s, 2H), 4.67 (q, 1H), 4.35 (d, 2H), 1.58 (d, 3H); m/z expected 333.1 found 334.3 (M+H)+.
4.1.2.21. 2-(2-Chloro-4-fluorophenoxy)propanoic acid (11c)
General synthesis D was followed using 2-chloro-4-fluorophenol (2.20 g, 15.0 mmol), to provide 2.82 g (86%) of product as an off-white powder that was used without further purification: Rf 0.28 (4:1 hexane:EtOAc w/2% AcOH); 1H-NMR (CDCl3) δ 8.95 (s, br, 1H), 7.14 (dd, 1H), 6.90 (d, 2H), 4.72 (q, 1H), 1.69 (d, 3H).
4.1.2.22. N-[3,4-(Methylenedioxy)benzyl]-2-(2-chloro-4-fluorophenoxy)propanamide (12c)
General synthesis E was followed using 2-(2-chloro-4-fluorophenoxy)propanoic acid (11c; 94 mg, 0.43 mmol) to provide 88 mg (63%) of product as a fluffy white solid: mp 95–97 °C; Rf 0.55 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.13 (dd, 1H), 6.97–6.85 (m, 3H), 6.76–6.68 (m, 3H), 5.94 (s, 2H), 4.68 (q, 1H), 4.45–4.32 (m, 2H), 1.62 (d, 3H); m/z expected 351.1 found 352.1 (M+H)+.
4.1.2.23. 2-(4-Chloro-2-fluorophenoxy)propanoic acid (11d)
General synthesis D was followed using 4-chloro-2-fluorophenol (1.36 g, 5.5 mmol), to provide 1.13 g (92%) of product as an off-white powder that was used without further purification: Rf 0.28 (4:1 hexane:EtOAc w/2% AcOH); 1H-NMR (CDCl3) δ 8.10 (s, 1H), 7.13 (dd, 1H), 7.03 (dq, 1H), 6.91 (t, 1H), 4.77 (q, 1H), 1.68 (d, 3H).
4.1.2.24. N-[3,4-(Methylenedioxy)benzyl]-2-(4-chloro-2-fluorophenoxy)propanamide (12d)
General synthesis E was followed using 2-(4-chloro-2-fluorophenoxy)propanoic acid (11d; 200 mg, 0.92 mmol) to provide 77 mg (24%) of product as an off-white powder: mp 98–101 °C; Rf 0.58 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.11 (dd, 1H), 7.04 (dd, 1H), 6.88 (t, 1H), 6.81 (s, br, 1H), 6.75–6.67 (m, 3H), 5.95 (s, 2H), 4.67 (q, 1H), 4.38 (d, 2H), 1.60 (d, 3H); m/z expected 351.1 found 352.1 (M+H)+.
4.1.2.25. 2-(2,4-Difluorophenoxy)propanoic acid (11e)
General synthesis D was followed using 2,4-difluorophenol (1.27 g, 5.5 mmol), to provide 1.03 g (92%) of product as an off-white crystalline solid that was used without further purification: Rf 0.26 (4:1 hexane:EtOAc w/2% AcOH); 1H-NMR (CDCl3) δ 10.66 (s, 1H), 7.02–6.94 (m, 1H), 6.91–6.83 (m, 1H), 6.81–6.75 (m, 1H), 7.73 (q, 1H), 1.67 (d, 3H).
4.1.2.26. N-[3,4-(Methylenedioxy)benzyl]-2-(2,4-difluorophenoxy)propanamide (12e)
General synthesis E was followed using 2-(2,4-difluorophenoxy)propanoic acid (11e; 200 mg, 0.99 mmol) to provide 200 mg (60%) of product as an off-white powder: mp 78–83 °C; Rf 0.53 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 6.97–6.69 (m, 7H), 5.95 (s, 2H), 4.63 (q, 1H), 4.45–4.33 (m, 2H), 1.59 (d, 3H); m/z expected 335.1 found 336.1 (M+H)+.
4.1.2.27. 2-(2-Chloro-4-methylphenoxy)propanoic acid (11f)
General synthesis D was followed using 2-chloro-4-methylphenol (1.45 g, 6.0 mmol), to provide 100 mg (8%) of product as an off-white crystalline solid that was used without further purification: 1H-NMR (CDCl3) δ 15.0–12.8 (s, br, 1H), 7.21 (d, 1H), 6.99 (dd, 1H), 6.83 (d, 1H), 4.75 (q, 1H), 2.28 (s, 3H), 1.69 (d, 3H).
4.1.2.28. N-[3,4-(Methylenedioxy)benzyl]-2-(2-chloro-4-methylphenoxy)propanamide (12f)
General synthesis E was followed using 2-(2-chloro-4-methylphenoxy)propanoic acid (11f; 95 mg, 0.44 mmol) to provide 112 mg (73%) of product as an off-white powder: mp 92–94 °C; Rf 0.53 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.18 (d, 1H), 7.05–6.98 (m, 2H), 6.82 (d, 1H), 6.75–6.68 (m, 3H), 5.94 (s, 2H), 4.71 (q, 1H), 4.38 (d, 2H), 2.28 (s, 3H), 1.62 (d, 3H); m/z expected 347.1 found 348.3 (M+H)+.
4.1.2.29. 2-(2-Chloro-4-cyanophenoxy)propanoic acid (11g)
General synthesis D was followed using 2-chloro-4-cyanophenol (2.30 g, 15.0 mmol), to provide 3.01 g (89%) of product as an off-white powder that was used without further purification: Rf 0.13 (4:1 hexane:EtOAc w/2% AcOH); 1H-NMR (CDCl3/CD3OD) δ 7.69 (d, 1H), 7.51 (dd, 1H), 6.90 (d, 2H), 4.91–4.79 (m, 2H), 1.73 (d, 3H).
4.1.2.30. N-[3,4-(Methylenedioxy)benzyl]-2-(2-chloro-4-cyanophenoxy)propanamide (12g)
General synthesis E was followed using 2-(2-chloro-4-cyanophenoxy)propanoic acid (11g; 97 mg, 0.43 mmol) to provide 97 mg (21%) of product as a white powder: mp 155–157 °C; Rf 0.32 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.68 (d, 1H), 7.54 (dd, 1H), 7.23–7.18 (m, 2H), 7.03–6.96 (m, 3H), 6.90 (s, br, 1H), 4.85 (q, 1H) 4.45 (d, 2H), 1.68 (d, 3H); m/z expected 358.1 found 359.1 (M+H)+.
4.1.2.31. 2-(2-Chloro-4-methoxyphenoxy)propanoic acid (11h)
General synthesis D was followed using 2-chloro-4-methoxyphenol (2.38 g, 15.0 mmol), to provide 3.18 g (92%) of product as a light pink powder that was used without further purification: Rf 0.25 (4:1 hexane:EtOAc w/2% AcOH); 1H-NMR (CDCl3) δ 9.21 (s, br, 1H), 6.95–6.87 (m, 2H), 6.73 (dd, 2H), 4.68 (q, 1H), 3.76 (s, 3H), 1.69 (d, 3H).
4.1.2.32. N-[3,4-(Methylenedioxy)benzyl]-2-(2-chloro-4-methoxyphenoxy)propanamide (12h)
General synthesis E was followed using 2-(2-chloro-4-methoxyphenoxy)propanoic acid (11h; 99 mg, 0.43 mmol) to provide 97 mg (67%) of product as a fluffy white solid: mp 89–92 °C; Rf 0.57 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.23–7.19 (m, 2H), 7.14 (s, br, 1H), 7.03–6.97 (m, 2H), 6.93 (d, 1H), 6.86 (d, 1H), 6.74 (dd, 1H), 4.65 (q, 1H), 4.52–4.39 (m, 2H), 3.76 (s, 3H), 1.60 (d, 3H); m/z expected 363.1 found 364.1 (M+H)+.
4.1.2.33. 2-(3,5-Dichloropyridin-2-yloxy)propanoic acid (15)
Ethyl (±)-lactate (19.8 g, 0.17 mol) was added to a suspension of sodium metal (3.91 g, 0.17 mol) in diglyme (20 mL). After the sodium metal was completely reacted, a solution of 2,3,5-trichloropyridine (19.9 g, 0.11 mol) in diglyme (100 mL) was added slowly to the above solution. The combined solution was then heated to 125 °C and allowed to stir at that temperature for 12 h. The mixture was then cooled to room temperature, and the remaining solids were filtered. The filtrate was evaporated under high vacuum (<5 mmHg) to provide a clear oil. The oil was dissolved in MeOH (200 mL), and a solution of potassium hydroxide (18.2 g, 0.45 mmol) in water was added. The solution was stirred at room temperature for 8 h, then excess MeOH was removed under vacuum. The remaining aqueous solution was acidified (pH 3) with aqueous HCl (1.0 M). The resulting solids were filtered, rinsed, and dried to provide 10.1 g (35%) of product as a white solid: mp 128–132 °C; Rf 0.35 (4:1 hexane:EtOAc w/2% AcOH); 1H-NMR (CDCl3) δ 8.18–8.14 (m, 2H), 5.23–5.15 (m, 1H), 1.55–1.52 (m, 3H).
