

Abstract
Cyclipostins are bicyclic lipophilic phosphate natural products. We report here that synthesized individual diastereomers of cyclipostins P and R have nanomolar IC50s toward hormone sensitive lipase (HSL). The less potent diastereomers of these compounds have 10-fold weaker IC50s. The monocyclic phosphate analog of cyclipostin P is nearly as potent as the bicyclic natural product. Bicyclic phosphonate analogs of both cyclipostins exhibit IC50s similar to those of the weaker diastereomer phosphates (about 400 nM). The monocyclic phosphonate analog of cyclipostin P has similar potency. A series of monocyclic phosphonate analogs in which a hydrophobic tail extends from the lactone side of the ring are considerably poorer inhibitors, with IC50s around 50 μM. Finally cyclophostin, a related natural product inhibitor of acetylcholinesterase (AChE) that lacks the hydrocarbon tail of cyclipostins, is not active against HSL. These results indicate a critical SAR for these compounds, the hydrophobic tail. The smaller lactone ring is not critical to activity, a similarity shared with cyclophostin and AChE. The HSL kinetics of inhibition for the cyclipostin P trans diastereomer were examined in detail. The reaction is irreversible with a KI of 40 nM and a rate constant for inactivation of 0.2 min−1. These results are similar to those observed for cyclophostin and AChE.
1. INTRODUCTION
The serine hydrolases constitute one of the largest classes of enzymes in biochemistry [1]. These biological catalysts share the classic α/β hydrolase fold structure and the well known catalytic triad composed of an acidic residue, His, and Ser. This last residue serves as the nucleophile in the attack on ester or amide substrates [2]. Among the serine hydrolases of therapeutic interest are acetylcholinesterase (AChE1), butyrylcholinesterase, and a number of lipases. For this reason, extensive literature has evolved from inhibition studies [3–5].
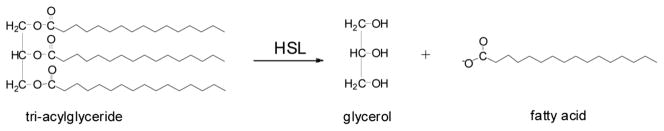
An interesting class of esterase inhibitors has emerged from the natural product literature. The basic structure is represented by cyclophostin 1a (Scheme 1), a bicyclic phosphate isolated from Streptomyces species strain DSM 13381 [6]. As we have recently characterized, this compound is a potent (nanomolar) irreversible inhibitor of AChE [7, 8]. Much like the chemical warfare agents Sarin and V/X [9], it targets the conserved Ser residue in the catalytic triad of this enzyme through phosphorylation. We recently demonstrated that bicyclic phosphonate analogs (2a, 2b) of cyclophostin are less potent AChE inhibitors than the natural product [8]. Monocyclic phosphonate analogs (large phosphate ring) are as potent as the corresponding bicyclic phosphonate.
Close structural relatives are the cyclipostins, long chain derivatives of cyclophostin (3a, 3b, 4a, 4b). These compounds isolated from bacteria were shown to be strong inhibitors of hormone sensitive lipase (HSL) [10]. This enzyme is able to catalyze the hydrolysis of mono-, di, and triacylglycerides to liberate fatty acids (Scheme 2) [11, 12]. Through insulin signaling, HSL becomes phosphorylated and translocates to lipid droplets in adipocytes [13]. Lipases are of wide interest due to their relevance to obesity and diabetes, the latter of which exhibits disturbances in both sugar and fat metabolism. There are only a few obesity drugs on the market. The most well known is Orlistat (Fig. 1), obtained from the catalytic hydrogenation of the natural product lipstatin, which targets the digestive gastric and pancreatic lipases [14]. Many more are/have been in clinical trials, and similarly a couple target lipases [3].
Fig. 1. Other Representative Lipase Inhibitors.
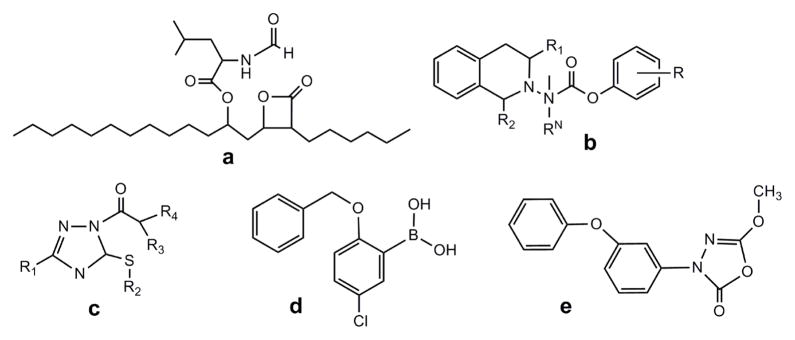
(a) pancreatic lipase inhibitor Orlistat [42]; HSL inhibitors (b) carbazates [18]; (c) carbamoyl triazole [17]; (d) boronic acids [19]; (e) 3-phenyl-5-alkoxy-1,3,4-oxadiazol-2-one [24, 25].
Scheme 1.
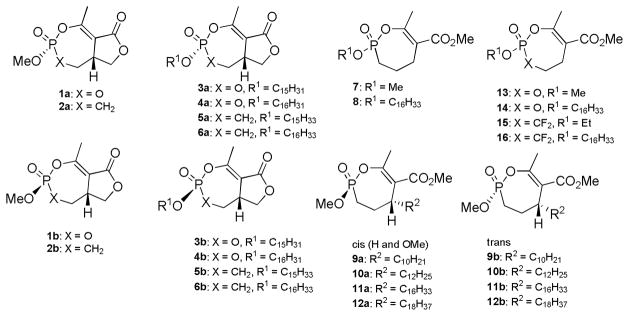
Scheme 2.
A number of low nanomolar HSL synthetic inhibitors have been reported (Fig. 1.; [15–24]). Very few detailed kinetic and mechanistic studies of HSL inhibitors exist [25].
Cyclipostins represent a unique niche among HSL inhibitors because they are natural products and, by virtue of being both lipophilic and electrophilic, are substrate mimic-mechanism-based inhibitors. The long hydrocarbon tails suggest a substrate-type binding mode. Cyclophostins share the cyclic phosphate electrophilic center of cyclipostins, which have already been shown to irreversibly modify the active site Ser of AChE [8] and microbial lipases [26]. We recently reported the synthesis of cyclipostin P (4a, 4b) [27]. Herein we characterize the activities for two cyclipostins and their analogs against hormone sensitive lipase. To our knowledge, this represents the most detailed study of HSL inhibitors reported to date.
