

Abstract
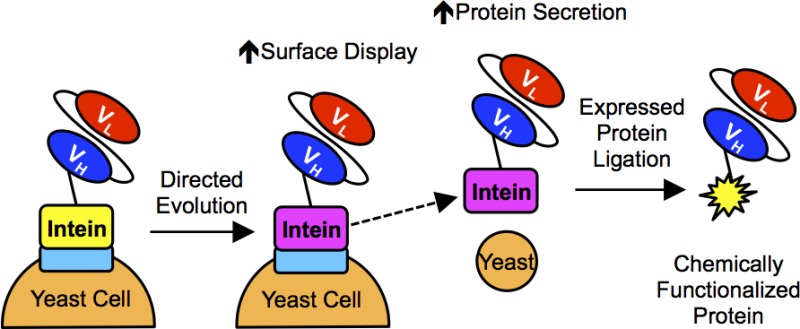
Expressing antibodies as fusions to the non-self-cleaving Mxe GyrA intein enables site-specific, carboxy-terminal chemical modification of the antibodies by expressed protein ligation (EPL). Bacterial antibody-intein fusion protein expression platforms typically yield insoluble inclusion bodies that require refolding to obtain active antibody-intein fusion proteins. Previously, we demonstrated that it was possible to employ yeast surface display to express properly folded single-chain antibody (scFv)-intein fusions, therefore permitting the direct small-scale chemical functionalization of scFvs. Here, directed evolution of the Mxe GyrA intein was performed to improve both the display and secretion levels of scFv-intein fusion proteins from yeast. The engineered intein was shown to increase the yeast display levels of eight different scFvs by up to 3-fold. Additionally, scFv- and green fluorescent protein (GFP)-intein fusion proteins can be secreted from yeast, and while fusion of the scFvs to the wild-type intein resulted in low expression levels, the engineered intein increased scFv-intein production levels by up to 30-fold. The secreted scFv- and GFP-intein fusion proteins retained their respective binding and fluorescent activities, and upon intein release, EPL resulted in carboxy-terminal azide functionalization of the target proteins. The azide-functionalized scFvs and GFP were subsequently employed in a copper-free, strain-promoted click reaction to site-specifically immobilize the proteins on surfaces, and it was demonstrated that the functionalized, immobilized scFvs retained their antigen binding specificity. Taken together, the evolved yeast intein platform provides a robust alternative to bacterial intein expression systems.
Therapeutic and biochemical properties of antibodies can be enhanced by custom chemical functionalization that enables modifications such as small molecule drug conjugation,1,2 PEGylation,3,4 and conjugation to nanoparticles.2,3,5 Expressed protein ligation (EPL) is one common approach to chemically modify proteins in a site-specific manner. In EPL, the target protein is expressed as a fusion partner to a non-self-cleaving intein such as Mxe GyrA.6−9 Intein-mediated protein splicing is activated by the addition of a thiol nucleophile that releases the target protein from the intein while simultaneously producing a carboxy-terminal thioester intermediate on the target protein. Subsequently, this carboxy-terminal thioester can be reacted with an appropriately functionalized amino-terminal cysteine to covalently attach a desired moiety to the carboxy-terminus of the target protein.
Non-self-cleaving intein fusion proteins are most often expressed in the cytoplasm of Escherichia coli.7,9−15 One disadvantage of cytoplasmic expression is the formation of insoluble inclusion bodies that contain inactive protein-intein fusions, therefore requiring solubilization of the inclusion bodies and refolding of the protein.7,9,10,14−16 Glutathione redox buffers that are typically used to refold disulfide-containing proteins such as antibodies can react with the thioester intermediate formed by the intein, thereby releasing it from the target protein and forming an unstable glutathione thioester on the carboxy-terminus of the target protein.10 This unstable glutathione thioester can subsequently be hydrolyzed leading to loss of the thioester functionality.7,10 Additionally, in vivo autocleavage of the intein has been observed during protein expression, resulting in up to 90% loss of the intein for some fusion proteins.17,18 These factors have combined to hamper antibody-intein fusion protein production using bacteria.6,7
Yeasts provide a possible alternative to bacterial expression systems, given their eukaryotic quality control machinery. Recently, scFvs were displayed as fusions to the Mxe GyrA intein on the surface of Saccharomyces cerevisiae.8 Contrasting with bacterial protein-intein fusion platforms, yeast-displayed scFv-intein protein fusions were properly folded and capable of engaging their antigenic targets. However, surface display levels of the scFvs were reduced by ∼40% when fused to intein compared to the unfused antibody. In addition, surface display of heterologous proteins is not ideally suited for protein production at a preparative scale since protein expression on the yeast surface is limited to ∼100 000 display constructs per yeast,19,20 producing on the order of 70 μg of scFv per liter of yeast culture,8 whereas baseline scFv secretion in yeast is in the multimilligram per liter range.21,22 The yeast display levels of scFv-intein proteins could potentially be improved via directed evolution, as has been previously reported for a variety of proteins.23,24 Moreover, improvements in yeast display often translate to improvements in secretion titer.25−27 While directed evolution approaches have been employed to engineer catalytic properties of inteins such as temperature, pH, and ligand dependence,28−31 intein-fusion protein expression levels have not been a target for improvement.
Therefore, in the current study, we sought to improve the production of scFv-intein fusion proteins both as displayed and secreted proteins. Directed evolution of the Mxe GyrA intein was employed as an scFv-intein fusion, and the yeast surface display levels of scFv-intein fusion proteins were restored to that of the unfused scFv. Furthermore, we demonstrated that the engineered intein dramatically improves secretion of scFv-intein fusion proteins from yeast and, since the secreted proteins are folded and active, the scFvs can be directly functionalized and site-specifically immobilized via EPL and click chemistry.
Methods
Yeast Strains and Plasmids
Saccharomyces cerevisiae strain EBY10019 (MATa AGA1::GAL1-AGA1::URA3 ura3–52 trp1 leu2Δ1 his3Δ200 pep4::HIS3 prb1Δ1.6R can1 GAL) was used for surface display, and strain YVH1032 (MATα PDI1::GAPDH-PDI1::LEU2 ura3–52 trp1 leu2Δ1 his3Δ200 pep4::HIS3 prb1Δ1.6R can1 GAL) was used for protein secretion. The unfused and non-self-cleaving Mxe GyrA intein-fused pCT4Re vectors8 were used as a backbone for surface display of the scFvs (Figure 1a). Constructs pCT4Re-4420, pCT4Re-4420-intein, pCT4Re-scFv2, pCT4Re-scFv2-intein, pCT4Re-GFP, and pCT4Re-GFP-intein were generated in a previous study.8 Anti-epidermal growth factor receptor mutant vIII (EGFRvIII) scFv, MR133 (GenBank accession number U76382), was synthesized by IDT DNA Technologies and subcloned into the pCT4Re constructs to create pCT4Re-MR1 and pCT4Re-MR1-intein. An scFv that binds the external domain of EGFR, 2224,34 was synthesized by Life Technologies based upon the sequence provided in patent US 20100009390 A135 and subcloned into the pCT4Re constructs to create pCT4Re-2224 and pCT4Re-2224-intein. RBE4 binding scFvs selected in a previous study36 were subcloned into the pCT4Re vectors to generate pCT4Re-scFvA, pCT4Re-scFvA-intein, pCT4Re-scFvD, pCT4Re-scFvD-intein, pCT4Re-scFvH, pCT4Re-scFvH-intein, and pCT4Re-scFv4S21, and pCT4Re-scFv4S21-intein. The pRS316-FLAG vector was created for protein secretion by inserting the constructs shown in Figure 1b into the pRS316-Gal vector37 between the GAL1–10 promoter and alpha factor terminator sequences to create unfused and intein-fused pRS316-FLAG vectors. The scFvs were subcloned into the pRS316-FLAG vectors to create pRS316-FLAG-4420, pRS316-FLAG-4420-intein, pRS316-FLAG-scFv2, pRS316-FLAG-scFv2-intein, pRS316-FLAG-GFP, pRS316-FLAG-GFP-intein, pRS316-FLAG-MR1, pRS316-FLAG-MR1-intein, and pRS316-FLAG-2224, pRS316-FLAG-2224-intein.
Figure 1.
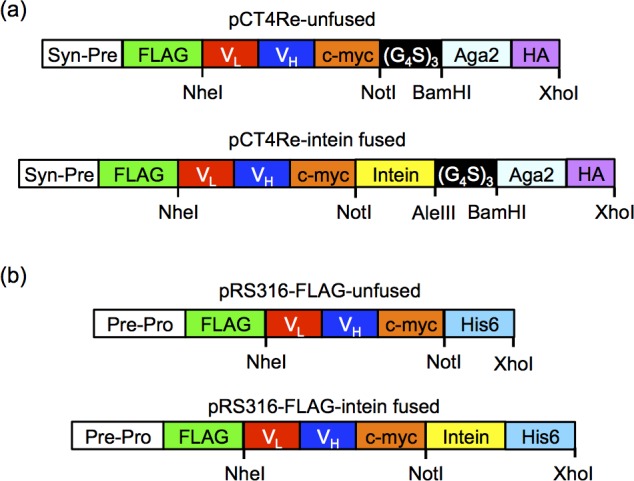
Surface display and secretion constructs. (a) In display construct pCT4Re, Aga2p is expressed at the carboxy-terminus to anchor the fusion protein to the yeast surface, while a FLAG epitope tag is expressed on the amino-terminus of the scFv or GFP to indicate full-length construct expression on the yeast surface. In the intein-containing display constructs, the non-self-cleaving Mxe GyrA intein is inserted between the carboxy-terminus of the scFv or GFP and the Aga2p surface anchor. (b) Secretion construct pRS316-FLAG is similar to the surface display construct, with a synthetic prepro leader sequence directing secretion and a six histidine epitope for purification.
