

Using integrated PET-MRI, Loggia et al. reveal elevated levels of the translocator protein (TSPO) in patients with chronic pain. As TSPO is a glial marker, these results provide the first demonstration of pain-related glial activation in humans. They also lend support to the pain-protective role of TSPO predicted by animal studies.
Keywords: chronic pain, 11C-PBR28, translocator protein (18kDa), TSPO, neuroinflammation, glia
Abstract
Although substantial evidence has established that microglia and astrocytes play a key role in the establishment and maintenance of persistent pain in animal models, the role of glial cells in human pain disorders remains unknown. Here, using the novel technology of integrated positron emission tomography-magnetic resonance imaging and the recently developed radioligand 11C-PBR28, we show increased brain levels of the translocator protein (TSPO), a marker of glial activation, in patients with chronic low back pain. As the Ala147Thr polymorphism in the TSPO gene affects binding affinity for 11C-PBR28, nine patient–control pairs were identified from a larger sample of subjects screened and genotyped, and compared in a matched-pairs design, in which each patient was matched to a TSPO polymorphism-, age- and sex-matched control subject (seven Ala/Ala and two Ala/Thr, five males and four females in each group; median age difference: 1 year; age range: 29–63 for patients and 28–65 for controls). Standardized uptake values normalized to whole brain were significantly higher in patients than controls in multiple brain regions, including thalamus and the putative somatosensory representations of the lumbar spine and leg. The thalamic levels of TSPO were negatively correlated with clinical pain and circulating levels of the proinflammatory citokine interleukin-6, suggesting that TSPO expression exerts pain-protective/anti-inflammatory effects in humans, as predicted by animal studies. Given the putative role of activated glia in the establishment and or maintenance of persistent pain, the present findings offer clinical implications that may serve to guide future studies of the pathophysiology and management of a variety of persistent pain conditions.
Using integrated PET-MRI, Loggia et al. reveal elevated levels of the translocator protein (TSPO) in patients with chronic pain. As TSPO is a glial marker, these results provide the first demonstration of pain-related glial activation in humans. They also lend support to the pain-protective role of TSPO predicted by animal studies.
Introduction
Until recently, chronic pain has been thought to arise primarily from neuronal dysfunction within nociceptive pathways of the nervous system (Hains and Waxman, 2006). In the last decade, however, a paradigm shift has occurred in the field of pain neurobiology. Animal studies have clearly demonstrated that microglia and astrocytes in the CNS, as well as neuro-glial interactions, play a key role in the establishment and maintenance of persistent pain (Tsuda et al., 2003; Watkins et al., 2007; Calvo et al., 2012; Ji et al., 2013). Both microglia and astrocytes respond to pathological events in CNS, such as strokes, trauma or neurodegenerative diseases, by undergoing a series of cellular responses collectively known as ‘glial activation’ (Gehrmann et al., 1995; Pekny et al., 2014). This response includes proliferation, morphological changes, increased or de novo expression of cell surface markers or receptors, and the production of cytokines and other inflammatory mediators. Normally, glial activation is an adaptive defensive mechanism that can contribute to handling acute stress, limiting tissue damage, and restoring homeostasis. However, when malfunctioning (and, in particular when it does not get resolved during the post-acute or early chronic stage after an injury event) (Rolls et al., 2009) glial activation can have deleterious effects, and turn into the primary pathogenic element (Pekny and Pekna, 2014). Several animal studies have now established that glial activation is a key contributing factor in persistent pain. It is well demonstrated that activated microglia and astrocytes can produce cytokines such as tumour necrosis factor alpha (TNFA) and interleukin 1 beta (IL1B), and these cytokines are thought to play an essential role in the pathogenesis of chronic pain (Watkins et al., 2007; Uceyler and Sommer, 2012). It has also been shown that TNFA and IL1B can directly modulate spinal cord synaptic transmission to induce central sensitization and enhance pain states (Kawasaki et al., 2008). Moreover, the intraspinal injection of activated glia produces tactile allodynia, a hallmark of neuropathic pain, in naive rats (Tsuda et al., 2003). Conversely, the injection of drugs inhibiting glial activation can inhibit, delay or reverse pain (Meller et al., 1994; Watkins et al., 1997; Guo et al., 2007; Okada-Ogawa et al., 2009). These observations, alongside other mounting evidence from laboratory models, suggest that chronic pain may result from gliopathy (Ji et al., 2013).
In humans, some observations suggest that activated glia may contribute to the pathophysiology of chronic pain. For instance, post-mortem immunohistochemical studies in the spinal cord of patients with complex regional pain syndrome (Del Valle et al., 2009) and HIV-related neuropathic pain (Shi et al., 2012), as well as CSF sampling in patients with fibromyalgia and chronic low back pain (LBP) (Brisby et al., 1999; Kadetoff et al., 2012), support a role for glia in chronic pain. Despite these observations, however, no study has yet demonstrated in vivo glial activation in humans suffering from chronic pain. Clinically, acknowledging the role of glial activation in pain disorders holds significant promise for the improved diagnostic accuracy of pain disorders. It may also provide a biological basis for the assessment of existing treatments, and the development of novel ones.
In this study we tested the hypothesis that patients with chronic pain demonstrate in vivo activation of brain glia. To assess this hypothesis, we imaged the brain of individuals diagnosed with chronic LBP as well as pain-free healthy volunteers using a Siemens 3 T integrated positron emission tomography/magnetic resonance imaging (PET/MRI) scanner and the recently developed PET radioligand, 11C-PBR28 (Brown et al., 2007; Briard et al., 2008). 11C-PBR28 binds to the translocator protein (18kDa) (TSPO), a protein upregulated in activated microglia and reactive astrocytes in animal models of pain (Hernstadt et al., 2009; Wei et al., 2013), and a putative imaging biomarker of inflammation (Cagnin et al., 2007). In addition, as evidence suggests that proinflammatory cytokines are secreted by glial cells in various animal models of pain (Ji et al., 2013), we assessed blood levels of interleukin 6 (IL6), IL1B and TNFA, and evaluated their association with our imaging findings.
Materials and methods
Study design
The study was conducted at the Athinoula A. Martinos Center for Biomedical Imaging at Massachusetts General Hospital. The protocol was approved by the Institutional Review Board and the Radioactive Drug Research Committee.
Subjects
Nineteen patients diagnosed with chronic LBP for at least 2 years (either with or without radicular pain complaints) and 25 healthy controls with no history of chronic pain were initially screened to participate in the study. Individuals were excluded if they had any PET/MRI contraindications (including pregnancy, metallic implants, claustrophobia), had a history of major medical disorders, or were on benzodiazepines or blood thinners (see Supplementary Table 1 for pain history and demographic information).
The Ala147Thr polymorphism in the TSPO gene predicts binding affinity for 11C-PBR28, with the Ala/Ala, Ala/Thr and Thr/Thr genotypes being associated with high, mixed and low affinity binding, respectively (Owen et al., 2012; Kreisl et al., 2013; Yoder et al., 2013). Thus, all participants were tested for this polymorphism and individuals with predicted low-binding affinity (Thr/Thr) were excluded. Given the effect of the TSPO polymorphism on binding affinity for 11C-PBR28, and the fact that the effects of sex and age on ligand binding are unknown, a matched-pairs design was adopted, in which each patient was matched to a TSPO polymorphism-, age- and sex-matched control. Of the 44 subjects initially screened, nine chronic LBP/control matching pairs (with two patients matching the same control) were identified among the initial pool, consisting of seven Ala/Ala and two Ala/Thr pairs, and five male and four female pairs, with a median age difference of 1 year (Supplementary Table 1).