4.1.2.34. N-[3,4-(Methylenedioxy)benzyl]-2-[(3,5-dichloropyridin-2-yl)oxy]propanamide (16)
To a solution of 2-(3,5-dichloropyridin-2-yloxy)propanoic acid (15; 102 mg, 0.43 mmol) in DMF (2 mL) were added 2-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate (HATU; 200 mg, 0.53 mmol), piperonylamine (85 µL, 0.66 mmol), and diisopropylethylamine (100 µL, 0.57 mmol). The resulting mixture was stirred at room temperature for 16 h, then poured into water (20 mL). The aqueous mixture was then stirred at room temperature until solids precipitated. The solids were filtered, rinsed with water, and dried to provide 122 mg (77%) of product as a white powder: mp 180–183 °C; Rf 0.65 (1:1 hexane:EtOAc); 1H-NMR (CDCl3) δ 8.01 (d, 1H), 7.67 (d, 1H), 6.75–6.67 (m, 4H), 5.94 (s, 2H), 5.50 (q, 1H), 4.46–4.32 (m, 2H), 1.65 (d, 3H); m/z expected 368.0 found 369.0 (M+H)+.
4.1.2.35. 2-[(2,4-Dichlorophenyl)thio]propanoic acid (19)
To a stirred solution of 2,4-dichlorothiophenol (17, 1.00 g, 5.6 mmol) in DMF (10 mL) were added K2CO3 (1.10 g, 8.0 mmol) and ethyl 2-bromopropanoate (1.0 mL, 7.7 mmol). The mixture was stirred at 50 °C for 16 h, then cooled to room temperature and filtered to remove the inorganic solids. The filtrate was evaporated under high vacuum (<1 mmHg) to provide a clear oil that was dissolved in absolute ethanol (25 mL). An aqueous solution of NaOH (2.0 M, 6.0 mL, 12 mmol) was added, and the combined solution heated to reflux for 4 h. The resulting mixture was cooled to room temperature and evaporated under reduced pressure to provide a residue that was dissolved in water (50 mL). The aqueous solution were acidified (pH 2) with aqueous HCl (2.0 M) to provide a precipitate that was filtered, rinsed with water (20 mL), and dried under vacuum to provide 1.28 g (91%) of the product as an off-white solid that was used without further purification: Rf 0.35 (4:1 hexane:EtOAc w/2% AcOH); 1H-NMR (CDCl3) δ 7.48–7.44 (m, 2H), 7.21 (dd, 1H), 3.86 (q, 1H), 1.52 (d, 3H).
4.1.2.36. N-[3,4-(Methylenedioxy)benzyl]-2-[(2,4-dichlorophenyl)thio]propanamide (20a)
To a solution of 2-[(2,4-dichlorophenyl)thio]propanoic acid (19, 108 mg, 0.43 mmol) in DMF (2 mL) were added 2-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate (HATU; 200 mg, 0.53 mmol), piperonylamine (86 mg, 0.57 mmol), and diisopropylethylamine (100 µL, 0.57 mmol). The resulting mixture was stirred at room temperature for 16 h, then poured into water (20 mL). The aqueous mixture was then stirred at room temperature until solids precipitated. The solids were filtered, rinsed with water, and dried to provide 106 mg (64%) of product as an off-white microcrystalline solid: mp 137–139 °C; Rf 0.67 (1:1 hexane:EtOAc); 1H-NMR (CDCl3) δ 7.36 (t, 1H), 7.15–7.12 (m, 2H), 6.77 (s, br, 1H), 6.68 (dd, 1H), 6.56–6.53 (m, 2H), 5.94 (s, 2H), 4.34 (dd, 1H), 4.20 (dd, 1H), 3.92 (q, 1H), 1.63–1.60 (m, 3H); m/z expected 383.0 found 384.0 (M+H)+.
4.1.2.37. N-(4-Fluorobenzyl)-2-[(2,4-dichlorophenyl)thio]propanamide (20b)
To a solution of 2-[(2,4-dichlorophenyl)thio]propanoic acid (19, 108 mg, 0.43 mmol) in DMF (2 mL) were added 2-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate (HATU; 200 mg, 0.53 mmol), 4-fluorobenzylamine (71 mg, 0.57 mmol), and diisopropylethylamine (100 µL, 0.57 mmol). The resulting mixture was stirred at room temperature for 16 h, then poured into water (20 mL). The aqueous mixture was then stirred at room temperature until solids precipitated. The solids were filtered, rinsed with water, and dried to provide 112 mg (73%) of product as fluffy white solid: mp 142–145 °C; Rf 0.66 (1:1 hexane:EtOAc); 1H-NMR (CDCl3) δ 7.36 (t, 1H), 7.19–7.12 (m, 2H), 7.06–7.01 (m, 2H), 6.97–6.91 (m, 2H), 6.82 (s, br, 1H), 4.39 (dd, 1H), 4.27 (dd, 1H), 3.93 (q, 1H), 1.62 (d, 3H); m/z expected 357.0 found 358.0 (M+H)+.
4.1.3.38. N-[3,4-(Methylenedioxy)benzyl]-2-[(2,4-dichlorophenyl)sulfonyl]propanamide (21a)
To a solution of N-[3,4-(methylenedioxy)benzyl]-2-[(2,4-dichlorophenyl)thio]propanamide (20a; 140 mg, 0.36 mmol) in CHCl3 (3 mL) was added m-chloroperoxybenzoic acid (204 mg, 1.18 mmol). The resulting solution was stirred at room temperature for 16 h, then diluted with additional CHCl3 (30 mL) and washed with saturated aqueous NaHCO3 (25 mL × 3) and brine (20 mL). The remaining organic solution was dried over MgSO4, filtered and evaporated to provide a residue that was recrystallized from hot CH2Cl2/hexane to provide 52 mg (34%) of product as a white powder: mp 135–137 °C; Rf 0.54 (1:1 hexanes:EtOAc); 1H-NMR (CDCl3) δ 7.85 (d, 1H), 7.57 (d, 1H), 7.35 (dd, 1H), 6.77–6.67 (m, 3H), 6.34 (s, br, 1H), 5.99–5.96 (m, 2H), 4.51–4.36 (m, 2H), 4.23 (dd, 1H), 1.59 (d, 3H); m/z expected 415.0 found 416.0 (M+H)+.
4.1.3.39. N-(4-Fluorobenzyl)-2-[(2,4-dichlorophenyl)sulfonyl]propanamide (21b)
To a solution of N-(4-fluorobenzyl)-2-[(2,4-dichlorophenyl)thio]propanamide (20b; 147 mg, 0.41 mmol) in CHCl3 (3 mL) was added m-chloroperoxybenzoic acid (230 mg, 1.33 mmol). The resulting solution was stirred at room temperature for 16 h, then diluted with additional CHCl3 (30 mL) and washed with saturated aqueous NaHCO3 (25 mL × 2) and brine (20 mL). The remaining organic solution was dried over MgSO4, filtered and evaporated to provide a residue that was recrystallized from hot CH2Cl2/hexane to provide 109 mg (68%) of product as a white powder: mp 125–127 °C; Rf 0.54 (1:1 hexanes:EtOAc); 1H-NMR (CDCl3) δ 7.85 (d, 1H), 7.57 (d, 1H), 7.34 (dd, 1H), 7.23–7.19 (m, 2H), 7.05–6.97 (m, 2H), 6.81 (s, br, 1H), 4.51–4.32 (m, 3H), 1.57 (d, 3H); m/z expected 389.0 found 390.1 (M+H)+.
4.1.3.40. N-[3,4-(Methylenedioxy)benzyl]-2-[(2,4-dichlorophenyl)amino]propanamide (23a)
To a solution of 2-[(2,4-dichlorophenyl)amino]propanoic acid (100 mg, 0.43 mmol) in DMF (2 mL) were added 2-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate (HATU; 200 mg, 0.53 mmol), piperonylamine (86 mg, 0.57 mmol), and diisopropylethylamine (100 µL, 0.57 mmol). The resulting mixture was stirred at room temperature for 16 h, then poured into water (20 mL). The aqueous mixture was then stirred at room temperature until solids precipitated. The solids were filtered, rinsed with water, and dried to provide 107 mg (68%) of product as a light brown solid: mp 134–136 °C; Rf 0.55 (1:1 hexane:EtOAc); 1H-NMR (CDCl3) δ 7.28 (d, 1H), 7.09 (dd, 1H), 6.82 (s, br, 1H), 6.72–6.61 (m, 3H), 6.45 (d, 1H), 5.93 (s, 2H), 4.45 (br s, 1H), 4.41–4.25 (m, 2H), 3.83–3.78 (m, 1H), 1.59 (d, 3H); m/z expected 366.1 found 367.1 (M+H)+.
4.1.3.41. N-(4-Fluorobenzyl)-2-[(2,4-dichlorophenyl)amino]propanamide (23b)
To a solution of 2-[(2,4-dichlorophenyl)amino]propanoic acid (100 mg, 0.43 mmol) in DMF (2 mL) were added 2-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate (HATU; 200 mg, 0.53 mmol), 4-fluorobenzylamine (71 mg, 0.57 mmol), and diisopropylethylamine (100 µL, 0.57 mmol). The resulting mixture was stirred at room temperature for 16 h, then poured into water (20 mL). The aqueous mixture was then stirred at room temperature until solids precipitated. The solids were filtered, rinsed with water, and dried to provide 75 mg (51%) of product as a light brown solid: mp 103–105 °C; Rf 0.54 (1:1 hexane:EtOAc); 1H-NMR (CDCl3) δ 7.29 (d, 1H), 7.16–7.07 (m, 3H), 7.01–6.93 (m, 2H), 6.88 (s, br, 1H), 6.45 (d, 1H), 4.47–4.32 (m, 3H), 3.86–3.78 (m, 1H), 1.62–1.58 (m, 3H); m/z expected 340.1 found 341.0 (M+H)−.
