

Abstract
Summary: MetMSLine represents a complete collection of functions in the R programming language as an accessible GUI for biomarker discovery in large-scale liquid-chromatography high-resolution mass spectral datasets from acquisition through to final metabolite identification forming a backend to output from any peak-picking software such as XCMS. MetMSLine automatically creates subdirectories, data tables and relevant figures at the following steps: (i) signal smoothing, normalization, filtration and noise transformation (PreProc.QC.LSC.R); (ii) PCA and automatic outlier removal (Auto.PCA.R); (iii) automatic regression, biomarker selection, hierarchical clustering and cluster ion/artefact identification (Auto.MV.Regress.R); (iv) Biomarker—MS/MS fragmentation spectra matching and fragment/neutral loss annotation (Auto.MS.MS.match.R) and (v) semi-targeted metabolite identification based on a list of theoretical masses obtained from public databases (DBAnnotate.R).
Availability and implementation: All source code and suggested parameters are available in an un-encapsulated layout on http://wmbedmands.github.io/MetMSLine/. Readme files and a synthetic dataset of both X-variables (simulated LC–MS data), Y-variables (simulated continuous variables) and metabolite theoretical masses are also available on our GitHub repository.
Contact: ScalbertA@iarc.fr
1 Introduction
Untargeted metabolite profiling is a promising approach to discover novel risk factors for chronic diseases and biomarkers for disease diagnosis (Wang et al., 2011; Ritchie et al., 2013). Liquid chromatography coupled to high-resolution mass spectrometry (LC–hrMS) instrumentation is being more routinely used for data-acquisition for metabolomic analyses in large-scale studies (e.g. >300 samples). Raw data need to be processed via computational tools to extract relevant information and meaningful biological conclusions.
Processing of LC–hrMS raw data is currently facilitated by softwares such as XCMS, MzMine, MetaboAnalyst and Maven (Smith et al. 2006; Pluskal et al. 2010; Clasquin et al. 2012; Xia et al. 2012). However, an automated software pipeline with minimal manual interaction for efficient, reproducible and objective large-scale metabolomic data analysis is also desirable. Software tools can systematically perform all downstream aspects of metabolomic analysis following peak-picking. Development of such a workflow in the R language can offer several advantages such as availability of modular packages for functional programming and graphics and the ease of accessibility of a GUI for non-specialists.
We have developed novel software, MetMSLine, coded in the R language which automates the process of untargeted metabolomic data analyses of large datasets from acquisition of data from LC–hrMS platforms through to unknown biomarker identification.
2 Results
An overview of data processing steps that are integrated by MetMSLine is shown in Figure 1. Use of the software via GUI requires a clear understanding of data processing steps in metabolomics.
Fig. 1.
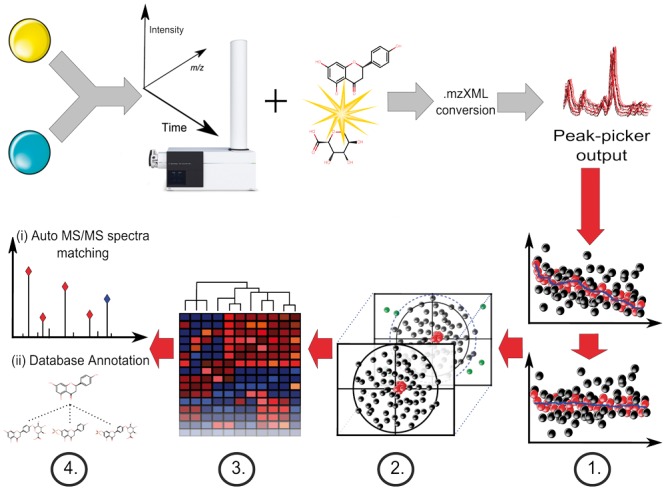
Data acquisition and MetMSLine data processing workflow (Steps 1–4). Sample preparation (e.g. urine dilution) is followed by untargeted MS and MS/MS data acquisition in sequence and peak picking softwares MetMSLine then performs sequentially: signal drift correction and pre-processing (Step 1), automatic PCA-based outlier removal (Step 2) (samples = black, QCs = red, outliers = green), automatic iterative regression based on continuous Y-variables supplied and cluster ion identification (Step 3) and final identification by data-dependent MS/MS and database matching (Step 4)
2.1 Step 1: Pre-processing of raw data matrix from XCMS
Large LC–hrMS metabolomics datasets contain unwanted variation introduced by MS signal drift/attenuation and multiplicative noise across the dynamic range. These effects can detrimentally impact biomarker discovery and MS features require rigorous quality assurance. PreProc.QC.LSC first zero-fills data, then if sample normalization is required the median fold change method can also be applied (Veselkov et al. 2011). PreProc.QC.LSC then uses the QC-based locally weighted scatter-plot smoothing method to alleviate the effects of signal drift (Dunn et al. 2011). The degree of smoothing is controlled by the smoother span value (e.g. f = 1/5), this argument sets the proportion of points used to smooth at each point. Data are then Log-transformed and finally features analytically stable across the regularly injected (every 5–10 true sample) pooled QCs are retained (e.g. RSD = 30, i.e. <30% relative standard deviation).