2. RESULTS AND DISCUSSION
2.1. Synthesis of Inhibitors
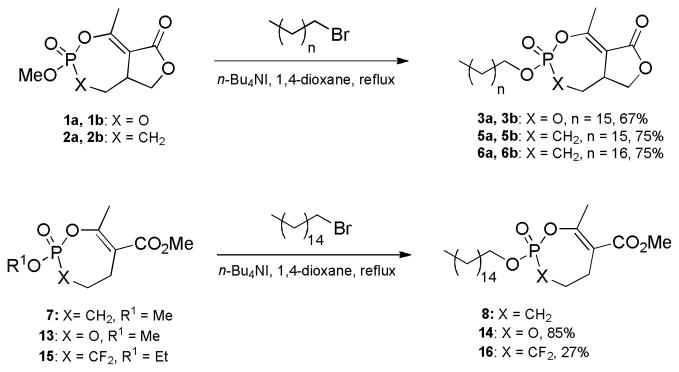
Cyclipostin R (3a, 3b) and the analogs used in this study were synthesized using our one-pot ester exchange process from (±)-cyclophostin 1a and its diastereomer 1b [27], the phosphonate analogs thereof 2a and 2b [7], and three monocyclic compounds 7, 13 and 15 (Scheme 3). Characterization data for the monocyclic compounds 13 and 15 appear in Supplemental Material. The syntheses these compounds will be reported in another article. Methyl esters of phosphates undergo transesterification cleanly in the presence of 5 mol% tetra-n-butylammonium iodide, but phosphonates and the α, α-difluorophosphonate ethyl ester 16 require 10 mol% for optimal conversion. Additional information can be found in Experimental and Supplementary Material.
Cyclophostin 1a 1b, cyclipostin P 4a, 4b and the phosphonate analogs of cyclophostin (5a, 5b, 6a, 6b) were prepared following previously published procedures [7, 27]. The synthesis of the monocyclic phosphonates (7, 8, 9a, 10a, 10b, 11a, 11b, 12a, 12b) also followed published procedures [8, 26].
Scheme 3.
2.2. Inhibition Studies with Cyclophostin and Cyclipostin
Table 1 summarizes all of the cyclophostin and cyclipostin analogs that were tested against recombinant rat HSL. The natural product and AChE inhibitor cyclophostin 1b is not potent against HSL (IC50 • 100 μM). The same is true of the synthetic isomer 1a and phosphonate isomer 2a. However, the cyclipostins with the same core cyclophostin structure with long carbon tails extending from the phosphate moiety (3–6) are more potent against HSL; a couple have IC50’s near 50 nM. This indicates that the long hydrocarbon tail of the phosphate moiety is a critical part of the inhibitor for successful inactivation of rat HSL. The most potent cyclipostin is cyclipostin P trans (4a) with an IC50 of 25 ± 15 nM.
Table 1.
Summary of HSL IC50s of Cyclipostins and Analogsa
| Compound | Structure | Tail | Diastereomer | IC50 [μM] | Published synthesis |
|---|---|---|---|---|---|
| Synthetic Cyclophostin 1a |
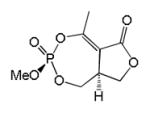
|
1 | Trans | >100 | [27] |
| Natural Cyclophostin 1b |
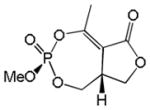
|
1 | Cis | >100 | [27] |
| Cyclophostin Phosphonate analog 2a |
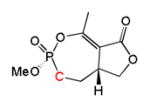
|
1 | Trans | 100 | [7] |
| Cyclipostin R 3a |
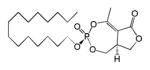
|
15 | Trans | 0.9±0.9 | This work |
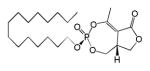
| Cyclipostin R 3b |
|
15 | Cis | 0.075±0.025 | This work |
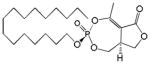
| Cyclipostin P 4a |
|
16 | Trans | 0.025±0.015 | [27] |
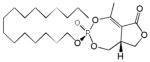
| Cyclipostin P 4b |
|
16 | Cis | 0.42±0.16 | [27] |
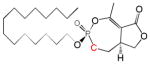
| Cyclipostin R Phosphonate 5a |
|
15 | Trans | 0.45 ± 0.25 | This work |
| Cyclipostin R Phosphonate 5b |
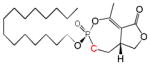
|
15 | Cis | 0.47 ± 0.38 | This work |
| Cyclipostin P Phosphonate 6a |
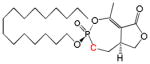
|
16 | Trans | 6.9 ± 2.4 | This work |
| Cyclipostin P Phosphonate 6b |
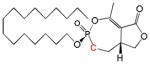
|
16 | Cis | 0.36 ± 0.34 | This work |
| 14 |
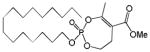
|
16 | 0.06±0.02 | This work | |
| 8 |
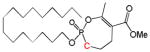
|
16 | 0.54±0.4 | [26] | |
| 16 |
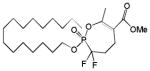
|
16 | >100 | This work | |
| 9aa |
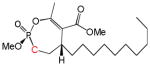
|
10 | Cis | 40 | [26] |
| 10a |
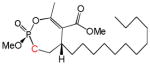
|
12 | Cis | 35 | [26] |
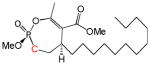
| 10b |
|
12 | Trans | 13.5 | [26] |
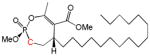
| 11a |
|
16 | Cis | ~60 | [26] |
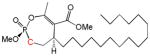
| 11b |
|
16 | Trans | ~10 | [26] |
| 12a |
|
18 | Cis | >100 | [26] |
| 12b |
|
18 | Trans | ~27 | [26] |
IC50s, the concentration at which the enzyme is inhibited 50% of the control activity, was obtained using triolein as a substrate as described in Experimental. Graphical abstract
2.3. Monocyclic and Phosphonate Analogs
Interestingly, phosphate 14, a monocyclic analog of cyclipostin P (4a, 4b) retains potency similar to the stronger diastereomer 4a of the natural product, indicating that like cyclophostin with AChE, the integrity of the smaller lactone ring is not critical to activity. To understand the importance of the phosphate itself, both bicyclic and monocyclic phosphonates were prepared and tested. Bicyclic phosphonate analogs of cyclipostins P and R (5a, 5b, 6a, and 6b) generally exhibit IC50s similar to those of the weaker diastereomer phosphates (about 400 nM). The exception is 6a, the trans cyclipostin P analog, which is about 10-fold less potent. The monocyclic phosphonate analog of cyclipostin P (8) exhibits potency similar to the weaker diastereomer of the natural product (4b). Interestingly, the activity difference between phosphates and phosphonates is smaller for HSL inhibitors than for AChE and cyclophostin (less than 10-fold vs. 100-fold; [28]). This further supports the contribution of the hydrocarbon chain to binding affinity.
2.4. Moving the Hydrophobic Tail
Another group of analogs extend hydrophobic tails of differing lengths (C10–C18) from the lactone side of the larger ring (instead of from the phosphate) (9a, 9b, 10a, 10b, 11a, 11b, 12a, 12b). Most of these are modest inhibitors of HSL, with IC50’s ranging from 13–60 μM. Among the compounds with chain lengths of 12–18 carbon atoms, 10b, 11b, 12b (trans) are generally 2–6 times more potent than the corresponding cis isomers (10a, 11a, 12a). Interestingly, these compounds are inhibitors of microbial lipolytic enzymes with marked stereoselectivity [26, 29].
2.5 Effect of Difluorination of Phosphonate Ring
If the inhibitor functions via electrophilicity, then introducing electron withdrawing groups into the phosph(on)ate ring should result in an increase in potency. To that end, fluorinated monocyclic monophosphate 16 was prepared. Interestingly, while its nonfluorinated relative 8 has a 0.5 μM IC50, the IC50 for 16 is over 100 μM. Clearly other factors, perhaps binding, affect inhibitor potency.