4.1.3.42. N-[3,4-(Methylenedioxy)benzyl]-2-(2,4-dichlorophenyl)-2-methylpropanamide (25a)
To a solution of 3-(2,4-dichlorophenyl)-2-methylpropanoic acid (100 mg, 0.43 mmol) in DMF (2 mL) were added 2-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate (HATU; 200 mg, 0.53 mmol), piperonylamine (86 mg, 0.57 mmol), and diisopropylethylamine (100 µL, 0.57 mmol). The resulting mixture was stirred at room temperature for 16 h, then poured into water (20 mL). The aqueous mixture was then stirred at room temperature until solids precipitated. The solids were filtered, rinsed with water, and dried to provide 113 mg (72%) of product as an off-white crystalline solid: mp 123–124 °C; Rf 0.69 (1:1 hexane:EtOAc); 1H-NMR (CDCl3) δ 7.33 (d, 1H), 7.16–7.09 (m, 2H), 6.70 (d, 1H), 6.56 (d, 1H), 6.50 (dd, 1H), 5.94 (s, 2H), 5.48 (s, br, 1H), 4.32 (dd, 1H), 4.13 (dd, 1H), 3.04–2.96 (m, 1H), 2.86–2.80 (m, 1H), 2.58–2.50 (m, 1H), 1.22 (d, 3H); m/z expected 365.1 found 366.1 (M+H)−.
4.1.3.43. N-(4-Fluorobenzyl)-2-(2,4-dichlorophenyl)-2-methylpropanamide (25b)
To a solution of 3-(2,4-dichlorophenyl)-2-methylpropanoic acid (100 mg, 0.43 mmol) in DMF (2 mL) were added 2-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate (HATU; 200 mg, 0.53 mmol), 4-fluorobenzylamine (71 mg, 0.57 mmol), and diisopropylethylamine (100 µL, 0.57 mmol). The resulting mixture was stirred at room temperature for 16 h, then poured into water (20 mL). The aqueous mixture was then stirred at room temperature until solids precipitated. The solids were filtered, rinsed with water, and dried to provide 75 mg (51%) of product as an off-white crystalline solid: mp 94–96 °C; Rf 0.68 (1:1 hexane:EtOAc); 1H-NMR (CDCl3) δ 7.34 (d, 1H), 7.15–7.08 (m, 2H), 7.00–6.92 (m, 4H), 5.50 (s, br, 1H), 4.40 (dd, 1H), 4.19 (dd, 1H), 3.03–2.96 (m, 1H), 2.87–2.81 (m, 1H), 2.61–2.53 (m, 1H), 1.24 (d, 3H); m/z expected 339.1 found 340.0 (M–H)−.
4.1.3.44. N-[1-(4-Fluorophenyl)ethyl]-2-(2,4-dichlorophenoxy)propanamide (27a)
General synthesis F was followed using α-methyl-4-fluorobenzylamine (70 mg, 0.50 mmol) to provide 18 mg (12%) of product as an off-white powder: mp 100–103 °C; Rf 0.58 (1:1 Hexane:EtOAc); 1H-NMR (DMSO-d6) δ 8.56, 8.49 (d, 1H), 7.59, 7.57 (d, 1H), 7.36–7.26 (m, 3H), 7.17–7.08 (m, 2H), 6.95, 6.92 (d, 1H), 4.98–4.85 (m, 1H), 4.82–4.75 (m, 1H), 1.47, 1.46 (d, 3H), 1.37, 1.34 (d, 3H) [∼35:65 mixture of diastereomers]; m/z expected 355.1 found 356.1 (M+H)+.
4.1.3.45. N-[(S)-1-(4-Fluorophenyl)ethyl]-(R)-2-(2,4-dichlorophenoxy)propanamide (27b)
General synthesis F was followed using (S)-α-methyl-4-fluorobenzylamine (70 mg, 0.50 mmol), with the exception that (R)-2-(2,4-dichlorophenoxy)propanoic acid was used, to provide 136 mg (89%) of product as a white powder: mp 128–130 °C; Rf 0.58 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.42 (d, 1H), 7.31–7.20 (m, 3H) 7.07–7.01 (m, 3H), 6.88 (d, 1H), 5.13–5.08 (m, 1H), 4.69 (q, 1H), 1.58 (d, 3H), 1.44 (d, 3H); m/z expected 355.1 found 356.1 (M+H)+.
4.1.3.46. N-[(R)-1-(4-Fluorophenyl)ethyl]-(R)-2-(2,4-dichlorophenoxy)propanamide (27c)
General synthesis F was followed using (R)-α-methyl-4-fluorobenzylamine (70 mg, 0.50 mmol), with the exception that (R)-2-(2,4-dichlorophenoxy)propanoic acid was used, to provide 132 mg (87%) of product as an off-white powder: mp 120–123 °C; Rf 0.58 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.39 (s, 1H), 7.17–7.14 (m, 3H), 6.98–6.92 (m, 3H), 6.78 (d, 1H), 5.11–5.06 (m, 1H), 4.71–4.64 (m, 1H), 1.64 (d, 3H), 1.51 (d, 3H); m/z expected 355.1 found 356.1 (M+H)+.
4.1.3.47. N-[2-(4-Fluorophenyl)propan-2-yl]-2-(2,4-dichlorophenoxy)propanamide (27d)
General synthesis F was followed using α-dimethyl-4-fluorobenzylamine (77 mg, 0.50 mmol) to provide 70 mg (44%) of product as a white powder: mp 105–106 °C; Rf 0.55 (2:1 Hexane:EtOAc); 1H-NMR (DMSO-d6) δ 8.26 (s, 1H), 7.57 (d, 1H), 7.40 (dt, 1H), 7.31 (dd, 2H), 7.06 (t, 2H), 6.98 (d, 1H), 4.81 (q, 1H), 1.55 (s, 3H), 1.53 (s, 3H), 1.46 (d, 3H); m/z expected 369.1 found 370.2 (M+H)+.
4.1.3.48. N-[1-(4-Fluorophenyl)cyclopropyl]-2-(2,4-dichlorophenoxy)propanamide (27e)
General synthesis F was followed using 1-(4-fluorophenyl)cyclopropylamine (76 mg, 0.50 mmol) to provide 131 mg (83%) of product as an off-white powder: mp 134–138 °C; Rf 0.66 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.25–7.15 (m, 4H), 6.98–6.92 (m, 2H), 6.81 (d, 1H), 4.63 (q, 1H), 1.59 (d, 3H), 1.27–1.18 (m, 4H); m/z expected 367.1 found 368.1 (M+H)+.
4.1.3.49. N-[1-(4-Fluorophenyl)propyl]-2-(2,4-dichlorophenoxy)propanamide (27f)
General synthesis F was followed using α-ethyl-4-fluorobenzylamine (77 mg, 0.50 mmol) to provide 56 mg (35%) of product as a white powder: mp 95–96 °C; Rf 0.41 (1:2 Hexane:EtOAc); 1H-NMR (DMSO-d6) δ 8.48 (dd, 1H), 7.57 (dd, 1H), 7.35–7.25 (m, 3H), 7.17–7.07 (m, 2H), 6.93 (dd, 1H), 4.84–4.77 (m, 1H), 4.73–4.61 (m, 1H), 1.73–1.65 (m, 2H), 1.47 (dd, 3H), 0.86–0.76 (m, 3H); m/z expected 369.1 found 370.2 (M+H)+.
4.1.3.50. N-[1-(4-Fluorophenyl)butyl]-2-(2,4-dichlorophenoxy)propanamide (27g)
General synthesis F was followed using α-propyl-4-fluorobenzylamine (84 mg, 0.50 mmol) to provide 142 mg (86%) of product as an off-white powder: mp 79–83 °C; Rf 0.68 (1:1 Hexane:EtOAc); 1H-NMR (DMSO-d6) δ 8.50, 8.46 (d, 1H), 7.59, 7.56 (d, 1H), 7.35–7.24 (m, 3H), 7.17–7.10 (m, 2H), 6.94, 6.88 (d, 1H), 4.83–4.73 (m, 2H), 1.73–1.58 (m, 2H), 1.48, 1.44 (d, 3H), 1.33–1.14 (m, 2H), 0.86, 0.83 (t, 3H) [∼60:40 mixture of diastereomers]; m/z expected 383.1 found 384.2 (M+H)+.
4.1.3.51. N-[1-(4-Fluorophenyl)-2-methylpropyl]-2-(2,4-dichlorophenoxy)propanamide (27h)
General synthesis F was followed using α-isopropyl-4-fluorobenzylamine (84 mg, 0.50 mmol) to provide 37 mg (22%) of product as a white powder: mp 107–108 °C; Rf 0.43 (2:1 Hexane:EtOAc); 1H-NMR (DMSO-d6) δ 8.43 (dd, 1H), 7.57 (dd, 1H), 7.35–7.08 (m, 5H), 6.90 (dd, 1H), 4.85–4.80 (m, 1H), 4.55–4.45 (m, 1H), 1.98 (quint, 1H), 1.44 (dd, 3H), 0.86 (dd, 3H), 0.68 (dd, 3H); m/z expected 383.1 found 384.2 (M+H)+.
4.1.3.52. N-[Cyclopropyl(4-fluorophenyl)methyl]-2-(2,4-dichlorophenoxy)propanamide (27i)
General synthesis F was followed using α-cyclopropyl-4-fluorobenzylamine (83 mg, 0.50 mmol) to provide 86 mg (52%) of product as a white powder: mp 109–110 °C; Rf 0.44 (2:1 Hexane:EtOAc); 1H-NMR (DMSO-d6) δ 8.68 (d, 1H), 7.58 (dd, 1H), 7.41–7.29 (m, 3H), 7.13 (q, 2H), 6.95 (dd, 1H), 4.81 (q, 1H), 4.18 (q, 1H), 1.48 (t, 3H), 1.24–1.11 (m, 1H), 0.53–0.41 (m, 2H), 0.36–0.33 (m, 1H), 0.28–0.26 (m, 1H); m/z expected 381.1 found 382.1 (M+H)+.