Screening visit
All participants considered potentially eligible after an initial phone screening were recruited to participate in a 2-h characterization and training visit. In this visit, venous blood was drawn in order to have all participants genotyped for the Ala147Thr TSPO polymorphism, and a urine test was performed to ensure that none of the subjects were on benzodiazepines, or taking illegal drugs. At the end of the visit, all participants completed the Beck Depression Inventory (Turner and Romano, 1984; Geisser et al., 1997) and the Hospital Anxiety and Depression Scale (Zigmond and Snaith, 1983). In addition, the patients completed the McGill Pain Questionnaire (MPQ), short form (Melzack, 1987).
Imaging visit
On a separate date, eligible participants were invited to participate in an imaging visit. At the beginning of the visit, venous blood was drawn to assess levels of circulating proinflammatory cytokines: IL6, IL1B and TNFA (Supplementary Table 2). These specific proinflammatory cytokines were assessed because they are secreted by glial cells in various animal models of pain (Ji et al., 2013).
Brain imaging was performed with a Siemens PET/MRI scanner (Catana et al., 2008) consisting of a dedicated brain avalanche photodiode-based PET scanner operating in the bore of a 3 T whole-body magnetic resonance scanner equipped with an 8-channel head coil. The use of integrated PET/MRI allowed us to collect structural MRI simultaneously with the PET data. Patients were scanned with 11C-PBR28, a recently developed TSPO radioligand that displays an 80-fold higher in vivo specific binding than the earlier generation TSPO radioligand 11C-(R)-PK11195 (Kreisl et al., 2010). 11C-PBR28 was produced in-house using a procedure modified from the literature (Imaizumi et al., 2007).
At the beginning of the imaging visit, we performed a series of magnetic resonance scans, including a multi-echo MPRAGE volume (repetition time/echo time 1/ echo time 2/ echo time 3/ echo time 4 = 2530/1.64/3.5/5.36/7.22 ms, flip angle = 7°, voxel size = 1 mm isotropic) for the purpose of anatomical localization, spatial normalization of the imaging data, as well as generation of attenuation correction maps (Izquierdo-Garcia et al., 2014). The radioligand was then injected as an intravenous bolus, with a median administered dose (interquartile range) of 11.2 (0.3) mCi for patients with chronic LBP and 11.2 (0.6) mCi for controls, and a median specific activity at time of injection of 2.7 (0.8) mCi/nmol for chronic LBP and 2.0 (1.2) mCi/nmol for controls (both values not significantly different across groups). PET data were acquired over the course of 90 min for all but two subjects (see below) and stored in list-mode format. During the imaging visit, all subjects except one rated their level of pain using a verbal 0–100 numerical ratings scale (pain: 0 = ‘no pain’, 100 = ‘most intense pain tolerable’). The remaining subject (a healthy volunteer) was scanned under a separate protocol, which did not call for ratings of pain before and after the functional runs (but was otherwise identical in the procedures and parameters of the imaging visit).
Data analysis
Using in-house software, standardized uptake values (SUV; i.e. mean radioactivity/injected dose/weight) were computed for each subject. SUVs have been previously used as a measure of TSPO expression in both humans (Hirvonen et al., 2012; Fujita et al., 2013) and rodents (Shao et al., 2013). SUVs were computed voxel-wise from the 60–90 min post-injection PET data, except for two subjects (one patient and one control) for whom SUVs were computed from 72–89 min and 60–86 min, respectively, because of unavailability of the full 60–90 min frame. The two subjects matching these subjects with incomplete data were reconstructed with identical frame onset and duration for the group analyses, to ensure that differences in the PET framing scheme would not affect our results. To maximize accuracy in the reconstruction, SUVs were generated in a two-step iterative process. First, a preliminary SUV image was created for each subject using an attenuation correction map (mu-map) computed from the MPRAGE in its native space (Izquierdo-Garcia et al., 2014). To account for motion that may have occurred between the time the MPRAGE and the 60–90 min PET data were acquired, a new mu-map was created from the MPRAGE registered to this temporary SUV map. This registration was performed using spmregister, a tool from the FreeSurfer suite (http://surfer.nmr.mgh.harvard.edu) (Dale et al., 1999) that performs a rigid-body registration of a functional volume to its relative FreeSurfer anatomical volume with Normalised Mutual Information using spm_coreg from the SPM suite (http://www.fil.ion.ucl.ac.uk/spm/) (Friston, 2003). A final SUV map was created using this new mu-map, now well in register with the 60–90 min PET data. SUV maps were then normalized to MNI space using non-linear registration (FNIRT, from the FSL suite; FMRIB's Software Library, version 4.1.9, www.fmrib.ox.ac.uk/fsl/) (Smith et al., 2004). Spatially-normalized SUV images were then spatially smoothed (full-width at half-maximum = 8 mm) to improve signal-to-noise ratio, and intensity-normalized to a mean of 1 (SUVRs) in order to account for global signal differences across subjects, such as those introduced by differences in the Ala147Thr polymorphism, which affects global binding affinity.
Although the subjects scanned were carefully selected from a larger sample size to match for genetic and demographic factors, the size of our final sample was relatively small. For this reason, imaging and behavioural group comparisons were performed using non-parametric testing. As glial activation has been postulated to spread transynaptically from the site of an injury, following the lesioned neural pathways (Banati, 2003), we hypothesized that in patients with chronic LBP glial activation would be detected in early pain processing regions, as they are most proximal to the pathological sites in the back (e.g. spinal cord/nerve roots). Thus, a thalamus-targeted region of interest analysis was first performed to test this regionally-specific hypothesis. In this analysis, a matched-pairs test (sign test) was performed on the mean SUVRs extracted from the voxels within the right and left thalamus labels of the Harvard-Oxford Subcortical Structural Atlas (Centre for Morphometric Analyses, http://www.cma.mgh.harvard.edu/fsl_atlas.html), thresholded at the arbitrary value of 30.
We then performed a whole-brain, voxel-wise, matched-pairs analysis with the purpose of identifying (i) which thalamic subregion would be driving the effect in the region of interest analyses (if any was detected); and (ii) additional regions of glial activation within the entire brain. This analysis was conducted using the non-parametric randomize tool from the FSL suite (Nichols and Holmes, 2002), with 10 000 permutations and 5 mm variance smoothing (which increases power with smaller sample sizes: http://fsl.fmrib.ox.ac.uk/fsl/fslwiki/Randomise/UserGuide). Finally, as exposure to opioids may also lead to a glial reaction per se (Watkins et al., 2009), we performed the same SUVR group analysis after the exclusion of the data from the two patients on opioids (and their matching controls; seven versus seven), and limited our search to significant clusters from the original analysis. In all cases, whole-brain voxel-wise group comparisons maps were thresholded using threshold-free cluster enhancement (Smith and Nichols, 2009), using a corrected threshold of P = 0.05. For both region of interest and whole-brain voxel-wise analyses, the comparison of nine patient-control pairs was repeated (for consistency sake) in two separate matched-pairs analyses, each using one of the two patients matched to the same control. In the voxel-wise analyses, we first performed a whole-brain search using the data from the patient that best matched the control in terms of age; subsequently the voxel-wise analyses were repeated using the data from the other patient, limiting our search to significant clusters from the first analysis.
Group comparison of behavioural and blood data was performed using the non-parametric sign test or, where applicable, the Fisher’s exact test. These analyses were performed using Statistica v.10 (StatSoft). For the exploratory assessment of the relation between imaging and behavioural/blood measures, it was necessary to assess the association between imaging and clinical parameters, adjusting for the TSPO polymorphism. We did this by using multiple regression analysis. To be consistent in our use of non-parametric methods, we used a non-parametric regression method, Generalized Additive Models. To display the relationship between variables in scatter plots, we computed residuals adjusting for the effect of genotype. We performed these analyses using the gam library in the R statistics package (http://www.R-project.org). Statistical significance was determined by thresholding at P = 0.05.