2.6. Stereochemical Differences Among Cyclipostins and Analogs
As summarized in Scheme 1, the presence of 2 stereocenters in both cyclophostin and cyclipostins structures give rise to two sets of diastereomers: cis refers to the isomers in which the phosphate ester chain and cyclic proton are on the same side of the ring system; trans refers to those isomers in which these groups are on opposing sides of the ring system.
The trans diastereomers of cyclipostin P 4a are the more potent, with an IC50 of 25 nM. The cis diastereomers of cyclipostin P 4b are 16 fold weaker. Interestingly, the opposite is true of cyclipostin R: in this case, the cis diastereomers 3b are similarly more potent than the trans 3a. Among the phosphonate analogs of cyclipostins P and R, the cis diastereomers of cyclipostin P analogs (6a, 6b) are more potent (360 nM, 20-fold better than trans). Interestingly, for the phosphonate analogs (5a, 5b) of cyclipostin R (3a, 3b), the IC50s for cis and trans are comparable. While it is difficult to easily rationalize why a one-carbon change in the hydrophobic tail would change diastereopreference by the enzyme, this behavior has precedent: The cis isomer of the C10 monocyclic phosphonate analog 9a is more potent against Rv183 lipase from Mycobacterium tuberculosis than the corresponding C10 trans analog 9b [26]. However, the trans isomer of the C12 analog 10b is more potent than the corresponding cis isomer 10a [26]. One possible rationalization is how hydrocarbon chains are accommodated in enzyme active sites. There is some precedent for chain length affecting the stereochemical behavior of lipases [30, 31]. Nevertheless, observing a difference in stereochemical behavior between compounds with tails of 15 and 16 carbon lengths is remarkable. Better understanding of this behavior would require a structure of HSL. Unfortunately, this enzyme has been refractory to structural biology efforts. While an HSL structural model has been constructed based on sequence homology between the crystallographically characterized brefeldin A esterase and the C-terminal domain of HSL [32], models with greater experimental support (and thus detail) would be necessary to meaningfully rationalize the IC50 patterns observed here.
2.7. Mechanism of Inhibition of Cyclipostin P Trans (4a)
As stated in the introduction, we have previously shown that cyclophostin inhibits AChE irreversibly [8]. Since the basic chemistry should be the same, we expect cyclipostin to inhibit HSL irreversibly. This clarification simplifies the kinetic analysis, since reversible and irreversible inhibitors are analyzed differently.
To confirm this, reaction timecourse data for cyclipostin P trans (4a) were fit to Eqn 1 to obtain kobs and k−2, the latter of which is the rate constant for the reverse reaction [8]. Sample data appear in Fig. 2A. In these fits at various inhibitor concentrations, k−2 averages to 0.001 min−1. Thus this inhibitor can be treated as irreversible.
Fig. 2. Kinetic Analysis with of Cyclipostin P Trans (4a).

A: Timecourse for HSL activity in the presence of 10 nM 4a, 80 nM HSL. Data were fit to Eqn 1 to demonstrate that k−2 is essentially zero. See text for details. B: Relative residual HSL activity as a function of cyclipostin P trans (4a) concentration. Data were fit to Eqn 2 to yield KI of 40 nM and k2 of 0.2 min−1.
To dissect this behavior, residual enzyme activities were obtained as a function of inhibitor concentration and incubation time for trans cyclipostin P (4a) (Fig. 2B). The irreversible inactivation mechanism in Scheme 5 [33] was utilized. The extent of reaction, as measured by the change in cpm for the liberated radioactive free fatty acid, decreases as a function of inhibitor concentration. This was fit to Eqn. 2 to obtain k2, the forward rate constant, and KI, the equilibrium constant for binding, for HSL and 4a. The values for these constants are 40 nM and 0.2 min−1, respectively. Dividing the former constant by the latter yields a first order rate constant for inactivation of 5 × 106 M−1min−1. Since we are unaware of any reports of this type of data for any other HSL inhibitor, we must compare to other enzymes. Indeed, this rate constant is comparable to that measured for cyclophostin [27] and tabun [8] toward human AChE. However, it is 1000-fold higher (faster) than that estimated from studies of the interactions of Orlistat with lipoprotein lipase [34].
It is possible that phosphonate analogs of cyclipostin P trans would display different kinetic details. Comparing AChE inhibitors cyclophostin (1a, 1b) and their phosphonate analogs (2a, 2b), k2 and k−2 were similar but KI values differed by 100-fold in parallel with the corresponding IC50s [27]. The HSL IC50’s of the cyclipostins (3a, 3b, 4a, 4b) and their phosphonate analogs tested here (3a, 3b, 4a, 4b) are generally more similar to one another (6 to 16-fold, excluding 6a). Thus more modest differences in kinetic parameters would be expected. It is not possible to perform analyses for additional compounds at this time.
In conclusion, we have prepared and evaluated a series of cyclipostins and their analogs toward hormone sensitive lipase. The most potent of these are active in the low nanomolar range and display irreversible kinetics. SAR studies indicate that the smaller lactone ring is not essential; however the lipophilic tail is critical to activity.
3. EXPERIMENTAL
3.1. Synthesis and Characterization of Organophosphates
The preparations of (1a, 1b, 2a, 4a, 4b, 8,16, 9a, 10a, 10b, 11a, 11b, 12a, 12b have been described previously [7, 26, 27].
3.1.1. Diastereomers of Cyclipostin R (3a, 3b)
To a solution cyclophostin diastereomer 1a, 1b (28.5 mg, 0.122 mmol) and 1-bromopentadecane (350 μL, 1.21 mmol) in dry 1,4-dioxane (0.5 mL) was added n-Bu4NI (2 mg, 0.005 mmol) at room temperature. The flask was immersed in an oil bath preheated at 110 °C until the reaction was complete (TLC and 31P NMR analysis). The solvent was removed under vacuum and the crude product was purified by column chromatography (SiO2, EtOAc/hexane) to give cyclipostin R 3a (16.6 mg) and the unnatural diastereomer 3b (9.5 mg) as white solids (67% overall).
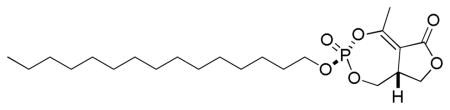
3.1.1.1. Cyclipostin R (3a, 3b). (±)-(3R(S),8aR(S))-5-methyl-3-(pentadecyloxy)-8,8a-dihydrofuro[3,4-e][1,3,2]dioxaphosphepin-6(1H)-one 3-oxide
IR (neat, ATR): 2916, 2850, 1749, 1671 cm−1; 1H NMR (300 MHz, CDCl3) δ 4.45 (1H, m), 4.35 (1H, td, JHH = 10.8 Hz, JHP = 6.2 Hz), 4.24 (2H, dt, JHP = JHH = 6.8 Hz), 4.15 (1H, ddd, JHP = 25.6 Hz, JHH = 11.2, 3.5 Hz), 3.80 (2H, m), 2.47 (3H, d, JHP = 1.9 Hz), 1.72 (2H, p, JHH = 6.8), 1.39 (2H, m), 1.26 (22H, br s), 0.88 (3H, t, JHH = 6.7 Hz); 13C NMR (75.4 MHz, CDCl3) δ 169.1, 161.8 (d, JCP = 7.7 Hz), 112.1 (d, JCP = 3.2 Hz), 70.4 (d, JCP = 6.3 Hz), 67.6 (d JCP =, 5.7 Hz), 64.4, 39.9, 32.1, 30.3 (d, JCP = 6.6 Hz), 29.90, 29.88, 29.85, 29.76, 29.68, 29.58, 29.26, 25.5, 22.9, 18.4 (d, JCP = 4.7 Hz), 14.3; 31P NMR (121.4 MHz, CDCl3) δ −8.55; HRMS (FAB, NBA, MH+) calcd for C22H40O6P: 431.2563, found: 431.2563.
