

Abstract
Fragile X-associated tremor/ataxia syndrome (FXTAS) is a late-onset neurodegenerative disorder associated with premutation alleles of the FMR1 gene that is characterized by progressive action tremor, gait ataxia, and cognitive decline. Recent studies of mitochondrial dysfunction in FXTAS have suggested that iron dysregulation may be one component of disease pathogenesis. We tested the hypothesis that iron dysregulation is part of the pathogenic process in FXTAS. We analyzed postmortem choroid plexus from FXTAS and control subjects, and found that in FXTAS iron accumulated in the stroma, transferrin levels were decreased in the epithelial cells, and transferrin receptor 1 distribution was shifted from the basolateral membrane (control) to a predominantly intracellular location (FXTAS). In addition, ferroportin and ceruloplasmin were markedly decreased within the epithelial cells. These alterations have implications not only for understanding the pathophysiology of FXTAS, but also for the development of new clinical treatments that may incorporate selective iron chelation.
Keywords: FXTAS, iron, Fragile X, neurodegeneration, FMR1, autism, premutation, CGG repeat, choroid plexus
Introduction
Fragile X-associated tremor/ataxia syndrome (FXTAS) is a late-onset neurodegenerative disorder associated with premutation alleles (55–200 CGG repeats) of the FMR1 gene; larger expansions (> 200 CGG repeats; full mutation) give rise to fragile X syndrome, the most common inherited form of cognitive impairment [1]. Carriers of premutation alleles are common in the general population, with an estimated frequency as high as 1 in 130 females and 1 in 250 males [2,3]. However, for reasons that are not currently understood, only about 40% of male carriers and 8–13% of female carriers will eventually develop FXTAS [4–6]. FXTAS is characterized by progressive action tremor, gait ataxia, cognitive decline, Parkinsonism, neuropathy, and autonomic dysfunction [7]. Central nervous system (CNS) pathology includes dystrophic white matter and intranuclear inclusions in neurons and astrocytes [8–10]. Although neurological symptoms of FXTAS have only been observed in adults, it is now clear that children carrying premutation alleles may also have forms of clinical involvement that include anxiety, attention deficit hyperactivity disorder, and autism spectrum disorders [11,12]. Additionally, premutation CGG-repeat expansions in the mouse Fmr1 gene have been shown to alter embryonic neocortical development [13]. There is significant oxidative stress in premutation neurons in culture, in addition to mitochondrial dysfunction [14,15]. Mitochondrial dysfunction is more severe in those with FXTAS compared to carriers without FXTAS, and there is documented iron dysregulation at the mitochondria in those with FXTAS [16]. Thus, FXTAS may actually be a late manifestation of pathogenic mechanisms that are operating throughout life in carriers of premutation alleles.
Iron is essential for many facets of cell metabolism, including transport of oxygen by hemoglobin, DNA synthesis, mitochondrial respiration, synthesis of neurotransmitters, and signal transduction in the CNS. For example, iron is an essential co-factor for tyrosine hydroxylase, and it required for the synthesis of myelin, and neurotransmitters such as dopamine, norepinephrine, and serotonin [17,18]. On the other hand, iron is a source of reactive oxygen species, cycling from ferric (Fe3+) to ferrous (Fe2+) and back to Fe3+, with the net conversion of the superoxide radical (O2−) and hydrogen peroxide (H2O2) to the highly reactive hydroxyl free radical (·OH), OH−, and O2 [19,20]. In other words, uncomplexed iron reacts with molecular oxygen to generate the reactive oxygen species that lead to oxidative stress. Thus, altered CNS iron metabolism is likely to initiate or contribute to the development of neurodegenerative diseases, such FXTAS. Iron dysregulation has been linked to a series of neurodegenerative disease, including Parkinson and Alzheimer diseases, amyotrophic lateral sclerosis, restless legs syndrome, and prion diseases [21]. Indeed, Parkinsonism and dementia are common in those with FXTAS [7,22], and restless legs syndrome is more common in those with the premutation compared to controls [23].
Iron and other elements enter the brain through one of two brain barriers, the blood-cerebrospinal fluid barrier (BCB) located at the choroid plexus, or the blood-brain barrier formed by the tight junctions of the brain capillary endothelium. Both barriers serve to protect the brain from toxic substances present in the blood. In the BCB, the epithelial cells in the choroid plexus form a monolayer with tight intercellular junctions that seal the transport pathway between the cells, so that substances entering the brain must use specialized cell transport systems. The basolateral side of the epithelial cells is in contact with capillaries located in the choroidal stroma, whereas the apical side is in contact with the cerebrospinal fluid (CSF). The choroid plexus synthesizes and secretes most of the components of the CSF, transports molecules from the circulation into the CSF, and eliminates toxic substances from the CSF. Therefore, any alteration in the transport function of the choroid plexus is likely to alter CSF composition and tissue homeostasis [24–27]. Iron in serum traverses the BCB after oxidation to Fe3+ by light ferritin and hephaestin. This process involves transferrin (Tf) and transferrin receptor 1 (TfR1), located on the basal side of the epithelial cells lining the BCB. Following TfR1-mediated transport, the Fe3+-Tf complex undergoes endocytosis with iron released from Tf within the endosome [28]. Divalent metal transporter 1 (DMT1) then facilitates the transport of iron out of the endosome into the cytoplasm [29]. Cytoplasmic iron is subsequently exported by ferroportin into the brain through the apical side of the epithelial cells [30,31]. Ferroportin mediates the release of iron in conjunction with ceruloplasmin, which must oxidize the iron transported by ferroportin before its release as Fe3+ into the extracellular medium [32,33]. Cytoplasmic iron can also be transferred to the mitochondria for use in heme and iron-sulfur cluster synthesis, or is stored in cytosolic ferritin.