Results
Higher brain TSPO levels in patients with chronic LBP
In the thalamic region of interest analysis, 11C-PBR28 SUVs, normalized to whole brain, (SUVRs) were significantly higher in patients with chronic LBP than controls (left thalamus: P ≤ 0.01; right thalamus: P ≤ 0.05; Fig. 1A). The voxel-wise distribution of thalamic SUVRs (Fig. 1B) revealed that in control subjects non-zero median voxel counts were observed only below values of 1.4, whereas in patients with chronic LBP a substantial number of voxels demonstrated values higher than 1.4 in both hemispheres (medians: 57.5 and 64 in the left and right thalamus, respectively).
Figure 1.
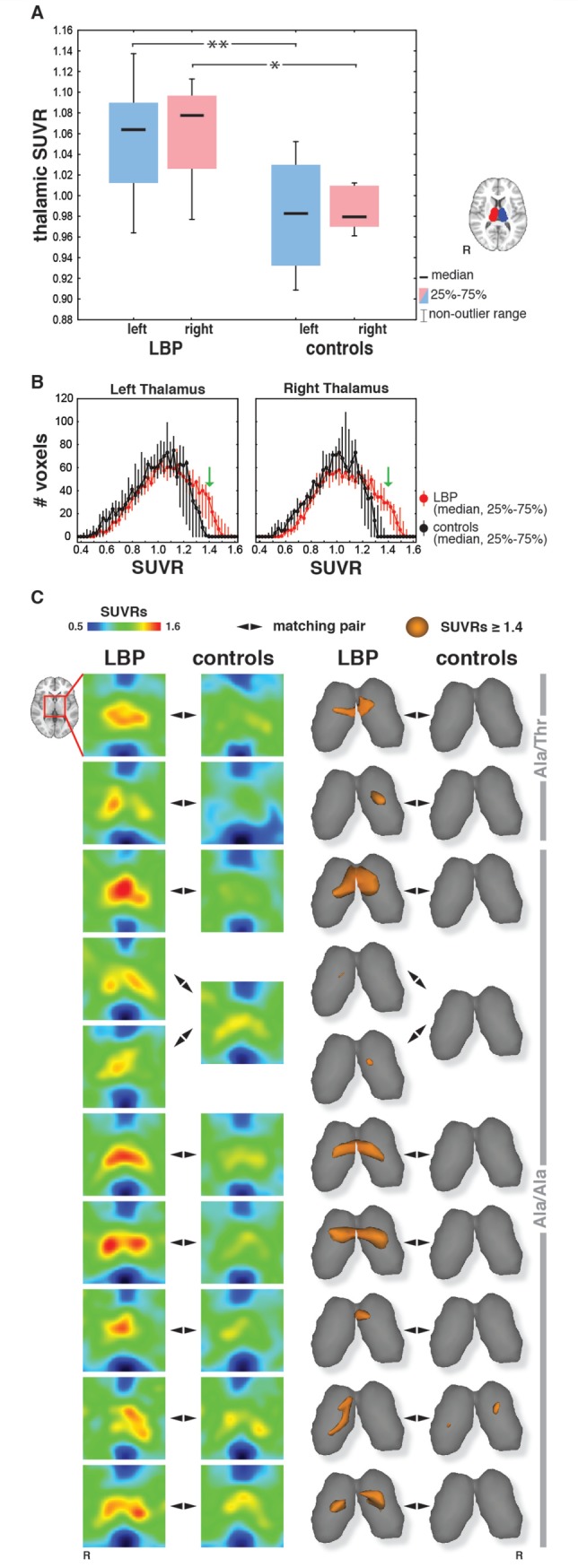
Evidence for glial activation in the thalamus of chronic LBP patients. (A) Boxplots are presented for the mean 11C-PBR28 SUVRs extracted for all 10 patients with chronic LBP and nine control subjects from the thalamic regions of interest (insert). The P-values refer to matched-pairs analyses (sign test) performed using nine chronic LBP-control matching pairs. The analyses were repeated twice, each time using one of the two patients matching the same control, with statistically significant results in both analyses. *P < 0.05, **P < 0.01. (B) Voxel-wise distribution of thalamic SUVRs, showing that patients with chronic LBP have a substantial number of voxels at values ≥ 1.4 (green arrows), whereas controls have a median voxel count of 0. (C) Individual thalamic SUVRs are presented as axial sections (left), and 3D rendering of values higher than the threshold of 1.4 (right). Each row displays SUVRs for each patient-control matched pair. TSPO polymorphism (Ala/Ala or Ala/Thr) is indicated.
The examination of individual thalamic SUVRs (Fig. 1C) shows that, strikingly, each patient exhibited higher SUVRs than his/her age- sex- and TSPO genotype-matched control in the thalamus. In all patients, the areas of maximal TSPO levels were consistently localized in dorsomedial subregions of the thalamus, as illustrated by the 3D rendering (Fig. 1C).
In the whole-brain voxel-wise analyses, SUVRs were significantly higher in thalamus, pre- and postcentral gyri and paracentral lobule (Fig. 2 and Supplementary Table 3). The peak group difference was observed in left thalamus, consistent with the mediodorsal nucleus. There were no brain regions for which the healthy controls showed statistically higher SUVRs than the patients with chronic LBP. When examined at the exploratory threshold of P < 0.01 uncorrected for multiple comparisons (Supplementary Fig. 1), additional regions demonstrated higher SUVRs in chronic LBP than controls, including insulae, middle cingulate cortex, ventromedial prefrontal cortex, posterior cingulate cortex, supplementary motor area and basal ganglia. Moreover, excluding the two patients on opioids (and their matching controls), yielded similar results (Supplementary Table 4). Thus, while exposure to opioids may also lead to a glial reaction (Watkins et al., 2009), our results do not seem to be confounded by opioid intake.
Figure 2.

Whole-brain voxel-wise analyses. (A) Median SUVR map from healthy controls (n = 9) and patients with chronic LBP (n = 10) are presented. Matched-pairs tests (nine versus nine) revealed significantly higher TSPO levels in patients, in thalamus, pre- and postcentral gyri, and paracentral lobule (P < 0.05 corrected for multiple comparisons; permutation testing, 10 000 permutations). As two patients were matching the same controls, the analyses were performed first using the patient best matching the control. A second analysis was performed using the other patient, limiting our search to significant clusters from the first analysis, with identical results. (B) Boxplots for each of the four regions demonstrating statistically higher SUVRs in patients are shown for illustrative purposes. postc. = postcentral; g. = gyrus; parac. lob. = postcentral lobule; prec. = precentral.
TSPO expression as a protective mechanism?
In the patients, 11C-PBR28 imaging metrics, corrected for genotype, were negatively associated with pain outcome measures (Fig. 3A–C) and circulating levels of IL6 (Fig. 3D). First, the number of thalamic voxels with a SUVR value > 1.4 was negatively associated with pain levels at the time of scan and the total score on the McGill Pain Questionnaire (P < 0.05), but not with IL6 or TNFA levels (P-values > 0.3). Second, the average SUVRs extracted from the thalamic cluster statistically significant in the voxel-wise analysis were negatively associated with levels of IL6 (P < 0.05), but not with TNFA levels (P = 0.38). The association of thalamic [11C]PBR28 SUVR with the McGill Pain Questionnaire total scores was negative, but did not reach statistical significance (P = 0.11). No association was observed between this imaging metric and pain levels during the scan (P = 0.25). In all subjects, IL1B levels were below the detection threshold, and therefore the association between imaging and IL1B data could not be assessed.
Figure 3.

Anti-inflammatory and anti-nociceptive role of TSPO. SUVRs were negatively associated with pain outcomes (A–C) and blood levels of interleukin-6 (D). The scatterplots show the residuals adjusting for the effect of genotype. MPQ = McGill Pain Questionnaire.