Yeast Growth and Induction
Yeast were transformed using the LiAc/ssDNA/PEG method.38 For surface display strain EBY100, transformants were selected on tryptophan and uracil deficient SD-CAA agar plates (20.0 g/L dextrose, 6.7 g/L yeast nitrogen base, 5.0 g/L casamino acids, 10.19 g/L Na2HPO4·7H2O, 8.56 g/L NaH2HPO4·H2O, 15 g/L agar). For secretion strain YVH10, transformants were selected on leucine and uracil deficient SD-2XSCAA + Trp agar plates (20 g/L dextrose, 6.7 g/L yeast nitrogenous base, 10.19 g/L Na2HPO4·7H2O, 8.56 g/L NaH2HPO4·H2O, 15 g/L agar 190 mg/L Arg, 108 mg/L Met, 52 mg/L Tyr, 290 mg/L Ile, 440 mg/L Lys, 200 mg/L Phe, 1260 mg/L Glu, 400 mg/L Asp, 480 mg/L Val, 220 mg/L Thr, 130 mg/L Gly, and 40 mg/L Trp, lacking leucine and uracil).
EBY100 yeast were grown in SD-CAA medium (20.0 g/L dextrose, 6.7 g/L yeast nitrogen base, 5.0 g/L casamino acids, 10.19 g/L Na2HPO4·7H2O, 8.56 g/L NaH2HPO4·H2O) until a culture density OD600 nm = 1.0 was reached. Surface display was induced by replacing the media with an equivalent volume of SG-CAA (20 g/L galactose replacing dextrose) for 20 h at 20 °C, 260 rpm. Yeast secretion strain YVH10 was grown in SD-2XSCAA + Trp (20 g/L dextrose, 6.7 g/L yeast nitrogenous base, 10.19 g/L Na2HPO4·7H2O, 8.56 g/L NaH2HPO4·H2O, 190 mg/L Arg, 108 mg/L Met, 52 mg/L Tyr, 290 mg/L Ile, 440 mg/L Lys, 200 mg/L Phe, 1260 mg/L Glu, 400 mg/L Asp, 480 mg/L Val, 220 mg/L Thr, 130 mg/L Gly, and 40 mg/L Trp, lacking leucine and uracil) at 30 °C, 260 rpm overnight. The following day, cultures were reset to an OD600 nm = 0.1, and grown for 72 h at 30 °C, 260 rpm. Yeast were induced by replacing the media with an equivalent volume of SG-2XSCAA + Trp (20 g/L galactose replacing dextrose) containing 0.1% w/v bovine serum albumin (BSA) and culturing the cells for 72 h at 20 °C and 260 rpm.
EGFR Cell Lines and Creation of Cell Lysates
A431 (ATCC) and U87-EGFRvIII (kindly provided by Dr. Donald O’Rourke and Dr. Gurpreet S. Kapoor, University of Pennsylvania, Department of Neurosurgery) cell lines were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM, Life Technologies) supplemented with 10% HyClone Cosmic Calf Serum (Thermo-Fisher) and 1× antibiotic/antimycotic (PSA, Gibco) at 37 °C and 5% CO2. To prepare for lysis, cells were grown to ∼90% confluence in 75 cm2 tissue culture-treated T-flasks and washed three times with PBS. Cells were lysed by the addition of ice-cold 1 mL lysis buffer, consisting of 1% v/v Triton X-100 (Thermo-Fisher), 2 mM EDTA, and 1× Complete Protease Inhibitor Cocktail (Roche). Cells were scraped from the flask using a cell scraper at 4 °C and collected into a microfuge tube. The lysed cells were rotated at 4 °C for 15 min and centrifuged for 30 min to remove insoluble cell debris. The clarified lysates were then used to label yeast or antibody-conjugated beads as described below.
Intein Library Construction
Mutagenesis of the Mxe GyrA for the initial library creation was performed by error-prone PCR39 of the pCT4Re-4420-intein construct containing intein using the nucleotide analogs 2′-deoxy-p-nucleoside-5′-triphosphate and 8-oxo-2′-deoxyguanosine-5′-triphosphate (TriLink Biotech) and primers (4420-intein-F 5′-CAGAACAAAAGCTTATTTCTGAAGAAGACTTGGCGGCCGCCGGCTGCATC-3′) and (4420-intein-R 5′-GGTGGTGGTGGTTCTGGTGGTGGTGGTTCTGGTGGTGGTGGTTCTGGATC-3′) that amplified the intein sequence but preserved the amino-terminal cysteine that is essential to protein splicing. The intein library was created by homologous recombination in EBY100 using the mutagenized intein PCR product and the NotI/AleIII linearized pCT4Re-4420-intein acceptor vector (Figure 1a). The initial library size was determined to be 2.5 × 107 clones by colony count. Twelve random yeast colonies were sequenced to determine an average nucleotide mutation rate of ∼1.7%.
The second intein library was created by shuffling the mutations of clones 202–03, 202–08, 202–12, 202–13, 505–05, and 505–11 through assembly of degenerate oligonucleotides.40 DNA oligonucleotides spanning the Mxe GyrA sequence were designed to contain the nucleotide base pair mutations at a 25:75 mutant/wild-type ratio. The intein gene was assembled from the oligonucleotides as previously described,41 and additional mutagenesis of the assembled gene was performed with error prone PCR. The library was created by homologous recombination as described above, and the initial library size was determined to be 3.2 × 107 clones by colony count. An average nucleotide mutation rate of 1.8% was determined by sequencing 22 of the yeast colonies.
Library Screening
The first intein library was screened via fluorescence activated cell sorting (FACS) in five rounds of enrichment. For the first round of FACS, 2 × 108 yeast from the initial library were labeled to detect FLAG tag expression using the flow cytometry procedure described below. Clones with the highest expression level (∼5%) were selected using a Becton Dickinson FACSVantage SE sorter (University of Wisconsin Comprehensive Cancer Center). Using yeast from the previous sort, rounds 2–4 were completed in a similar manner. For the fifth round, yeast clones exhibiting both high construct expression and 4–4–20 activity were selected by labeling for the FLAG tag and binding to FITC-dextran. From the second intein library, 1.5 × 108 cells were labeled to detect FLAG tag expression and FITC-dextran binding, and clones with the highest expression level and binding (5%) were selected. Four additional rounds of FACS were performed, each time enriching the pool from the previous sort for high expression and FITC-dextran binding.
Individual clones were isolated by plating the final library pools on selective media (SD-CAA) and selecting single colonies for characterization. Plasmids were recovered from the yeast with the ZymoPrep Yeast Plasmid Miniprep II Kit (Zymo Research), and clones were sequenced with the following primers: mxe4420seq_F (5′ TCTGTGAAAGGCAGATTCACCA3′) and mxe4420seq_R (5′ACAAAGAGTACGGCGTCGATT3′). Clones were retransformed into parent strain EBY100 for subsequent analysis.
Flow Cytometry
To determine surface display expression levels, the following anti-FLAG immunolabeling steps were performed at 4 °C prior to flow cytometry analysis. Induced EBY100 yeast were incubated with an anti-FLAG rabbit polyclonal antibody (Sigma–Aldrich, diluted 1:500 in PBS containing 0.1% BSA, PBS-BSA) for 30 min and washed once with PBS-BSA. Secondary antibody labeling was performed by incubating with anti-rabbit Alexa 488 (Life Technologies, diluted 1:500 PBS-BSA), anti-rabbit PE (Sigma-Aldrich, diluted 1:45 in PBS-BSA), or anti-rabbit allophycocyanin (APC) (Life Technologies, diluted 1:500 in PBS-BSA) for 30 min, followed by a final wash with PBS-BSA. To evaluate 4–4–20 binding activity, yeast were incubated with 10 μM fluorescein isothiocyanate-functionalized dextran in PBS-BSA (FITC-dextran, Sigma-Aldrich) for 30 min at 4 °C followed by washing once with PBS-BSA prior to flow cytometry analysis. Activity of surface-displayed scFv2 and 2224 was evaluated by incubating yeast with purified human EGFR isolated from A431 cells by immunoaffinity chromatography42 (4 μg/mL in PBS-BSA) for 1 h at 4 °C, followed by washing once with PBS-BSA. Yeast were next incubated with anti-EGFR mouse antibody cocktail Ab-12 (Lab Vision Corporation, diluted 1:200 in PBS-BSA) for 30 min, washed once with PBS-BSA, and labeled with anti-mouse PE (Sigma-Aldrich, diluted 1:40 in PBS-BSA) for 30 min followed by a final wash with PBS-BSA. Binding of MR1 to EGFRvIII was evaluated by yeast display immunoprecipitation (YDIP).43 Yeast were incubated with undiluted U87-EGFRvIII lysates in PBS containing 1% v/v Triton-X-100 (PBS-TX) for 1 h at 4 °C, followed by washing once with PBS-TX and anti-EGFR primary and secondary antibody labeling steps as performed for scFv2 and 2224. GFP activity was evaluated by measuring the GFP fluorescence of the yeast at 488 nm excitation. The yeast cell fluorescence was measured using a FACSCalibur flow cytometer (Becton Dickinson), and the geometric mean fluorescence intensities of the protein displaying populations were quantified with the FlowJo software package to determine relative display levels and activities.