In the current work, we tested the hypothesis that iron transport from the circulation to the CSF is dysregulated and is part of the pathogenic process in FXTAS. Using postmortem choroid plexus tissue from patients with FXTAS, we found that iron accumulates in the choroid plexus, and that levels and distribution of key iron-binding proteins are altered in FXTAS. These data suggest a potential alteration of iron metabolism in the CNS.
Results
We obtained choroid plexus from the brains of 8 FXTAS subjects (7 males and 1 female) and 9 controls (8 males and 1 female). The median number of CGG repeats for the FXTAS group was 84 (range, 65–106 repeats). The median age at time of death was 73.8 years (range, 52–87 years) for the FXTAS group and 67.3 years (range, 57–82 years) for the controls (p=0.2).
Iron accumulates in choroid plexus of FXTAS subjects
We used random samples of choroid plexus to study iron localization using Perl’s method for iron staining, which detects iron bound to the hemosiderin that is localized to the stroma of the choroid plexus [34]. We found iron deposits in the stroma of the choroid plexus in most control and FXTAS subjects. The percentage of stromal area occupied by iron deposits was higher in FXTAS (3.41% ± 0.009) than in control (0.68% ± 0.001), p = 0.02. Some of the iron deposits were located in psammoma bodies within the stroma (Fig. 1); psammoma bodies are concentric, lamellated, calcified structures that appear in the choroid plexus and are associated with aging [35]. The majority of subjects in the current cohorts possessed psammoma bodies (5/9 controls; 5/8 FXTAS subjects). In our subjects, there was no relationship between the presence of psammoma bodies and age or group (control, FXTAS). These data indicate that there is an increase in iron deposition in the choroid plexus of FXTAS patients compared to controls (Figs. 1 and 2).
Fig. 1.
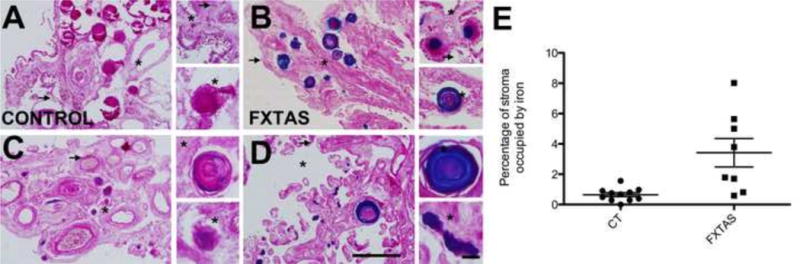
Iron deposition (blue) in control (A, C) and FXTAS (B, D) subjects. Asterisks are located within the stroma and arrows point to the apical surface of epithelial cells. Scale bars: 500 and 50 μm. E. Assessment of iron deposition in the stroma of the choroid plexus FXTAS and control subjects.
Fig. 2.
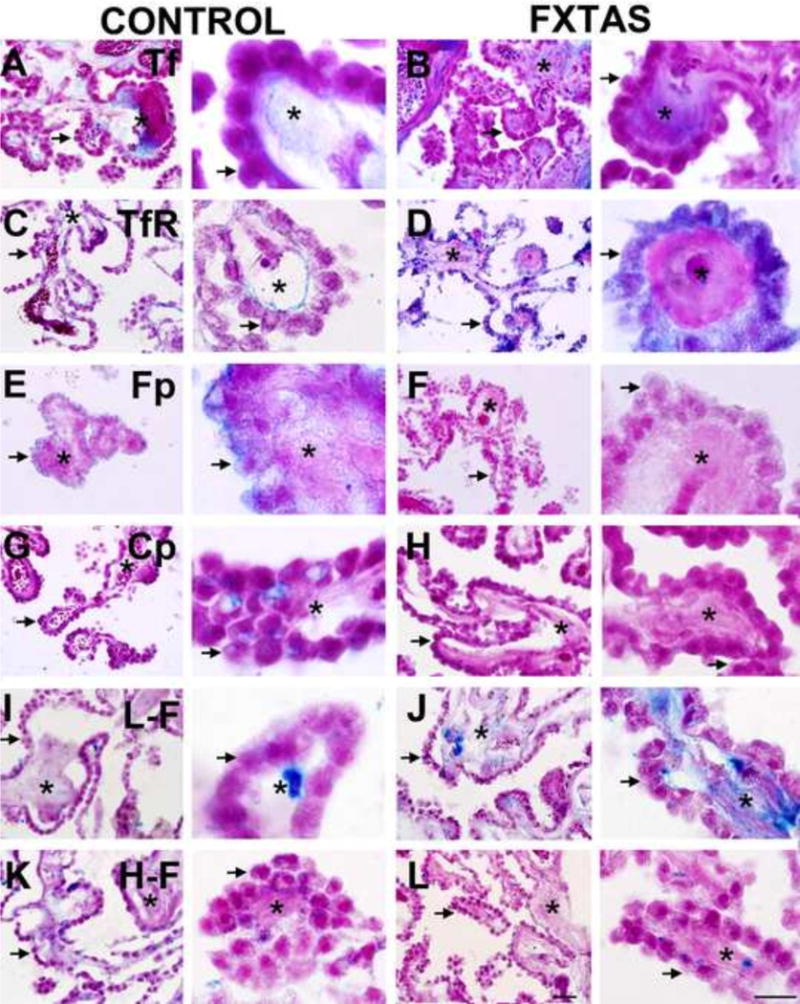
Iron binding protein expression in control and FXTAS subjects: (A, B) Tf: Transferrin; (C,D) TfR: Transferrin Receptor 1 ; (E, F) Fp: Ferroportin; (G, H) Cp: Ceruloplasmin; (I, J) L-F Light Ferritin; (K, L); H-F: Heavy Ferritin. Each image includes and asterisk located within the stroma and an arrow pointing to the apical surface of epithelial cells. Scale bars: 500 and 50 μm.