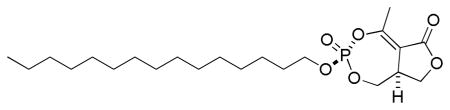
3.1.1.2. (±)-(3R(S),8aS(R))-5-methyl-3-(pentadecyloxy)-8,8a-dihydrofuro[3,4-e][1,3,2]dioxaphosphepin-6(1H)-one 3-oxide
IR (neat, ATR): 2914, 2847, 1745, 1666, 1289, 1242, 1204, 1070, 1034 cm−1; 1H NMR (300 MHz, CDCl3): δ 4.46 (1H, t, JHH = 9.3 Hz), 4.29 (1H, m), 4.18 (2H, m), 4.04 (2H, m), 3.79 (1H, dd, JHH = 9.6, 5.7 HZ), 2.44 (3H, d, JHP = 1.8 Hz), 1.73 (2H, p, JHH = 6.7 Hz), 1.26 (24H, br s), 0.89 (3H, t, JHH = 6.6 Hz); 13C NMR (75.4 MHz, CDCl3): δ 169.2, 161.6 (d, JCP = 10.5 Hz), 111.0 (d, JCP = 2.6 Hz), 69.9 (d, JCP = 6.3 Hz), 69.5 (d, JCP = 7.6 Hz), 64.7, 38.6, 32.1, 30.4 (d, JCP = 6.5 Hz), 29.90, 29.88, 29.84, 29.76, 29.67, 29.58, 29.3, 25.5, 18.1 (d, JCP = 5.9 Hz), 14.3; 31P NMR (121.4 MHz, CDCl3): δ −12.4; HRMS (FAB, NBA, MH+) calcd for C22H40O6P: 431.2563, found: 431.2570.
3.1.2. Phosphonate analog of Cyclipostin R and Its Diastereomer (5a, 5b)
To a solution of phosphonate analogs 2a and 2b (17.8 mg, 0.077 mmol) and 1-bromopentadecane (220 μL, 0.76 mmol) in dry 1,4-dioxane (390 μL) was added n-Bu4NI (3 mg, 0.008 mmol). The flask was immersed in an oil bath preheated to 110 °C until the reaction was complete (TLC, 31P NMR analysis). The solvent was removed under vacuum and the crude product was purified by column chromatography (SiO2, EtOAc/ hexane) giving the phosphonate analog of cyclipostin R (13.4 mg) and its diastereomer (8.9 mg) as a white solid and white semisolid, respectively (75% overall).
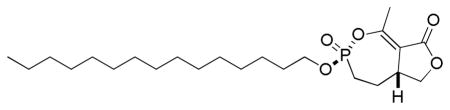
3.1.2.1. (±)-(3R(S),5aR(S))-1-methyl-3-(pentadecyloxy)-4,5,5a,6-tetrahydrofuro[3,4-e][1,2]oxaphosphepin-8(3H)-one 3-oxide
IR (neat, ATR): 2915, 2849, 1742, 1662, 1246, 1036 cm−1; 1H NMR (300 MHz, CDCl3): δ 4.50 (1H, t, JHH = 9.3 Hz), 4.19 (2H, m), 3.85 (1H, dd, JHH = 9.2, 6.6 Hz), 3.32 (1H, m), 2.45 (3H, s), 2.32 (1H, m), 1.89–2.13 (3H, m), 1.72 (2H, p, JHH = 6.9), 1.26 (24H, br s), 0.88 (3H, t, JHH = 6.6 Hz); 13C NMR (75.4 MHz, CDCl3): δ 170.1 (d, JCP = 1.6 Hz), 161.0 (d, JCP = 7.2 Hz), 114.5 (d, JCP = 3.8), 69.9, 66.8 (d, JCP = 7.3 Hz), 39.1 (d, JCP = 1.0 Hz), 32.1, 30.5 (d, JCP = 6.1 Hz), 29.89, 29.88, 29.86, 29.85, 29.83, 29.75, 29.7, 29.6, 29.3, 26.8 (d, JCP = 136.4 Hz), 26.6 (d, JCP = 6.9 Hz), 25.6, 22.9, 19.0 (d, JCP = 2.3 Hz), 14.3; 31P NMR (121.4 MHz, CDCl3): δ 23.4; HRMS (FAB, NBA, MH+) calcd for C23H42O5P: 429.2770, found: 429.2777
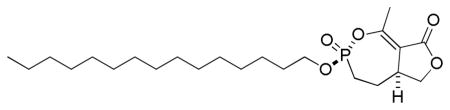
3.1.2.2. (±)-(3R(S),5aS(R))-1-methyl-3-(pentadecyloxy)-4,5,5a,6-tetrahydrofuro[3,4-e][1,2]oxaphosphepin-8(3H)-one 3-oxide
IR (neat, ATR): 2915, 2848, 1742, 1672, 1246, 1198, 1040, 1011 cm−1; 1H NMR (300 MHz, CDCl3): δ 4.50 (1H, t, JHH = 9.3 Hz), 4.24 (1H, ddt, JHH = 8.3 Hz, JHP = JHH = 7.7 Hz), 4.06 (1H, ddt, JHH = 9.9 Hz, JHP = JHH = 6.9 Hz), 3.84 (1H, dd, JHH = 9.2, 6.5 Hz), 3.40 (1H, m), 2.43 (3H, d, JHP = 1.7 Hz), 1.80–2.40 (4H, m), 1.70 (2H, p, JHH = 6.9 Hz), 1.26 (24H, br s), 0.88 (3H, t, JHH = 6.7 Hz); 13C NMR (75.4 MHz, CDCl3): δ 170.3, 160.5 (d, JCP = 9.6 Hz), 113.8 (d, JCP = 3.0 Hz), 69.9, 67.0 (d, JCP = 7.0 Hz), 38.6 (d, JCP = 2.0 Hz), 32.1, 29.89, 29.87, 29.83, 29.76, 29.68, 29.57, 29.3, 27.0 (d, JCP = 7.5 Hz), 26.6 (d, JCP = 134.5 Hz), 25.6, 22.9, 18.7 (d, JCP = 3.5 Hz), 14.3; 31P NMR (121.4 MHz, CDCl3): δ 19.9; HRMS (FAB, NBA, MNa+) calcd for C23H41O5PNa: 451.2589, found: 451.2601.
3.1.3. Phosphonate Analog of Cyclipostin P and Its Diastereomer (6a, 6b)
To a solution of phosphonate analog 2a, 2b (10.7 mg, 0.046 mmol) and 1-bromohexadecane (140 μL, 0.46 mmol) in dry 1,4-dioxane (230 μL) was added n-Bu4NI (1.7 mg, 0.0046 mmol). The flask was immersed in an oil bath preheated to 110 °C until the reaction was complete (TLC, 31P NMR analysis). The solvent was removed under vacuum and the crude product was purified by column chromatography (SiO2, EtOAc/ hexane) giving the phosphonate analog of cyclipostin P (7.8 mg) and its diastereomer (5.5 mg) as a white solid and white semisolid, respectively (75% overall).