Protein Purification
Following YVH10 growth and induction at the 50 mL scale, the yeast supernatant containing the secreted proteins was separated from the yeast by centrifugation and dialyzed against Tris-buffered saline (TBS, 25 mM Tris, 150 mM NaCl, 2 mM KCl, pH 7.9). The purification column was loaded with 750 μL Ni-NTA agarose (Qiagen) and equilibrated with 10 mL of bind buffer (TBS with 5 mM imidazole) prior to loading the dialyzed yeast supernatant. The column was subsequently washed with 15 mL of bind buffer followed by 3 mL of wash buffer (TBS with 20 mM imidazole), and the proteins were eluted with 2 mL TBS containing 250 mM imidazole.
SDS-PAGE and Western Blotting
Protein samples were reduced and denatured by boiling in LDS sample buffer (Life Technologies) containing 1 mM 2-mercaptoethanol for 10 min prior to resolution on 4–12% Bis-Tris gels (Life Technologies). Under these conditions, no additional intein cleavage above that of the 20-h MESNA reaction is observed. Proteins were subsequently transferred to a nitrocellulose membrane for Western blot analysis. Detection of FLAG tagged proteins was performed by probing the membranes with anti-FLAG M2 mouse monoclonal antibody (Sigma-Aldrich, diluted 1:3000) followed by anti-mouse HRP conjugate (Sigma-Aldrich, diluted 1:2000). To detect biotinylated proteins, membranes were probed with anti-biotin mouse monoclonal antibody Ab-2 clone BTN.4 (Lab Vision Corporation, diluted 1:500) followed by anti-mouse HRP conjugate. Membranes were developed using Clarity Western ECL Substrate (Bio-Rad) and imaged with the ChemiDoc XRS+ system (Bio-Rad). Unsaturated band intensities were measured with the Image Lab Software (Bio-Rad) to quantify the relative protein amounts.
Fluorescein Quench Assay and GFP Activity
The Kd value for secreted 4–4–20 and 4–4–20–202–08 was calculated by fluorescein quenching as previously described.32,44 Yeast supernatants containing the soluble proteins were dialyzed against TBS prior to analysis. Fluorescein (Sigma–Aldrich) was added stepwise to 1 mL of the dialyzed supernatant, and the resulting fluorescence at 514 nm was monitored using a Fluoromax-3 Spectrofluorometer (Horiba) and an excitation wavelength of 492 nm. The fluorescence intensities were fitted to an equilibrium binding model to determine the concentration and Kd of the 4–4–20 proteins.
Secreted GFP activity was determined by measuring the emission spectrum of purified samples at 488 nm excitation with the Fluoromax-3 spectrofluorometer, and the area under the curve was calculated. Anti-FLAG quantitative Western blotting was performed to determine relative GFP expression levels, and the fluorescence intensity was divided by expression level to calculate specific activity.
Intein-Mediated Release and EPL
The ability to release the scFvs and GFP from the display construct in an intein-dependent manner was also evaluated as previously described.8 Briefly, yeast displaying the intein-linked constructs were incubated with 50 mM 2-mercapthoethanesulfonic acid (MESNA, Sigma-Aldrich) in TBS for 45 min at room temperature (RT) to release a mixture of scFvs and scFv-intein-Aga2p fusion proteins. The yeast were subsequently removed from the reaction mixture by centrifugation, and the supernatant containing MESNA and the released proteins was allowed to react for 20 h to complete release of the scFvs from intein. The released proteins were subjected to anti-FLAG Western blot analysis. Expressed protein ligation (EPL) with a biotinylated cysteine peptide was also performed as previously described.8 Following the 45 min reaction of the yeast with the MESNA solution, the released proteins were separated from the yeast by centrifugation and 1 mM Bio-P1 peptide was added (synthesized by the University of Wisconsin Biotechnology Center, Sequence: NH2-CDPEK(Bt)DS-CONH2). The combined release and EPL reaction was allowed to proceed for 20 h at RT, and the proteins were analyzed with an anti-biotin Western blot.
For release and functionalization of the secreted scFvs and GFP, 100 μL of 1 M MESNA was added to 900 μL of purified scFv- or GFP-intein (∼5–300 mg fusion protein/L) and the reaction was allowed to proceed for 20 h at RT prior to anti-FLAG Western blot analysis. To generate azide-functionalized proteins, cysteine azide (Anaspec) was added to a final concentration of 5 mM during a combined 20-h release and EPL reaction. The proteins were subsequently dialyzed with TBS to remove unreacted cysteine azides prior to performing the immobilization reactions.
Protein Immobilization via Strain-Promoted Click Chemistry
The following protein immobilization and incubation steps were performed at RT with gentle rotation. Dibenzocyclooctyne (DBCO)-fuctionalized agarose (10 μL, Click Chemistry Tools) was blocked with 500 μL DBCO blocking buffer (PBS with 2% w/v BSA and 1% Tween-20) for 1 h. The blocking buffer was removed, and 200 μL of the azide-modified proteins were added to the beads for 2 h. The beads were subsequently washed twice with PBS-BSA and once with DBCO blocking buffer. To evaluate 4–4–20 binding to fluorescein, the antibody-linked beads were incubated in 10 μM FITC-dextran in PBS-BSA for 30 min followed by washing three times with PBS-BSA. Activity of the EGFR scFv was assayed by incubating the antibody-linked agarose beads with 200 μL of undiluted A431 cell lysates or U87-EGFRvIII cell lysates for 1 h. The beads were washed twice with PBS-TX and once with DBCO blocking buffer before incubation with 200 μL anti-EGFR antibody cocktail Ab-12 (1:200 dilution in DBCO blocking buffer) for 30 min. The antibody-linked beads were subsequently washed twice with PBS-BSA and once with DBCO blocking buffer, followed by incubating with 200 μL anti-mouse Alexa 488 antibody (1:500 dilution in DBCO blocking buffer) for 30 min. The beads were washed three times with PBS-BSA. Beads were imaged with an Olympus IX70 fluorescence microscope, and the fluorescence intensities of the beads at 509 nm were measured using a Tecan Infinite M1000 fluorescent microplate reader with an excitation wavelength of 488 nm.
Results
Intein Library Generation and Screening
Evolution of the non-self-cleaving Mxe GyrA intein was performed with the primary screening criterion being increased yeast display of an scFv-intein fusion. The anti-fluorescein scFv (4–4–20) construct was employed as the fusion partner since intein fusion decreased yeast display by 40% compared with unfused 4–4–20 display,8 offering a convenient screening pressure of improved yeast display (Figures 1a and 2a). For the first round of directed evolution, random mutagenesis was selectively targeted to the intein moiety, and upon recombination with unmutated 4–4–20, a library of ∼2.5 × 107 4–4–20-intein fusion mutants was generated. The library was enriched for clones with elevated full-length surface expression (FLAG epitope tag) through four rounds of fluorescence activated cell sorting (FACS), followed by one additional round of FACS that ensured retention of 4–4–20 binding activity by using fluorescein labeling in addition to FLAG epitope labeling (Figure 2a). A 1.7-fold increase in display and fluorescein binding compared to the wild-type intein was observed in this final sorted pool (Figure 2b, compare panels i and ii), and the display levels of the 4–4–20-intein fusion were restored to that of the unfused 4–4–20 protein (Figure 2b, panel iv).
Figure 2.