Transferrin levels in choroid plexus are decreased in FXTAS
To further investigate potential iron dysregulation in the brains of subjects with FXTAS, we performed immunostaining with antibodies against several iron-binding proteins. Using ImageJ, we first analyzed the distribution and the amount of Tf expression. Tf staining was intracellular, and was also present in the stroma of the choroid plexus (Fig. 3 A, B). We observed a significant decrease in the amount of Tf in the epithelial cells of FXTAS subjects (17.89 ± 6.80 sem, control; 3.25 ± 1.97, FXTAS; p= 0.05), (Fig. 4A).
Fig. 3.
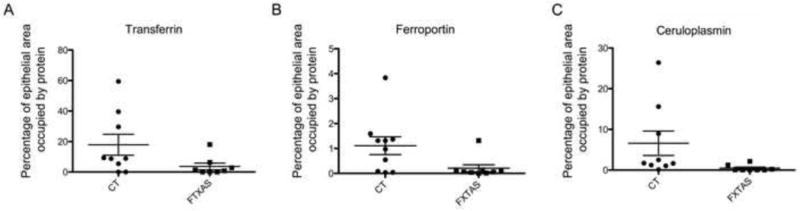
Assessment of iron-binding protein expression in the epithelial cells of FXTAS and control subjects.
Fig. 4.
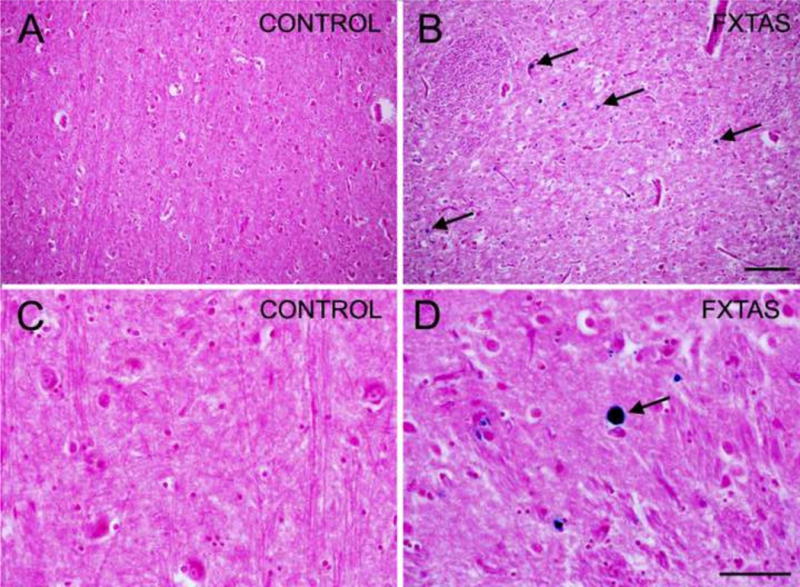
Iron deposition within the putamen (blue) in FXTAS (A, C) and control (B, D) subjects. Arrows point to iron deposits. Scale bars: 250 and 50 μm.
We next quantified TfR1 expression. We determined that TfR1 was both associated with the basolateral membrane of the epithelial cells and within the cytoplasm (Fig. 3 C, D). In the basolateral membrane, TfR1 formed a thin, uniform monolayer of puncta. We did not find any difference between FXTAS and control groups for the number and size of TfR1 puncta (p = 0.19). However, we observed that in control subjects, TfR1 was mostly localized to the basolateral membrane, and only occasionally intracellular, while most of the TfR1 staining in FXTAS subjects was located within the cytoplasmic compartment. These results indicate that, while the number of TfR1 puncta did not change, most of the TfR1 molecules re-localized from the membrane to the cytoplasm in FXTAS. This change in location of TfR1 is likely to play a role in determining the amount of iron within the choroidal stroma that can bind TfR1 in the membrane of the epithelial cells, and therefore may have an effect on the transport of iron through the BCB into the brain.
Ferroportin and ceruloplasmin levels in choroid plexus are decreased in FXTAS
In conjunction with ceruloplasmin, which generates Fe3+, ferroportin mediates the release of ferric iron from epithelial cells into the CSF [36]. Ferroportin was observed primarily in the apical membrane of the epithelial cells, while ceruloplasmin was located within the cytoplasm (Fig. 3 E–F); none of these proteins was present within the stroma. The amount of ferroportin was markedly decreased for FXTAS subjects (0.05 ± 0.01, FXTAS; 1.22 ± 0.37, control; p = 0.01, Fig. 4B). The amount of ceruloplasmin was also decreased in FXTAS subjects (0.40 ± 0.25, FXTAS; 6.58 ± 02.99 controls; p = 0.05, Fig. 4C).
Light-chain ferritin and heavy-chain ferritin levels did not change in the choroid plexus FXTAS
We next quantified levels of light-chain ferritin (L–F). Light-chain ferritin was present both in the choroidal stroma and in the epithelial cells. The stromal labeling was distributed relatively uniformly, while the epithelial labeling was confined to single pools within the cytoplasm (Fig. 2E, F). In the subjects with psammoma bodies within the stroma, L–F was localized to the rings of the outer periphery. There was not a significant change in the amount of L–F deposits within the epithelial cells of FXTAS subjects compared to controls (p = 0.21, Fig. 2 I, J).
Heavy-chain ferritin (H–F), associated with iron transport, was distributed within the stroma with higher density in the area between the basal membrane of the epithelial cells and the stromal capillaries. H–F had a granular and as in the case of L–F, H–F localized to rings of the outer periphery of the psammoma bodies. There was no significant change in the amount of H-F deposits within the epithelial cells of FXTAS subjects compared to controls (p = 0.54, Fig. 2 K, L).