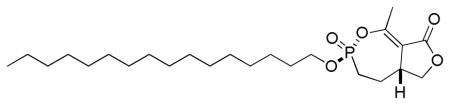
3.1.3.1. (±)-(3R(S),5aR(S))-3-(hexadecyloxy)-1-methyl-4,5,5a,6-tetrahydrofuro[3,4-e][1,2]oxaphosphepin-8(3H)-one 3-oxide
IR (neat, ATR): 2918, 2852, 1742, 1661, 1246, 1040 cm−1; 1H NMR (300 MHz, CDCl3): δ 4.50 (1H, t, JHH = 9.3 Hz), 4.20 (2H, m), 3.86 (1H, dd, JHH = 9.2, 6.6 Hz), 3.34 (1H, m), 2.46 (3H, s), 2.26–2.38 (1H, m), 1.89–2.13 (3H, m), 1.72 (2H, p, JHH = 6.9 Hz), 1.26 (26H, br s), 0.88 (3H, t, JHH = 6.7 Hz); 13C NMR (75.4 MHz, CDCl3): δ 170.1, 161.0 (d, JCP = 7.0 Hz), 114.5 (d, JCP = 4.0 Hz), 69.9, 66.8 (d, JCP = 7.1 Hz), 39.1, 32.1, 30.5, 29.89, 29.87, 29.84, 29.77, 29.70, 29.6, 29.3, 26.8 (d, JCP = 136.0 Hz), 26.6 (d, JCP = 7.0 Hz), 25.6, 22.9, 19.0 (d, JCP = 2.6 Hz), 14.3; 31P NMR (121.4 MHz, CDCl3): δ 23.4; HRMS (FAB, NBA, MH+) calcd for C24H44O5P: 443.2926, found: 443.2935.
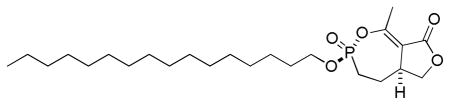
3.1.3.2. (±)-(3R(S),5aS(R))-3-(hexadecyloxy)-1-methyl-4,5,5a,6-tetrahydrofuro[3,4-e][1,2]oxaphosphepin-8(3H)-one 3-oxide
IR (neat, ATR): 2916, 2848, 1749, 1669, 1242, 1201, 1043, 1011; 1H NMR (300 MHz, CDCl3): δ 4.51 (1H, t, JHH = 9.1 Hz), 4.24 (1H, ddt, JHH = 9.8 Hz, JHH = JHP = 7.2 Hz), 4.06 (1H, ddt, JHH = 10.0 Hz, JHH = JHP = 7.0 Hz), 3.84 (1H, dd, JHH = 9.1, 6.4 Hz), 3.41 (1H, m), 2.43 (3H, s), 1.80–2.49 (4H, m), 1.70 (2H, p, JHH = 6.8 Hz), 1.32 (2H, m), 1.26 (24H, br s), 0.88 (3H, t, JHH = 6.6 Hz). 13C NMR (75.4 MHz, CDCl3): δ 170.3, 160.5 (d, JCP = 9.8 Hz), 113.8 (d, JCP = 3.0 Hz), 70.0, 67.1 (d, JCP = 7.1 Hz), 38.6 (d, JCP = 2.2 Hz), 32.1, 30.6 (d, JCP = 5.9 Hz), 29.92, 29.90, 29.88, 29.87, 29.84, 29.76, 29.68, 29.58, 29.3, 27.0 (d, JCP = 7.9 Hz), 26.6 (d, JCP = 134.7 Hz), 25.6, 22.9, 18.7 (d, JCP = 3.2 Hz), 14.3; 31P NMR (121.4 MHz, CDCl3): δ 20.0; HRMS (FAB, NBA, MH+) calcd for C24H44O5P: 443.2926, found: 443.2919
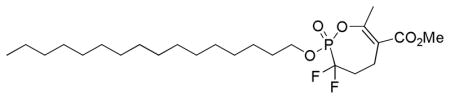
3.1.4. Methyl 3,3-difluoro-2-(hexadecyloxy)-7-methyl-2,3,4,5-tetrahydro-1,2-oxaphosphepine-6-carboxylate 2-oxide (16)
To a solution of cyclic phosphonate 15 (26 mg, 0.091 mmol) in dry 1,4-dioxane (0.45 mL) was added 1-bromohexadecane (280 μL, 0.92 mmol) and n-Bu4NI (3.4 mg, 0.0092 mmol). The flask was immersed in an oil bath preheated to 110 °C until the reaction was complete (TLC, 31P NMR analysis). The mixture was cooled to room temperature and concentrated in vacuo, and the residue was chromatographed on C18 reverse phase silica gel (CH3OH/H2O) to give 16 (12 mg, 27%) as a white solid. IR (Neat, ATR) 2963, 2917, 2850, 1717, 1661, 1292, 1271, 1095, 1011 cm−1; 1H NMR (300 MHz, CDCl3): δ 4.34 (2H, dt, JHH = JHP = 7.0 Hz), 3.79 (3H, s), 2.65–2.79 (1H, m), 2.45–2.59 (1H, m), 2.40 (3H, s), 2.17–2.39 (2H, m), 1.78 (2H, p, JHH = 6.9 Hz), 1.26 (26H, br s), 0.89 (3H, t, JH,H = 6.8 Hz); 13C NMR (75.4 MHz, CDCl3): δ 167.4, 159.2 (d, JCP = 8.1 Hz), 120.4 (td, JCF = 261.7 Hz, JCP = 205.1 Hz), 120.1 (d, JCP = 5.6 Hz), 69.9 (d, JCP = 7.0 Hz), 52.3, 35.3 (td, JCF = 20.5 Hz, JCP = 11.4 Hz), 32.152, 30.6 (d, JCP = 5.6 Hz), 29.90, 29.88, 29.84, 29.74, 29.66, 29.57, 29.2, 25.4, 22.9, 21.4, 20.2 (dd, JCF = 9.1 Hz, 3.5 Hz), 14.3; 31P NMR (121.4 MHz, CDCl3): δ 1.4 (dd, JPF = 111.2 Hz, 100.6 Hz); 19F NMR (282.2 MHz, CDCl3): δ −109.2 (dd, JFF = 290.1 Hz, JFP = 111.2 Hz), −106.1 (dd, JFF = 290.1 Hz, JFP = 100.3 Hz); HRMS (FAB, NBA, MH+) calcd for C24H44F2O5P: 481.2894, found 481.2900.