Directed evolution of the Mxe GyrA intein. (a) For directed evolution round 1, the Mxe GyrA intein library was created by random mutagenesis and recombined into the pCT4Re-4420 construct. The library was screened in four rounds of enrichment for improved FLAG tag expression via FACS. A fifth round of enrichment selected for both improved FLAG tag expression and comensurate increases in fluorescein binding. Individual clones were isolated and screened for intein activity by the addition of MESNA, which releases the scFv from the display construct when an active intein is present. For directed evolution round 2, the round 1 clones were shuffled and mutagenized prior to screening for increased display levels. (b) Flow cytometry dot plots depicting expression and binding activity of scFv-intein clones and pools on the yeast surface. Geometric mean flurescence intensity (MFI) of the FLAG signal for the entire displaying population is shown to allow comparison. In addition, a sample sort gate is shown to illustrate the enrichment. Panel i, wild-type intein fusion; panel ii, round 1 final selected pool; panel iii, round 2 final selected pool, panel iv, unfused 4–4–20 scFv; panel v, round 1 202–08 intein mutant. (c) The MFI of the displaying population was quantified and normalized to the wild-type 4–4–20-intein contruct to compare the relative expression levels (FLAG) and activity (fluorescein binding) of the unfused 4–4–20 construct, wild-type intein construct, and the 202–08 intein mutant. Activity per molecule is expressed as the ratio of fluorescein binding to FLAG expression level. Plotted are the means ± SD from three independent yeast transformants. Statistically significant improvements over the wild-type intein construct were determined by an unpaired Student’s t-test (*p < 0.05; **p < 0.01; NS, not significant p > 0.05). Display data for other individual intein mutants are compiled in Table 1. (d) Quantitative anti-FLAG Western blotting was performed to determine the relative amount of 4–4–20 released from the yeast surface in the MESNA reaction. Plotted are means ± SD for three independent reactions originating from three independent yeast surface display transformants. Next to the bar graph are the triplicate Western blot data at the cleaved scFv size of ∼30 kDa. A small amount of the uncleaved, scFv-intein product appears at a size of ∼90 kDa due to its fusion to glycosylated Aga2p. The double asterisk represents a statistically significant increase in 4–4–20 release for clone 202–08 (p < 0.01) as determined by an unpaired Student’s t-test. (e) The crystal structure of the Mxe GyrA intein (pdb ID: 1AM2(64)) is shown with the mutations found in the 202–08 intein highlighted. A flexible loop missing from the crystal structure is denoted by a dotted line and the structure on the right was rotated 90°.
Individual intein clones were next isolated and evaluated for display levels and intein activity. Since mutations to the Mxe GyrA intein could potentially inhibit intein activity,45 the clones were first screened for activity by examining 4–4–20 release from the 4–4–20-intein fusion construct by reaction with a sulfur nucleophile, MESNA. The wild-type intein catalyzes an N- to S-acyl shift at the amino-terminal cysteine of the intein, forming a thioester that is susceptible to a nucleophilic attack. Reaction with a nucleophile, such as MESNA, releases 4–4–20 from the intein and the yeast display construct while simultaneously appending a carboxy-terminal thioester onto 4–4–20 (Figure 3c).8 Because MESNA also reduces the disulfide bonds between Aga1p and Aga2p on the yeast surface (Figure 2a),8 4–4–20 release from the intein could not be measured inline with the screen by flow cytometry. Instead, intein activity was determined for each individual clone via anti-FLAG Western blotting (shown in Figure 2d for clone 202–08). Six mutated intein clones exhibited an increase in surface display over the wild-type intein and retained their cleavage activity (Table 1). In an attempt to further improve 4–4–20-intein fusion display levels, these six clones were shuffled and additionally mutated to create a library containing ∼3.2 × 107 clones for a second round of directed evolution (see Methods for details). In the second round of evolution, the library was again screened for elevated display levels and fluorescein binding over five rounds of FACS. Characterization of the final pool demonstrated an increase in display levels compared to the wild-type intein, but display level was not significantly greater than that achieve through the first round of directed evolution (Figure 2b, iii), as also confirmed by evaluation of individual clones (Supporting Information Table 1).
Figure 3.

Analysis of surface displayed scFv- and GFP-202–08 fusions. (a) Surface display levels of unfused, wild-type intein fused or 202–08 intein fused scFvs and GFP were analyzed by flow cytometry. The MFI of the FLAG-positive yeast populations was quantified, and all were normalized to the 4–4–20 construct containing the wild-type intein. Reported are the means ± SD of three independent yeast transformants. Statistical analysis was performed by an unpaired Student’s t-test (*p < 0.05; **p < 0.01; NS, not significant p > 0.05). (b) ScFv and GFP per molecule activity was evaluated by detecting binding to the scFv antigens at saturating ligand concentrations or by measuring GFP fluorescence. Activity per molecule was determined by calculating the ratio of the geometric means for activity (binding or fluorescence) to FLAG expression levels and normalizing to the unfused construct lacking intein. Plotted are the means ± SD from three independent yeast transformants, with statistical significance determined by an unpaired Student’s t-test (*p < 0.05; **p < 0.01; NS, not significant p > 0.05) (c) For intein-mediated protein release, MESNA reacts to release the scFv or GFP from the display construct and append a carboxy-terminal thioester. For EPL functionalization, the carboxy-terminal thioester reacts with a biotinylated peptide containing an amino-terminal cysteine to covalently link the scFv or GFP to the biotin by an amide bond. (d) Products of the reaction depicted in panel c resolved and analyzed by Western blotting to detect release of the scFv or GFP (∼30 kDa) from the 202–08 intein construct using an anti-FLAG antibody (F) or biotin functionalization via EPL with an anti-biotin antibody (B). A small amount of uncleaved scFv-intein-Aga2p product can be seen in the anti-FLAG Western blot between ∼80 kDa and 100 kDa due to the glycosylation of Aga2p.
Table 1. Intein Mutations and Surface Display Levels.
| amino
acid |
|||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 21 | 33 | 50 | 51 | 74 | 105 | 107 | 110 | 112 | 114 | 117 | 118 | 124 | 129 | 144 | 158 | 160 | 164 | 168 | 190 | 191 | fold increasea | statistical significanceb | |
| wild-type | I | I | L | F | N | I | R | F | V | C | F | A | F | Y | H | D | R | A | S | I | T | 1.0 ± 0.0 | |
| 202–03 | D | R | R | G | 1.4 ± 0.1 | ** | |||||||||||||||||
| 202–08 | L | V | C | S | L | L | G | T | 1.8 ± 0.2 | ** | |||||||||||||
| 202–12 | T | P | A | C | 1.5 ± 0.3 | * | |||||||||||||||||
| 202–13 | T | S | G | T | Q | 1.3 ± 0.2 | NS | ||||||||||||||||
| 505–05 | T | R | V | 1.3 ± 0.1 | ** | ||||||||||||||||||
| 505–11 | C | P | M | 1.7 ± 0.1 | ** | ||||||||||||||||||
Fold increase relative to the wild-type intein as fusions to 4–4–20, mean ± SD from three independent yeast transformants.
Statistical analysis was perfomed by an unpaired Student’s t-test, with double asterisks representing p < 0.01, single asterisks representing p < 0.05, and NS designating that differences are not significant (p > 0.05).
Surface Display Characterization of the 202–08 Intein
Clone 202–08 from round 1 of directed evolution was selected for further characterization based upon its elevated display levels, retention of protein splicing activity in the presence of MESNA, and its capability to also significantly elevate secretion of scFv-intein fusions (discussed below). The 202–08 intein contained eight amino acid mutations (Table 1, Figure 2e) and increased the surface display level of 4–4–20 1.8-fold compared to the wild-type intein fusion (Figure 2b, v), making its display level comparable to that of the unfused 4–4–20 protein (Figure 2c). Furthermore, the fluorescein binding per molecule of 4–4–20 was unchanged by fusion to 202–08 (Figure 2c). The retention of 202–08 catalytic activity was confirmed by examining the relative amount of 4–4–20 cleaved from the yeast surface display construct in a MESNA release reaction. Quantitative Western blotting demonstrated a 2.6-fold increase in the amount of 4–4–20 released from yeast with clone 202–08 compared to the wild-type intein (Figure 2d), consistent with the increased surface display levels mediated by the 202–08 intein (Figure 2c).
Next, the generalizability of the 202–08 intein mutant was evaluated by testing effects on display and activity after its fusion to GFP and a cohort of 7 additional scFvs. The tested scFvs included three epidermal growth factor receptor (EGFR)-binding scFvs, scFv2,46 MR1,33 and 2224,34,35 and a panel of brain endothelial-binding scFvs, scFvA, scFvD, scFvH, and 4S2136 that collectively exhibit a range of unfused expression levels on the yeast surface (Figure 3a). The expression level of GFP was unchanged upon fusion to wild-type intein as previously reported,8 while scFv fusion to the wild-type intein generally decreased construct expression levels ∼25–50%, regardless of unfused display efficiency (Figure 3a). The lone exception was 2224, where both the unfused and wild-type intein-fused forms exhibited similar, low display levels (Figure 3a). When each scFv or GFP was instead expressed as a fusion to the 202–08 intein, display was uniformly improved compared to the wild-type intein fusion reaching levels similar to or greater than that of the unfused protein (Figure 3a). Next, the activity of GFP- and EGFR-specific scFv-intein fusions was evaluated to ensure the 202–08 intein did not have deleterious effects on the specific activity of its fusion partner. Much like the case of 4–4–20, GFP fluorescence activity was not altered by fusion with the 202–08 intein (Figure 3b). Interestingly, compared with unfused scFv, fusion to 202–08 yielded small increases in per molecule EGFR binding for scFv2 and MR1, while 2224 exhibited more substantial 1.5-fold increases in binding to its EGFR ligand (Figure 3b).