Iron accumulates in the putamen in FXTAS
We collected tissue from the putamen of the 67 y/o case of FXTAS and the 79 y/o control case, performed Perl staining for iron and observed that while the control case hardly had any iron in the putamen, the FXTAS case presented with numerous iron deposits that varied in size up to 15 μm in diameter (Fig. 4).
Discussion
Overall, we found that (1) iron accumulated in the stroma of the choroid plexus (i.e., external to the CSF and to the epithelial cells comprising the BCB); (2) Tf expression decreased in the epithelial cells of the choroid plexus; (3) TfR1 distribution was altered, shifting to the cytoplasm from the basolateral membrane; (4) Ferroportin and ceruloplasmin were markedly decreased within the epithelial cells, (5) light-chain ferritin and heavy-chain ferritin levels did not show any change in distribution or quantity in the epithelial cells, and (6) there is potential iron accumulation within the putamen, consistent with recent multimodal imaging analysis of subcortical gray matter [37].
Iron deficiency in the central nervous system is known to cause motor impairment and cognitive deficits [21,38–40], traits that are present in FXTAS. In addition, brain iron deficiency has been reported to cause fatigue, irritability, lethargy, listlessness, reduced concentration, hypoactivity, reduced intellectual functioning, and hyperactivity in children, traits that are present in many individuals with FXTAS and in some carriers without FXTAS [7]. To study the state of iron dysregulation in FXTAS, we examined the transport of iron from the circulation to the CSF though the BCB. We focused our study on the expression of iron and iron-binding proteins in the choroid plexus, the site of iron transport across the BCB. Using Perl’s method for iron staining, we found an increase in the iron bound to hemosiderin deposits in the stroma of the choroid plexus in FXTAS subjects. Ferritin is responsible for normal iron storage, whereas hemosiderin is a degradation product of ferritin, and is prominent during conditions of iron overload [41]. Therefore, our finding of increased iron bound to hemosiderin indicates an iron overload within the stroma of the choroid plexus in the FXTAS brain. These data suggest that there is an alteration in the transport of iron from general circulation into the CSF, and further suggests the possibility of altered iron transport and metabolism in the FXTAS brain. In agreement with these results, we observed that in the pair of cases where we examined the putamen, there was an increased in iron deposition within the putamen of FXTAS compared to control, a nucleus of the basal ganglia involved in motor symptoms in neurodegenerative diseases. In this regard, we have previously published evidence of mitochondrial dysfunction involving iron dysregulation and accumulation in FXTAS, and the use of multimodal imaging analysis of subcortical gray matter, where there is evidence from DWI of iron deposition [37,42].
To further understand the alteration of iron transport into the CSF, we examined the levels and distribution of iron-binding proteins in the choroid plexus of control and FXTAS brains. Transferrin is the major conduit for iron delivery to neurons and, when dysregulated or deficient, is known to play an important role in neurodegenerative diseases [28], likely reflecting altered mobilization and transport of iron. Apotransferrin binds with high affinity to iron in circulation, forming holotransferrin, which binds to TfR1 expressed by endothelial cells in the blood-brain barrier and epithelial cells in the BCB [43]. The holotransferrin complex is then endocytosed into the endothelial/epithelial cells, ending up in the endosomal/lysosomal compartment, where the acid environment dissociates iron from holotransferrin, followed by iron transport to the cytosol [44]. Once iron is released, apotransferrin is then recycled to the cell membrane. Our data indicate that intracellular levels of transferrin are decreased in choroidal epithelial cells in FXTAS, suggesting a decrease in the transport function of transferrin, and subsequently, decreased iron import into the CSF.
Our results also show that TfR1 is localized to the basolateral membrane and within the cytoplasm of choroidal epithelial cells; however, we noted different expression patterns in control and FXTAS plexuses. In control brains, TfR1 was expressed primarily in the basolateral membrane, but was expressed predominately within the cell in the FXTAS tissue. This redistribution of TfR1 indicates that more TfR1 is internalized into the cytoplasm, and that less TfR1 is available to bind extracellular iron in FXTAS. Decreased membrane TfR1 would be expected to reduce iron intake into the cell, and, in the presence of normal circulating iron in blood, would produce an accumulation of iron bound to hemosiderin in the stroma, as observed in FXTAS subjects; the result is a decreased iron concentration within epithelial cells.
Since iron-binding proteins are transcriptionally repressed by low iron concentration [45], we would predict decreased expression levels for the iron-binding proteins, which transport iron from epithelial cells into the CSF, in FXTAS. Indeed, our data show a decrease of ferroportin and ceruloplasmin in FXTAS brains compared to control.