2.2 Step 2: Removal of outliers
The next function performs automated removal of outliers in the pre-processed data based on expansion of the Hotellings T2 distribution ellipse. The argument ‘out.tol’ (outlier tolerance) controls the proportional expansion of the ellipse (e.g. 1.1 or a 10% proportional expansion). Any samples within the first and second component PCA score plot beyond this expanded ellipse are removed and the PCA model recalculated. Assuming outliers are detected Auto.PCA performs two rounds of outlier removal and saves details of outliers removed along with corresponding samples from the Y-variable data table supplied in the parent directory in .csv format.
2.3 Step 3: Multivariate regression
Auto.MV.Regress utilizes continuous Y-variables to regress to the pre-processed MS dataset. Auto.MV.Regress creates a subfolder for each Y-variable supplied, then identifies potential biomarkers based on a user-defined correlation threshold (e.g. Corr.thresh = 0.3) and below a multiple testing corrected P-value (P = 0.01) and both scatterplots and box and whisker plots are generated. Potential biomarkers above the threshold are hierarchically clustered and ‘X–Y’ and ‘X–X’ heatmaps generated. Inter-feature clustering (X–X) is used to identify cluster ions from a list of 88 isotope, adduct, fragment and co-metabolite mass shifts.
2.4 Step 4 (i): MS/MS matching for biomarker structure elucidation
As an LC–hrMS platform can acquire MS/MS data with precise masses, we coded a function to use the MS/MS data for the identification of metabolites. This function matches potential biomarkers identified by Auto.MV.Regress to MS/MS fragmentation spectra by a retention time window (ret = 10 s) and mass tolerance (Frag.ppm = 20). Auto.MS.MS.match calculates the precursor (in blue on plot) to fragment (in red on plot) and inter-fragment mass differences, and labels where available the neutral losses/fragments commonly encountered in MS/MS spectra.
2.5 Step 4 (ii): Compound annotation using exact mass matching
The final function utilizes targeted lists of experiment-specific anticipated metabolites (in .csv format) provided by the user to annotate the unknown biomarkers. DBAnnotate optionally calculates from the targeted lists of anticipated metabolites, expected m/z of both typical phase II conjugates and electrospray adducts. DBAnnotate matches against all iterations of these potential theoretical masses below user-defined mass tolerances (MassAcc = 10 ppm) and returns an aggregated result table.
3 Conclusion
MetMSLine presents a complete data processing method; it is easy to use as a GUI and should be very beneficial to researchers to rapidly process large-scale LC–hrMS dataset. It potentially requires minimal manual interaction with the software, when compared with the high-manual interaction required by commonly used softwares for LC–hrMS datasets such as MetaboAnalyst, MAVEN, apLCMS, MzMine and IDEOM (Yu et al. 2009; Pluskal et al. 2010; Clasquin et al. 2012; Creek et al. 2012; Xia et al. 2012). The rapidity of the process allows great scope for parameter optimization and the subsequent ability to dedicate more time to result interpretation.
Funding
This work was supported by the European Union (NutriTech and the European Cancer Platform (EUROCAN) [grant No. 289511, 260791]).
Conflict of interest: none declared.
References
- Clasquin M.F., et al. (2012) LC–MS data processing with MAVEN: a metabolomic analysis and visualization engine. Curr. Protoc. Bioinform., 3, 14.11.1–14.11.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creek D.J., et al. (2012) IDEOM: an Excel interface for analysis of LC–MS-based metabolomics data. Bioinformatics, 28, 1048–1049. [DOI] [PubMed] [Google Scholar]
- Dunn W.B., et al. (2011) Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protocols, 6, 1060–1083. [DOI] [PubMed] [Google Scholar]
- Pluskal T., et al. (2010) MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics, 11, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie S.A., et al. (2013) Low-serum GTA-446 anti-inflammatory fatty acid levels as a new risk factor for colon cancer. Int. J. Cancer, 132, 355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C.A., et al. (2006) XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem., 78, 779–787. [DOI] [PubMed] [Google Scholar]
- Veselkov K.A. (2011) Optimized preprocessing of ultra-performance liquid chromatography/mass spectrometry urinary metabolic profiles for improved information recovery. Anal. Chem., 83, 5864–5872. [DOI] [PubMed] [Google Scholar]
- Wang Z., et al. (2011) Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature, 472, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J.G., et al. (2012) MetaboAnalyst 2.0-a comprehensive server for metabolomic data analysis. Nucleic Acids Res., 40, W127–W133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T.W., et al. (2009) apLCMS-adaptive processing of high-resolution LC/MS data. Bioinformatics, 25, 1930–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]