Discussion
Our study demonstrates the occurrence of glial activation, as measured by an increase in 11C-PBR28 binding, in the brain of patients with chronic pain. Increased tracer binding was observed most prominently in the thalamus, and with remarkable across-subject consistency (Fig. 1C).
Within the primary somatosensory and motor cortices (S1/M1) the SUVRs were higher in the putative sensorimotor representations of the lumbar spine [in the postcentral gyrus; Boendermaker et al. (2014), and leg (in the paracentral lobule); Loggia et al. (2012)]. This spatial pattern is consistent with the majority of patients suffering from pain in the lower back and leg(s), and is suggestive of somatotopically organized glial activation in S1/M1 (which in turn is also consistent with the observation that spinal glial activation generally follows somatotopic boundaries after unilateral spared nerve injury in rats) (Beggs and Salter, 2007).
In the last decade, animal research has led to the increased recognition of the importance of glial cells (such as microglia and astrocytes), and their interaction with neuronal cells, in the pathogenesis of pain conditions (Tsuda et al., 2003; Watkins et al., 2007; Calvo et al., 2012; Ji et al., 2013). For instance, nerve injury induces a profound activation and proliferation of spinal microglia (Liu et al., 2000; Beggs and Salter, 2007; Echeverry et al., 2008; Beggs et al., 2012; Calvo and Bennett, 2012) and the upregulation of a variety of receptors in these cells, such as the adenosine triphosphate (ATP) receptor P2RX4 (also known as P2X4) (Tsuda et al., 2003) and the chemokine receptor CX3CR1 (Verge et al., 2004), which induce hyperalgesia. Activated microglial cells produce inflammatory mediators, including proinflammatory cytokines and brain-derived neurotrophic factor (BDNF; Coull et al., 2005; Ji and Suter, 2007) that activate or sensitize nociceptive neurons. The intrathecal injection of activated microglia produces tactile allodynia in naive rats, suggesting that microglial activation is sufficient to induce pain sensitization (Tsuda et al., 2003). Finally, pharmacological inhibition of microglial activation prevents or delays neuropathic pain (Raghavendra et al., 2003; Ledeboer et al., 2005). Similar to microglia, astrocytes have been shown to have a role in the induction and maintenance of pain sensitization (Ji et al., 2006; Ren and Dubner, 2010; Ji et al., 2013). For instance, in trigeminal models of inflammatory hyperalgesia (Guo et al., 2007) or following trigeminal nerve injury (Okada-Ogawa et al., 2009), astrocytes exhibit hypertrophy, and express enzymes, such as nitric oxide synthase (Meller et al., 1994), as well as inflammatory mediators, such as the proinflammatory cytokine IL1B (Guo et al., 2007) and chemokines such as CXCL2 (Chen et al., 2014), that contribute to hyperalgesia and allodynia. Thermal and mechanical hyperalgesia are inhibited or attenuated by the injection of agents that disrupt either astroglial function (such as fluorocitrate, a glial metabolic inhibitor) (Meller et al., 1994; Watkins et al., 1997; Guo et al., 2007; Okada-Ogawa et al., 2009), or the action of glial products (such as IL1 receptor antagonists) (Watkins et al., 1997). Taken together, these studies demonstrate that microglia and astrocytes play an important role in the pathogenesis of persistent pain in animals.
Although a plethora of animal studies has demonstrated that glial cells are involved in the establishment and maintenance of persistent pain, no study has previously demonstrated in vivo glial activation in humans suffering from chronic pain. Our observations have several potential implications. Firstly, they provide a rationale for exploring the role of glia as therapeutic target for chronic pain. In animals, drugs that reduce glial activation (e.g. propentofylline and minocycline) have been found to potently inhibit proinflammatory cytokines, thereby suppressing the development of neuropathic pain (Mika, 2008; Leblanc et al., 2011). Importantly, some of these molecules are already FDA approved to treat human conditions of different aetiologies and testing for new, chronic pain-related indications would therefore be immediately possible. Two recent clinical trials (8- and 12-weeks long, respectively) have indicated that low-dose naltrexone (LDN) may have a clinically beneficial impact on fibromyalgia pain (Younger and Mackey, 2009; Younger et al., 2013). As LDN is thought to produce anti-inflammatory effects mainly by antagonizing the activity of glial cells (Mattioli et al., 2010), these studies suggest that glial modulators may be beneficial for fibromyalgia and perhaps other subgroups of chronic pain patients. On the other hand, it should be noted that some clinical trials have had negative results. A recent clinical trial assessing the efficacy of propentophylline to reduce pain in post-herpetic neuralgia was negative (Landry et al., 2012). Another study suggested that perioperative minocycline administration did not improve persistent pain after lumbar discectomy (Martinez et al., 2013). However, methodological concerns with these studies (Watkins et al., 2012) limit the significance of these negative study outcomes. In particular, the duration of trial design was unusually short in both studies. In the former study, propentophylline was administered for 4 weeks, and in the latter minocycline was administered peri-operatively for only 8 days, in both cases a significantly shorter duration than 12 weeks, as is more typically adopted in clinical trials. Moreover, as studies have shown that it is easier to prevent than reverse neuropathic pain using glial modulators (Raghavendra et al., 2003), longer trial durations may be needed to achieve therapeutic efficacy (Watkins et al., 2012). Other factors may also contribute to explain the negative results in these trials, including the dosage and potential interaction with other drugs or food intake (which may not have allowed for the drug to reach CNS sites at meaningful levels), or the choice of patient population (particularly for the propentofylline trial, as the preclinical evidence in support of a role for glia in post-herpetic neuralgia is more tenuous than in other chronic pain conditions) (Watkins et al., 2012).
The possibility to image pain-related glial activation in vivo, which we document in the present study, may help to identify patients most likely to benefit from this therapeutic approach, and to identify optimal treatment duration or dosage. Given the putative role of activated glia in many challenging issues associated with pain management, such as the induction of opioid-induced hyperalgesia and tolerance (Eidson and Murphy, 2013; Ferrini et al., 2013), the present findings offer clinical implications that may serve to guide future studies of the pathophysiology and management of a variety of persistent pain conditions.
In this study, glial activation was assessed using brain levels of TSPO, formerly called peripheral benzodiazepine receptor (Banati et al., 1997; Papadopoulos et al., 2006). As experimental animal models and human post-mortem studies of CNS disorders have reliably shown concomitant and co-localized increases in TSPO expression and markers for activated astrocytes and/or microglia, TSPO expression is widely acknowledged as a marker of glial activation in CNS injury and disease. For instance, co-localization of glial activation and TSPO upregulation was observed in rodent models of experimental autoimmune encephalomyelitis, in rodent models of multiple sclerosis, as well as in human multiple sclerosis lesions (Vowinckel et al., 1997; Banati et al., 2000; Chen et al., 2004; Chen and Guilarte, 2006; Ji et al., 2008; Cosenza-Nashat et al., 2009; Abourbeh et al., 2012), in non-human primate models of HIV encephalitis (Venneti et al., 2007; Cosenza-Nashat et al., 2009), as well as in human HIV encephalitis (Cosenza-Nashat et al., 2009), in both human post-mortem and experimental rodent models of ischaemia (Rojas et al., 2007; Cosenza-Nashat et al., 2009; Martin et al., 2010) and Alzheimer’s disease (Ji et al., 2008; Cosenza-Nashat et al., 2009; Gulyas et al., 2009), and in rodent models of ethanol and trimethyl neurotoxicity (Kuhlmann and Guilarte, 2000; Maeda et al., 2007). More pertinent to our study, TSPO was upregulated in spinal astrocytes and microglia in Complete Freund’s Adjuvant (CFA)-induced monoarthritis of the tibio-tarsal joint (Hernstadt et al., 2009) and following L5 spinal nerve ligation pain (Wei et al., 2013). Given these observations, the increased 11C-PBR28 levels we observed in patients with chronic pain can be interpreted as evidence of glial activation. Interestingly, the involvement of glial subtypes in neuroinflammatory responses seems to depend on the time course of disease. In several animal models, initial TSPO upregulation following acute CNS insult is accompanied by a predominantly microglial response that typically peaks and begins dissipating several days to weeks after injury. This rapid microglial response is paralleled by a delayed but steadily increasing astrocytic component (Kuhlmann and Guilarte, 2000; Chen et al., 2004; Chen and Guilarte, 2006; Martin et al., 2010; Liu et al., 2014). Human post-mortem data seem to corroborate this phase-dependent glial contribution: in acute multiple sclerosis lesions, microglia and macrophages represent most TSPO+ cells, whereas astrocytes are the dominant TSPO+ cells in chronic, silent lesions (Cosenza-Nashat et al., 2009). Because our patients suffered from years of pain, it is plausible that astrocytes provided a significant contribution to the increased PET signal observed in our data.