Altered iron metabolism is a common feature of neurodegenerative diseases, including neurodegeneration with brain iron accumulation disorders (NBIA), Parkinson’s (PD), Alzheimer’s (AD), and Huntington’s diseases; amyotrophic lateral sclerosis; multiple sclerosis; restless legs syndrome; Friedreich ataxia; and prion-mediated disorders such as Creutfeldt-Jakob disease [21,38–40,46]. The most common of the NBIA is panthothenate-associated neurodegeneration (PKAN) that is characterized by progressive dystonia and parkinsonism associated with intellectual decline [47]. Perl’s staining of PKAN tissue shows a widespread perivascular deposition of iron largely confined to the globus pallidus. Restless legs syndrome, a disease that shares motor symptoms in common with FXTAS and occurs more frequently in premutation carriers than control, has also been linked to an alteration in iron transport into the brain [48]. Huntington’s disease that is characterized by an alteration of iron metabolism within the brain, also shares motor symptoms in common with FXTAS [49,50]. To our knowledge there is only one report of iron alteration within the choroid plexus in the diseases above, in restless leg syndrome. However this report indicated that iron was reduced in the epithelial cells of choroid plexus and brain microvasculature [51]. Based on our current results, iron accumulation in FXTAS may share, at least in part, mechanisms of iron dysfunction that are operating in these neurodegenerative diseases. What is generally unknown about neurodegenerative disorders that accumulate iron is whether iron dysregulation is a cause or consequence of the disease. Processes that alter the transcription and/or translation of iron regulatory proteins (IRP) or iron binding proteins (IBP) will result in altered iron metabolism, and therefore in the level of oxidative stress, itself a risk factor for neurodegeneration. We hypothesize that the observed dysregulation of iron processing is secondary to the RNA toxicity present in carriers that leads to FXTAS [7,52]. We further hypothesize that expanded CGG transcripts in the FMR1 gene may sequester one or more IRP/IBPs, thus impairing their functions and resulting in altered processing of iron homeostasis and ultimately in the neuropathology of FXTAS. A similar process takes place in PD and AD, where Lewy bodies – rich in α-synuclein protein – and neuritic plaques, – rich in β-amyloid – present with iron deposits. Accordingly, follow-on investigations should determine whether, in FXTAS, there is an accumulation of iron in the brain microvasculature similar to PKAN, or a decrease of iron as in restless legs syndrome.
In summary, we show that, in the choroid plexus of FXTAS subjects, there is decreased epithelial transferrin, altered intracellular distribution of TfR1, and decreased ferroportin and ceruloplasmin expression (Fig. 8). Together, these observations provide evidence of altered iron transport at the BCB in FXTAS. This alteration has implications for understanding the pathophysiology of FXTAS, and for the development of new clinical treatments.
Experimental Procedure
Sample collection
FXTAS and control tissue was obtained from the FXTAS brain repository at the University of California, Davis, School of Medicine. All human tissue specimens were obtained through consented autopsies with institutional review board approval. We used tissue from 8 FXTAS subjects and 9 control subjects, where the control group consisted of tissue samples from subjects who lacked any significant neurological history at the time of death. Additional control tissue was obtained from the Neuropathology Core of the Alzheimer’s Disease Center at the University of California, Davis, Health System. All tissue was fixed. We did not have access to fresh tissue.
DNA analysis
DNA was isolated from human brain tissue using the Gentra Puregene genomic DNA purification kit (Qiagen, Valencia, CA), which routinely yields 5–15 μg of genomic DNA per sample. CGG-repeat lengths were determined following PCR amplification of the repeat region using a standard protocol with 100 ng of genomic DNA, utilizing the Expand Long Template PCR System (Roche Diagnostics Corporation, Indianapolis, IN) and primers C3 (5′-TGTTTACACCCGCAGCGGGCCGGGGGTTC) and F (5′-AGCCCCGCACTTCCACCACCAGCTCCTCCA). PCR reactions were resolved by agarose gel electrophoresis and then sized by regression relative to the HiLo DNA Marker (Bionexus, Inc, Oakland, CA) using the Alpha Innotech Fluorchem 8900 BioImaging System running AlphaEaseFC software (ProteinSimple, Santa Clara, CA).
Perl’s method
Samples were washed in PBS for 5 min, immersed in 30% sucrose for two days, then frozen and sectioned at 14μm on a cryostat. Sections were washed in distilled water for 5 min, immersed in potassium ferrocyanide solution for 20 min (5% K4Fe(CN)6.3H2O), washed in distilled water for 5 min, immersed in nuclear fast red for 7 min, washed in distilled water for 5 min, dehydrated with ethanol, and cleared with xylene. Sections were coverslipped with permount (Fisher, Pittsburgh, PA). Upon addition of potassium ferrocyanide, the deposits of iron present in the brain in the ferric state form insoluble, blue ferric ferrocyanide molecules [34].
Immunostaining
Antigen retrieval was performed with 1x diva solution (heat-induced epitope retrieval (HIER) buffer; Biocare Medical, Concord, CA) at 80–110°C (15 min), followed by 0.1N HCl (7 min), 20% glacial acetic acid (30 sec), levamisole (Vector Laboratories Inc., Burlingame, CA) + 0.025% Tween 20 (Fisher) for 30 min to 1 hr, primary antibody (specified below, 5% BSA + 10% donkey serum, 0.0025% Tween 20, 4°C, 15 hr), secondary antibody (below, same diluent, 1:100, 1 hr, room temperature), and Vector Blue Alkaline Phosphatase Substrate (Vector Laboratories Inc., Burlingame, CA) for 10 min, counterstained with Neutral Red, dehydrated with ethanol, cleared with xylene, and mounted with permount. Washes were performed between each step with TBS buffer. For TfR1 and mitoferritin, the HCl/glacial acetic acid step was skipped. Primary antibodies: rabbit anti-transferrin (1:500, Ab9538, Abcam, Cambridge, MA), rabbit anti-light ferritin (1:1000, Ab69090, Abcam), rabbit anti-heavy ferritin (1:100, Ab75972 RabMabs, Cambridge, MA), mouse anti-ceruloplasmin (1:100, 611488, BD Transduction Labs, San Jose, CA), CD71 (1:50, 10084-2-AP, Proteintech, Chicago, IL), goat anti-ferroportin (1:50, SC-49668, Santa Cruz, Dallas, TX), and mitochondrial ferritin (1:100, Ab75973, RabMabs, Ab75973). Secondary antibodies: donkey anti goat/mouse/rabbit-AP (1:100, Jackson Immuno Research, West Grove, PA), mouse (115-056-006), goat (705-056-147) and rabbit (711-056-152). To quantify the amount of iron and proteins in each sample we used image J, and determined the percentage of epithelial cell area occupied by iron or iron related protein.