Protein Release and EPL for 202–08 Intein Fusions
After demonstrating that intein clone 202–08 improved surface display of multiple scFvs and GFP, the intein cleavage activity was next confirmed. Yeast displaying 202–08 intein fusion proteins were reacted with MESNA to release the scFvs or GFP from the display construct, thereby generating scFv- and GFP-thioester proteins (Figure 3c). Western blotting with an anti-FLAG antibody demonstrated nearly quantitative release of each of the scFvs and GFP from the 202–08 fusion display construct (Figure 3d). The installation of the carboxy-terminal thioester functionality produced by intein-mediated release was confirmed by subjecting the MESNA-released scFvs and GFP to an EPL reaction with a biotinylated peptide possessing an amino-terminal cysteine (Figure 3d). Anti-biotin Western blotting demonstrated successful biotinylation of the scFvs and GFP (Figure 3d), indicating that the engineered 202–08 intein produces carboxy-terminal thioesters capable of EPL.
Secretion of scFv-intein Fusion Proteins
Next, yeast secretion constructs were designed to flank scFv or GFP inserts with the FLAG epitope tag at the amino-terminus and a six histidine epitope tag at the carboxy-terminus to permit protein detection (before or after intein release) and purification, respectively (Figure 1b). Similar to the surface display experiments, secretion of unfused scFv or GFP was compared directly to the secretion of the same protein as a fusion to the amino-terminus of the wild-type or 202–08 intein (Figure 1b). Along with GFP, four scFvs (4–4–20 and the EGFR-binding scFvs, scFv2, MR1, and 2224) were examined in the protein secretion studies. When the scFvs were produced as fusions to wild-type intein, quantitative Western blotting analysis demonstrated substantial decreases in scFv secretion, ranging from 75% (MR1) to 99% (scFv2) reduction compared to the unfused scFv, while GFP expression did not decrease when fused to wild-type intein (Figure 4a). However, as observed with surface display, secreting the scFvs and GFP as fusions to the evolved 202–08 intein substantially improved the protein production compared to the wild-type intein fusion (Figure 4a). Expression of MR1 and 4–4–20 increased 3- and 10-fold, respectively, compared to the wild-type intein fusion to achieve secretion levels that were comparable to the unfused protein (Figure 4a). Fusion of 202–08 to scFv2 and 2224 increased secretion ∼30-fold and ∼3-fold over the wild-type intein fusions, respectively, although expression of these scFvs was not fully restored to the unfused protein level (Figure 4a). Furthermore, even though the GFP fusion to the wild-type intein did not decrease secretion compared to the unfused GFP, expression when fused to 202–08 was modestly improved (∼1.5 fold) over that of the wild-type intein (Figure 4a).
Figure 4.

Secretion of scFv and GFP intein fusion proteins. (a) Yeast supernatants containing scFv or GFP fused to the wild-type intein or 202–08 intein were subjected to anti-FLAG quantitative Western blotting and compared to the unfused target protein. Values are normalized to the level of the 4–4–20–202–08 fusion to determine relative amounts. The absolute secretion titer of the 4–4–20–202–08 fusion protein is 3.1 mg/L as determined in panel b. Reported are the means ± SD from three independent yeast transformants. Statistical significance was determined by an unpaired Student’s t-test (*p < 0.05; **p < 0.01; NS, not significant p > 0.05). Western blot of supernatant samples used for the quantitation of relative 4–4–20 protein secretion is shown below the bar graph. (b) An equilibrium binding curve was generated by fluorescein quenching to compare the Kd of unfused 4–4–20 and 4–4–20 fused to 202–08. A sample curve for each of the proteins is shown, and the mean ± SD for the fitted parameters of Kd value and 4–4–20 concentration were obtained by fitting quench curves generated from supernatants resulting from three independent yeast transformants. From the molar concentrations of 4–4–20, the average mass concentration of the 4–4–20 component was calculated to be 1.6 mg/L of yeast culture for both the unfused and the intein-fused 4–4–20 (corresponding to 3.1 mg/L for the full 4–4–20–202–08 fusion protein) The Kd and 4–4–20 concentrations were statistically indistinguishable, as determined by an unpaired Student’s t-test (p > 0.05). (c) GFP activity was determined by calculating the ratio of fluorescence to FLAG expression levels and normalizing to the unfused construct lacking intein. The mean ± SD results from three independent yeast transformants. The fluorescence per molecule of unfused GFP and 202–08 fused GFP was statistically indistinguishable, as determined by an unpaired Student’s t-test (**p > 0.05). (d) The catalytic activity of 202–08 was examined by reacting secreted and purified proteins with MESNA and evaluating cleaved yield after standard 20 h reaction. Anti-FLAG Western blotting demonstrates between 70% (2224) and 99% (MR1) release of the target protein from the 202–08 intein in the presence of MESNA.
Next, the activities of secreted scFv-intein fusion proteins were examined both from fusion partner and intein perspectives. First, 4–4–20 scFv and GFP activity was quantitatively evaluated using the secreted 4–4–20 and GFP intein fusion proteins (functionality of anti-EGFR scFvs evaluated as immobilized proteins below). The equilibrium binding affinity of 4–4–20 fused to 202–08 was measured in order to ensure that the antibody component of the fusion protein was folded and functional. Monitoring the fluorescence quench upon binding of fluorescein to 4–4–20 allowed determination of the equilibrium dissociation constant, Kd, of the 4–4–20–202–08 fusion protein to be 1.5 ± 0.4 nM, making it statistically indistinguishable (p > 0.05) from that of the unfused 4–4–20 protein (1.9 ± 0.5 nM) (Figure 4b). The activity of GFP fused to 202–08 was assessed by measuring its fluorescence per molecule and was shown to be identical to that of the unfused GFP (p > 0.05) (Figure 4c). Next, the intein-mediated release of the scFv or GFP from the 202–08 intein was evaluated by reacting the secreted and purified scFv or GFP fusion proteins with MESNA. All four scFvs along with GFP were released from the intein with efficiencies ranging from 70 to 99%, thus demonstrating that the 202–08 intein component is active when produced as a soluble fusion protein (Figure 4d). Similar release efficiencies were observed for wild-type intein fusion proteins indicating that the engineered intein did not affect the cleaved scFv or GFP yields (Supporting Information Figure 1).
Immobilization of scFv and GFP via Strained Cycloaddition Reaction
Next, by employing EPL functionalization techniques,8,13 the scFvs and GFP were chemically functionalized to enable covalent immobilization of the proteins onto surfaces. The secreted and purified scFv- and GFP-202–08 intein fusion proteins were reacted with MESNA in the presence of cysteine azide, thereby releasing the scFv or GFP from the intein and installing a carboxy-terminal azide onto the protein (Figure 5a). The azide-modified scFvs and GFP were subsequently reacted with dibenzocyclooctyne (DBCO)-functionalized agarose beads to immobilize the proteins via strain-promoted azide–alkyne cycloaddition (SPAAC) (Figure 5a). In this way, GFP-azide protein was immobilized on the beads and yielded roughly 40-fold more GFP fluorescence than beads reacted with the control thioester functionalized GFP, indicating specific SPAAC-mediated immobilization of active GFP protein (Figure 5b). Similarly, immobilization and activity of 4–4–20 was confirmed by specificity of fluorescein binding to beads loaded with 4–4–20-azide, but not EGFR-specific scFv2-azide (Figure 5c). Finally, beads reacted with azide-functionalized EGFR scFvs were shown to bind their antigens from whole cell lysates that contained either wild-type EGFR or mutant EGFR vIII. ScFv2 and scFv2224 recognize epitopes conserved on both wild-type34,35,46 and vIII EGFR isoforms,34 while MR1 is a vIII-specific scFv.33 Accordingly, if beads decorated with scFv2, 2224, or MR1 were incubated with cell lysates containing the EGFR vIII mutant, they all bound EGFR vIII as expected, while the anti-fluorescein 4–4–20 scFv exhibited negligible nonspecific binding to the cell lysates (Figure 5d). When incubated with wild-type EGFR-containing A431 cell lysates, beads loaded with scFv2-azide and 2224-azide again exhibited a clear binding signal. In contrast, beads loaded with MR1 exhibited a marked, 85% reduction in A431-derived EGFR binding signal compared to that generated from EGFR vIII cell lysates, indicating a clear preference for MR1 binding to the EGFR vIII mutant (Figure 5d). Taken together, each of the scFvs retained antigen-specific binding activity after being produced as secreted protein-intein fusions, EPL reaction, and SPAAC immobilization.
Figure 5.