4.1.3.53. N-[1-(4-Fluorophenyl)pentyl]-2-(2,4-dichlorophenoxy)propanamide (27j)
General synthesis F was followed using α-butyl-4-fluorobenzylamine (91 mg, 0.50 mmol) to provide 112 mg (65%) of product as a white powder: mp 100–106 °C; Rf 0.70 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.43–7.38 (m, 1H), 7.28–7.19 (m, 2H + CHCl3), 7.14–7.09 (m, 2H), 7.03 (t, 1H), 6.97–6.87 (m, 2.4H), 6.76 (d, 0.6H), 4.90 (q, 1H), 4.75–4.60 (m, 1H), 1.84–1.44 (m, 5H), 1.36–1.07 (m, 4H), 0.91–0.78 (m, 3H); m/z expected 397.1 found 398.2 (M+H)+.
4.1.3.54. N-[Cyclobutyl(4-fluorophenyl)methyl]-2-(2,4-dichlorophenoxy)propanamide (27k)
General synthesis F was followed using α-cyclobutyl-4-fluorobenzylamine (90 mg, 0.50 mmol) to provide 51 mg (32%) of product as an off-white powder: mp 103–104 °C; Rf 0.47 (1:2 Hexane:EtOAc); 1H-NMR (DMSO-d6) δ 8.42 (t, 1H), 7.59–7.56 (m, 1H), 7.34–7.24 (m, 3H), 7.11 (q, 2H), 6.90 (dd, 1H), 4.82–4.67 (m, 2H), 2.67–2.50 (m, 1H), 2.05–1.95 (m, 1H), 1.95–1.65 (m, 5H), 1.45 (dd, 3H); m/z expected 395.1 found 396.2 (M+H)+.
4.1.3.55. N-[1-(4-Fluorophenyl)-2-hydroxyethyl]-2-(2,4-dichlorophenoxy)propanamide (27l)
General synthesis F was followed using α-hydroxymethyl-4-fluorobenzylamine (78 mg, 0.50 mmol) to provide 49 mg (31%) of product as a white powder: mp 98–102 °C; Rf 0.29 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.46–7.40 (m, 2H), 7.32–7.15 (m, 1H), 7.23–7.13 (m, 2H), 7.10–6.96 (m, 2H), 6.91–6.80 (m, 1H), 5.10–5.04 (m, 1H), 4.76–4.69 (m, 1H), 3.92–3.82 (m, 2H), 2.32–2.26 (m, 1H), 1.68–1.59 (m, 3H); m/z expected 371.1 found 372.2 (M+H)+.
4.1.3.56. N-[Cyano(4-fluorophenyl)methyl]-2-(2,4-dichlorophenoxy)propanamide (27m)
General synthesis F was followed using α-cyano-4-fluorobenzylamine (75 mg, 0.50 mmol) to provide 120 mg (76%) of product as a light tan powder: mp 112–116 °C; Rf 0.70 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.51–7.47 (m, 1H), 7.42–7.35 (m, 3H), 7.26–7.16 (m, 2H), 7.12–7.06 (m, 1H), 6.90–6.82 (m, 1H), 6.09 (d, 1H), 4.79–4.71 (m, 1H), 1.68–1.61 (m, 3H); m/z expected 366.0 found 367.2 (M+H)+.
4.1.3.57. N-[Methoxycarbonyl(4-fluorophenyl)methyl]-2-(2,4-dichlorophenoxy)propanamide (27n)
General synthesis F was followed using methyl 2-amino-2-(4-fluorophenyl)acetate (92 mg, 0.50 mmol) to provide 170 mg (99%) of product as an off-white powder: mp 84–97 °C; Rf 0.68 (1:1 Hexane:EtOAc); 1H-NMR (DMSO-d6) δ 9.00, 8.98 (d, 1H), 7.60, 7.57 (d, 1H), 7.4–.41 (m, 2H), 7.34, 7.31 (dd, 1H), 7.2–.19 (m, 2H), 7.03, 6.94 (d, 1H), 5.47, 5.45 (d, 1H), 4.9–.89 (m, 1H), 3.63 (s, 3H), 1.50, 1.47 (d, 3H) [∼50:50 mixture of diastereomers]; m/z expected 399.0 found 400.1 (M+H)+.
4.1.3.58. N-[Carboxy(4-fluorophenyl)methyl]-2-(2,4-dichlorophenoxy)propanamide (27o)
To a solution of N-[methoxycarbonyl(4-fluorophenyl)methyl]-2-(2,4-dichlorophenoxy)propanamide (27o; 150 mg 0.38 mmol) in dioxane (3 mL) was added a solution of lithium hydroxide in water (1.0 M, 3.0 mL, 3.0 mmol). The combined solution was stirred at room temperature for 1 h. The solvent was then removed under vacuum, and the residue dissolved in water (20 mL). The aqueous solution was acidified with 2.0 M aqueous HCl solution (pH 1). The resulting mixture was extracted with CH2Cl2 (25 mL × 3), and the combined extracts were dried over MgSO4, filtered, and evaporated to provide 110 mg (76%) of product as a white powder: mp 160–175 °C; Rf 0.08 (1:1 Hexane:EtOAc); 1H-NMR (DMSO-d6) δ 8.84 (dd, 1H), 7.57 (dd, 1H), 7.4–.39 (m, 2H), 7.3–.17 (m, 3H), 7.00 (dd, 1H) 5.34 (dd, 1H), 4.96 (hex, 1H), 1.47 (t, 3H); m/z expected 385.0 found 386.1 (M+H)+.
4.1.3.59. N-(4-Fluorophenyl)-2-(2,4-dichlorophenoxy)propanamide (28a)
General synthesis G was followed using 4-fluoroaniline (56 mg, 0.50 mmol) to provide 93 mg (66%) of product as a fluffy white solid: mp 102–104 °C; Rf 0.78 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 8.55 (s, br, 1H), 7.5–.53 (m, 2H), 7.45 (d, 1H), 7.23 (dd, 1H), 7.0–.01 (m, 2H), 6.93 (d, 1H), 4.80 (q, 1H), 1.71 (d, 3H); m/z expected 327.0 found 328.0 (M+H)+.
4.1.3.60. N-(4-Fluorophenethyl)-2-(2,4-dichlorophenoxy)propanamide (28b)
General synthesis G was followed using 4-fluorophenethylamine (70 mg, 0.50 mmol) to provide 93 mg (82%) of product as an off-white powder: mp 129–131 °C; Rf 0.64 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.38 (d, 1H), 7.16 (dd, 1H), 7.1–.05 (m, 2H), 6.9–.91 (m, 2H), 6.77 (d, 1H), 6.63 (s, br, 1H), 4.64 (q, 1H), 3.6–.48 (m, 2H), 2.79 (td, 2H), 1.56 (d, 3H); m/z expected 355.1 found 356.1 (M+H)+.
4.1.3.61. N-[3-(4-Fluorophenyl)propyl]-2-(2,4-dichlorophenoxy)propanamide (28c)
General synthesis G was followed using 3-(4-fluorophenyl)propylamine (77 mg, 0.50 mmol) to provide 65 mg (41%) of product as a white crystalline solid: mp 112–116 °C; Rf 0.49 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.41 (d, 1H), 7.19 (dd, 1H), 7.1–.06 (m, 2H), 6.9–.92 (m, 2H), 6.48 (d, 1H), 6.71 (s, br, 1H), 4.68 (q, 1H), 3.3–.27 (m, 2H), 2.58 (t, 2H), 1.8–.77 (m, 2H), 1.60 (d, 3H); m/z expected 369.1 found 370.2 (M+H)+.
4.1.3.62. N-(4-Fluorobenzyl)-N-methyl-2-(2,4-dichlorophenoxy)propanamide (28d)
To a solution of 2-(2,4-dichlorophenoxy)propionic acid (235 mg, 1.0 mmol) in DMF were added 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI; 250 mg, 1.3 mmol), hydroxybenzotriazole (HOBt; 176 mg, 1.3 mmol), N-(4-fluorobenzyl)methylamine (168 mg, 1.2 mmol), and triethylamine (303 mg, 3.0 mmol). The solution was stirred at room temperature for 16 h, then poured into mixture of ice cold water (5 mL) and saturated aqueous NaHCO3 (5 mL). The organic suspension was extracted with EtOAc (20 mL), and the organic extract was washed with water (20 mL), brine (20 mL) and dried over Na2SO4. The organic extract was evaporated to provide a crude oil which was purified by flash chromatography on silica gel with 0–25% EtOAc/hexane. Product-containing fractions were pooled and evaporated to provide 262 mg (73%) of product as a clear, colorless oil: Rf 0.40 (20% EtOAc/hexane); 1HNMR (CDCl3): δ 7.38 (d, 1H), 7.2–.32 (m, 1H), 7.06–7.18 (m, 2H), 6.8–.98 (m, 3H), 4.9–.08 (m, 1H), 4.5–.78 (m, 2H), 2.8–.02 (m, 3H), 1.6–.72 (m, 3H); m/z expected 355.1 found 356.1 (M+H)+.
4.1.3.63. 4-Fluorobenzyl 2-(2,4-dichlorophenoxy)propanoate (29a)
To a solution of 2-(2,4-dichlorophenoxy)propionic acid (235 mg, 1.0 mmol) in DMF were added 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI; 250 mg, 1.3 mmol), hydroxybenzotriazole (HOBt; 176 mg, 1.3 mmol), 4-fluorobenzyl alcohol (152 mg, 1.2 mmol), and triethylamine (303 mg, 3.0 mmol). The solution was stirred at room temperature for 16 h, then poured into mixture of ice cold water (5 mL) and saturated aqueous NaHCO3 (5 mL). The organic suspension was extracted with EtOAc (20 mL), and the organic extract was washed with water (20 mL), brine (20 mL) and dried over Na2SO4. The organic extract was evaporated to provide a crude oil which was purified by flash chromatography on silica gel with 0–25% EtOAc/hexane. Product-containing fractions were pooled and evaporated to provide 260 mg (73%) of product as a clear, colorless oil: Rf 0.42 (20% EtOAc/hexane); 1HNMR (CDCl3): δ 7.36 (d, 1H), 7.2–.28 (m, 2H), 6.9–.08 (m, 3H), 6.68 (d, 1H), 5.14 (q, 2H), 4.76 (q, 1H), 1.66 (d, 3H); m/z expected 342.0 found 342.1 (GC-MS).