Statistical analysis
The amount of protein expression was compared between brains from control and FXTAS age-matched subjects using paired t-test (GraphPad Prism software). A p value of 0.05 was used for statistical significance.
Fig. 5.
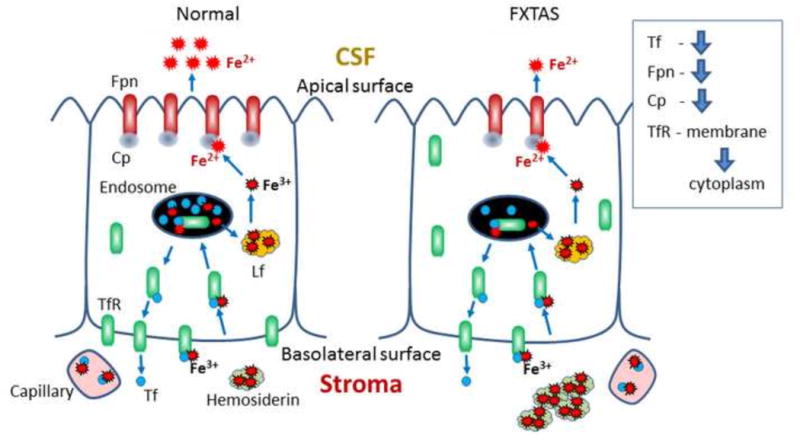
Scheme depicting distribution and amount of iron and iron-binding proteins in control and FXTAS choroid plexus. Tf: Transferrin, Fpn: Ferroportin, Cp: Ceruloplasmin, Lf: Light ferritin, TfR: Transferrin Receptor.
Table 1.
Clinical characteristics of postmortem cases, including number of CGG repetitions, age, gender, postmortem interval, and cause of death.
| Diagnosis | Repetitions | Age | Gender | PMI (h) | Cause of death |
|---|---|---|---|---|---|
| FXTAS | 79 | 52 | F | NK | Multiple Sclerosis |
| FXTAS | 97 | 58 | M | 4 | Multiple Sclerosis |
| FXTAS | 78 | 75 | M | 22 | Prostate cancer |
| FXTAS | 71 | 77 | M | 24 | NK |
| FXTAS | 106 | 78 | M | NK | Respiratory death |
| FXTAS | 81 | 79 | M | 7 | Choking |
| FXTAS | NK | 85 | M | 80 | Respiratory death |
| FXTAS | 65 | 87 | M | NK | Respiratory death |
| Control | NA | 57 | M | 23 | Pulmonary disease |
| Control | NA | 58 | M | 21 | Cirrhosis |
| Control | NA | 60 | M | 14 | Bladder Cancer |
| Control | NA | 61 | M | 37 | Myocardiac infarction |
| Control | NA | 67 | M | 72 | Adenocarcinoma |
| Control | NA | 70 | M | NK | Pulmonary disease |
| Control | NA | 72 | M | 36 | Cardiovascular disease |
| Control | NA | 79 | M | 20 | Cardiovascular disease |
| Control | NA | 82 | F | 122 | Cardiovascular disease |
NA: Not applicable. NK: Not Known.
Highlights.
In the choroid plexus of FXTAS subjects there is: Increase of iron deposits.
Decreased of epithelial transferrin, and decreased ferroportin and ceruloplasmin expression.
Altered intracellular distribution of TfR1.
Altered iron transport at the blood-cerebrospinal fluid barrier in FXTAS.
Iron-chelating drugs may help to inhibit the progression of FXTAS.
Acknowledgments
The authors wish to acknowledge support from the National Institutes of Health (MH094681; HD040661; HD036071), and Shriners Hospitals. The authors acknowledge the support of the UC Davis National Center for Advancing Translational Sciences, National Institutes of Health, through grant number UL1 TR000002. The authors also wish to thank the families who have graciously supported our research effort.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hagerman RJ, Hall DA, Coffey S, Leehey M, Bourgeois J, et al. Treatment of fragile X-associated tremor ataxia syndrome (FXTAS) and related neurological problems. Clin Interv Aging. 2008;3:251–262. doi: 10.2147/cia.s1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kogan CS, Turk J, Hagerman RJ, Cornish KM. Impact of the Fragile X mental retardation 1 (FMR1) gene premutation on neuropsychiatric functioning in adult males without fragile X-associated Tremor/Ataxia syndrome: a controlled study. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:859–872. doi: 10.1002/ajmg.b.30685. [DOI] [PubMed] [Google Scholar]
- 3.Tassone F, Hagerman R. The fragile X-associated tremor ataxia syndrome. Results Probi Cell Differ. 2012;54:337–357. doi: 10.1007/978-3-642-21649-7_18. [DOI] [PubMed] [Google Scholar]
- 4.Coffey SM, Cook K, Tartaglia N, Tassone F, Nguyen DV, et al. Expanded clinical phenotype of women with the FMR1 premutation. Am J Med Genet A. 2008;146A:1009–1016. doi: 10.1002/ajmg.a.32060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodriguez-Revenga L, Madrigal I, Pagonabarraga J, Xuncla M, Badenas C, et al. Penetrance of FMR1 premutation associated pathologies in fragile X syndrome families. Eur J Hum Genet. 2009;17:1359–1362. doi: 10.1038/ejhg.2009.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jacquemont S, Hagerman RJ, Leehey MA, Hall DA, Levine RA, et al. Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. JAMA. 2004;291:460–469. doi: 10.1001/jama.291.4.460. [DOI] [PubMed] [Google Scholar]
- 7.Hagerman R, Hagerman P. The other face of the fragile X gene: Advances in clinical and molecular understanding of the FMR1 premutation and FXTAS. Lancet Neurol. 2013 doi: 10.1016/S1474-4422(13)70125-X. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greco CM, Tassone F, Garcia-Arocena D, Tartaglia N, Coffey SM, et al. Clinical and neuropathologic findings in a woman with the FMR1 premutation and multiple sclerosis. Arch Neurol. 2008;65:1114–1116. doi: 10.1001/archneur.65.8.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greco CM, Berman RF, Martin RM, Tassone F, Schwartz PH, et al. Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS) Brain. 2006;129:243–255. doi: 10.1093/brain/awh683. [DOI] [PubMed] [Google Scholar]
- 10.Greco CM, Hagerman RJ, Tassone F, Chudley AE, Del Bigio MR, et al. Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain. 2002;125:1760–1771. doi: 10.1093/brain/awf184. [DOI] [PubMed] [Google Scholar]