Although TSPO is a marker of activated glia, a phenomenon thought to be responsible for the amplification of pain signals within the CNS (Ji et al., 2013), TSPO expression itself has been shown in animals to exert inhibitory effects on neuroinflammation (Wei et al., 2013; Bae et al., 2014; Wang et al., 2014), and to promote recovery from neuropathic pain (Wei et al., 2013), likely through the stimulation of steroidogenesis (Batarseh and Papadopoulos, 2010; Wei et al., 2013). In fact, studies suggest that one of the functions of TSPO in activated glia is to limit the magnitude of inflammatory responses after their initiation (Wang et al., 2014). Our observation that 11C-PBR28 SUVRs negatively correlate with levels of the circulating proinflammatory cytokine IL6 and pain further corroborate the hypothesis that TSPO expression has anti-inflammatory and pain-protective effects. The negative correlations found in our study therefore support the contention that TSPO ligands may be a novel therapeutic target for the treatment of pathological pain (Wei et al., 2013), as previously suggested for a variety of conditions (Rupprecht et al., 2009, 2010). As TSPO expression is upregulated in activated glia and was found to negatively correlate with peripheral cytokines and pain in our study, low levels of TSPO might be interpreted differently between groups. For example, in healthy volunteers, low levels could simply reflect low levels of glial activation. On the other hand, patients with chronic pain exhibiting lower TSPO levels may have impairments in TSPO expression in activated glia, and therefore in their ability to limit the glial responses after their initiation. Clearly, additional studies are required to elucidate the relationship between 11C-PBR28 and peripheral markers of inflammation, particularly as peripheral cytokine levels are often found to be decoupled from those within the CNS (Bromander et al., 2012).
Future studies, including some currently already underway in our laboratory, will need to determine whether different pain populations present differences in the spatial distribution of glial activation. The discovery of ‘glial signatures’ of chronic pain states might lead to the identification of objective imaging markers that could (i) complement the patient’s subjective assessment and other measures (e.g. quantitative sensory testing) to guide clinical practice; and (ii) reduce the patient heterogeneity which has traditionally led to poor signal-to-noise ratios in most clinical drug evaluation studies (Gomez-Mancilla et al., 2005). Finally, as glial cells respond to very subtle changes in their microenvironment that even precede pathological changes that are detectable histologically (de Vries et al., 2006; Cagnin et al., 2007), glial activation might be an early marker of the alterations that have been shown to occur in the brains of chronic pain patients (Tracey and Bushnell, 2009). This might allow early identification of individuals at risk of transitioning from acute to chronic pain, thus optimizing treatment strategies.
In sum, our findings demonstrate a role of glia in human pain disorders, support the role of the assessment of glial activation and TSPO expression in selective brain areas as an imaging marker and potential treatment target for chronic pain disorders in humans.
Acknowledgements
We would like to thank Patricia McCarthy, Nancy Shearer and the Massachusetts General Hospital Clinical Research Centre for providing nursing support, Drs Douglas Greve, Marc Normandin, Nicolas Guehl and Jean Logan for help with data processing, Dr Julie Price for helpful comments, Dr Nazem Atassi for sharing imaging resources, Drs Randy Gollub and Alexander Guimaraes for their help with obtaining informed consent, Ms Jiaxuan (Jessie) Wang for her support with the study logistics and Ms Ekaterina Protsenko for editorial assistance.
Funding
This project was supported by Harvard Catalyst Advanced Imaging Pilot Grant (J.M.H.), 1R21NS087472-01A1 (M.L.L.) 1UL1TR001102-01, 8UL1TR000170-05, from the National Centre for Advancing Translational Science, and 1UL1RR025758-04 from the National Centre for Research Resources, Harvard Clinical and Translational Science Centre, and financial contributions from Harvard University and its affiliated academic healthcare centres. The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard Catalyst, Harvard University and its affiliated academic healthcare centres, or the National Institutes of Health. The authors have no conflicts of interest or competing financial interests to declare.
Supplementary material
Supplementary material is available at Brain online.
Glossary
Abbreviations
- LBP
low back pain
- SUV
standardized uptake value
References
- Abourbeh G, Theze B, Maroy R, Dubois A, Brulon V, Fontyn Y, et al. Imaging microglial/macrophage activation in spinal cords of experimental autoimmune encephalomyelitis rats by positron emission tomography using the mitochondrial 18 kDa translocator protein radioligand [(1)(8)F]DPA-714. J Neurosci. 2012 doi: 10.1523/JNEUROSCI.2900-11.2012. 25; 32: 5728–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae KR, Shim HJ, Balu D, Kim SR, Yu SW. Translocator protein 18 kDa negatively regulates inflammation in microglia. J Neuroimmune Pharmacol. 2014;9:424–37. doi: 10.1007/s11481-014-9540-6. [DOI] [PubMed] [Google Scholar]
- Banati RB, Myers R, Kreutzberg GW. PK (‘peripheral benzodiazepine')—binding sites in the CNS indicate early and discrete brain lesions: microautoradiographic detection of [3H]PK11195 binding to activated microglia. J Neurocytol. 1997;26:77–82. doi: 10.1023/a:1018567510105. [DOI] [PubMed] [Google Scholar]
- Banati RB, Newcombe J, Gunn RN, Cagnin A, Turkheimer F, Heppner F, et al. The peripheral benzodiazepine binding site in the brain in multiple sclerosis: quantitative in vivo imaging of microglia as a measure of disease activity. Brain. 2000;123(Pt 11):2321–37. doi: 10.1093/brain/123.11.2321. [DOI] [PubMed] [Google Scholar]
- Banati RB. Neuropathological imaging: in vivo detection of glial activation as a measure of disease and adaptive change in the brain. Br Med Bull. 2003;65:121–31. doi: 10.1093/bmb/65.1.121. [DOI] [PubMed] [Google Scholar]
- Batarseh A, Papadopoulos V. Regulation of translocator protein 18 kDa (TSPO) expression in health and disease states. Mol Cell Endocrinol. 2010;327:1–12. doi: 10.1016/j.mce.2010.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beggs S, Salter MW. Stereological and somatotopic analysis of the spinal microglial response to peripheral nerve injury. Brain Behav Immun. 2007;21:624–33. doi: 10.1016/j.bbi.2006.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beggs S, Trang T, Salter MW. P2X4R+ microglia drive neuropathic pain. Nat Neurosci. 2012;15:1068–73. doi: 10.1038/nn.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boendermaker B, Meier ML, Luechinger R, Humphreys BK, Hotz-Boendermaker S. The cortical and cerebellar representation of the lumbar spine. Hum Brain Mapp. 2014;35:3962–71. doi: 10.1002/hbm.22451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brisby H, Olmarker K, Rosengren L, Cederlund CG, Rydevik B. Markers of nerve tissue injury in the cerebrospinal fluid in patients with lumbar disc herniation and sciatica. Spine (Phila Pa 1976) 1999;24:742–6. doi: 10.1097/00007632-199904150-00003. [DOI] [PubMed] [Google Scholar]