4.1.3.64. 4-Fluorophenethyl 2-(2,4-dichlorophenoxy)propanoate (29b)
To a solution of 2-(2,4-dichlorophenoxy)propanoic acid (100 mg, 0.43 mmol) in DMF (2 mL) were added 2-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate (HATU; 200 mg, 0.53 mmol), 2-(4-fluorophenyl)ethanol (64 µL, 0.50 mmol), and diisopropylethylamine (100 µL, 0.57 mmol). The resulting mixture was stirred at room temperature for 2 days, then poured into water (20 mL). The aqueous mixture was then extracted with EtOAc (20 mL × 3), and the combined organic extracts were dried over MgSO4, filtered, and evaporated to provide a yellow oil. The oil was subjected to flash chromatography on silica gel with 0–30% EtOAc/hexane. Product-containing fractions were pooled and evaporated to provide 120 mg (75%) of product as a clear, colorless oil: Rf 0.79 (1:1 EtOAc/hexane); 1H-NMR (CDCl3) δ 7.37 (d, 1H), 7.1–.05 (m, 3H), 6.95 (t, 2H), 6.64 (d, 1H), 4.67 (q, 1H), 4.3–.31 (m, 2H), 2.89 (t, 2H), 1.61 (d, 3H); m/z expected 356.0 found 356.1 (GC-MS).
4.1.3.65. N'-(4-Fluorophenyl)-2-(2,4-dichlorophenoxy)-propanehydrazide (30a)
To a solution of 2-(2,4-dichlorophenoxy)propanoic acid (100 mg, 0.43 mmol) in DMF (2 mL) were added 2-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate (HATU; 200 mg, 0.53 mmol), 4-fluorophenylhydrazine (63 mg, 0.50 mmol), and diisopropylethylamine (100 µL, 0.57 mmol). The resulting mixture was stirred at room temperature for 16 h, then poured into water (20 mL). The aqueous mixture was then stirred at room temperature until solids precipitated. The solids were filtered, rinsed with water, and dried to provide 124 mg (84%) of product as an off-white crystalline solid: mp 112–117 °C; Rf 0.54 (1:1 hexane:EtOAc); 1H-NMR (CDCl3) δ 8.39 (d, br, 1H), 7.45 (d, 1H), 7.23 (dd, 1H), 6.9–.89 (m, 3H), 6.7–.72 (m, 2H), 6.04 (d, 1H), 4.85 (q, 1H), 1.67 (d, 3H); m/z expected 342.0 found 343.1 (M+H)+.
4.1.3.66. N'-(4-Fluorobenzyl)-2-(2,4-dichlorophenoxy)-propanehydrazide (30b)
To a solution of 2-(2,4-dichlorophenoxy)propanoic acid (500 mg, 2.1 mmol) in DMF (10 mL) were added 2-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate (HATU; 1.0 g, 2.6 mmol), 4-fluorobenzylhydrazine (0.70 g, 5.0 mmol), and diisopropylethylamine (0.50 mL, 5.7 mmol). The resulting mixture was stirred at room temperature for 16 h, then poured into water (200 mL). The aqueous mixture was then stirred at room temperature until solids precipitated. The solids were filtered, rinsed with water, and dried to provide 725 mg (96%) of product as a white crystalline solid: mp 123–126 °C; Rf 0.44 (1:1 hexane:EtOAc); 1H-NMR (CDCl3) δ 7.93 (s, br, 1H), 7.36 (d, 1H), 7.2–.23 (m, 2H), 7.17 (dd, 1H), 7.03–6.95 (m, 2H), 6.79 (d, 1H), 4.72 (q, 1H), 3.9–.89 (m, 2H), 1.59 (d, 3H); m/z expected 356.0 found 357.1 (M+H)+.
4.1.3.67. N-[(4-Fluorobenzyl)oxy]-2-(2,4-dichlorophenoxy)propanamide (31)
To a solution of 2-(2,4-dichlorophenoxy)propanoic acid (100 mg, 0.43 mmol) in DMF (2 mL) were added 2-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate (HATU; 200 mg, 0.53 mmol), O-(4-fluorobenzyl)hydroxylamine (71 mg, 0.50 mmol), and diisopropylethylamine (100 µL, 0.57 mmol). The resulting mixture was stirred at room temperature for 16 h, then poured into water (20 mL). The aqueous mixture was then stirred at room temperature until solids precipitated. The solids were filtered, rinsed with water, and dried to provide 130 mg (84%) of product as an off-white crystalline solid: mp 148–153 °C; Rf 0.54 (1:1 hexane:EtOAc); 1H-NMR (CDCl3) δ 8.99 (s, br, 1H), 7.3–.32 (m, 3H), 7.18 (dd, 1H), 7.0–.01 (m, 2H), 6.82 (d, 1H), 4.9–.85 (m, 2H), 4.73 (q, 1H), 1.61 (d, 3H); m/z expected 357.0 found 358.1 (M+H)+.
4.1.3.68. N-Methoxy-N-methyl-2-(2,4-dichlorophenoxy)propanamide (32)
To a solution of 2-(2,4-dichlorophenoxy)propanoic acid (300 mg, 1.28 mmol) in DMF (5 mL) were added 2-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate (HATU; 582 mg, 1.53 mmol), N,O-dimethylhydroxylamine hydrochloride (149 mg, 1.53 mmol), and diisopropylethylamine (0.56 mL, 1.66 mmol). The resulting mixture was stirred at room temperature for 16 h, then poured into water (30 mL). The aqueous mixture was then extracted with CH2Cl2 (20 mL × 3), and the combined organic extracts were dried over MgSO4, filtered, and evaporated to provide a yellow oil. The oil was subjected to flash chromatography on silica gel with 0–30% EtOAc/hexane. Product-containing fractions were pooled and evaporated to provide 295 mg (83%) of product as a light yellow oil that was used without further purification: 1H-NMR (CDCl3) δ 7.37 (d, 1H), 7.13 (dd, 1H), 6.83 (d, 2H), 5.08 (q, 1H), 3.71 (s, 3H), 3.22 (s, 3H), 1.63 (d, 3H); m/z expected 277.0 found 278.0 (M+H)+.
4.1.3.69. 1-(4-Fluorophenyl)-4-(2,4-dichlorophenoxy)-pentan-3-one (33)
To a solution of N-Methoxy-N-methyl-2-(2,4-dichlorophenoxy)propanamide (32; 246 mg, 0.88 mmol) in dry THF (2 mL) maintained at 0 °C was added a solution of 4-fluorophenethylmagnesium bromide in THF (0.5 M, 5.3 mL, 2.7 mmol), dropwise over 5 min. The resulting solution was stirred at 0 °C for 90 min, then warmed to room temperature over 1 h. The reaction was poured into saturated aq. NH4Cl, (20 mL) and this mixture was extracted with CH2Cl2 (20 mL × 3). The combined organic extracts were dried over MgSO4, filtered, and evaporated to provide a yellow residue. The residue was subjected to flash chromatography on silica gel with 0–25% EtOAc/hexane. Product-containing fractions were pooled and evaporated to provide 123 mg (41%) of product as a clear, colorless oil: Rf 0.61 (1:1 EtOAc/hexane); 1H-NMR (CDCl3) δ 7.39 (d, 1H), 7.1–.07 (m, 3H), 6.9–.90 (m, 2H), 6.60 (d, 1H), 4.59 (q, 1H), 3.0–.94 (m, 1H), 2.8–.77 (m, 3H), 1.45 (d, 3H); m/z expected 340.0 found 340.1 (GC-MS).
4.1.3.70. N-(Benzyl)-2-(2,4-dichlorophenoxy)propanamide (34a)
General synthesis H was followed using benzylamine (54 mg, 0.50 mmol) to provide 91 mg (68%) of product as a white powder: mp 121–123 °C; Rf 0.77 (EtOAc); 1H-NMR (DMSO-d6) δ 8.60 (t, 1H), 7.59 (d, 1H), 7.3–.18 (m, 6H), 7.00 (d, 1H), 4.83 (q, 1H), 4.30 (d, 2H), 1.50 (d, 3H); m/z expected 323.0 found 323.9 (M+H)+.
4.1.3.71. N-(3-Fluorobenzyl)-2-(2,4-dichlorophenoxy)propanamide (34b)
General synthesis H was followed using 3-fluorobenzylamine (63 mg, 0.50 mmol) to provide 118 mg (80%) of product as a fluffy white solid: mp 106–107 °C; Rf 0.67 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.39 (d, 1H), 7.3–.24 (m, 1H), 7.20 (dd, 1H), 7.01–6.90 (m, 4H), 6.86 (d, 1H), 4.75 (q, 1H), 4.49 (q, 2H), 1.65 (d, 3H); m/z expected 341.0 found 342.0 (M+H)+.