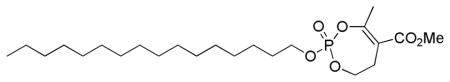
3.1.5. Methyl 2-(hexadecyloxy)-4-methyl-6,7-dihydro-1,3,2-dioxaphosphepine-5-carboxylate 2-oxide (14)
To a solution of phosphate 13 (11 mg, 0.047 mmol) and 1-bromohexadecane (175 μL, 0.473 mmol) in dry 1,4-dioxane (235 μL) was added n-Bu4NI (1 mg, 0.003 mmol). The flask was immersed in an oil bath preheated to 110 °C until the reaction was complete (TLC, 31P NMR analysis). The solvent was removed under vacuum and the crude product was purified by column chromatography (SiO2, EtOAc/ hexane) giving 14 (18 mg, 85%) as a colorless oil. IR (neat, NaCl): 2924, 2854, 1723, 1645, 1301, 1104, 1011 cm−1; 1H NMR (300 MHz, CDCl3): δ 4.37 (1H, m), 4.17 (3H, m), 3.77 (3H, s), 3.00 (1H, ddd, JHH = 15.5, 8.8, 3.2 Hz), 2.84 (1H, ddd, JHH = 15.6, 5.9, 3.6 Hz), 1.71 (2H, p, JHH = 6.7 Hz), 1.26 (26H, br s), 0.88 (3H, t, JHH = 6.7 Hz); 13C NMR (75.4 MHz, CDCl3): δ 167.3 (d, JCP = 2.0 Hz), 161.1 (d, JCP = 9.6 Hz), 115.6 (d, JCP = 4.1 Hz), 69.3 (d, JCP = 6.6 Hz), 68.3 (d, JCP = 6.6 Hz), 52.2, 32.1, 30.5 (d, JCP = 6.6 Hz), 29.90, 29.87, 29.84, 29.76, 29.69, 29.55, 29.3, 28.5, 25.6, 22.9, 20.5 (d, JCP = 4.0 Hz), 14.3; 31P NMR (121.4 MHz, CDCl3): δ −10.8; HRMS (FAB, NBA, MH+) calcd for C23H44O6P: 447.2876, found 447.2864.
3.2. Enzyme Studies
3.2.1. Protein Expression
The Spodoptera frugiperda (Sf9) cells were grown in TNM-FH medium supplemented with 10% heat inactivated FBS, 100 units/ml penicillin G, and 100 μg/ml streptomycin at 27°C. The cells were maintained in T-150 flasks (Fisher Scientific) and subcultured in 1:3 ratio whenever they reached 90% confluency.
The rat hormone-sensitive lipase gene was a kind gift from Dr. Fredric Kraemer. The gene was cloned into a pcDNA3 vector using HindIII and XbaI sites. The HSL gene was subcloned in the pAcHLT-A baculovirus transfer vector (BD Biosciences), having N-terminal 6X-His Tag gene at a SmaI blunt cloning site. The pAcHLT-A-HSL construct was transformed into TOP10F′ E. coli competent cells for large scale DNA production.
Recombinant baculovirus containing the HSL gene was prepared using the HSL gene from the pAcHLT-HSL plasmid and the linearized Baculogold DNA (BD Biosciences). The transfection into Sf9 insect cells was accomplished by utilizing the CaII-phosphate precipitation method using a BaculoGold Transfection Kit (BD Biosciences). The successful formation of recombinant baculovirus containing the HSL gene was confirmed by Western Blot analysis using an anti-rabbit HSL polyclonal antibody as the primary antibody (Cayman Chemicals Ltd.), and anti-rabbit horseradish peroxidase (Promega) as the secondary antibody on the virus-infected cell lysate [35, 36].
For high titer rat HSL baculovirus production, transfected SF9 cells were left to grow for 144 hours (6 days). The media from transfected SF9 cells was collected and sent to Allele Biotechnology for virus amplification (1 e8 pfu/ml). For protein production, 4 e7 SF9 cells in T-150 flasks (Fisher) (70 % confluent) were infected with recombinant baculovirus at 8 m.o.i. SF9 cells were collected after 72 hour post infection and centrifuged at 1,500 g for 20 minutes and stored at −80°C.
3.2.2. Protein Purification
In a protocol adapted from the literature [32], cell pellet was homogenized in 75 mM Tris, 10% glycerol, 1 mM DTT and 1% C13E12 (poly (ethylene glycol)(12) tridecyl) (Sigma) at pH 8.0 at 4°C and supplemented with 0.2 μM bestatin hydrochloride, 0.01 μM E-64 and 10 μM phosphoramidon disodium salt to inhibit proteolytic degradation. The solution volume was adjusted to 15 ml of buffer per gram of pellet. The resulting suspension was sonicated on ice using a Branson Sonifier 250 sonicator for 2 min at setting 2, then 2 min at setting 4 with 30 sec intervals between each minute of sonication. This solution was centrifuged 11,000 g at 4°C for 30 min. Supernatant containing 6X-His tag rat HSL incubated Ni-NTA beads for 1.5 hour at 4 °C. This suspension was washed twice with 1 mL of elution buffer containing 75 mM Tris, 0.3 M NaCl, 1 mM DTT, 10 % glycerol, 0.01 % C8E4 (tetrathylene glycol monooctyl ether) (Sigma) pH 8.0 to wash away nonspecifically bound proteins. Pure HSL was eluted with an imidazole gradient (5–400 mM) in elution buffer. Fractions were collected and evaluated by SDS PAGE (10%). The most concentrated and most pure fractions were combined and dialyzed using a Slide-A-lyzer (Pierce) against 1 L of elution buffer without imidazole. The concentration of recombinant rat HSL was measured using a Shimadzu UV-1800 spectrophotometer with an extinction coefficient of 52090 M−1 cm−1 at 280 nm [37].
3.2.3. HSL Lipase Assay
Oleic acid release from triolein embedded in liposomes was used to monitor activity of HSL. In protocol adapted from the literature [38, 39], liposomes consisted of 200 μM phosphatidylcholine and phosphatidylinositol (3:1 w/w) and 500 μM cold triolein and 120 μL of 3H triolein per 20 mL. All of these components were dried from chloroform under vacuum before resuspension in assay buffer (25 mM Tris, 150 mM NaCl, 0.2 mM EDTA, and 1 mM β-mercaptoethanol, pH7.4). Sonication was performed on ice for 2 min at setting 2 and 2 minutes at setting 4 with 30 second pause between each minute. 0.2 % BSA was added to the reaction.
Reactions were performed at 37°C with HSL (20–100 nM as needed) and followed for 30 min by removing 120 μL of the reaction mixture at various time points (either every 5 or 10 min for 6–8 total points) and adding 50 μL stop buffer (50 mM each of K2CO3, K3BO3, KOH, pH 10). Liberated, radioactively labeled oleic acid was extracted twice with 100 μL of methanol: chloroform: heptanes (1.41: 1.25:1, v/v) [38]. The aqueous layer was then assessed by scintillation counting. These data were used to calculate a raw rate (cpm/min).
3.2.4. Inhibition Studies
Inhibitor solutions were prepared similar to those above for triolein. A separate liposome solution was prepared for each inhibitor concentration used. For IC50 determinations, enzyme was preincubated for 30 min on ice with inhibitor-embedded liposomes of the same component concentrations (PC, PI and inhibitor) as in the assay. Triolein was omitted in the preincubation mixture. At 10 min intervals (6–8 timepoints), the reaction mixture was assayed. IC50 was determined as amount of inhibitor required to reduce rat HSL activity to 50 %. Each IC50 was performed in triplicate.
3.2.5. Determination of Rate Constants for Binding and Inactivation
We began with the general reaction scheme:
Scheme 4.