Strain-promoted click chemistry immobilization. (a) Secreted and purified scFv and GFP proteins fused to the 202–08 intein were released with MESNA to form scFv- and GFP-thioesters. The carboxy-terminal thioesters were subsequently reacted with a cysteine azide via EPL to install an azido group onto the protein. To immobilize the proteins on surfaces, the scFv- and GFP-azide proteins were reacted with DBCO-functionalized agarose beads in a strain promoted click chemistry reaction. (b) Fluorescent microscope images of GFP fluorescence associated with beads reacted with GFP-azide or nonazido GFP (GFP-thioester). Relative protein immobilization was quantified by measuring total bead fluorescence and normalizing to the azide-GFP loaded beads. The mean ± SD of three independent immobilization reactions is plotted. Statistical significance was determined by an unpaired Student’s t-test (**p < 0.01) (c) Binding of fluorescein to beads reacted with azide functionalized 4–4–20 was analyzed and compared to beads reacted azide-linked scFv2. FITC-dextran binding was quantified by measuring the fluorescence intensity of the beads, and the fluorescence was normalized to the 4–4–20-linked sample. Three independent immobilization reactions were carried out to obtain the mean ± SD values. An unpaired Student’s t-test was performed to determine statistical significance (**p < 0.01) (d) Immobilized EGFR scFv activity was assessed by EGFR capture from cell lysates. Fluorescent microscopy images were employed to demonstrate EGFR capture and EGFR isoform specificity. A431 cells express wild-type EGFR while U87 cells are transfected to express the EGFR vIII isoform. ScFv activity was quantified by measuring the resulting fluorescence intensity of the beads, and the fluorescence value was normalized to the signal originating from the U87-EGFRvIII lysate binding to the respective scFv. The fluorescence value for the negative control, 4–4–20, was normalized to the signal originating from the U87-EGFRvIII binding to MR1. The mean ± SD of three independent immobilization reactions is plotted. Statistical significance was determined by an unpaired Student’s t-test (**p < 0.01).
Discussion
Producing antibodies as fusion partners to the non-self-cleaving Mxe GyrA intein enables site-specific, bioorthogonal chemical protein modification, thereby enabling antibody conjugation to desired small molecules, proteins, or surfaces. Through directed evolution, we have engineered the Mxe GyrA intein to increase the amount of scFv-intein fusion proteins displayed on the yeast surface by ∼1.5- to 3-fold, thus increasing the amount of chemically functionalized protein obtained via intein-linked yeast surface display. Importantly, the engineered 202–08 intein clone was shown to be generalizable by increasing the surface display of GFP and eight different scFvs Furthermore, we demonstrated that the engineered intein improves secretion of scFv-intein fusion proteins by ∼3- to 30-fold over the wild-type intein. Finally, secreted scFvs could be directly modified via EPL, immobilized onto surfaces using SPAAC, and employed to bind their respective antigens.
While previous studies have employed rational design to improve Mxe GyrA production levels by reducing in vivo autocleavage16,18 or by reducing intein size,16 we instead employed directed evolution to achieve this goal. The surface display levels of scFv-intein fusions are generally 25–50% reduced compared to the unfused scFv, thus providing a screening pressure for improved intein clones. Although the screen employed intein fusion to the anti-fluorescein scFv, 4–4–20, intein clone 202–08 increased surface display of seven additional scFvs that exhibited a range of display levels as unfused proteins. For many different scFvs, 202–08 returned surface display of scFv-intein fusions back to unfused levels and this unfused display level appeared to be the ceiling for 4–4–20 expression, given the inability to achieve further expression increases in a second round of directed evolution. However, the display levels of two of the tested proteins, GFP and 2224, fused to the 202–08 intein did exceed that of the respective unfused proteins, and the 2224 scFv had an improved EGFR-specific binding capacity, indicating beneficial folding and processing effects of the intein fusion partner. These two proteins also did not demonstrate a decrease in expression upon fusion to the wild-type intein, and so the “chaperone-like” effects of the 202–08 may be limited to proteins that are better equipped to handle intein fusion. It has previously been reported that surface display levels often correlate with secretion levels,21,25−27 and that modest elevation in surface display can lead to substantial increases in protein secretion.47 Similarly, in this study, fairly modest display improvements produced by 202–08 resulted in substantial secretion improvements. For two of the scFvs, 4–4–20 and MR1, the 202–08 fusion increased expression 10- and 3-fold, respectively, to restore the secretion level to that of the unfused protein. Although unfused protein secretion levels were not restored for all of the tested scFvs, substantial increases in secretion were still obtained. As a result of 202–08 fusion, scFv production levels using the basal low-copy expression vector were estimated to range from 90 μg to 1.6 mg per liter of yeast culture for the antibodies tested here (6 mg/L for GFP), which is consistent with typical scFv yields in yeast,22 and greatly improves upon that for wild-type intein fusions (30 to 250 μg/L). In addition, much like the 202–08 intein fusion yeast surface display levels, the 202–08 fusion secretion levels tend to track reasonably well with those of the unfused proteins, suggesting that the engineered intein has minimized the detrimental effects of intein fusion on protein secretion. Thus, the 202–08 intein should be generally compatible with fusion protein partners that are successfully produced in Saccharomyces cerevisiae.
Non-self-cleaving intein fusion proteins have traditionally been expressed in the cytoplasm of E. coli, where they are often produced as insoluble inclusion bodies, thus requiring protein solubilization and refolding in order to obtain active protein.7,10,14−16,18 In addition to requiring postproduction processing to produce active intein-fusion proteins, the refolding process can result in thioester hydrolysis, thus preventing or substantially reducing subsequent EPL functionalization of the target protein.7,10 One possibility to circumvent refolding issues in bacteria would be targeting of fusion proteins to the periplasm, where the oxidizing environment enables the formation of disulfide bonds and can potentially provide advantages for protein folding. This approach was successful with a single-domain antibody (sdAb) fused to the Mxe GyrA intein6 but has not yet been demonstrated for a broad panel of antibody fusion partners. In addition, since some antibodies are still expressed as unfolded aggregates in the periplasm,48−50 while others simply cannot be expressed,50,51 periplasmic expression of antibody-intein fusions may have limitations. Thus, as an alternative, expressing scFv-intein fusion proteins in a eukaryotic organism such as yeast could be beneficial. Indeed, by employing the evolved 202–08 intein, a panel of active scFv- and GFP-intein fusion proteins could be displayed or secreted from yeast and directly functionalized via EPL without any solubilization or refolding steps. In addition, expression levels in yeast and bacteria are often quite similar when comparing the same scFvs,52 and 202–08 intein fusion expression levels for the more well-expressed scFvs tested were similar to the reported ∼2–5 mg/L levels for scFv- and sdAb-intein fusion proteins using bacteria.6,7 Thus, the yeast-based 202–08 intein system, with its combination of reasonable fusion protein yields and proper fusion protein folding, represents a competitive alternative to bacterial intein expression systems.
While we observed near complete release of the scFv or GFP with surface displayed intein fusion proteins, the secreted protein cleavage efficiencies ranged from 70 to 99% depending upon the fusion partner. The evolved 202–08 intein did not appear to affect cleavage efficiency compared with the wild-type intein, and these cleavage efficiencies are consistent with those observed for bacterially produced proteins.7,16,53 Furthermore, the release could possibly be enhanced by optimizing the carboxy-terminal residue of the target protein, which has previously been shown to impact the cleavage efficiency.53,54 Regardless, the small amount of uncleaved material is not chemically functionalized and would not impact many downstream applications like antibody immobilization, but the uncleaved material could be removed by depletion via histidine tag purification if desired.
The directed evolution process revealed that several different combinations of mutations led to improvements in scFv-intein surface display levels, and that no single mutation dominated either round of directed evolution (Table 1 and Supporting Information Table 1). A large percentage of the mutations (44%) found in the round 1 clones were within or in close proximity to the flexible loop of the Mxe GyrA intein that could not be resolved by crystallography (residues 112–129). Specifically, for 202–08, two of its eight mutations (F117L, F124L) fell within the flexible loop, while three other mutations occurred near the amino-terminus of the loop (I105V, R107C, F110S) (Figure 2c). Thus, it appears that modifications in and around the flexible loop may be key to improving fusion protein expression. This finding is also supported by a recent study where a smaller Mxe GyrA intein was created by deleting residues 107–160 (including the flexible loop) and replacing the deletion with a short glycine-serine linker. This smaller intein variant led to a 1.2-fold increase in intein-peptide fusion production in E. coli.16
The secreted, EPL-functionalized scFvs and GFP were shown to be compatible with strain-promoted click chemistry, thus demonstrating the utility of intein fusion protein production in yeast. A carboxy-terminal azide was installed via EPL, and using SPAAC, active scFv and GFP were site-specifically immobilized on beads decorated with a strained alkyne. Previously we had demonstrated the compatibility of yeast displayed scFv-intein fusions with one of the most widely used forms of click chemistry, copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC).8,55 CuAAC requires the addition of copper, a reducing reagent, and a stabilizing ligand. In contrast, SPAAC enables the direct immobilization of azide-conjugated proteins without a copper catalyst, reducing reagent, or stabilizing ligand. Not only does SPAAC simplify the conjugation process, but it also prevents issues associated the copper catalyst, such as protein precipitation12,56,57 and toxicity.58,59 Thus, the ability to employ these scFvs in SPAAC reactions offers many potential applications such as the generation of antibody-drug conjugates60,61 and targeted nanoparticles.62,63 In conclusion, directed evolution of the Mxe GyrA intein has permitted the extension of EPL and click chemistry modification techniques to scFvs secreted from yeast, thereby providing a viable alternative to bacterial expression systems and a facile method to chemically functionalize antibodies and other proteins.