4.1.3.72. N-(2-Fluorobenzyl)-2-(2,4-dichlorophenoxy)propanamide (34c)
General synthesis H was followed using 2-fluorobenzylamine (63 mg, 0.50 mmol) to provide 89 mg (61%) of product as an off-white powder: mp 121–122 °C; Rf 0.69 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.37 (d, 1H), 7.2–.23 (m, 2H), 7.15 (dd, 1H), 7.1–.99 (m, 3H), 6.81 (d, 1H), 4.71 (q, 1H), 4.54 (q, 2H), 1.62 (d, 3H); m/z expected 341.0 found 342.0 (M+H)+.
4.1.3.73. N-(4-Chlorobenzyl)-2-(2,4-dichlorophenoxy)propanamide (34d)
General synthesis H was followed using 4-chlorobenzylamine (71 mg, 0.50 mmol) to provide 110 mg (71%) of product as a white powder: mp 115–116 °C; Rf 0.63 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.38 (d, 1H), 7.29 (dd, 2H), 7.2–.14 (m, 3H), 7.00 (s, br, 1H), 6.85 (d, 1H), 4.73 (q, 1H), 4.45 (q, 2H), 1.64 (d, 3H); m/z expected 357.0 found 358.0 (M+H)+.
4.1.3.74. N-(3-Chlorobenzyl)-2-(2,4-dichlorophenoxy)propanamide (34e)
General synthesis H was followed using 3-chlorobenzylamine (71 mg, 0.50 mmol) to provide 128 mg (83%) of product as a fluffy white solid: mp 140–142 °C; Rf 0.64 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.39 (d, 1H), 7.25 (d, 2H), 7.2–.18 (m, 2H), 7.1–.09 (m, 1H), 7.01 (s, br, 1H), 6.86 (d, 1H), 4.75 (q, 1H), 4.52 (dd, 1H), 4.41 (dd, 1H), 1.65 (d, 3H); m/z expected 357.0 found 358.0 (M+H)+.
4.1.3.75. N-(2-Chlorobenzyl)-2-(2,4-dichlorophenoxy)propanamide (34f)
General synthesis H was followed using 2-chlorobenzylamine (71 mg, 0.50 mmol) to provide 37 mg (24%) of product as a white powder: mp 113–115 °C; Rf 0.65 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.37 (d, 1H), 7.3–.30 (m, 2H), 7.2–.21 (m, 2H), 7.1–.11 (m, 2H), 6.80 (d, 1H), 4.71 (q, 1H), 4.57 (t, 2H), 1.63 (d, 3H); m/z expected 357.0 found 357.9 (M+H)+.
4.1.3.76. N-(4-Methylbenzyl)-2-(2,4-dichlorophenoxy)propanamide (34g)
General synthesis H was followed using 4-methylbenzylamine (61 mg, 0.50 mmol) to provide 68 mg (47%) of product as an off-white powder: mp 90–93 °C; Rf 0.69 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.37 (d, 1H), 7.17 (dd, 1H), 7.12 (s, 4H), 6.92 (s, br, 1H), 6.84 (d, 1H), 4.72 (q, 1H), 4.44 (d, 2H), 2.33 (s, 3H), 1.63 (d, 3H); m/z expected 337.1 found 338.0 (M+H)+.
N4.1.3.77. -(3-Methylbenzyl)-2-(2,4-dichlorophenoxy)propanamide (34h)
General synthesis H was followed using 3-methylbenzylamine (61 mg, 0.50 mmol) to provide 37 mg (26%) of product as a fluffy white solid: mp 113–114 °C; Rf 0.64 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.37 (d, 1H), 7.2–.17 (m, 2H), 7.08 (br d, 1H), 7.01 (d, 2H), 6.94 (br s, 1H), 6.85 (d, 1H), 4.74 (q, 1H), 4.5–.38 (m, 2H), 2.32 (s, 3H), 1.65 (d, 3H); m/z expected 337.1 found 338.0 (M+H)+.
4.1.3.78. N-(2-Methylbenzyl)-2-(2,4-dichlorophenoxy)propanamide (34i)
General synthesis H was followed using 2-methylbenzylamine (61 mg, 0.50 mmol) to provide 65 mg (45%) of product as a white powder: mp 128–130 °C; Rf 0.64 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.37 (d, 1H), 7.2–.15 (m, 5H), 6.85 (d, 2H), 4.73 (q, 1H), 4.48 (d, 2H), 2.29 (s, 3H), 1.64 (d, 3H); m/z expected 337.1 found 338.0 (M+H)+.
4.1.3.79. N-(4-Methoxybenzyl)-2-(2,4-dichlorophenoxy)propanamide (34j)
General synthesis H was followed using 4-methoxybenzylamine (69 mg, 0.50 mmol) to provide 62 mg (41%) of product as a white powder: mp 76–78 °C; Rf 0.78 (EtOAc); 1H-NMR (CDCl3) δ 7.36 (d, 1H), 7.2–.14 (m, 3H), 6.9–83 (m, 4H), 4.72 (q, 1H) 4.42 (d, 2H), 3.80 (s, 3H), 1.63 (d, 3H); m/z expected 353.1 found 354.0 (M+H)+.
4.1.3.80. N-(3-Methoxybenzyl)-2-(2,4-dichlorophenoxy)propanamide (34k)
General synthesis H was followed using 3-methoxybenzylamine (69 mg, 0.50 mmol) to provide 109 mg (72%) of product as an off-white fluffy solid: mp 119–120 °C; Rf 0.79 (EtOAc); 1H-NMR (CDCl3) δ 7.38 (d, 1H), 7.2–.17 (m, 2H), 6.97 (s, br, 1H), 6.8–.76 (m, 4H), 4.72 (q, 1H) 4.47 (dd, 2H), 3.78 (s, 3H), 1.65 (d, 3H); m/z expected 353.1 found 354.0 (M+H)+.
4.1.3.81. N-(2-Methoxybenzyl)-2-(2,4-dichlorophenoxy)propanamide (34l)
General synthesis H was followed using 2-methoxybenzylamine (70 mg, 0.50 mmol) to provide 116 mg (77%) of product as a white solid: mp 100–103 °C; Rf 0.36 (20% EtOAc/hexane); 1H-NMR (DMSO-d6) δ 8.33 (t, 1H), 7.60 (d, 1H), 7.34 (dd, 1H), 7.23 (t, 1H), 6.9–.08 (m, 3H), 6.86 (t, 1H), 4.86 (q, 1H), 4.26 (d, 2H), 3.78 (s, 3H), 1.50 (d, 3H); m/z expected 353.1 found 354.1 (M+H)+.
4.1.3.82. N-(3,4-Dimethoxybenzyl)-2-(2,4-dichlorophenoxy)propanamide (34m)
General synthesis H was followed using 3,4-dimethoxybenzylamine (84 mg, 0.50 mmol) to provide 135 mg (82%) of product as a white solid: mp 127–128 °C; Rf 0.71 (EtOAc); 1H-NMR (CDCl3) δ 7.37 (d, 1H), 7.18 (dd, 1H), 6.94 (br s, 1H), 6.85 (d, 1H), 6.8–.75 (m, 3H), 4.73 (q, 1H) 4.42 (t, 2H), 3.87 (s, 3H), 3.83 (s, 3H), 1.64 (d, 3H); m/z expected 383.1 found 384.1 (M+H)+.
4.1.3.83. N-(2,4-Dimethoxybenzyl)-2-(2,4-dichlorophenoxy)propanamide (34n)
General synthesis H was followed using 2,4-dimethoxybenzylamine (84 mg, 0.50 mmol) to provide 120 mg (73%) of product as a white powder: mp 117–119 °C; Rf 0.50 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.36 (d, 1H), 7.1–.07 (m, 3H), 6.74 (d, 1H), 6.4–.39 (m, 2H), 4.65 (q, 1H), 4.38 (q, 2H), 3.79 (s, 3H), 3.73 (s, 3H), 1.60 (d, 3H); m/z expected 383.1 found 384.0 (M+H)+.
4.1.3.84. N-(Benzothiophene-5-yl)-2-(2,4-dichlorophenoxy)propanamide (34o)
General synthesis H was followed using 5-aminomethylbenzothiophene (82 mg, 0.50 mmol) to provide 97 mg (59%) of product as a beige powder: mp 156–159 °C; Rf 0.67 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.81 (d, 1H), 7.65 (s, 1H), 7.46 (d, 1H), 7.36 (d, 1H), 7.24 (dd, 1H + CHCl3), 7.2–.15 (m, 2H), 7.02 (br s, 1H), 6.86 (d, 1H), 4.75 (q, 1H), 4.6–.53 (m, 2H), 1.65 (d, 3H); m/z expected 379.0 found 380.1 (M+H)+.
4.1.3.85. N-[(Benzofuran-5-yl)methyl]-2-(2,4-dichlorophenoxy)propanamide (34p)
General synthesis HI was followed using 5-aminomethylbenzofuran (74 mg, 0.50 mmol) to provide 86 mg (55%) of product as a beige powder: mp 125–132 °C; Rf 0.69 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.62 (s, 1H), 7.4–.36 (m, 3H), 7.1–.14 (m, 2H), 6.99 (s, br, 1H), 6.85 (d, 1H), 6.72 (s, 1H), 4.74 (q, 1H), 4.6–.50 (m, 2H), 1.65 (d, 3H); m/z expected 363.0 found 364.1 (M+H)+.
4.1.3.86. N-[(Benzimidazol-5-ylmethyl])-2-(2,4-dichlorophenoxy)propanamide (34q)
General synthesis HI was followed using 5-aminomethylbenzimidazole (74 mg, 0.50 mmol) to provide 43 mg (27%) of product as a colorless glass: mp 71–77 °C; Rf 0.17 (1 EtOAc); 1H-NMR (CDCl3) δ 8.07 (s, 1H), 7.59 (d, 1H), 7.51 (s, 1H), 7.33 (d, 1H), 7.1–.14 (m, 3H), 6.84 (d, 1H), 5.08 (s, br, 1H), 4.73 (q, 1H), 4.6–.55 (m, 2H), 1.63 (d, 3H); m/z expected 363.1 found 363.9 (M+H)+.