- Briard E, Zoghbi SS, Imaizumi M, Gourley JP, Shetty HU, Hong J, et al. Synthesis and evaluation in monkey of two sensitive 11C-labeled aryloxyanilide ligands for imaging brain peripheral benzodiazepine receptors in vivo. J Med Chem. 2008;51:17–30. doi: 10.1021/jm0707370. [DOI] [PubMed] [Google Scholar]
- Bromander S, Anckarsater R, Kristiansson M, Blennow K, Zetterberg H, Anckarsater H, et al. Changes in serum and cerebrospinal fluid cytokines in response to non-neurological surgery: an observational study. J Neuroinflammation. 2012;9:242. doi: 10.1186/1742-2094-9-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AK, Fujita M, Fujimura Y, Liow JS, Stabin M, Ryu YH, et al. Radiation dosimetry and biodistribution in monkey and man of 11C-PBR28: a PET radioligand to image inflammation. J Nucl Med. 2007;48:2072–9. doi: 10.2967/jnumed.107.044842. [DOI] [PubMed] [Google Scholar]
- Cagnin A, Kassiou M, Meikle SR, Banati RB. Positron emission tomography imaging of neuroinflammation. Neurotherapeutics. 2007;4:443–52. doi: 10.1016/j.nurt.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo M, Bennett DL. The mechanisms of microgliosis and pain following peripheral nerve injury. Exp Neurol. 2012;234:271–82. doi: 10.1016/j.expneurol.2011.08.018. [DOI] [PubMed] [Google Scholar]
- Calvo M, Dawes JM, Bennett DL. The role of the immune system in the generation of neuropathic pain. Lancet Neurol. 2012;11:629–42. doi: 10.1016/S1474-4422(12)70134-5. [DOI] [PubMed] [Google Scholar]
- Catana C, Procissi D, Wu Y, Judenhofer MS, Qi J, Pichler BJ, et al. Simultaneous in vivo positron emission tomography and magnetic resonance imaging. Proc Natl Acad Sci USA. 2008;105:3705–10. doi: 10.1073/pnas.0711622105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosenza-Nashat M, Zhao ML, Suh HS, Morgan J, Natividad R, Morgello S, et al. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol Appl Neurobiol. 2009;35:306–28. doi: 10.1111/j.1365-2990.2008.01006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MK, Baidoo K, Verina T, Guilarte TR. Peripheral benzodiazepine receptor imaging in CNS demyelination: functional implications of anatomical and cellular localization. Brain. 2004;127(Pt 6):1379–92. doi: 10.1093/brain/awh161. [DOI] [PubMed] [Google Scholar]
- Chen G, Park CK, Xie RG, Berta T, Nedergaard M, Ji RR. Connexin-43 induces chemokine release from spinal cord astrocytes to maintain late-phase neuropathic pain in mice. Brain. 2014;137(Pt 8):2193–209. doi: 10.1093/brain/awu140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MK, Guilarte TR. Imaging the peripheral benzodiazepine receptor response in central nervous system demyelination and remyelination. Toxicol Sci. 2006;91:532–9. doi: 10.1093/toxsci/kfj172. [DOI] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–21. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- Dale AM, Fischl B, Sereno MI. Cortical surface-based analysis. I. Segmentation and surface reconstruction. Neuroimage. 1999;9:179–94. doi: 10.1006/nimg.1998.0395. [DOI] [PubMed] [Google Scholar]
- Del Valle L, Schwartzman RJ, Alexander G. Spinal cord histopathological alterations in a patient with longstanding complex regional pain syndrome. Brain Behav Immun. 2009;23:85–91. doi: 10.1016/j.bbi.2008.08.004. [DOI] [PubMed] [Google Scholar]
- Echeverry S, Shi XQ, Zhang J. Characterization of cell proliferation in rat spinal cord following peripheral nerve injury and the relationship with neuropathic pain. Pain. 2008;135:37–47. doi: 10.1016/j.pain.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Eidson LN, Murphy AZ. Blockade of Toll-like receptor 4 attenuates morphine tolerance and facilitates the pain relieving properties of morphine. J Neurosci. 2013;33:15952–63. doi: 10.1523/JNEUROSCI.1609-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrini F, Trang T, Mattioli TA, Laffray S, Del'Guidice T, Lorenzo LE, et al. Morphine hyperalgesia gated through microglia-mediated disruption of neuronal Cl(-) homeostasis. Nat Neurosci. 2013;16:183–92. doi: 10.1038/nn.3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friston K. Introduction: experimental design and statistical parametric mapping. In: Frackowiak RSJ, Ashburner JT, Penny WD, Zeki S, Friston K, Frith CD, et al., editors. Human brain function. London, UK: Academic Press; 2003. [Google Scholar]
- Fujita M, Mahanty S, Zoghbi SS, Ferraris Araneta MD, Hong J, Pike VW, et al. PET reveals inflammation around calcified Taenia solium granulomas with perilesional edema. PLoS One. 2013;8:e74052. doi: 10.1371/journal.pone.0074052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrmann J, Matsumoto Y, Kreutzberg GW. Microglia: intrinsic immuneffector cell of the brain. Brain Res Brain Res Rev. 1995;20:269–87. doi: 10.1016/0165-0173(94)00015-h. [DOI] [PubMed] [Google Scholar]
- Geisser ME, Roth RS, Robinson ME. Assessing depression among persons with chronic pain using the Center for Epidemiological Studies-Depression Scale and the Beck Depression Inventory: a comparative analysis. Clin J Pain. 1997;13:163–70. doi: 10.1097/00002508-199706000-00011. [DOI] [PubMed] [Google Scholar]
- Gomez-Mancilla B, Marrer E, Kehren J, Kinnunen A, Imbert G, Hillebrand R, et al. Central nervous system drug development: an integrative biomarker approach toward individualized medicine. NeuroRx. 2005;2:683–95. doi: 10.1602/neurorx.2.4.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyas B, Makkai B, Kasa P, Gulya K, Bakota L, Varszegi S, et al. A comparative autoradiography study in post mortem whole hemisphere human brain slices taken from Alzheimer patients and age-matched controls using two radiolabelled DAA1106 analogues with high affinity to the peripheral benzodiazepine receptor (PBR) system. Neurochem Int. 2009;54:28–36. doi: 10.1016/j.neuint.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Guo W, Wang H, Watanabe M, Shimizu K, Zou S, LaGraize SC, et al. Glial-cytokine-neuronal interactions underlying the mechanisms of persistent pain. J Neurosci. 2007;27:6006–18. doi: 10.1523/JNEUROSCI.0176-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hains BC, Waxman SG. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J Neurosci. 2006;26:4308–17. doi: 10.1523/JNEUROSCI.0003-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernstadt H, Wang S, Lim G, Mao J. Spinal translocator protein (TSPO) modulates pain behavior in rats with CFA-induced monoarthritis. Brain Res. 2009;1286:42–52. doi: 10.1016/j.brainres.2009.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirvonen J, Kreisl WC, Fujita M, Dustin I, Khan O, Appel S, et al. Increased in vivo expression of an inflammatory marker in temporal lobe epilepsy. J Nuclear Med. 2012;53:234–40. doi: 10.2967/jnumed.111.091694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imaizumi M, Kim HJ, Zoghbi SS, Briard E, Hong J, Musachio JL, et al. PET imaging with 11C-PBR28 can localize and quantify upregulated peripheral benzodiazepine receptors associated with cerebral ischemia in rat. Neurosci Lett. 2007;411:200–5. doi: 10.1016/j.neulet.2006.09.093. [DOI] [PubMed] [Google Scholar]