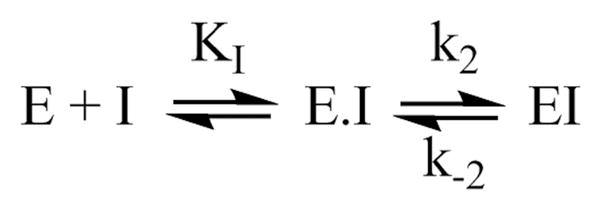
where KI represents the dissociation equilibrium constant for the inhibitor, k2 is the forward rate constant for inactivation of the enzyme and k−2 is the reverse rate constant for inactivation. In many irreversible inhibition reactions, k−2 is neglected [33]. To determine if that was appropriate here, kinetic data at a few representative inhibitor concentrations were fit to the equation:
| (1) |
where At equals the fraction of enzymatic activity remaining following pre-incubation time (t), A0 represents the enzymatic activity at time zero (i.e., the initial activity), and kobs is the observed rate of decay. k−2 fit to essential zero (Fig. 2), which simplifies the model to [40, 41]:
Scheme 5.
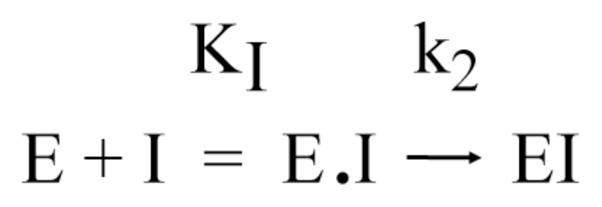
In this analysis, HSL was not preincubated with inhibitor on ice, but was added immediately to substrate-inhibitor mixture at 37° C and timepoints taken every 3–5 minutes for 30 minutes. As inhibitor concentration is increased, the extent of reaction (as measured from scintillation counting of extracted samples) decreases. This is converted to a relative normalized activity and can be expressed as a function of k2 and KI:
| (2) |
Data were fit to this equation using Kaliedagraph to yield KI and k2.
Supplementary Material
Acknowledgments
This work was supported in part by the NIH GM067596-01.
Footnotes
AChE, acetylcholinesterase; HSL, hormone sensitive lipase.
Supplementary Material. Supplementary data associated with this article can be found, in the online version.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kidd D, Liu Y, Cravatt BF. Profiling serine hydrolase activities in complex proteomes. Biochemistry. 2001;40:4005–4015. doi: 10.1021/bi002579j. [DOI] [PubMed] [Google Scholar]
- 2.Dodson G, Wlodawer A. Catalytic triads and their relatives. Trends in Biochem Sci. 1998;23:347–352. doi: 10.1016/s0968-0004(98)01254-7. [DOI] [PubMed] [Google Scholar]
- 3.Birari RB, Bhutani KK. Pancreatic lipase inhibitors from natural sources: unexplored potential. Drug Discovery Today. 2007;12:879–889. doi: 10.1016/j.drudis.2007.07.024. [DOI] [PubMed] [Google Scholar]
- 4.Zhou X, Wang XB, Wang T, Kong LY. Design, synthesis, and acetylcholinesterase inhibitory activity of novel coumarin analogues. Bioorg Med Chem. 2008;16:8011–8021. doi: 10.1016/j.bmc.2008.07.068. [DOI] [PubMed] [Google Scholar]
- 5.Musial A, Bajda M, Malawska B. Recent developments in cholinesterases inhibitors for Alzheimer’s disease treatment. Curr Med Chem. 2007;14:2654–2679. doi: 10.2174/092986707782023217. [DOI] [PubMed] [Google Scholar]
- 6.Kurokawa T, Suzuki K, Hayaoka T, Nakagawa T. Cyclophostin, acetylcholinesterase inhibitor from Streptomyces lavendulae. J Antibiotics. 1993;46:1315–1318. doi: 10.7164/antibiotics.46.1315. [DOI] [PubMed] [Google Scholar]
- 7.Bandyopadhyay S, Dutta S, Spilling C, Dupureur C, Rath N. Synthesis and Biological Evaluation of a Phosphonate Analog of the Natural Acetyl Cholinesterase Inhibitor Cyclophostin. J Org Chem. 2008;73:8386–8391. doi: 10.1021/jo801453v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dutta S, Malla R, Bandyopadhyay S, Spilling C, Dupureur CM. Synthesis and Kinetic Analysis of Some Phosphonate Analogs of Cyclophostin as Inhibitors of Human Acetylcholinesterase. Bioorg Med Chem. 2010;18:2265–2274. doi: 10.1016/j.bmc.2010.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh M, Kaur M, Kukreja H, Chugh R, Silakari O, Singh D. Acetylcholinesterase inhibitors as Alzheimer therapy: from nerve toxins to neuroprotection. Eur J Med Chem. 2013;70:165–188. doi: 10.1016/j.ejmech.2013.09.050. [DOI] [PubMed] [Google Scholar]
- 10.Vertesy L, Beck B, Bronstrup M, Ehrlich K, Kurz M, Muller G, Schummer D, Seibert G. Cyclipostins, Novel hormone-sensitive lipase inhibitors from Streptomyces sp. DSM 13381. II. Isolation, structure elucidation and biological properties. J Antibiot (Tokyo) 2002;55:480–494. doi: 10.7164/antibiotics.55.480. [DOI] [PubMed] [Google Scholar]
- 11.Osterlund T. Structure-function relationships of hormone-sensitive lipase. Eur J Biochem/ FEBS. 2001;268:1899–1907. doi: 10.1046/j.1432-1327.2001.02097.x. [DOI] [PubMed] [Google Scholar]
- 12.Yeaman SJ. Hormone-sensitive lipase--new roles for an old enzyme. Biochem J. 2004;379:11–22. doi: 10.1042/BJ20031811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walther TC, Farese JRV. Lipid Droplets and Cellular Lipid Metabolism. Ann Rev Biochem. 2012;81:687–714. doi: 10.1146/annurev-biochem-061009-102430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tiss A, Miled N, Verger R, Gargouri Y, Abousalham A. Digestive Lipases Inhibition: an In vitro Study. In: Müller G, Petry S, editors. Lipases and phospholipases in drug development. Wiley-VCH; Weinheim: 2004. pp. 155–193. [Google Scholar]
- 15.Lowe DB, Magnuson S, Qi N, Campbell AM, Cook J, Hong Z, Wang M, Rodriguez M, Achebe F, Kluender H, Wong WC, Bullock WH, Salhanick AI, Witman-Jones T, Bowling ME, Keiper C, Clairmont KB. In vitro SAR of (5-(2H)-isoxazolonyl) ureas, potent inhibitors of hormone-sensitive lipase. Bioorg Med Chem Lett. 2004;14:3155–3159. doi: 10.1016/j.bmcl.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 16.Slee DH, Bhat AS, Nguyen TN, Kish M, Lundeen K, Newman MJ, McConnell SJ. Pyrrolopyrazinedione-based inhibitors of human hormone-sensitive lipase. J Med Chem. 2003;46:1120–1122. doi: 10.1021/jm020460y. [DOI] [PubMed] [Google Scholar]
- 17.Ebdrup S, Sorensen LG, Olsen OH, Jacobsen P. Synthesis and structure-activity relationship for a novel class of potent and selective carbamoyl-triazole based inhibitors of hormone sensitive lipase. J Med Chem. 2004;47:400–410. doi: 10.1021/jm031004s. [DOI] [PubMed] [Google Scholar]
- 18.de Jong JC, Sorensen LG, Tornqvist H, Jacobsen P. Carbazates as potent inhibitors of hormone-sensitive lipase. Bioorg Med Chem Lett. 2004;14:1741–1744. doi: 10.1016/j.bmcl.2004.01.038. [DOI] [PubMed] [Google Scholar]