4.1.3.87. N-[(Indol-5-yl)methyl]-2-(2,4-dichlorophenoxy)propanamide (34r)
General synthesis H was followed using 5-aminomethylindole (73 mg, 0.50 mmol) to provide 75 mg (48%) of product as a light brown crystalline solid: mp 91–95 °C; Rf 0.56 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.21 (s, br, 1H), 7.48 (d, 1H), 7.3–.32 (m, 2H), 7.24 (dd, 1H), 7.16 (dd, 1H), 7.06 (dd, 1H), 6.93 (s, br, 1H), 6.84 (d, 1H), 6.5–.50 (m, 1H), 4.73 (q, 1H), 4.6–.50 (m, 2H), 1.65 (d, 3H); m/z expected 362.1 found 363.1 (M+H)+.
4.1.3.88. N-[(Indol-6-yl)methyl]-2-(2,4-dichlorophenoxy)propanamide (34s)
General synthesis H was followed using 6-aminomethylindole (73 mg, 0.50 mmol) to provide 71 mg (46%) of product as a beige powder: mp 120–123 °C; Rf 0.53 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 8.21 (s, br, 1H), 7.58 (d, 1H), 7.35 (d, 1H), 7.2–.14 (m, 3H), 6.98 (dd, 1H), 6.84 (dd, 1H), 6.5–.52 (m, 1H), 4.73 (q, 1H), 4.6–.09 (m, 2H), 1.64 (d, 3H); m/z expected 362.1 found 363.1 (M+H)+.
4.1.3.89. N-[(Indol-4-yl)methyl]-2-(2,4-dichlorophenoxy)propanamide (34t)
General synthesis H was followed using 4-aminomethylindole (73 mg, 0.50 mmol) to provide 81 mg (52%) of product as a pale pink powder: mp 107–111 °C; Rf 0.55 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 8.28 (s, br, 1H), 7.38 (d, 1H), 7.35 (d, 1H), 7.21 (t, 1H), 7.2–.09 (m, 2H), 6.9–.95 (m, 2H), 6.80 (d, 1H), 6.5–.52 (m, 1H), 4.7–.70 (m, 3H), 1.64 (d, 3H); m/z expected 362.1 found 363.1 (M+H)+.
4.1.3.90. N-[(Indol-2-yl)methyl]-2-(2,4-dichlorophenoxy)propanamide (34u)
General synthesis IH was followed using 2-aminomethylindole (73 mg, 0.50 mmol) to provide 93 mg (60%) of product as a pink glass: mp 95–102 °C; Rf 0.54 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 8.88 (s, br, 1H), 7.57 (d, 1H), 7.3–.33 (m, 2H), 7.1–.05 (m, 3H), 6.81 (d, 1H), 6.36 (s, 1H), 4.73 (q, 1H), 4.56 (d, 2H), 1.61 (d, 3H); m/z expected 362.1 found 363.2 (M+H)+.
4.1.3.91. N-[(1-Methylindol-5-yl)methyl]-2-(2,4-dichlorophenoxy)propanamide (34v)
General synthesis H was followed using 5-aminomethyl-1-methylindole (80 mg, 0.50 mmol) to provide 144 mg (89%) of product as a white powder: mp 146–152 °C; Rf 0.67 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 7.46 (d, 1H), 7.34 (d, 1H), 7.2–.25 (m, 1H), 7.15 (dd, 1H), 7.1–.05 (m, 2H), 6.91 (s, br, 1H), 6.84 (d, 1H), 6.43 (q, 1H), 4.72 (q, 1H), 4.5–.55 (m, 2H), 3.78 (s, 3H), 1.64 (d, 3H); m/z expected 376.1 found 377.1 (M+H)+.
4.1.3.92. N-[(Pyrrolo[2,3-b]pyridin-5-yl)methyl]-2-(2,4-dichlorophenoxy)propanamide (34w)
General synthesis H was followed using 5-aminomethylpyrrolo[2,3-b]pyridine (74 mg, 0.50 mmol) to provide 61 mg (39%) of product as a white powder: mp 198–201 °C; Rf 0.19 (1:1 Hexane:EtOAc); 1H-NMR (DMSO-d6) δ 11.58 (s, br, 1H), 8.63 (t, 1H), 8.09 (d, 1H), 7.71 (s, 1H), 7.59 (d, 1H), 7.45 (t, 1H), 7.27 (dd, 1H), 6.96 (d, 1H), 6.38 (dd, 1H), 4.80 (q, 1H), 4.37 (d, 2H), 1.48 (d, 3H); m/z expected 363.1 found 364.4 (M+H)+.
4.1.3.93. N-[(6-Fluoropyridin-3-yl)methyl]-2-(2,4-dichlorophenoxy)propanamide (34x)
General synthesis H was followed using 3-aminomethyl-6-fluoropyridine (63 mg, 0.50 mmol) to provide 52 mg (35%) of product as an off-white solid: mp 55–59 °C; Rf 0.36 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 8.03 (d, 1H), 7.7–.72 (m, 2H), 7.45 (dd, 1H), 6.94 (dd, 1H), 6.86 (d, 1H), 4.74 (q, 1H), 4.47 (d, 2H), 1.62 (d, 3H); m/z expected 342.0 found 343.1 (M+H)+.
4.1.3.94. N-[(5-Fluoropyridin-2-yl)methyl]-2-(2,4-dichlorophenoxy)propanamide (34y)
General synthesis H was followed using 2-aminomethyl-5-fluoropyridine (63 mg, 0.50 mmol) to provide 101 mg (68%) of product as a white powder: mp 78–82 °C; Rf 0.30 (1:1 Hexane:EtOAc); 1H-NMR (CDCl3) δ 8.38 (s, 1H), 7.76 (s, br, 1H), 7.4–.34 (m, 2H), 7.2–.15 (m, 2H), 6.86 (d, 1H), 4.75 (q, 1H), 4.58 (d, 2H), 1.64 (d, 3H); m/z expected 342.0 found 343.1 (M+H)+.
4.1.3.95. (R)-2-[(3,5-dichloropyridin-2-yl)oxy]butanoic acid (36)
To a solution of 3,5-dichloropyridin-2-one (18.3 g , 111 mmol) and triphenyl phosphine (38.0 g, 145 mmol in dry DMF (120 mL) was added (S)-methyl 2-hydroxybutanoate (17.1 g, 145 mmol). The solution was cooled to −20 °C (ice/salt bath) and diisopropyl azodicarboxylate (DIAD; 29.3 g, 145 mmol) was added dropwise over 45 min at −20 °C. After addition, the resulting solution was warmed to room temperature and was stirred at that temperature for 16 h. The solvent was then removed under vacuum, and the residue was subjected to flash chromatography on silica gel with 0–15% EtOAc/hexane. Product-containing fractions were pooled and evaporated to provide a pale yellow oil that was dissolved in MeOH (100 mL). A solution of potassium hydroxide (8.40 g, 150 mmol) in water (20 mL) was added to the methanolic solution at −20 °C over 10 min. The solution was warmed to room temperature over 2 h, then the solvent was removed under reduced pressure, and the residue diluted with water (150 mL). The aqueous solution was washed with Et2O (150 mL × 2), then the aqueous solution was acidified (pH 4) with aqueous HCl (3.0 M). The resulting suspension was extracted with EtOAc (150 mL × 6), and the combined organic extracts were dried over MgSO4, filtered, and evaporated to provide 21.0 g (80%) of product as a white solid: mp 87–90 °C; Rf 0.38 (4:1 hexane:EtOAc w/2% AcOH); 1H-NMR (CDCl3) δ 11.0–10.0 (s, br, 1H), 7.94 (dd, 1H), 7.67 (dd, 1H), 5.15 (t, 1H), 2.08 (quint, 2H), 1.13 (t, 3H).
4.1.3.96. (R)-N-[(Benzothiophene-5-yl)methyl]-2-[(3,5-dichloropyridin-2-yl)oxy]butanamide (37a)
General synthesis I was followed using 5-aminomethylbenzothiophene (98 mg, 0.60 mmol) to provide 89 mg (45%) of product as a white powder: mp 110–119 °C; Rf 0.73 (1:1 hexane:EtOAc); 1H-NMR (CDCl3) δ 7.99 (d, 1H), 7.80 (d, 1H), 7.65 (d, 2H), 7.45 (d, 1H), 7.26 (d, 1H), 7.20 (dd, 1H), 6.69 (s, br, 1H), 5.47 (t, 1H), 4.60 (dd, 2H), 2.1–.06 (m, 2H), 1.03 (t, 3H); m/z expected 394.0 found 395.1 (M+H)+.
4.1.3.97. (R)-N-[(Indol-5-yl)methyl]-2-[(3,5-dichloropyridin-2-yl)oxy]butanamide (37b)
General synthesis I was followed using 5-aminomethylindole (86 mg, 0.60 mmol) to provide 52 mg (28%) of product as an off-white powder: mp 86–88 °C; Rf 0.34 (3% MeOH/CH2Cl2); 1H-NMR (CDCl3) δ 8.23 (s, br, 1H), 7.98 (d, 1H), 7.64 (d, 1H), 7.47 (s, 1H), 7.32 (d, 1H), 7.22 (t, 1H), 7.05 (dd, 1H), 6.61 (s, br, 1H), 6.50 (t, 1H), 5.46 (t, 1H), 4.56 (d, 2H), 2.0–.12 (m, 2H), 1.02 (t, 3H); m/z expected 377.1 found 378.3 (M+H)+.