- 11.Farzin F, Perry H, Hessl D, Loesch D, Cohen J, et al. Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. J Dev Behav Pediatr. 2006;27:S137–144. doi: 10.1097/00004703-200604002-00012. [DOI] [PubMed] [Google Scholar]
- 12.Chonchaiya W, Au J, Schneider A, Hessl D, Harris SW, et al. Increased prevalence of seizures in boys who were probands with the FMR1 premutation and co-morbid autism spectrum disorder. Hum Genet. 2012;131:581–589. doi: 10.1007/s00439-011-1106-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cunningham CL, Martinez Cerdeno V, Navarro Porras E, Prakash AN, Angelastro JM, et al. Premutation CGG-repeat expansion of the Fmrl gene impairs mouse neocortical development. Hum Mol Genet. 2011;20:64–79. doi: 10.1093/hmg/ddq432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaplan ES, Cao Z, Hulsizer S, Tassone F, Berman RF, et al. Early mitochondrial abnormalities in hippocampal neurons cultured from Fmrl pre-mutation mouse model. J Neurochem. 2012;123:613–621. doi: 10.1111/j.1471-4159.2012.07936.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cao Z, Hulsizer S, Tassone F, Tang HT, Hagerman RJ, et al. Clustered burst firing in FMR1 premutation hippocampal neurons: amelioration with allopregnanolone. Hum Mol Genet. 2012;21:2923–2935. doi: 10.1093/hmg/dds118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ross-Inta C, Omanska-Klusek A, Wong S, Barrow C, Garcia-Arocena D, et al. Evidence of mitochondrial dysfunction in fragile X-associated tremor/ataxia syndrome. Biochem J. 2010;429:545–552. doi: 10.1042/BJ20091960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beard JL, Wiesinger JA, Connor JR. Pre- and postweaning iron deficiency alters myelination in Sprague-Dawley rats. Dev Neurosci. 2003;25:308–315. doi: 10.1159/000073507. [DOI] [PubMed] [Google Scholar]
- 18.Beard J, Erikson KM, Jones BC. Neonatal iron deficiency results in irreversible changes in dopamine function in rats. J Nutr. 2003;133:1174–1179. doi: 10.1093/jn/133.4.1174. [DOI] [PubMed] [Google Scholar]
- 19.Jomova K, Valko M. Importance of iron chelation in free radical-induced oxidative stress and human disease. Curr Pharm Des. 2011;17:3460–3473. doi: 10.2174/138161211798072463. [DOI] [PubMed] [Google Scholar]
- 20.Nunez MT, Urrutia P, Mena N, Aguirre P, Tapia V, et al. Iron toxicity in neurodegeneration. Biometals. 2012;25:761–776. doi: 10.1007/s10534-012-9523-0. [DOI] [PubMed] [Google Scholar]
- 21.Batista-Nascimento L, Pimentel C, Menezes RA, Rodrigues-Pousada C. Iron and neurodegeneration: from cellular homeostasis to disease. Oxid Med Cell Longev. 2012;2012:128647. doi: 10.1155/2012/128647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tassone F, Greco CM, Hunsaker MR, Seritan AL, Berman RF, et al. Neuropathological, clinical and molecular pathology in female fragile X premutation carriers with and without FXTAS. Genes Brain Behav. 2012;11:577–585. doi: 10.1111/j.1601-183X.2012.00779.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Summers S, Cogswell J, Goodrich J, Mu Y, Nguyen D, et al. Fatigue and Body Mass Index in the Fragile X Premutation Carrier. Clinical Genetics. 2014 doi: 10.1111/cge.12249. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Engelhardt B, Sorokin L. The blood-brain and the blood-cerebrospinal fluid barriers: function and dysfunction. Semin Immunopathol. 2009;31:497–511. doi: 10.1007/s00281-009-0177-0. [DOI] [PubMed] [Google Scholar]
- 25.Johanson CE, Duncan JA, 3rd, Klinge PM, Brinker T, Stopa EG, et al. Multiplicity of cerebrospinal fluid functions: New challenges in health and disease. Cerebrospinal Fluid Res. 2008;5:10. doi: 10.1186/1743-8454-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Emerich DF, Skinner SJ, Borlongan CV, Vasconcellos AV, Thanos CG. The choroid plexus in the rise, fall and repair of the brain. Bioessays. 2005;27:262–274. doi: 10.1002/bies.20193. [DOI] [PubMed] [Google Scholar]
- 27.Mesquita SD, Ferreira AC, Sousa JC, Santos NC, Correia-Neves M, et al. Modulation of iron metabolism in aging and in Alzheimer’s disease: relevance of the choroid plexus. Front Cell Neurosci. 2012;6:25. doi: 10.3389/fncel.2012.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luck AN, Mason AB. Transferrin-mediated cellular iron delivery. Curr Top Membr. 2012;69:3–35. doi: 10.1016/B978-0-12-394390-3.00001-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohgami RS, Campagna DR, McDonald A, Fleming MD. The Steap proteins are metalloreductases. Blood. 2006;108:1388–1394. doi: 10.1182/blood-2006-02-003681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moos T, Rosengren Nielsen T. Ferroportin in the postnatal rat brain: implications for axonal transport and neuronal export of iron. Semin Pediatr Neurol. 2006;13:149–157. doi: 10.1016/j.spen.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 31.Rouault TA, Cooperman S. Brain iron metabolism. Semin Pediatr Neurol. 2006;13:142–148. doi: 10.1016/j.spen.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 32.Harris ED. Ceruloplasmin and iron: vindication after 30 years. Nutrition. 1999;15:72–74. doi: 10.1016/s0899-9007(98)00144-0. [DOI] [PubMed] [Google Scholar]