Acknowledgments
This work was funded by National Institutes of Health grant R01 CA108467 and National Science Foundation grant CBET-1403350. The authors recognize the University of Wisconsin Comprehensive Cancer Center (UWCCC) Flow Cytometry Laboratory, a Shared Service of the UWCCC, funded by the UWCCC Support Grant P30 CA014520. Dr. D. O’Rourke and Dr. G. Kapoor at University of Pennsylvania Department of Neurosurgery generously provided the U87-EGFRvIII cell lines.
Supporting Information Available
Table of intein mutations and surface display levels for directed evolution round 2 and a Western blot of the MESNA release reaction for the secreted 4–4–20 and GFP as wild-type intein fusion proteins. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Adams G.; Shaller C.; Chappell L.; Wu C.; Horak E.; Simmons H.; Litwin S.; Marks J.; Weiner L.; Brechbiel M. (2000) Delivery of the α-emitting radioisotope bismuth-213 to solid tumors via single-chain Fv and diabody molecules. Nucl. Med. Biol. 27, 339–346. [DOI] [PubMed] [Google Scholar]
- Kuimova M. K.; Bhatti M.; Deonarain M.; Yahioglu G.; Levitt J. A.; Stamati I.; Suhling K.; Phillips D. (2007) Fluorescence characterisation of multiply-loaded anti-HER2 single chain Fv-photosesitizer conjugates suitable for photodynamic therapy. Photochem. Photobiol. Sci. 6, 933–939. [DOI] [PubMed] [Google Scholar]
- Nielsen U. B.; Kirpotin D. B.; Pickering E. M.; Hong K.; Park J. W.; Refaat Shalaby M.; Shao Y.; Benz C. C.; Marks J. D. (2002) Therapeutic efficacy of anti-ErbB2 immunoliposomes targeted by a phage antibody selected for cellular endocytosis. Biochim. Biophys. Acta, Mol. Cell Res. 1591, 109–118. [DOI] [PubMed] [Google Scholar]
- Natarajan A.; Xiong C. Y.; Albrecht H.; DeNardo G. L.; DeNardo S. J. (2005) Characterization of site-specific ScFv PEGylation for tumor-targeting pharmaceuticals. Bioconjugate Chem. 16, 113–121. [DOI] [PubMed] [Google Scholar]
- Lu R. M.; Chang Y. L.; Chen M. S.; Wu H. C. (2011) Single chain anti-c-Met antibody conjugated nanoparticles for in vivo tumor-targeted imaging and drug delivery. Biomaterials 32, 3265–3274. [DOI] [PubMed] [Google Scholar]
- Reulen S. W.; van Baal I.; Raats J. M.; Merkx M. (2009) Efficient, chemoselective synthesis of immunomicelles using single-domain antibodies with a C-terminal thioester. BMC Biotechnol. 9, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sydor J. R.; Mariano M.; Sideris S.; Nock S. (2002) Establishment of intein-mediated protein ligation under denaturing conditions: C-terminal labeling of a single-chain antibody for biochip screening. Bioconjugate Chem. 13, 707–712. [DOI] [PubMed] [Google Scholar]
- Marshall C. J.; Agarwal N.; Kalia J.; Grosskopf V. A.; McGrath N. A.; Abbott N. L.; Raines R. T.; Shusta E. V. (2013) Facile chemical functionalization of proteins through intein-linked yeast display. Bioconjugate Chem. 24, 1634–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayers B.; Blaschke U. K.; Camarero J. A.; Cotton G. J.; Holford M.; Muir T. W. (1999) Introduction of unnatural amino acids into proteins using expressed protein ligation. Pept. Sci. 51, 343–354. [DOI] [PubMed] [Google Scholar]
- Bastings M. M. C.; Van Baal I.; Meijer E.; Merkx M. (2008) One-step refolding and purification of disulfide-containing proteins with a C-terminal MESNA thioester. BMC Biotechnol. 8, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias D. R.; Cheng Z.; Tsourkas A. (2010) An intein-mediated site-specific click conjugation strategy for improved tumor targeting of nanoparticle systems. Small 6, 2460–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia J.; Raines R. T. (2006) Reactivity of intein thioesters: Appending a functional group to a protein. ChemBioChem 7, 1375–1383. [DOI] [PubMed] [Google Scholar]
- Lin P. C.; Ueng S. H.; Tseng M. C.; Ko J. L.; Huang K. T.; Yu S. C.; Adak A. K.; Chen Y. J.; Lin C. C. (2006) Site-specific protein modification through CuI-catalyzed 1,2,3-triazole formation and its implementation in protein microarray fabrication. Angew. Chem. 118, 4392–4396. [DOI] [PubMed] [Google Scholar]
- Valiyaveetil F. I.; MacKinnon R.; Muir T. W. (2002) Semisynthesis and folding of the potassium channel KcsA. J. Am. Chem. Soc. 124, 9113–9120. [DOI] [PubMed] [Google Scholar]
- Guo C.; Li Z.; Shi Y.; Xu M.; Wise J. G.; Trommer W. E.; Yuan J. (2004) Intein-mediated fusion expression, high efficient refolding, and one-step purification of gelonin toxin. Protein Expression Purif. 37, 361–367. [DOI] [PubMed] [Google Scholar]
- Albertsen L.; Shaw A. C.; Norrild J. C.; Strømgaard K. (2013) Recombinant production of peptide C-terminal α-amides using an engineered intein. Bioconjugate Chem. 24, 1883–1894. [DOI] [PubMed] [Google Scholar]
- Wood R. J.; Pascoe D. D.; Brown Z. K.; Medlicott E. M.; Kriek M.; Neylon C.; Roach P. L. (2004) Optimized conjugation of a fluorescent label to proteins via intein-mediated activation and ligation. Bioconjugate Chem. 15, 366–372. [DOI] [PubMed] [Google Scholar]
- Cui C.; Zhao W.; Chen J.; Wang J.; Li Q. (2006) Elimination of in vivo cleavage between target protein and intein in the intein-mediated protein purification systems. Protein Expression Purif. 50, 74–81. [DOI] [PubMed] [Google Scholar]
- Boder E. T.; Wittrup K. D. (1997) Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 15, 553–557. [DOI] [PubMed] [Google Scholar]
- Chao G.; Lau W. L.; Hackel B. J.; Sazinsky S. L.; Lippow S. M.; Wittrup K. D. (2006) Isolating and engineering human antibodies using yeast surface display. Nat. Protoc. 1, 755–768. [DOI] [PubMed] [Google Scholar]
- Wentz A. E.; Shusta E. V. (2008) Enhanced secretion of heterologous proteins from yeast by overexpression of ribosomal subunit RPP0. Biotechnol. Prog. 24, 748–756. [DOI] [PubMed] [Google Scholar]
- Huang D.; Shusta E. V. (2006) A yeast platform for the production of single-chain antibody-green fluorescent protein fusions. Appl. Environ. Microbiol. 72, 7748–7759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shusta E. V.; Holler P. D.; Kieke M. C.; Kranz D. M.; Wittrup K. D. (2000) Directed evolution of a stable scaffold for T-cell receptor engineering. Nat. Biotechnol. 18, 754–759. [DOI] [PubMed] [Google Scholar]
- Starwalt S. E.; Masteller E. L.; Bluestone J. A.; Kranz D. M. (2003) Directed evolution of a single-chain class II MHC product by yeast display. Protein Eng., Des. Sel. 16, 147–156. [DOI] [PubMed] [Google Scholar]
- Shusta E. V.; Kieke M. C.; Parke E.; Kranz D. M.; Wittrup K. D. (1999) Yeast polypeptide fusion surface display levels predict thermal stability and soluble secretion efficiency. J. Mol. Biol. 292, 949–956. [DOI] [PubMed] [Google Scholar]
- Piatesi A.; Howland S. W.; Rakestraw J. A.; Renner C.; Robson N.; Cebon J.; Maraskovsky E.; Ritter G.; Old L.; Wittrup K. D. (2006) Directed evolution for improved secretion of cancer–testis antigen NY-ESO-1 from yeast. Protein Expression Purif. 48, 232–242. [DOI] [PubMed] [Google Scholar]
- Rakestraw J. A.; Sazinsky S. L.; Piatesi A.; Antipov E.; Wittrup K. D. (2009) Directed evolution of a secretory leader for the improved expression of heterologous proteins and full-length antibodies in Saccharomyces cerevisiae. Biotechnol. Bioeng. 103, 1192–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam E.; Perler F. B. (2002) Development of a positive genetic selection system for inhibition of protein splicing using mycobacterial inteins in Escherichia coli DNA gyrase subunit A. J. Mol. Microbiol. Biotechnol. 4, 479–488. [PubMed] [Google Scholar]
- Cann I. K.; Amaya K. R.; Southworth M. W.; Perler F. B. (2004) Bacteriophage-based genetic system for selection of nonsplicing inteins. Appl. Environ. Microbiol. 70, 3158–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidler M. P.; Tan C.; Bellaiche Y.; Cherry S.; Häder S.; Gayko U.; Perrimon N. (2004) Temperature-sensitive control of protein activity by conditionally splicing inteins. Nat. Biotechnol. 22, 871–876. [DOI] [PubMed] [Google Scholar]
- Buskirk A. R.; Ong Y. C.; Gartner Z. J.; Liu D. R. (2004) Directed evolution of ligand dependence: Small-molecule-activated protein splicing. Proc. Natl. Acad. Sci. U.S.A. 101, 10505–10510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shusta E. V.; Raines R. T.; Plückthun A.; Wittrup K. D. (1998) Increasing the secretory capacity of Saccharomyces cerevisiae for production of single-chain antibody fragments. Nat. Biotechnol. 16, 773–777. [DOI] [PubMed] [Google Scholar]
- Lorimer I. A.; Keppler-Hafkemeyer A.; Beers R. A.; Pegram C. N.; Bigner D. D.; Pastan I. (1996) Recombinant immunotoxins specific for a mutant epidermal growth factor receptor: Targeting with a single chain antibody variable domain isolated by phage display. Proc. Natl. Acad. Sci. U.S.A. 93, 14815–14820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Drummond D. C.; Zou H.; Hayes M. E.; Adams G. P.; Kirpotin D. B.; Marks J. D. (2007) Impact of single-chain Fv antibody fragment affinity on nanoparticle targeting of epidermal growth factor receptor-expressing tumor cells. J. Mol. Biol. 371, 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks J. D., Zhou Y. (2010) Mutant antibodies with high affinity for EGFR. U.S. Patent 20100009390.