- Izquierdo-Garcia D, Hansen AE, Förster S, Benoit D, Schachoff S, Fürst S, et al. An SPM8-based approach for attenuation correction combining segmentation and non-rigid template formation: application to simultaneous PET/MR brain imaging. J Nucl Med. 2014;55:1825–30. doi: 10.2967/jnumed.113.136341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Berta T, Nedergaard M. Glia and pain: Is chronic pain a gliopathy? Pain. 2013;154(Suppl 1):S10–28. doi: 10.1016/j.pain.2013.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Kawasaki Y, Zhuang ZY, Wen YR, Decosterd I. Possible role of spinal astrocytes in maintaining chronic pain sensitization: review of current evidence with focus on bFGF/JNK pathway. Neuron Glia Biol. 2006;2:259–69. doi: 10.1017/S1740925X07000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji B, Maeda J, Sawada M, Ono M, Okauchi T, Inaji M, et al. Imaging of peripheral benzodiazepine receptor expression as biomarkers of detrimental versus beneficial glial responses in mouse models of Alzheimer's and other CNS pathologies. J Neurosci. 2008;28:12255–67. doi: 10.1523/JNEUROSCI.2312-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Suter MR. p38 MAPK, microglial signaling, and neuropathic pain. Mol Pain. 2007;3:33. doi: 10.1186/1744-8069-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadetoff D, Lampa J, Westman M, Andersson M, Kosek E. Evidence of central inflammation in fibromyalgia-increased cerebrospinal fluid interleukin-8 levels. J Neuroimmunol. 2012;242:33–8. doi: 10.1016/j.jneuroim.2011.10.013. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci. 2008;28:5189–94. doi: 10.1523/JNEUROSCI.3338-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreisl WC, Fujita M, Fujimura Y, Kimura N, Jenko KJ, Kannan P, et al. Comparison of [(11)C]-(R)-PK 11195 and [(11)C]PBR28, two radioligands for translocator protein (18 kDa) in human and monkey: Implications for positron emission tomographic imaging of this inflammation biomarker. Neuroimage. 2010;49:2924–32. doi: 10.1016/j.neuroimage.2009.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreisl WC, Jenko KJ, Hines CS, Lyoo CH, Corona W, Morse CL, et al. A genetic polymorphism for translocator protein 18 kDa affects both in vitro and in vivo radioligand binding in human brain to this putative biomarker of neuroinflammation. J Cereb Blood Flow Metab. 2013;33:53–8. doi: 10.1038/jcbfm.2012.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlmann AC, Guilarte TR. Cellular and subcellular localization of peripheral benzodiazepine receptors after trimethyltin neurotoxicity. J Neurochem. 2000;74:1694–704. doi: 10.1046/j.1471-4159.2000.0741694.x. [DOI] [PubMed] [Google Scholar]
- Landry RP, Jacobs VL, Romero-Sandoval EA, DeLeo JA. Propentofylline, a CNS glial modulator does not decrease pain in post-herpetic neuralgia patients: in vitro evidence for differential responses in human and rodent microglia and macrophages. Exp Neurol. 2012;234:340–50. doi: 10.1016/j.expneurol.2011.11.006. [DOI] [PubMed] [Google Scholar]
- Leblanc BW, Zerah ML, Kadasi LM, Chai N, Saab CY. Minocycline injection in the ventral posterolateral thalamus reverses microglial reactivity and thermal hyperalgesia secondary to sciatic neuropathy. Neurosci Lett. 2011;498:138–42. doi: 10.1016/j.neulet.2011.04.077. [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Sloane EM, Milligan ED, Frank MG, Mahony JH, Maier SF, et al. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain. 2005;115:71–83. doi: 10.1016/j.pain.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Liu L, Rudin M, Kozlova EN. Glial cell proliferation in the spinal cord after dorsal rhizotomy or sciatic nerve transection in the adult rat. Exp Brain Res. 2000;131:64–73. doi: 10.1007/s002219900273. [DOI] [PubMed] [Google Scholar]
- Liu X, Li W, Dai L, Zhang T, Xia W, Liu H, et al. Early repeated administration of progesterone improves the recovery of neuropathic pain and modulates spinal 18kDa-translocator protein (TSPO) expression. J Steroid Biochem Mol Biol. 2014;143:130–40. doi: 10.1016/j.jsbmb.2014.02.017. [DOI] [PubMed] [Google Scholar]
- Loggia ML, Edwards RR, Kim J, Vangel MG, Wasan AD, Gollub RL, et al. Disentangling linear and nonlinear brain responses to evoked deep tissue pain. Pain. 2012;153:2140–51. doi: 10.1016/j.pain.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda J, Higuchi M, Inaji M, Ji B, Haneda E, Okauchi T, et al. Phase-dependent roles of reactive microglia and astrocytes in nervous system injury as delineated by imaging of peripheral benzodiazepine receptor. Brain Res. 2007;1157:100–11. doi: 10.1016/j.brainres.2007.04.054. [DOI] [PubMed] [Google Scholar]
- Martin A, Boisgard R, Theze B, Van Camp N, Kuhnast B, Damont A, et al. Evaluation of the PBR/TSPO radioligand [(18)F]DPA-714 in a rat model of focal cerebral ischemia. J Cereb Blood Flow Metab. 2010;30:230–41. doi: 10.1038/jcbfm.2009.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez V, Szekely B, Lemarie J, Martin F, Gentili M, Ben Ammar S, et al. The efficacy of a glial inhibitor, minocycline, for preventing persistent pain after lumbar discectomy: a randomized, double-blind, controlled study. Pain. 2013;154:1197–203. doi: 10.1016/j.pain.2013.03.028. [DOI] [PubMed] [Google Scholar]
- Mattioli TA, Milne B, Cahill CM. Ultra-low dose naltrexone attenuates chronic morphine-induced gliosis in rats. Mol Pain. 2010;6:22. doi: 10.1186/1744-8069-6-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meller ST, Dykstra C, Grzybycki D, Murphy S, Gebhart GF. The possible role of glia in nociceptive processing and hyperalgesia in the spinal cord of the rat. Neuropharmacology. 1994;33:1471–8. doi: 10.1016/0028-3908(94)90051-5. [DOI] [PubMed] [Google Scholar]
- Melzack R. The short-form McGill Pain questionnaire. Pain. 1987;30:191–7. doi: 10.1016/0304-3959(87)91074-8. [DOI] [PubMed] [Google Scholar]
- Mika J. Modulation of microglia can attenuate neuropathic pain symptoms and enhance morphine effectiveness. Pharmacol Rep. 2008;60:297–307. [PubMed] [Google Scholar]
- Nichols TE, Holmes AP. Nonparametric permutation tests for functional neuroimaging: a primer with examples. Hum Brain Mapp. 2002;15:1–25. doi: 10.1002/hbm.1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada-Ogawa A, Suzuki I, Sessle BJ, Chiang CY, Salter MW, Dostrovsky JO, et al. Astroglia in medullary dorsal horn (trigeminal spinal subnucleus caudalis) are involved in trigeminal neuropathic pain mechanisms. J Neurosci. 2009;29:11161–71. doi: 10.1523/JNEUROSCI.3365-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen DR, Yeo AJ, Gunn RN, Song K, Wadsworth G, Lewis A, et al. An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J Cereb Blood Flow Metab. 2012;32:1–5. doi: 10.1038/jcbfm.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos V, Baraldi M, Guilarte TR, Knudsen TB, Lacapere JJ, Lindemann P, et al. Translocator protein (18kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol Sci. 2006;27:402–9. doi: 10.1016/j.tips.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Pekny M, Pekna M. Astrocyte reactivity and reactive astrogliosis: costs and benefits. Physiol Rev. 2014;94:1077–98. doi: 10.1152/physrev.00041.2013. [DOI] [PubMed] [Google Scholar]