- 19.Ebdrup S, Jacobsen P, Farrington AD, Vedso P. Structure-activity relationship for aryl and heteroaryl boronic acid inhibitors of hormone-sensitive lipase. Bioorg Med Chem. 2005;13:2305–2312. doi: 10.1016/j.bmc.2004.12.042. [DOI] [PubMed] [Google Scholar]
- 20.Ebdrup S, Refsgaard HH, Fledelius C, Jacobsen P. Synthesis and structure-activity relationship for a novel class of potent and selective carbamate-based inhibitors of hormone selective lipase with acute in vivo antilipolytic effects. J Med Chem. 2007;50:5449–5456. doi: 10.1021/jm0607653. [DOI] [PubMed] [Google Scholar]
- 21.Schoenafinger K, Petry S, Mueller G, Baringhaus K-H. Substituted 3-phenyl-5-alkoxi-1,3,4-oxdiazol-2-one and use thereof for inhibiting hormone-sensitive lipase. WO 0166531 PCT Int Appl. 2001
- 22.Beltrandelrio H, Jacobsen P, DeJong J. Preparation of (3,4-dihydro-1H-isoquinolin-2-yl)carbamates for treating disorders where a decreased level of plasma FFA is desired. WO 0187843 PCT Int Appl. 2001
- 23.Petry S, Schoenafinger K, Mueler G, Baringhaus K-H. Substituted 3-phenyl-5-alkoxi-1,3,4-oxdiazol-2-ones and their use as lipase inhibitors. WO 0117981 PCT Appl. 2001
- 24.Ben Ali Y, Chahinian H, Petry S, Muller G, Lebrun R, Verger R, Carrière F, Mandrich L, Rossi M, Manco G, Sarda L, Abousalham A. Use of an inhibitor To identify members of the hormone-sensitive lipase family. Biochemistry. 2006;45:14183–14191. doi: 10.1021/bi0613978. [DOI] [PubMed] [Google Scholar]
- 25.Ben Ali Y, Verger R, Carriere F, Petry S, Muller G, Abousalham A. The molecular mechanism of human hormone-sensitive lipase inhibition by substituted 3-phenyl-5-alkoxy-1,3,4-oxadiazol-2-ones. Biochimie. 2012;94:137–145. doi: 10.1016/j.biochi.2011.09.028. [DOI] [PubMed] [Google Scholar]
- 26.Point V, Malla R, Diomande S, Martin B, Delorme V, Carriere F, Canaan S, Rath N, Spilling C, Fo Cavalier J. Synthesis and kinetic evaluation of cyclophostin and cyclipostins phosphonate analogs as selective and potent inhibitors of microbial lipases. J Med Chem. 2012;55:10204–10219. doi: 10.1021/jm301216x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malla RK, Bandyopadhyay S, Spilling CD, Dutta S, Dupureur CM. The first total synthesis of (+/−)-cyclophostin and (+/−)-cyclipostin P: inhibitors of the serine hydrolases acetyl cholinesterase and hormone sensitive lipase. Org Lett. 2011;13:3094–3097. doi: 10.1021/ol200991x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dutta S. Chemistry and Biochemistry. Univ. Missouri; St. Louis: 2010. Inhibition studies of serine hydrolases by cyclic phosphates and phosphonates. [Google Scholar]
- 29.Point V, Malla R, Carriere F, Canaan S, Spilling CD, Cavalier J. Enantioselective inhibition of microbial lipolytic enzymes by nonracemic monocyclic enolphosphonate analogues of cyclophostin. J Med Chem. 2013;56:4393–4401. doi: 10.1021/jm4000787. [DOI] [PubMed] [Google Scholar]
- 30.Mannesse ML, Cox RC, Koops BC, Verheij HM, de Haas GH, Egmond MR, van der Hijden HT, de Vlieg J. Cutinase from Fusarium solani pisi hydrolyzing triglyceride analogues. Effect of acyl chain length and position in the substrate molecule on activity and enantioselectivity. Biochemistry. 1995;34:6400–6407. doi: 10.1021/bi00019a020. [DOI] [PubMed] [Google Scholar]
- 31.Ottosson J, Hult K. Influence of acyl chain length on the enantioselectivity of Candida antarctica lipase B and its thermodynamic components in kinetic resolution of sec-alcohols. J Mol Cat B: Enzym. 2001;11:1025–1028. [Google Scholar]
- 32.Ben Ali Y, Chahinian H, Petry S, Muller G, Carriere F, Verger R, Abousalham A. Might the kinetic behavior of hormone-sensitive lipase reflect the absence of the lid domain? Biochemistry. 2004;43:9298–9306. doi: 10.1021/bi049479o. [DOI] [PubMed] [Google Scholar]
- 33.Kitz R, Wilson IB. Esters of methanesulfonic acid as irreversible inhibitors of acetylcholinesterase. J Biol Chem. 1962;237:3245–3249. [PubMed] [Google Scholar]
- 34.Lookene A, Skottova N, Olivecrona G. Interactions of lipoprotein lipase with the active-site inhibitor tetrahydrolipstatin (Orlistat) Eur J Biochem/ FEBS. 1994;222:395–403. doi: 10.1111/j.1432-1033.1994.tb18878.x. [DOI] [PubMed] [Google Scholar]
- 35.Kropp TJ, Richardson RJ. Mechanism of aging of mipafox-inhibited butyrylcholinesterase. Chem Res Toxicol. 2007;20:504–510. doi: 10.1021/tx600310y. [DOI] [PubMed] [Google Scholar]
- 36.Kraemer FB, Shen WJ. Hormone-sensitive lipase: control of intracellular tri-(di-)acylglycerol and cholesteryl ester hydrolysis. J Lipid Res. 2002;43:1585–1594. doi: 10.1194/jlr.r200009-jlr200. [DOI] [PubMed] [Google Scholar]
- 37.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Science. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Belfrage P, Vaughan M. Simple liquid-liquid partition system for isolation of labeled oleic acid from mixtures with glycerides. J Lipid Res. 1969;10:341–344. [PubMed] [Google Scholar]
- 39.Fredrikson G, Stralfors P, Nilsson NO, Belfrage P. Hormone-sensitive lipase of rat adipose tissue. Purification and some properties. J Biol Chem. 1981;256:6311–6320. [PubMed] [Google Scholar]
- 40.Walker MC, Kurumbail RG, Kiefer JR, Moreland KT, Koboldt CM, Isakson PC, Seibert K, Gierse JK. A three-step kinetic mechanism for selective inhibition of cyclo-oxygenase-2 by diarylheterocyclic inhibitors. Biochem J. 2001;357:709–718. doi: 10.1042/0264-6021:3570709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Callan OH, So OY, Swinney DC. The kinetic factors that determine the affinity and selectivity for slow binding inhibition of human prostaglandin H synthase 1 and 2 by indomethacin and flurbiprofen. J Biol Chem. 1996;271:3548–3554. doi: 10.1074/jbc.271.7.3548. [DOI] [PubMed] [Google Scholar]
- 42.Barbier P, Schneider F. Syntheses of Tetrahydrolipstatin and Absolute Configuration of Tetrahydrolipstatin and Lipstatin. Helvetica Chimica Acta. 1987;70:196–202. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.