- 33.Anderson GJ, Wang F. Essential but toxic: controlling the flux of iron in the body. Clin Exp Pharmacol Physiol. 2012;39:719–724. doi: 10.1111/j.1440-1681.2011.05661.x. [DOI] [PubMed] [Google Scholar]
- 34.McManus Mowry. Staining methods: histological and histochemical. Medical Division of Harper and Brothers; New York, NY: 1960. [Google Scholar]
- 35.Jovanovic I, Ugrenovic S, Vasovic L, Petrovic D, Cekic S. Psammoma bodies – friends or foes of the aging choroid plexus. Med Hypotheses. 2010;74:1017–1020. doi: 10.1016/j.mehy.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 36.Kono S. Aceruloplasminemia. Curr Drug Targets. 2012;13:1190–1199. doi: 10.2174/138945012802002320. [DOI] [PubMed] [Google Scholar]
- 37.Wang JY, Hagerman RJ, Rivera SM. A multimodal imaging analysis of subcortical gray matter in fragile X premutation carriers. Mov Disord. 2013 doi: 10.1002/mds.25473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kruer MC, Boddaert N, Schneider SA, Houlden H, Bhatia KP, et al. Neuroimaging features of neurodegeneration with brain iron accumulation. AJNR Am J Neuroradiol. 2012;33:407–414. doi: 10.3174/ajnr.A2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schneider SA, Bhatia KP. Syndromes of neurodegeneration with brain iron accumulation. Semin Pediatr Neurol. 2012;19:57–66. doi: 10.1016/j.spen.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 40.Dusek P, Schneider SA. Neurodegeneration with brain iron accumulation. Curr Opin Neurol. 2012;25:499–506. doi: 10.1097/WCO.0b013e3283550cac. [DOI] [PubMed] [Google Scholar]
- 41.Brittenham GM. New advances in iron metabolism, iron deficiency, and iron overload. Curr Opin Hematol. 1994;1:101–106. [PubMed] [Google Scholar]
- 42.Napoli E, Ross-Inta C, Wong S, Omanska-Klusek A, Barrow C, et al. Altered zinc transport disrupts mitochondrial protein processing/import in fragile X-associated tremor/ataxia syndrome. Hum Mol Genet. 2011;20:3079–3092. doi: 10.1093/hmg/ddr211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng W, Monnot AD. Regulation of brain iron and copper homeostasis by brain barrier systems: implication in neurodegenerative diseases. Pharmacol Ther. 2012;133:177–188. doi: 10.1016/j.pharmthera.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leitner DF, Connor JR. Functional roles of transferrin in the brain. Biochim Biophys Acta. 2012;1820:393–402. doi: 10.1016/j.bbagen.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 45.Han J, Day JR, Connor JR, Beard JL. Gene expression of transferrin and transferrin receptor in brains of control vs. iron-deficient rats. Nutr Neurosci. 2003;6:1–10. [PubMed] [Google Scholar]
- 46.Kell DB. Towards a unifying, systems biology understanding of large-scale cellular death and destruction caused by poorly liganded iron: Parkinson’s, Huntington’s, Alzheimer’s, prions, bactericides, chemical toxicology and others as examples. Arch Toxicol. 2010;84:825–889. doi: 10.1007/s00204-010-0577-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kruer MC. The neuropathology of neurodegeneration with brain iron accumulation. Int Rev Neurobiol. 2013;110:165–194. doi: 10.1016/B978-0-12-410502-7.00009-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Earley CJ, Connor JR, Beard JL, Malecki EA, Epstein DK, et al. Abnormalities in CSF concentrations of ferritin and transferrin in restless legs syndrome. Neurology. 2000;54:1698–1700. doi: 10.1212/wnl.54.8.1698. [DOI] [PubMed] [Google Scholar]
- 49.Grootveld M, Halliwell B. An aromatic hydroxylation assay for hydroxyl radicals utilizing high-performance liquid chromatography (HPLC). Use to investigate the effect of EDTA on the Fenton reaction. Free Radie Res Commun. 1986;1:243–250. doi: 10.3109/10715768609051634. [DOI] [PubMed] [Google Scholar]
- 50.Thomas C, Mackey MM, Diaz AA, Cox DP. Hydroxyl radical is produced via the Fenton reaction in submitochondrial particles under oxidative stress: implications for diseases associated with iron accumulation. Redox Rep. 2009;14:102–108. doi: 10.1179/135100009X392566. [DOI] [PubMed] [Google Scholar]
- 51.Connor JR, Ponnuru P, Wang XS, Patton SM, Allen RP, et al. Profile of altered brain iron acquisition in restless legs syndrome. Brain. 2011;134:959–968. doi: 10.1093/brain/awr012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sellier C, Freyermuth F, Tabet R, Tran T, He F, et al. Sequestration of DROSHA and DGCR8 by expanded CGG RNA repeats alters microRNA processing in fragile X-associated tremor/ataxia syndrome. Cell Rep. 2013;3:869–880. doi: 10.1016/j.celrep.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]