4.1.3.98. (R)-N-[1-(4-Fluorophenyl)-2-hydroxyethyl]-2-[(3,5-dichloropyridin-2-yl)oxy]butanamide (37c)
General synthesis I was followed using α-hydroxymethyl-4-fluorobenzylamine (93 mg, 0.60 mmol) to provide 143 mg (74%) of product as a white powder (as an inseparable mixture of diastereomers): mp 192–194 °C; Rf 0.32 (3% MeOH/CH2Cl2); 1H-NMR (DMSO-d6) δ 8.46, 8.34 (d, 1H), 8.18, 8.14 (d, 1H), 8.10, 8.06 (d, 1H), 7.06–7.40 (m, 4H), 5.0–.16 (m, 1H), 4.92 (s, 1H), 4.7–.83 (m, 1H), 3.5–.58 (m, 2H), 1.8–.92 (m, 2H), 0.9–.03 (m, 3H); m/z expected 386.1 found 387.3 (M+H)+.
4.2. Biology
4.2.1. T3SS-mediated secretion assay
Inhibition of T3SS-mediated secretion of an ExoS-βLA fusion protein into the culture medium by P. aerguinosa strain MDM973 was detected by measuring the hydrolysis of the chromogenic β-lactamase substrate nitrocefin in clear 96-well microplates as described previously.29 Absorbance at 490 nm was measured and percent inhibition was calculated relative to drug-free (0%) and uninduced (i.e. no ExoS-βLA is secreted; 100%) controls.
4.2.2. T3SS-mediated translocation assay and cytotoxicity
CHO cells were incubated with P. aeruginosa and compound as described previously,29 except that P. aeruginosa strain PA99U33 was used. Cell death was measured by LDH release to detect T3SS effector ExoU-mediated lysis of CHO cells. Percent cell death (expressed as percent LDH release) was calculated relative to that of the uninfected control (0%), and that of cells infected with P. aeruginosa unprotected by test compound (100%). The same assay was run in parallel with inhibitor and CHO cells, but without P. aeruginosa cells, to evaluate the CHO cell cytotoxicity (CC50) of each compound.
Supplementary Material
Scheme 7.
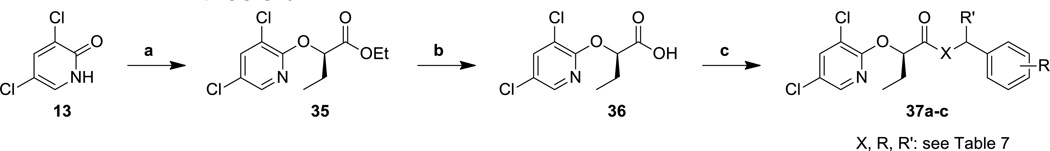
Synthesis of phenoxyacetamide T3SS inhibitors with multiple optimizations.
Reagents and conditions: (a) ethyl (S)-hydroxybutanoate, DIAD, PPh3, THF, 20 °C; (b) KOH, MeOH/H2O, 20 °C; (c) amine, HATU, DIPEA, DMF, 20 °C.
Acknowledgements
We would like to thank Tommy Tashjian and Deb Mills for assistance in performing biological assays. The research reported here was supported in part by the National Institute of Allergy and Infectious Disease of the National Institutes of Health under award AI068185. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1.Alanis AJ. Arch Med Res. 2005;36:697. doi: 10.1016/j.arcmed.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 2.Christina Tobin CTK. Nat Rev Microbiol. 2013;11:146. [Google Scholar]
- 3.Falagas ME, Bliziotis IA. Int J Antimicrob Agents. 2007;29:630. doi: 10.1016/j.ijantimicag.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 4.Wenzel RP. N Engl J Med. 2004;351:523. doi: 10.1056/NEJMp048093. [DOI] [PubMed] [Google Scholar]
- 5.Charles PG, Grayson ML. Med J Aust. 2004;181:549. doi: 10.5694/j.1326-5377.2004.tb06444.x. [DOI] [PubMed] [Google Scholar]
- 6.Norrby SR, Nord CE, Finch R. Lancet Infect Dis. 2005;5:115. doi: 10.1016/S1473-3099(05)01283-1. [DOI] [PubMed] [Google Scholar]
- 7.Cooper MA, Shlaes D. Nature. 2011;472:32. doi: 10.1038/472032a. [DOI] [PubMed] [Google Scholar]
- 8.Shlaes DM. Curr Opin Pharmacol. 2003;3:470. doi: 10.1016/j.coph.2003.04.003. [DOI] [PubMed] [Google Scholar]
- 9.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Nat Rev Drug Discov. 2007;6:29. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 10.Clatworthy AE, Pierson E, Hung DT. Nat Chem Biol. 2007;3:541. doi: 10.1038/nchembio.2007.24. [DOI] [PubMed] [Google Scholar]
- 11.Alksne LE, Projan SJ. Curr Opin Biotechnol. 2000;11:625. doi: 10.1016/s0958-1669(00)00155-5. [DOI] [PubMed] [Google Scholar]
- 12.Baron C, Coombes B. Infect Disord Drug Targets. 2007;7:19. doi: 10.2174/187152607780090685. [DOI] [PubMed] [Google Scholar]
- 13.Baron C. Curr Opin Microbiol. 2010;13:100. doi: 10.1016/j.mib.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 14.Durand E, Verger D, Rego AT, Chandran V, Meng G, Fronzes R, Waksman G. Infect Disord Drug Targets. 2009;9:518. doi: 10.2174/187152609789105722. [DOI] [PubMed] [Google Scholar]
- 15.Coburn B, Sekirov I, Finlay BB. Clin Microbiol Rev. 2007;20:535. doi: 10.1128/CMR.00013-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Engel J, Balachandran P. Curr Opin Microbiol. 2009;12:61. doi: 10.1016/j.mib.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 17.Hauser AR. Nat Rev Microbiol. 2009;7:654. doi: 10.1038/nrmicro2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roy-Burman A, Savel RH, Racine S, Swanson BL, Revadigar NS, Fujimoto J, Sawa T, Frank DW, Wiener-Kronish JP. J Infect Dis. 2001;183:1767. doi: 10.1086/320737. [DOI] [PubMed] [Google Scholar]
- 19.El Solh AA, Akinnusi ME, Wiener-Kronish JP, Lynch SV, Pineda LA, Szarpa K. Am J Respir Crit Care Med. 2008;178:513. doi: 10.1164/rccm.200802-239OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rello J, Mariscal D, March F, Jubert P, Sanchez F, Valles J, Coll P. Am J Respir Crit Care Med. 1998;157:912. doi: 10.1164/ajrccm.157.3.9703014. [DOI] [PubMed] [Google Scholar]
- 21.Keyser P, Elofsson M, Rosell S, Wolf-Watz H. J Intern Med. 2008;264:17. doi: 10.1111/j.1365-2796.2008.01941.x. [DOI] [PubMed] [Google Scholar]
- 22.Francois B, Luyt CE, Dugard A, Wolff M, Diehl JL, Jaber S, Forel JM, Garot D, Kipnis E, Mebazaa A, Misset B, Andremont A, Ploy MC, Jacobs A, Yarranton G, Pearce T, Fagon JY, Chastre J. Crit Care Med. 2012;40:2320. doi: 10.1097/CCM.0b013e31825334f6. [DOI] [PubMed] [Google Scholar]
- 23.Milla CE, Chmiel JF, Accurso FJ, VanDevanter DR, Konstan MW, Yarranton G, Geller DE. Pediatr Pulmonol. 2013;49:650. doi: 10.1002/ppul.22890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Warrener P, Varkey R, Bonnell JC, DiGiandomenico A, Camara M, Cook K, Peng L, Zha J, Chowdury P, Sellman B, Stover CK. Antimicrob Agents Chemother. 2014;58:4384. doi: 10.1128/AAC.02643-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duncan MC, Linington RG, Auerbuch V. Antimicrob Agents Chemother. 2012;56:5433. doi: 10.1128/AAC.00975-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kauppi AM, Nordfelth R, Hagglund U, Wolf-Watz H, Elofsson M. Adv Exp Med Biol. 2003;529:97. doi: 10.1007/0-306-48416-1_17. [DOI] [PubMed] [Google Scholar]
- 27.Kauppi AM, Nordfelth R, Uvell H, Wolf-Watz H, Elofsson M. Chem Biol. 2003;10:241. doi: 10.1016/s1074-5521(03)00046-2. [DOI] [PubMed] [Google Scholar]
- 28.Wang D, Zetterstrom CE, Gabrielsen M, Beckham KS, Tree JJ, Macdonald SE, Byron O, Mitchell TJ, Gally DL, Herzyk P, Mahajan A, Uvell H, Burchmore R, Smith BO, Elofsson M, Roe AJ. J Biol Chem. 2011;286:29922. doi: 10.1074/jbc.M111.233858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aiello D, Williams JD, Majgier-Baranowska H, Patel I, Peet NP, Huang J, Lory S, Bowlin TL, Moir DT. Antimicrob Agents Chemother. 2010;54:1988. doi: 10.1128/AAC.01598-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bowlin NO, Williams JD, Knoten CA, Torhan MC, Tashjian TF, Li B, Aiello D, Mecsas J, Hauser AR, Peet NP, Bowlin TL, Moir DT. Antimicrob Agents Chemother. 2014;58:2211. doi: 10.1128/AAC.02795-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leung CS, Leung SS, Tirado-Rives J, Jorgensen WL. J Med Chem. 2012;55:4489. doi: 10.1021/jm3003697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Torhan MC, Peet NP, Williams JD. Tetrahedron Lett. 2013;54:3926. [Google Scholar]
- 33.Shaver CM, Hauser AR. Infect Immun. 2004;72:6969. doi: 10.1128/IAI.72.12.6969-6977.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.