- Wang X. X.; Cho Y. K.; Shusta E. V. (2007) Mining a yeast library for brain endothelial cell-binding antibodies. Nat. Methods 4, 143–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackel B. J.; Huang D.; Bubolz J. C.; Wang X. X.; Shusta E. V. (2006) Production of soluble and active transferrin receptor-targeting single-chain antibody using Saccharomyces cerevisiae. Pharm. Res. 23, 790–797. [DOI] [PubMed] [Google Scholar]
- Gietz R. D.; Schiestl R. H. (2007) High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2, 31–34. [DOI] [PubMed] [Google Scholar]
- Zaccolo M.; Williams D. M.; Brown D. M.; Gherardi E. (1996) An approach to random mutagenesis of DNA using mixtures of triphosphate derivatives of nucleoside analogues. J. Mol. Biol. 255, 589–603. [DOI] [PubMed] [Google Scholar]
- Ness J. E.; Kim S.; Gottman A.; Pak R.; Krebber A.; Borchert T. V.; Govindarajan S.; Mundorff E. C.; Minshull J. (2002) Synthetic shuffling expands functional protein diversity by allowing amino acids to recombine independently. Nat. Biotechnol. 20, 1251–1255. [DOI] [PubMed] [Google Scholar]
- Stemmer W. P.; Crameri A.; Ha K. D.; Brennan T. M.; Heyneker H. L. (1995) Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides. Gene 164, 49–53. [DOI] [PubMed] [Google Scholar]
- Wiepz G.; Guadaramma A.; Fulgham D.; Bertics P. (2006) Purification and assay of kinase-active EGF receptor from mammalian cells by immunoaffinity chromatography. Methods Mol. Biol. 327, 25. [DOI] [PubMed] [Google Scholar]
- Cho Y. K.; Chen I.; Wei X.; Li L.; Shusta E. V. (2009) A yeast display immunoprecipitation method for efficient isolation and characterization of antigens. J. Immunol. Methods 341, 117–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boder E. T.; Midelfort K. S.; Wittrup K. D. (2000) Directed evolution of antibody fragments with monovalent femtomolar antigen-binding affinity. Proc. Natl. Acad. Sci. U.S.A. 97, 10701–10705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanelli A.; Shekhtman A.; Cowburn D.; Muir T. W. (2004) Semisynthesis of a segmental isotopically labeled protein splicing precursor: NMR evidence for an unusual peptide bond at the N-extein-intein junction. Proc. Natl. Acad. Sci. U.S.A. 101, 6397–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyland S.; Beerli R. R.; Barbas C. F.; Hynes N. E.; Wels W. (2003) Generation and functional characterization of intracellular antibodies interacting with the kinase domain of human EGF receptor. Oncogene 22, 1557–1567. [DOI] [PubMed] [Google Scholar]
- Wentz A. E.; Shusta E. V. (2007) A novel high-throughput screen reveals yeast genes that increase secretion of heterologous proteins. Appl. Environ. Microbiol. 73, 1189–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J. Q.; Li Q. M.; Zhou J. Y.; Zhang G. P.; Yang Y. Y.; Xing G. X.; Zhao D.; You S. Y.; Zhang C. Y. (2006) Efficient recovery of the functional IP10-scFv fusion protein from inclusion bodies with an on-column refolding system. Protein Expression Purif. 45, 168–174. [DOI] [PubMed] [Google Scholar]
- Kipriyanov S. M.; Moldenhauer G.; Little M. (1997) High level production of soluble single chain antibodies in small-scale Escherichia coli cultures. J. Immunol. Methods 200, 69–77. [DOI] [PubMed] [Google Scholar]
- Verma R.; Boleti E.; George A. (1998) Antibody engineering: Comparison of bacterial, yeast, insect, and mammalian expression systems. J. Immunol. Methods 216, 165–181. [DOI] [PubMed] [Google Scholar]
- Tsumoto K.; Ogasahara K.; Ueda Y.; Watanabe K.; Yutani K.; Kumagai I. (1995) Role of Tyr residues in the contact region of anti-lysozyme monoclonal antibody HyHEL10 for antigen binding. J. Biol. Chem. 270, 18551–18557. [DOI] [PubMed] [Google Scholar]
- Miller K. D.; Weaver-Feldhaus J.; Gray S. A.; Siegel R. W.; Feldhaus M. J. (2005) Production, purification, and characterization of human scFv antibodies expressed in Saccharomyces cerevisiae, Pichia pastoris, and Escherichia coli. Protein Expression Purif. 42, 255–267. [DOI] [PubMed] [Google Scholar]
- Evans T. C.; Benner J.; Xu M. Q. (1998) Semisynthesis of cytotoxic proteins using a modified protein splicing element. Protein Sci. 7, 2256–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southworth M. W.; Amaya K.; Evans T. C.; Xu M. Q.; Perler F. B. (1999) Purification of proteins fused to either the amino or carboxy terminus of the Mycobacterium xenopi gyrase A intein. Biotechniques 27, 110–114116, 118–120.. [DOI] [PubMed] [Google Scholar]
- Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. (2002) A stepwise huisgen cycloaddition process: Copper (I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. 114, 2708–2711. [DOI] [PubMed] [Google Scholar]
- Kalia J.; Raines R. T. (2010) Advances in bioconjugation. Curr. Org. Chem. 14, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speers A. E.; Adam G. C.; Cravatt B. F. (2003) Activity-based protein profiling in vivo using a copper(I)-catalyzed azide-alkyne [3+2] cycloaddition. J. Am. Chem. Soc. 125, 4686–4687. [DOI] [PubMed] [Google Scholar]
- Hong V.; Steinmetz N. F.; Manchester M.; Finn M. G. (2010) Labeling live cells by copper-catalyzed alkyne–azide click chemistry. Bioconjugate Chem. 21, 1912–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agard N. J.; Prescher J. A.; Bertozzi C. R. (2004) A strain-promoted [3+2] azide–alkyne cycloaddition for covalent modification of biomolecules in living systems. J. Am. Chem. Soc. 126, 15046–15047. [DOI] [PubMed] [Google Scholar]
- Dennler P.; Chiotellis A.; Fischer E.; Brégeon D.; Belmant C.; Gauthier L.; Lhospice F.; Romagne F.; Schibli R. (2014) Transglutaminase-based chemo-enzymatic conjugation approach yields homogeneous antibody–drug conjugates. Bioconjugate Chem. 25, 569–578. [DOI] [PubMed] [Google Scholar]
- Thomas J. D.; Cui H.; North P. J.; Hofer T.; Rader C.; Burke T. R. Jr. (2012) Application of strain-promoted azide–alkyne cycloaddition and tetrazine ligation to targeted Fc-drug conjugates. Bioconjugate Chem. 23, 2007–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo M.; Sommaruga S.; Mazzucchelli S.; Polito L.; Verderio P.; Galeffi P.; Corsi F.; Tortora P.; Prosperi D. (2012) Site-specific conjugation of ScFvs antibodies to nanoparticles by bioorthogonal strain-promoted alkyne–nitrone cycloaddition. Angew. Chem. 124, 511–514. [DOI] [PubMed] [Google Scholar]
- Kotagiri N.; Li Z.; Xu X.; Mondal S.; Nehorai A.; Achilefu S. (2014) Antibody quantum dot conjugates developed via copper-free click chemistry for rapid analysis of biological samples using a microfluidic microsphere array system. Bioconjugate Chem. 25, 1272–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klabunde T.; Sharma S.; Telenti A.; Jacobs W. R.; Sacchettini J. C. (1998) Crystal structure of GyrA intein from Mycobacterium xenopi reveals structural basis of protein splicing. Nat. Struct. Mol. Biol. 5, 31–36. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.