- Pekny M, Wilhelmsson U, Pekna M. The dual role of astrocyte activation and reactive gliosis. Neurosci Lett. 2014;565:30–8. doi: 10.1016/j.neulet.2013.12.071. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga F, DeLeo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther. 2003;306:624–30. doi: 10.1124/jpet.103.052407. [DOI] [PubMed] [Google Scholar]
- Ren K, Dubner R. Interactions between the immune and nervous systems in pain. Nat Med. 2010;16:1267–76. doi: 10.1038/nm.2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas S, Martin A, Arranz MJ, Pareto D, Purroy J, Verdaguer E, et al. Imaging brain inflammation with [(11)C]PK11195 by PET and induction of the peripheral-type benzodiazepine receptor after transient focal ischemia in rats. J Cereb Blood Flow Metab. 2007;27:1975–86. doi: 10.1038/sj.jcbfm.9600500. [DOI] [PubMed] [Google Scholar]
- Rolls A, Shechter R, Schwartz M. The bright side of the glial scar in CNS repair. Nat Rev Neurosci. 2009;10:235–41. doi: 10.1038/nrn2591. [DOI] [PubMed] [Google Scholar]
- Rupprecht R, Papadopoulos V, Rammes G, Baghai TC, Fan J, Akula N, et al. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat Rev Drug Discov. 2010;9:971–88. doi: 10.1038/nrd3295. [DOI] [PubMed] [Google Scholar]
- Rupprecht R, Rammes G, Eser D, Baghai TC, Schule C, Nothdurfter C, et al. Translocator protein (18 kD) as target for anxiolytics without benzodiazepine-like side effects. Science. 2009;325:490–3. doi: 10.1126/science.1175055. [DOI] [PubMed] [Google Scholar]
- Shao X, Wang X, English SJ, Desmond T, Sherman PS, Quesada CA, et al. Imaging of carrageenan-induced local inflammation and adjuvant-induced systemic arthritis with [(11)C]PBR28 PET. Nucleic Med Biol. 2013;40:906–11. doi: 10.1016/j.nucmedbio.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Gelman BB, Lisinicchia JG, Tang SJ. Chronic-pain-associated astrocytic reaction in the spinal cord dorsal horn of human immunodeficiency virus-infected patients. J Neurosci. 2012;32:10833–40. doi: 10.1523/JNEUROSCI.5628-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM, Jenkinson M, Woolrich MW, Beckmann CF, Behrens TE, Johansen-Berg H, et al. Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage. 2004;23(Suppl 1):S208–19. doi: 10.1016/j.neuroimage.2004.07.051. [DOI] [PubMed] [Google Scholar]
- Smith SM, Nichols TE. Threshold-free cluster enhancement: addressing problems of smoothing, threshold dependence and localisation in cluster inference. Neuroimage. 2009;44:83–98. doi: 10.1016/j.neuroimage.2008.03.061. [DOI] [PubMed] [Google Scholar]
- Vowinckel E, Reutens D, Becher B, Verge G, Evans A, Owens T, et al. PK11195 binding to the peripheral benzodiazepine receptor as a marker of microglia activation in multiple sclerosis and experimental autoimmune encephalomyelitis. J Neurosci Res. 1997;50:345–53. doi: 10.1002/(SICI)1097-4547(19971015)50:2<345::AID-JNR22>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- de Vries EF, Dierckx RA, Klein HC. Nuclear imaging of inflammation in neurologic and psychiatric disorders. Curr Clin Pharmacol. 2006;1:229–42. doi: 10.2174/157488406778249334. [DOI] [PubMed] [Google Scholar]
- Tracey I, Bushnell MC. How neuroimaging studies have challenged us to rethink: is chronic pain a disease? J Pain. 2009;10:1113–20. doi: 10.1016/j.jpain.2009.09.001. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, et al. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424:778–83. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- Turner JA, Romano JM. Self-report screening measures for depression in chronic pain patients. J Clin Psychol. 1984;40:909–13. doi: 10.1002/1097-4679(198407)40:4<909::aid-jclp2270400407>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Uceyler N, Sommer C. Cytokine-related and histological biomarkers for neuropathic pain assessment. Pain Manag. 2012;2:391–8. doi: 10.2217/pmt.12.28. [DOI] [PubMed] [Google Scholar]
- Venneti S, Wang G, Wiley CA. Activated macrophages in HIV encephalitis and a macaque model show increased [3H](R)-PK11195 binding in a PI3-kinase-dependent manner. Neurosci Lett. 2007;426:117–22. doi: 10.1016/j.neulet.2007.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verge GM, Milligan ED, Maier SF, Watkins LR, Naeve GS, Foster AC. Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. Eur J Neurosci. 2004;20:1150–60. doi: 10.1111/j.1460-9568.2004.03593.x. [DOI] [PubMed] [Google Scholar]
- Wang M, Wang X, Zhao L, Ma W, Rodriguez IR, Fariss RN, et al. Macroglia-microglia interactions via TSPO signaling regulates microglial activation in the mouse retina. J Neurosci. 2014;34:3793–806. doi: 10.1523/JNEUROSCI.3153-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Johnson KW. Commentary on Landry et al.: “Propentofylline, a CNS glial modulator, does not decrease pain in post-herpetic neuralgia patients: in vitro evidence for differential responses in human and rodent microglia and macrophages". Exp Neurol. 2012;234:351–3. doi: 10.1016/j.expneurol.2012.01.006. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Ledeboer A, Wieseler-Frank J, Milligan ED, Maier SF. Norman Cousins Lecture. Glia as the “bad guys”: implications for improving clinical pain control and the clinical utility of opioids. Brain Behav Immun. 2007;21:131–46. doi: 10.1016/j.bbi.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Rice KC, Maier SF. The “toll” of opioid-induced glial activation: improving the clinical efficacy of opioids by targeting glia. Trends Pharmacol Sci. 2009;30:581–91. doi: 10.1016/j.tips.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Martin D, Ulrich P, Tracey KJ, Maier SF. Evidence for the involvement of spinal cord glia in subcutaneous formalin induced hyperalgesia in the rat. Pain. 1997;71:225–35. doi: 10.1016/s0304-3959(97)03369-1. [DOI] [PubMed] [Google Scholar]
- Wei XH, Wei X, Chen FY, Zang Y, Xin WJ, Pang RP, et al. The upregulation of translocator protein (18 kDa) promotes recovery from neuropathic pain in rats. J Neurosci. 2013;33:1540–51. doi: 10.1523/JNEUROSCI.0324-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younger J, Mackey S. Fibromyalgia symptoms are reduced by low-dose naltrexone: a pilot study. Pain Med. 2009;10:663–72. doi: 10.1111/j.1526-4637.2009.00613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younger J, Noor N, McCue R, Mackey S. Low-dose naltrexone for the treatment of fibromyalgia: findings of a small, randomized, double-blind, placebo-controlled, counterbalanced, crossover trial assessing daily pain levels. Arthritis Rheum. 2013;65:529–38. doi: 10.1002/art.37734. [DOI] [PubMed] [Google Scholar]
- Yoder KK, Nho K, Risacher SL, Kim S, Shen L, Saykin AJ. Influence of TSPO genotype on 11C-PBR28 standardized uptake values. J Nuclear Med. 2013;54:1320–2. doi: 10.2967/jnumed.112.118885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zigmond AS, Snaith RP. The hospital anxiety and depression scale. Acta Psychiatr Scand. 1983;67:361–70. doi: 10.1111/j.1600-0447.1983.tb09716.x. [DOI] [PubMed] [Google Scholar]